

Abstract
Background
The ongoing coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is a global public health concern due to relatively easy person-to-person transmission and the current lack of effective antiviral therapy. However, the exact molecular mechanisms of SARS-CoV-2 pathogenesis remain largely unknown.
Methods
Genome-wide screening was used to establish intraviral and viral-host interactomes. Quantitative proteomics was used to investigate the peripheral blood mononuclear cell (PBMC) proteome signature in COVID-19.
Findings
We elucidated 286 host proteins targeted by SARS-CoV-2 and >350 host proteins that are significantly perturbed in COVID-19-derived PBMCs. This signature in severe COVID-19 PBMCs reveals a significant upregulation of cellular proteins related to neutrophil activation and blood coagulation, as well as a downregulation of proteins mediating T cell receptor signaling. From the interactome, we further identified that non-structural protein 10 interacts with NF-κB-repressing factor (NKRF) to facilitate interleukin-8 (IL-8) induction, which potentially contributes to IL-8-mediated chemotaxis of neutrophils and the overexuberant host inflammatory response observed in COVID-19 patients.
Conclusions
Our study not only presents a systematic examination of SARS-CoV-2-induced perturbation of host targets and cellular networks but it also reveals insights into the mechanisms by which SARS-CoV-2 triggers cytokine storms, representing a powerful resource in the pursuit of therapeutic interventions.
Funding
National Key Research and Development Project of China, National Natural Science Foundation of China, National Science and Technology Major Project, Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, Shanghai Science and Technology Commission, Shanghai Municipal Health Commission, Shanghai Municipal Key Clinical Specialty, Innovative Research Team of High-level Local Universities in Shanghai, Interdisciplinary Program of Shanghai Jiao Tong University, SII Challenge Fund for COVID-19 Research, Chinese Academy of Sciences (CAS) Large Research Infrastructure of Maintenance and Remolding Project, and Chinese Academy of Sciences Key Technology Talent Program.
Keywords: COVID-19, SARS-CoV-2, interactome, proteomics, IL-8, nsp10, NKRF
Graphical Abstract
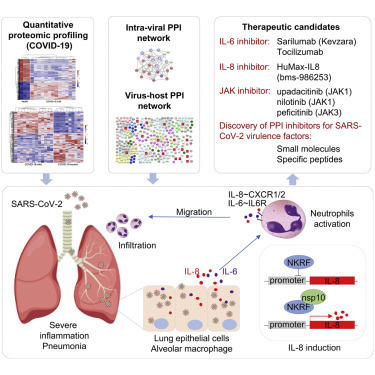
Context and Significance
The COVID-19 pandemic is caused by SARS-CoV-2, but little is known about the functions of its viral proteins. The authors characterized the SARS-CoV-2 intraviral and virus-host interaction networks in human cells and identified 286 potential host targets. Quantitative proteomic analysis revealed elevated levels of IL-6 and IL-8 in PBMCs collected from severe COVID-19 patients compared with mild ones, and the functional annotation of differentially expressed proteins implicate pathways involved in neutrophil activation, T cell receptor signaling, and the coagulation cascade. Combining virus-host interactome with COVID-19 proteomic analysis, the authors found that nsp10 interacts with NKRF to mediate IL-8 expression, providing a potential molecular mechanism for SARS-CoV-2-induced cytokine storm and marking it as a possible emerging therapeutic target.
Li et al. establish the SARS-CoV-2 intraviral and virus-host protein-protein interaction networks and reveal the overall proteome signature in PBMCS from COVID-19 patients by quantitative proteomic analysis. Validation assays indicate that nsp10, which interacted with NKRF, is involved in IL-8 production.
Introduction
In December 2019, severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2, previously known by the provisional name “2019 novel coronavirus” [2019-nCoV] assigned by the World Health Organization [WHO]), was first isolated from a cluster of patients with pneumonia. This novel strain of coronavirus has been identified as the causative agent for the epidemic of acute respiratory distress syndrome (also referred to as coronavirus disease 2019 [COVID-19]) in Wuhan, China.1, 2, 3 Since then, the COVID-19 outbreak has rapidly swept all of the provinces of China and has disseminated globally to 216 countries and territories through travel.4 On January 31, 2020, the WHO declared SARS-CoV-2 a “Public Health Emergency of International Concern.”5 As of June 25, 2020, the current pandemic of SARS-CoV-2 has caused >9 million confirmed infection cases, including 479,133 deaths in 216 countries and territories, calling for extended efforts to understand the molecular drivers of its immune evasion and pathogenesis.
COVID-19 is a potential zoonotic disease with a low to moderate mortality rate (2%–5%) and is primarily transmitted through droplet and direct contact with infected individuals or incubation carriers.6 COVID-19 patients develop dynamic innate and adaptive immune responses to SARS-CoV-2.7, 8, 9 SARS-CoV-2 has a preferential tropism for airway epithelial cells mediated through the accessibility of the angiotensin-converting enzyme 2 (ACE2) receptor.10, 11, 12, 13 The common clinical manifestations of fever and cough in conjunction with laboratory results of progressively increasing neutrophil counts and leukocytopenia in serum samples taken from COVID-19 patients at various stages of the illness indicate uncontrolled neutrophil extravasation and activation.14, 15, 16 Neutrophils are the most abundant type of white blood cells in human blood and serve as the first responders in the line of defense against acute infection.17 Chemical signals such as interleukin-8 (IL-8) and IL-6 stimulate neutrophil chemotaxis and recruitment to the site of infection, where they congregate and cause the release of more pro-inflammatory mediators that in turn activate additional inflammatory signals, leading to cytokine storms that may progress to vital organ failure.18 A large number of macrophages and neutrophils were found in alveolar cavities in COVID-19 fatalities,19 suggesting their roles in cytokine storms. While uncontrolled neutrophil activity and aberrant cytokine response contribute to SARS-CoV-2 pathogenesis in COVID-19, how SARS-CoV-2 causes these immunological changes, especially the enhancement of neutrophil chemotaxis and activation, remains largely undetermined.
SARS-CoV-2 is a single-stranded positive-sense RNA virus and belongs to the β-coronavirus family consisting of many animal species and human pathogens including SARS-CoV and Middle East respiratory syndrome (MERS)-CoV. The ∼30-kb non-segmented genome of SARS-CoV-2 encodes 14 major open reading frames (ORFs), which are further processed into 4 structural proteins (glycoprotein spike [S], membrane [M], envelope [E], and nucleocapsid [N]), 16 non-structural proteins (nsp1–nsp16), and at least 8 accessory proteins (ORF3a, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF9c, and ORF10).20 , 21 Similar to SARS-CoV, the spike protein of SARS-CoV-2 directly interacts with human ACE2 expressed on human airway epithelia to facilitate virus entry.10, 11, 12, 13 However, host targets for other viral proteins remain largely unknown, which limits our understanding of immune evasion and pathogenesis of SARS-CoV-2, further impeding antiviral drug development and the design of therapeutic approaches.
Results
Genome-wide Screens for SARS-CoV-2 Intraviral Protein-Protein Interactions
Limited by their relatively small genomes, viral proteins form different combinations of protein complexes to fulfill a variety of critical functions during the viral life cycle, making the investigation of intraviral protein-protein interactions (PPIs) a priority. To systematically characterize the intraviral PPI networks, both genome-wide yeast two-hybrid (Y2H) screens and co-immunoprecipitation (coIP) experiments with SARS-CoV-2 genes were performed. All of the predicted viral genes were synthesized based on the SARS-CoV-2 Wuhan-Hu-1 isolate (GenBank: MN_908947). The entire library was cloned into the Y2H DNA-binding domain (pACT2) and activation domain (pGBTK7) vectors and then transformed into the Y2HGold yeast strain (Clontech). Nsp3 was truncated into two smaller fragments in this experiment since large genes tend to yield fewer interactions in the Y2H assay. All of the possible pairwise combinations were examined independently for the activation of the reporter genes HIS3 and ADE2 in triplicate. We captured 19 PPIs from the Y2H screens—nsp15-nsp2, ORF7b-nsp1, N-nsp4, ORF3a-nsp4, ORF7b-nsp7, nsp7-nsp8, nsp12-nsp8, nsp7-nsp9, nsp14-nsp10, nsp16-nsp10, ORF6-nsp14, E-E, N-E, ORF3a-E, ORF9b-E, N-N, nsp7-ORF7a, nsp8-ORF8, and ORF9b-ORF9b (Figure S1). Most intraviral PPIs characterized by Y2H were confirmed by coIP (Figure S2A). The coIP assay resulted in the identification of an additional 39 PPIs (Figures S1 and S2B). Some viral proteins, such as M, N, E, nsp2, nsp5, nsp8, ORF6, ORF7a, ORF7b, ORF9b, and ORF10, displayed self-association (Figure 1 A), suggesting that their dimer or oligomer forms may carry out important functions during infection. In total, we characterized 58 distinct intraviral PPIs among 28 SARS-CoV-2 genes that are potentially involved in virus replication and can be expected to be important to immune evasion and viral pathogenesis (Figure 1A).
Figure 1.
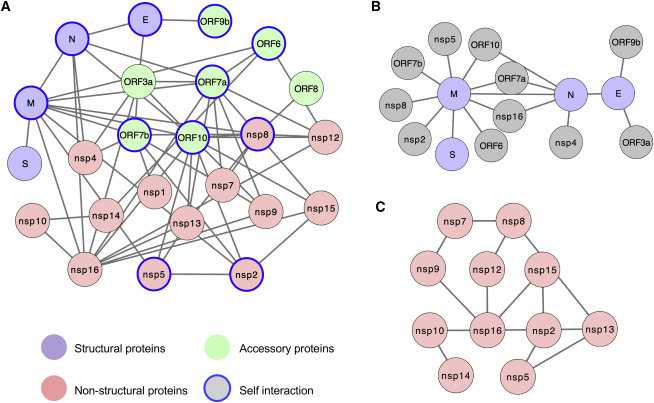
SARS-CoV-2 Intraviral Protein-Protein Interaction Network
(A) SARS-CoV-2 intraviral interactions based on Y2H screens and coIP experiments as described in Figure S1. Different types of viral proteins are labeled with the indicated colors. Self-associated viral proteins are circled in blue.
(B) Interactions between SARS-CoV-2 structural proteins and other viral proteins.
(C) Interaction network among SARS-CoV-2 non-structural proteins.
See also Figures S1 and S2.
Two independent intraviral PPI networks of closely related SARS-CoV have been previously established with Y2H or mammalian 2H systems, identifying 72 and 40 intraviral PPIs, respectively.22 , 23 Due to the amino acid sequence similarity, 20 intraviral PPIs are overlapped between SARS-CoV-2 and SARS-CoV—nsp2-nsp15, nsp7-nsp8, nsp7-nsp9, nsp8-nsp12, nsp8-nsp13, nsp10-nsp14, nsp10-nsp16, nsp10-nsp16, nsp16-N, nsp16-ORF7a, M-N, M-ORF6, E-ORF9b, and ORF6-ORF7a, as well as self-associations of nsp2, nsp5, nsp8, E, N, and ORF9b—suggesting that these intraviral PPIs play critical roles for maintaining fundamental functions in the SARS coronavirus family.
Among the 4 classical structural proteins of SARS-CoV-2, 3 PPIs were detected (Figure 1B); these interactions of structural proteins may be critical in the organization of viral particles during viral replication and assembly. We also found 8 PPIs between structural proteins and accessory proteins (E-ORF3a, E-ORF9b M-ORF6, M-ORF7a, M-ORF7b, M-ORF10, N-ORF7a, and N-ORF10) and 6 PPIs between structural proteins and non-structural proteins (nsp2, nsp4, nsp5, nsp8, and nsp16) (Figure 1B), which are potentially involved in viral structural protein processing, modification, and trafficking, supporting their importance in viral particle assembly and egress.
While structural proteins are components of the capsid and the envelope of the virus, several nsps, such as nsp3, nsp5, nsp12, nsp14, and nsp16, have associated enzymatic activities that are essential for coronavirus RNA replication and viral protein translation. In addition, nsps can function as virulence factors that inhibit the immune system defenses of the host. We speculate that the relatively large number of nsp interactions detected (12) may imply a crucial role for nsps in the formation of multimeric complexes during the viral life cycle (Figure 1C). In fact, nsp7 and nsp8 support the RNA-dependent RNA polymerase function of nsp12.24, 25, 26 Similar to SARS-CoV,27 , 28 SARS-CoV-2 nsp10 may facilitate the methyltransferase activities of nsp14 and nsp16 by nsp10-nsp14 and nsp10-nsp16 associations, respectively. While the functional importance of most intraviral PPIs remains less well understood, our network provides a powerful resource to dissect the role of these viral proteins in mediating the synthesis, assembly, and processing of viral proteins for SARS-CoV-2. The disruption of these intraviral PPIs by repurposing small molecule or peptide drugs that target specific epitopes can be a treatment strategy against COVID-19.29
Genome-wide Proteomic Screen for SARS-CoV-2 Viral-Host Protein-Protein Interactions
SARS-CoV-2 infection can lead to imbalanced host immune responses such as diminished type I and type III interferon (IFN) signaling, as well as strong chemotactic and inflammatory response in infected cell lines, animal models, and COVID-19 patients,7 , 8 , 30, 31, 32, 33 suggesting that this virus must encode components to modulate cellular signaling pathways and shape its unique immune signature. To better understand the roles of these viral proteins during the virus life cycle, it is pivotal to characterize the host targets of SARS-CoV-2 in human cells. We overexpressed the plasmids encoding each SARS-CoV-2 encoded gene with N-terminal 3xFLAG epitope in HEK293 cells and purified each SARS-CoV-2 protein complex by affinity purification (AP). Co-purified cellular proteins were subsequently analyzed by liquid chromatography and tandem mass spectrometry (AP-LC-MS/MS). With statistical analysis by the MiST scoring algorithm (MiST score > 0.8, spectral count > 8, fold change > 5),34 286 cellular proteins were identified as interacting with SARS-CoV-2 proteins, resulting in 295 high-confidence interactions (Figures 2 and S3A; Table S1). The host cellular pathways that are the most likely affected by SARS-CoV-2 viral proteins are ATP biosynthesis and metabolic processes (M), mRNA transport (N), melanoma differentiation-associated protein 5 (MDA5) and retinoic acid-inducible gene I (RIG-I) RNA sensing signaling (nsp1), nucleotide-excision repair (nsp4), protein methylation and alkylation (nsp5), translation initiation (nsp9), cellular amino acid metabolic process (nsp10), reactive oxygen species (ROS) metabolic process (nsp14), Golgi to plasma membrane transport (nsp16), endoplasmic reticulum stress (ORF3a), and mRNA transport (ORF6) (Figures S3B and S3C), which is consistent with the proteome analysis of SARS-CoV-2 infected cells.30
Figure 2.

SARS-CoV-2-Human Protein-Protein Interaction Network
Network representation of the high-confidence SARS-CoV-2-host interactome in HEK293 cells. SARS-CoV-2 bait proteins and interacting host proteins are labeled with red squares and circles, respectively. The subcellular localization of host proteins is labeled with the indicated colors. Proteins with known functions in inflammation and immune responses or ubiquitination pathway are circled in purple or red, respectively.
It is important to note that our AP-LC-MS/MS analysis shares ∼16% overlap (45 targets) with the SARS-CoV-2-host protein interactions recently identified by the Krogan laboratory using 2xStrep-tagged viral proteins isolated from HEK293T/17 cells.35 Some cellular targets characterized by both interactomes include G3BP1/G3BP2 (anti-viral stress granule proteins),36 RAE1 (an IFN-inducible mRNA nuclear export protein),37 TBK1 (TANK binding kinase 1; essential kinase for type I IFN pathway),38 TRIM59 (ubiquitin E3 involved in antiviral response),39 and SigmaR1 (involved in lipid remodeling and ER stress response).35 The identification of common SARS-CoV-2 protein and human protein interactions suggest that these cellular processes are vital molecular targets of SARS-CoV-2 infection. PB28, an agonist of Sigma receptor, has potent anti-SARS-CoV-2 activity in cell culture models.35
Notably, our AP-LC-MS/MS analysis uncovered additional host targets that are pertinent to inflammatory and innate immune responses (Figure 2) and may help contextualize the unique immune signature identified in SARS-CoV-2-infected cells.7 , 9 Key viral-cellular PPIs that may influence the pathogenesis of SARS-CoV-2 include interactions between nuclear factor-κB-repressing factor (NKRF) and nsp10, TBK1 and ORF6, TANK and ORF6, TRAF2 and ORF6, and KIT and ORF3a. NKRF is a transcriptional repressor and controls the induction of IL-8 to stimulate neutrophil chemotaxis and recruitment to the site of infection.40 , 41 Nsp10 may regulate IL-8 levels by targeting NKRF and shapes the unique immune signature in COVID-19. TBK1 plays critical roles for type I IFN and NF-κB pathways upon viral infection: it phosphorylates the key transcriptional factors, IFN regulatory factor 3 (IRF3) and IRF7, to activate IFN promoters38; it also forms a ternary complex with TANK and tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2) to facilitate NF-κB activation.42 ORF6 may interact with the TBK1-TANK-TRAF2 ternary complex to modulate both type I IFN and NF-κB pathways upon infection, facilitating SARS-CoV-2 to escape from host innate immune surveillance. The tyrosine-protein kinase KIT has been shown to facilitate the activation of signal transducers and activators of transcription (STATs),43 such as STAT1, STAT3, and STAT5, which are involved in signaling transduction upon IFN or IL-6 stimulation. ORF3a may regulate IFN or IL-6 signaling pathways by association with KIT.
Several viral proteins that form intraviral PPIs (Figure 1A) are associated with host ubiquitin pathway components, such as interaction partners ORF7a-UBE2J1 (E2 ligase), ORF3a-TRIM59 (E3 ligase), and ORF6-RNF20, RNF40, RFWD3, and MYCBP2 (E3 ligase), suggesting the potential modulation of the host ubiquitin system by these viral protein complexes (Figure 2). Our observations provide an overview of host proteins and signaling pathways that are modulated by SARS-CoV-2 viral proteins, and these PPIs may serve as potential druggable targets toward the development of new antiviral drugs or repurposing of existing drugs.
Global Proteome Profile Changes in Peripheral Blood Mononuclear Cells (PBMCs) Isolated from COVID-19 Patients
To elucidate the mechanism and effect of SARS-CoV-2 infection in patients, we determined the global proteome profile changes in PBMCs samples taken from healthy donors compared with PBMCs collected from COVID-19 patients with mild or severe symptoms. The PBMC samples were obtained from the Shanghai Public Health Clinical Center and include 6 healthy donors, 22 COVID-19 patients with mild symptoms (COVID-19-MS), and 13 COVID-19 patients with severe symptoms (COVID-19-SS) (Table S2). PBMCs were harvested from the leftover blood samples and all of the patients were confirmed negative for influenza A virus. Following the Guideline of Novel Coronavirus Laboratory Safety (Edition 2), PBMCs were collected, lysed in denaturing buffer (STAR Methods) at the Shanghai Public Health Clinical Center, and sent for proteome analysis at the National Facility for Protein Science in Shanghai.
A total of 6,739 proteins were identified in our study, 4,274 of which could be quantified in all of the PMBCs samples. Proteins with <3 quantitation values in each category did not pass the filtering threshold and were excluded from the quantification and statistical analysis. A total of 5,521 proteins were quantified according to criteria and included in global proteomic profiling to reveal a list of 726 proteins found to be differentially expressed (|log2 [fold change]| ≥ 0.58, adjusted p ≤ 0.01, n ≥ 3) in the PBMCs of COVID-19-MS compared with those of healthy donors; 333 proteins showed a statistically significant increase, while 393 proteins showed a reduction in expression when comparing COVID-19-MS with control (Figures 3 A, S4A, and S4B; Table S3). Consistent with the animal infection model,7 , 9 while some IFN-stimulated genes (ISGs) such as OAS3, MX2, and IFIT5 were upregulated, there was no obvious type I IFN response or overall strong induction of ISGs, suggesting that SARS-CoV-2-encoded components may evade cellular IFN response to facilitate virus replication (Figures 3A, 3B, and S4C). Functional analysis by Cluster Profiler revealed that the underexpressed proteins were enriched in specific cellular biological processes, including negative regulation of megakaryocyte differentiation, blood coagulation, hemostasis, and negative regulation of immune system processes (Figures 3C and S4C). Analysis of the global proteomic changes reveals significantly reduced levels of PF4, GP1BA, SERPINA5, HBB, HBD, C1QBP, HRG, and RAC1 in the COVID-19-MS PBMC cohort (Figure 3B). Platelet factor 4 (PF4 or CXCL4) could neutralize the anticoagulant effect during platelet aggregation and serve as a chemotactic signal for neutrophils and monocytes.44 , 45 Glycoprotein Ib platelet subunit α (GP1BA) is a surface membrane protein on platelets and is involved in the formation of platelet plugs.46 Serpin peptidase inhibitor, clade A (SERPINA5), a serine protease inhibitor, acts as a procoagulant and inflammatory factor and plays hemostatic roles in blood plasma.47 , 48 Complement C1q binding protein (C1QBP), a mitochondrial complement component binding protein, is involved in the inflammatory response to infections.49, 50, 51 Ras-related C3 botulinum toxin substrate 1 (RAC1) is a plasma membrane-associated small GTPase that plays a general role in the platelet procoagulant response.52 RAC1 also directly interacts with SigmaR1, a potential therapeutic target for COVID-19.35 Overall, our functional analysis reveals the complexity of host responses to SARS-CoV-2 infection in mildly symptomatic COVID-19 patients, highlighting the abnormal regulations in blood coagulation and hemostasis.
Figure 3.
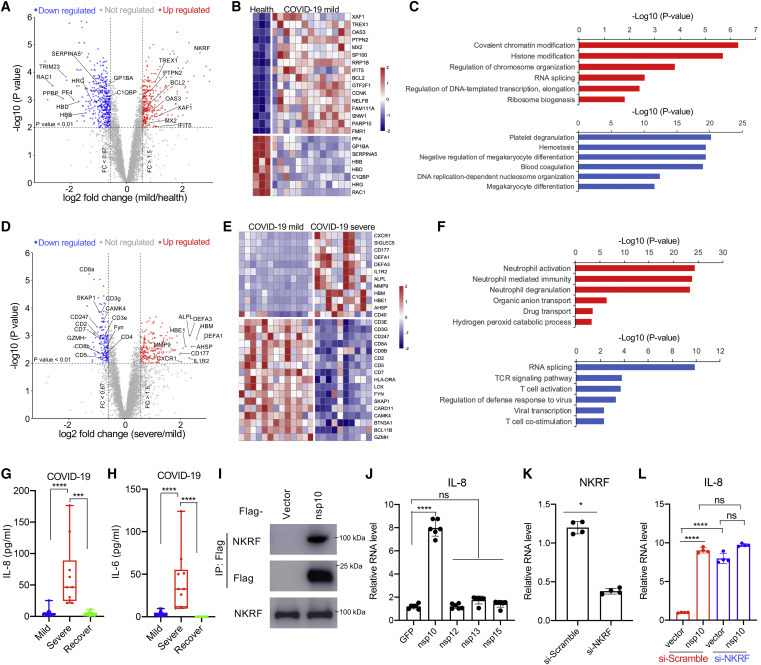
Proteome Profile Change in PBMCs of COVID-19 Patients Suggests a Potential Role for the nsp10-NKRF-IL-8 Axis for SARS-CoV-2 Pathogenesis
(A–C) Comparative proteomic analysis of COVID-19 cases with mild symptoms (N = 13) compared to healthy controls (N = 3).
(A) In the volcano plot, each circle represents a protein that was quantifiable in at least 3 biological replicates. Significantly up- or downregulated proteins (adjusted p ≤ 0.01; |log2(fold change)| ≥ 0.58) are shown in red and blue, respectively.
(B) Heatmap of the differential expressed proteins (adjusted p ≤ 0.01; |log2(fold change)| ≥ 0.58) that play important roles in immune responses.
(C) Gene Ontology analysis shows the enrichment of biological processes with significantly up- (red bar) or downregulated (blue bar) proteins.
(D–F) Comparative proteomic analysis of COVID-19 cases with severe symptoms (N = 10) compared to mild symptoms (N = 13).
(D) Each circle represents a protein that was quantifiable in at least 3 biological replicates in the volcano plot. Significantly up- or downregulated proteins (adjusted p ≤ 0.01; |log2(fold change)| ≥ 0.58) are shown in red and blue, respectively. Immune response-related differentially expressed proteins are downregulated and labeled.
(E) Heatmap of the differential expressed proteins (adjusted p ≤ 0.01; |log2(fold change)| ≥ 0.58) that play important roles in neutrophil activation or TCR signaling.
(F) Gene Ontology analysis shows the enrichment of biological processes with significantly up- (red bar) or downregulated (blue bar) proteins.
(G and H) The levels of IL-8 (G) and IL-6 (H) in PBMCs from COVID-19 patients with mild or severe symptoms or recovered patients (viral RNA negative) were determined by fluorescence-activated cell sorting (FACS) analysis. Means ± SEMs; ***p < 0.001 and ****p < 0.0001 by 1-way ANOVA with Bonferroni’s post hoc test.
(I) nsp10 interaction with endogenous NKRF in cells. HEK293T cells were transfected with FLAG-nsp10 or vector control. CoIP and immunoblot were performed at 48 h post-transfection with the indicated antibodies.
(J) Stable expression of nsp10 from SARS-CoV-2 significantly promotes the mRNA level of IL-8 in the lung epithelial cell line. Means ± SEMs; n = 6; non-significant (ns) and ****p < 0.0001 by 1-way ANOVA with Bonferroni’s post hoc test.
(K and L) The promotion of IL-8 mRNA level by nsp10 depends on NKRF. A549-vector or A549-NKRF stable cells were transfected with small interfering-Scramble (si-Scramble) or si-NKRF. NKRF knockdown efficiency by specific siRNA was confirmed (K) and the IL-8 mRNA level was examined (L) by qPCR. Means ± SEMs; n = 4; *p < 0.05 by Student’s t test in (K). ns and ****p < 0.0001 by 1-way ANOVA with Bonferroni’s post hoc test in (L).
Next, we compared the proteome change between PBMCs isolated from COVID-19-SS and COVID-19-MS patients. Among 6,235 quantified proteins (n ≥ 3), 366 proteins were found to be significantly differentially expressed (|log2 [fold change]| ≥ 0.58, adjusted p ≤ 0.01) between the PMBCs of COVID-19-SS and COVID-19-MS. A total of 209 of these proteins were upregulated and 157 of these proteins were downregulated in COVID-19-SS as compared to COVID-19-MS (Figures 3D, 3E, S4D, and S4E; Table S3). Functional analysis by Cluster Profiler revealed that these upregulated genes are highly enriched in pathways associated with neutrophil activation, neutrophil degranulation, gas transport, bicarbonate transport, myeloid cell differentiation, and blood coagulation (Figures 3F and S4F). The expression levels of neutrophil surface receptors (CXCR1, Siglec5, and CD177) and neutrophil granule contents (DEFA1) were dramatically upregulated in COVID-19-SS (Figures 3D and 3E), which is consistent with the elevated neutrophil population in COVID-19 patients.14 , 16 Consistent with RNA sequencing results from COVID-19 patients,7 , 9 , 32 these differentially expressed proteins support a model by which SARS-CoV-2 triggers cytokine release, such as IL-6 and IL-8, resulting in uncontrolled infiltration and activation of neutrophils to the sites of infection, followed by eventual progression to cytokine storm and respiratory distress. Laboratory results reveal increased neutrophils counts to ∼90% of the white blood cell count as COVID-19 progresses to critical stages.14, 15, 16 The elevation of IL-8 and IL-6 but not IL-17 in PMBCs collected from severe symptomatic COVID-19 patients indicates critical roles for IL-8 and IL-6 in the severe inflammatory response induced by SARS-CoV-2 infection (Figures 3G, 3H, and S4H). In addition, a cluster of genes related to blood coagulation was dramatically upregulated in COVID-19-SS as compared to COVID-19-MS (Figure S4G), supporting the hypothesis that the abnormal blood clotting contributes to de ath in some severe COVID-19 patients.53
As compared to COVID-19-MS, the downregulated proteins of COVID-19-SS are highly enriched in T cell activation, T cell receptor (TCR) signaling, viral transcription, viral gene expression, translation initiation, and lymphocyte co-stimulation (Figures 3F and S4F). The expression of TCR subunits (CD3e, CD3g, and CD247), T cell surface molecules (CD4, CD8a, CD8b, CD2, CD5, and CD7), major histocompatibility complex (MHC) class II molecules (HLA-DRA), TCR signaling kinases and adaptors (LCK, FYN, SKAP1, CARD11, CAMK4, and BTN3A1), and T cell differentiation and survival (BCL11B) were dramatically diminished in COVID-19-SS compared to COVID-19-MS (Figures 3D and 3E), suggesting a failure of T cell activation and function. These results demonstrate that severe COVID-19 patients undergo a functional decline in adaptive immunity, which correlates with their lower T cell population.14, 15, 16
SARS-CoV-2 nsp10 Targets NKRF to Facilitate IL-8 Production
From the quantitative proteome analysis of PBMCs, it is clear that SARS-CoV-2 triggers IL-8 expression and increases neutrophil count, which may mediate the manifestation of acute respiratory distress syndrome in COVID-19 patients. Therefore, we hypothesized that SARS-CoV-2 virulence factors may potentiate or regulate the induction of IL-8 during infection, leading to the uncontrolled infiltration and activation of neutrophils. From our virus-host interactome, we discovered that NKRF interacts with SARS-CoV-2 nsp10 (Figures 2 and S3A). CoIP further confirmed that nsp10 binds to endogenous NKRF in cells (Figure 3I). Notably, nsp10 did not bind to NKRF in the Y2H assay (Figure S4I), suggesting that this interaction is either not directly a PPI or additional post-translational modifications are required for this interaction. Individually expressed nsp10 facilitated IL-8 induction in lung epithelial A549 cells, while individually expressed nsp12, nsp13, or nsp15 had little or no effect (Figure 3J), suggesting that nsp10 specifically promotes IL-8 induction. The levels of IL-1β and TNF-α were not significantly affected by nsp10 (Figures S4J and S4K), indicating that nsp10 does not affect NK-κB signaling transduction overall. Since NKRF is a transcriptional repressor that inhibits IL-8 induction by competing with NF-κB for IL-8 promoter binding,40 , 41 we hypothesize that nsp10 targets NKRF to modulate IL-8 expression. With specific small interfering RNA (siRNA) transfection, the NKRF level was diminished in A549 cells (Figures 3K and S4L), resulting in an elevated induction of IL-8 (Figure 3L), confirming the critical role of NKRF in IL-8 production. Nsp10 increased the IL-8 expression level in cells treated with scrambled siRNA (Figure 3L, red bar), while it has almost no effect on IL-8 in cells treated with NKRF-specific siRNAs, as compared to vector control (Figure 3L, blue bar), indicating that nsp10 specifically targets NKRF to regulate IL-8 induction.
Similar to other coronaviruses, SARS-CoV-2 has undergone rapid adaptive changes against evolutionary pressures since its isolation.16 , 54 Phylogenetic and genetic analyses55 of 160 public SARS-CoV-2 sequences reveal 137 amino acid substitutions in 23 viral proteins, with 2 mutations (A61G and F68L) occurring on nsp10 with very low variation rates during evolution (1/160 for each site) (Figures S4M and S4N; Table S4), which indicates nsp10 has remained relatively stable during evolution. Accordingly, these 2 nsp10 mutations did not affect its interaction with NKRF in cells (Figure S4O). Furthermore, although there are 4 amino acid variations between nsp10SARS-CoV and nsp10SARS-CoV-2 (Figure S4P), nsp10SARS-CoV remains tightly associated with NKRF (Figure S4Q), indicating that these 4 sites are not essential for nsp10-NKRF binding. Therefore, our data suggest that nsp10 is a potential stable drug target to curb SARS-CoV-2-mediated IL-8 activation and subsequent recruitment of neutrophils.
Discussion
Establishing viral-viral and virus-host PPI networks is of high priority to understand the host response to SARS-CoV-2 infection and guide the design of potential drug candidates for COVID-19. Thus far, limited data have been obtained describing these PPI networks, preventing our understanding of virus immune evasion and pathogenesis. Using systematic approaches, we characterized 58 intraviral PPIs and 295 virus-host PPIs for SARS-CoV-2. From our observation, most intraviral PPIs identified by Y2H were confirmed by coIP and more PPIs were detected by coIP than Y2H in yeast. This is common for most viruses because cellular proteins or post-translational modifications may be required for the intraviral complex formation. Due to the sequence similarity, 20 of 58 SARS-CoV-2 intraviral PPIs overlap with SARS-CoV,22 , 23 such as nsp7-nsp8, nsp8-nsp12, nsp10-nsp14, nsp10-nsp16, and nsp14-nsp16, indicating the general roles of these PPIs for coronavirus replication. The additional 38 intraviral PPIs may provide evidence for the difference between SARS-CoV and SARS-CoV-2, not only for virus replication efficiency but also for the unique modulation of cellular signaling pathways.
An independent SARS-CoV-2-host interactome was recently generated by the Krogan group35 in HEK293T/17 cells with 2xStrep tag purification. Although different tag purifications from different cell lines could generate variability on the binding partners, our SARS-CoV-2-host interactome shares 45 key host targets with the interactome from the Krogan group, such as TBK1, G3BP1, G3BP2, RAE1, TRIM59, and SigmaR1, which are involved in host innate immune responses to virus infection. Since they lack the specific antibodies for viral protein immunoprecipitation, the detection of endogenous virus-host PPIs in the context of virus remains unavailable. Some signal peptides and viral co-factors may also affect the expression and localization of viral protein in cells, which potentially changes the binding partners of viral proteins in physiological conditions. Some true positive PPIs may have been discarded during analysis, since we omitted ribosomal proteins and proteins bound to more than three viral targets during MiST analysis. With MiST statistical analysis,34 we obtained 295 additional high-confident PPIs using parameters with MiST scores > 0.8, average peptides >8 from 2 independent biological replicates, and fold changes >5 as compared to GFP control. While nsp10-NKRF interaction was not captured by the Krogan group,35 we have confirmed that nsp10 is associated with endogenous NKRF in HEK293T cells, suggesting that our dataset could serve as a valuable additional resource for host targets that are potentially involved in SARS-CoV-2 immune evasion and pathogenesis. In addition, we evaluated the effect of nsp10 on IL-8 induction, highlighting the potential role of nsp10 in SARS-CoV-2 pathogenesis. Further functional analysis of nsp10 in the context of SARS-CoV-2 infection is in progress in our lab. Therefore, a combination list of intraviral and SARS-CoV-2-host PPI networks could serve as a powerful resource to dissect the molecular mechanism for each SARS-CoV-2 viral protein, outlining the unique feature of SARS-CoV-2-mediated pathogenesis at the molecular level.
Although recent RNA sequencing analysis of infected cell lines, infected animal models, and patient samples have improved our understanding of host response to SARS-CoV-2 infection,7 , 8 , 30, 31, 32, 33 there is still a lack of direct evidence at the protein level. Our quantitative proteomic analysis using PBMC samples derived from both mild and severe COVID-19 patients begins to address this gap in knowledge. Since our viral-host interactome analysis indicates that SARS-CoV-2 viral proteins may modulate and evade host innate immune response by interacting with key kinases and adaptors of the immune signaling pathway, it is not surprising that COVID-19 patients fail to mount robust antiviral IFN responses. From our quantitative proteomic analysis, neutrophilia, leukocytopenia, and hypercoagulation were observed in PBMCs of COVID-19-SS patients as compared to COVID-19-MS patients, supporting the hypothesis that imbalanced inflammatory cytokine production, such as IL-8 and IL-6 dysregulation, contributes to the overactivation of neutrophils and regulation of the bloodclotting cascade.16 , 56 These molecular signatures could serve as diagnostic markers to prognosticate the severity of SARS-CoV-2 infection. Further proteomic analysis with sorted cells will specifically address which cell types may play critical roles in shaping these clinical signatures.
Overall, our study integrated virus-host interaction network and quantitative proteome approaches to identify host targets of SARS-CoV-2 viral proteins, demonstrating that SARS-CoV-2 evolved multiple mechanisms to modulate fundamental host cellular processes and evade the host immune system. To our knowledge, this is the first study to characterize the PBMC proteome profile in both mild and severe cases of COVID-19 infection, and it contextualizes the overall clinical features of COVID-19 patients at the protein level. These systematic analyses not only generate groundwork for future SARS-CoV-2 research on the nature of this virus but they also provide valuable targets for specific drug development or repurposing. With these approaches, we identified nsp10 as the potential virulence factor for SARS-CoV-2 through the interaction with NKRF to facilitate IL-8 induction and potentially lead to uncontrolled infiltration and activation of neutrophils (Figure 4 ). Specific agents targeting IL-8 and IL-6 or the downstream JAK kinases could be repurposed as suitable drug candidates for clinical evaluation. These small-molecule inhibitors (i.e., IL-6 or IL-8 receptor antagonists) or agents blocking virulence factor PPIs would have enormous value as antivirulence therapies in the treatment of SARS-CoV-2 infections (Figure 4). Follow-up work is required to tease out the detailed mechanisms involved and translate the therapeutic relevance of these findings in targeting the cytokine storm in COVID-19.
Figure 4.
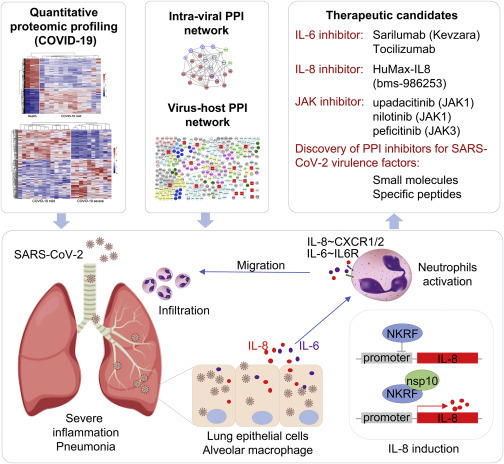
Models of Nsp10-Mediated SARS-CoV-2 Pathogenesis and Potential Design or Repurposing for Specific Inhibitors
Limitations of Study
The experiment design for viral-host PPIs in this study may not provide the exact physiological environment in terms of protein processing, accessory proteins, and so forth, and hence additional validation in a more physiologically relevant system, such as during SARS-CoV-2 infection, would be needed. Our study provides a preliminary picture of the PPI network, but further validation is needed to confirm these findings and explore their functional significance.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-GAPDH | Cell Signaling | Cat #2118L; RRID: AB_561053 |
| Mouse monoclonal anti-Flag | Sigma | Cat #F1804; RRID: AB_262044 |
| Rabbit polyclonal anti-Flag | Sigma | Cat #F7425; RRID: AB_439687 |
| Mouse monoclonal anti-HA | Biolegend | Cat #901503; RRID: AB_2565005 |
| Rabbit polyclonal anti-HA | Biolegend | Cat #923502; RRID: AB_2565438 |
| Rabbit polyclonal anti-NKRF | Abcam | Cat #ab168829 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Protease inhibitor cocktail | Roche | Cat #4693159001 |
| Phosphatase inhibitor PhosSTOP | Roche | Cat #4906845001 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat #74104 |
| qScript One-Step qRT-PCR kit | Quanta Biosciences | Cat #95057-050 |
| BCA Protein Assay Kit | Thermo Fisher Scientific | Cat #23227 |
| Lipofectamine 3000 Transfection Kit | Thermo Fisher Scientific | Cat #3000015 |
| Lipofectamine RNAiMAX Kit | Thermo Fisher Scientific | Cat #13778075 |
| Deposited Data | ||
| Proteome data | This paper | IPX0002285000 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat #CRL-11268 |
| HEK293 | ATCC | Cat #CCL-1573 |
| A549 | ATCC | Cat #CCL-185 |
| Oligonucleotides | ||
| Human TNFα forward primer for qPCR | CTCCAGGCGGTGCTTGTTC | NA |
| Human TNFα reverse primer for qPCR | GGCTACAGGCTTGTCACTCG | NA |
| Human IL-8 forward primer for qPCR | TCTTGCACAAATATTTGATGC | NA |
| Human IL-8 reverse primer for qPCR | CCACTGTGCCTTGGTTTC | NA |
| Human NKRF forward primer for qPCR | GTAAACATGCAGCTGCCGAC | NA |
| Human NKRF reverse primer for qPCR | CGTGCACACGGGATTTGAAG | NA |
| Human GAPDH forward primer for qPCR | GAGTCAACGGATTTGGTCGT | NA |
| Human GAPDH reverse primer for qPCR | TTGATTTTGGAGGGATCTCG | NA |
| Human IL-1β forward primer for qPCR | CTCGCCAGTGAAATGATGGCT | NA |
| Human IL-1β reverse primer for qPCR | GTCGGAGATTCGTAGCTGGAT | NA |
| Recombinant DNA | ||
| pEF-3xHA-SARS-2 gene | This paper | NA |
| pLVX-3xFlag-SARS-2 gene-P2A-TagRFP | This paper | NA |
| pACT2-SARS-CoV-2 gene | This paper | NA |
| pGBTK7-SARS-CoV-2 gene | This paper | NA |
| pLVX-3xFlag-NKRF-P2A-TagRFP | This paper | NA |
| pGBTK7-vBcl2 | Liang et al.59 | NA |
| pACT2-ORF55 | Liang et al.59 | NA |
| Human NKRF siRNA | GenePharma | Cat #A10001 |
| Software and Algorithms | ||
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Cytoscape | Cytoscape Software | https://cytoscape.org/ |
| MiST algorithm | Verschueren et al.34 | https://modbase.compbio.ucsf.edu/mist/ |
| ClusterProfiler v3.14.3 | Yu et al.60 | NA |
| MEGA | MEGA software | https://www.megasoftware.net/ |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact Qiming Liang (liangqiming@shsmu.edu.cn).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with completed Materials Transfer Agreement.
Data and Code Availability
The datasets generated during this study were deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository61 with the dataset identifier IPX0002285000.
Experimental Model and Subject Details
Ethics statement
This study was approved by the Ethics Committee of Shanghai Public Health Clinical Center (#YJ-2020-S052-02). The proteomic analysis of PBMC samples was performed on existing samples collected during standard diagnostic tests, posing no extra burden to patients.
Cell lines, plasmids, and antibodies
HEK293 (ATCC, #CCL-1573), HEK293T (ATCC, #CRL-11268), and A549 cells (ATCC, #CCL-185) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO-BRL) containing 4 mM glutamine and 10% FBS. Vero cells were cultured in DMEM with 5% FBS and 4 mM glutamine. Transient transfections were performed with Lipofectamine 3000 (Thermo Fisher Scientific, #3000015). SARS-CoV-2 genes were synthesized as DNA fragments (Sangon Biotech) and these expression constructs were amplified by PCR and cloned into pLVX-3xFlag-MCS-P2A-tagRFP (puro), pEF-MCS-3xHA, pGBTK7, or pACT2 vectors. Mutations of nsp10 were generated using the QuikChange site-directed mutagenesis kit (Stratagene). All constructs were sequenced using an ABI PRISM 377 automatic DNA sequencer to verify 100% correspondence with the original sequence.
Method Details
Purification of SARS-CoV-2 viral protein complexes
HEK293 were transfected with 3xFlag-tagged plasmids encoding each SARS-CoV-2 protein by Lipofectamine 3000 (Thermo Fisher Scientific, #L3000015). 3x107 HEK293 cells were harvested in 10 mL lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% Nonidet P40, 10% glycerol, phosphatase inhibitors, and protease inhibitors) at 72 h post-transfection, followed by centrifugation and filtration (0.45 μm) to remove debris. Cell lysates were precleared with 100 μL protein A/G resin and then incubated with 25 μL anti-Flag M2 resin (Sigma, #F2426) for 12 h at 4°C on a rotator. The resin was washed 5 times and transferred to a spin column with 40 μL of 3 X Flag peptide (Sigma, #F4799) for 1 h at 4°C on a rotator. The purified complexes were sent for mass spectrometry analysis for protein-protein interaction networks at the National Facility for Protein Science in Shanghai.
Virus-host Interaction network analysis
The samples (purified complexes) were precipitated by TCA and resolved in 8 M urea, and then treated with 5 mM TCEP and 10 mM IAA to reduce the disulfide bonds and alkylate the resulting thiol groups, sequentially. The mixture was digested for 16 h at 37°C by trypsin at an enzyme-to-substrate ratio of 1:50 (wt/wt). The trypsin-digested peptides were desalted by monospin C18 and loaded on a home-made 40 cm-long pulled-tip analytical column (75 μm ID x 360 μm OD, ReproSil-Pur C18-AQ 1.9 μm resin, Dr. Maisch GmbH), connected to a Thermo Easy-nLC1000 HPLC system. The samples were analyzed with a 120 min-HPLC gradient from 0 to 100% of buffer B (0.1% formic acid in 80% acetonitrile) at 300 nL/min:0-1 min, 5%–10% B;1-96 min, 10%–40% B; 97-104min, 40%–60% B; 104-105min, 60%–100% B; 105-120min, 100% B. The eluted peptides were ionized and directly introduced into a Q-Exactive mass spectrometer using a nano-spray source with a distal 2.0-kV spray voltage. Survey full-scan MS spectra (from m/z 300–1,800) was acquired in the Orbitrap analyzer with resolution r = 70,000 at m/z 200. One acquisition cycle included one full-scan MS spectrum followed by top 20 MS/MS events, sequentially generated on the first to the twentieth most intense ions selected from the full MS spectrum at a 28% normalized collision energy. The acquired MS/MS data were analyzed against a UniProtKB Human database (database released on Sept. 30, 2018) containing SARS-CoV-2 viral proteins using Maxquant V1.6.10.43. To accurately estimate peptide probabilities and false discovery rates (FDR), we used a decoy database containing the reversed sequences of all the proteins appended to the target database. FDR was set at 0.01. Mass tolerance for precursor ions was set at 20 ppm. Trypsin was defined as a cleavage enzyme and the maximal number of missed cleavage sites was set at 3. Carbamidomethylation (+57.02146) of cysteine was considered as a static modification. Methionine oxidation was set as variable modifications. For AP-MS analysis, proteins with spectral counts no more than 8 and fold change no more than 5 were excluded as background. MiST algorithm was used to calculate the statistical significance of the dataset.34 To screening the high-confidence interactions, the MiST score setting at 0.8 was used to filter the interacting host targets. The ribosome proteins were also removed from the list due to their high background for protein purification. Finally, the interactome network was generated by Cytoscape software (https://cytoscape.org/).
Proteomic study of PBMCs from COVID-19 patients
Preparation of PMBC cells from patients
The information of 35 patients with confirmed SARS-CoV-2 infection (13 COVID-19 patients with severe symptoms and 22 COVID-19 patients with mild symptoms) and 6 health donors as control are listed in Table S2, including age (47-73), sex, and symptoms. This study followed the Guideline of Novel Coronavirus Laboratory Safety (Edition 2) and was approved by the Shanghai Public Health Clinical Center (#YJ-2020-S052-02). The peripheral venous whole blood samples were collected from all patients and PBMCs were prepared using density-gradient centrifugation followed by washing with cold PBS twice. The cell pellets were lysed by 8M Urea in 20mM Tris-HCl (pH 8.0) with protein inhibitor cocktail and phosphor-STOP cocktail (Roche) to denature the proteins and sonicated twice on ice to break the protein-protein or DNA-protein interactions. All the procedures were carried out within the certified laboratory for studies of infectious materials. For all the sample sets, individual patient PBMC samples of each category (health, MS or SS) were pooled into several pools to achieve the minimal starting amount of total protein.62 Basically, protein samples were combined to generate 3 control sample pools from 6 healthy donors (one sample pool was excluded from the list for the sample quality issue), 13 mild-case sample pools from 22 COVID-19-MS patients, and 10 severe-case sample pools from 13 COVID-19-SS patients according to the protein concentrations of all the samples due to the limited protein amounts extracted from the variable PBMCs of each case (Table S2). The resulted lysates were considered individual cases and subjected to protein digestion for the following proteomic experiments.
Protein Digestion
The protein concentration was measured, and equal amount of proteins were used for tryptic digestion. TCEP [tris(2-carboxyethyl) phosphine, final concentration is 5 mM] (Thermo Scientific) and iodoacetamide (final concentration is 10mM) (Sigma) for reduction and alkylation were added to the solution and incubated at room temperature for 30 minutes, respectively. The protein mixture was diluted four times and digested with LysC at 1:50 (w/w) for two hours and then with Trypsin at 1:40 (w/w) (Promega) at 37°C overnight. The tryptic-digested peptides soups were desalted by MonoSpin™ C18 (GL Science) and the eluents were dried out in speed vacuum. The peptides mixtures were finally dissolved in HEPES buffer (100 mM pH 8.0).
TMT labeling
The peptide concentrations were measured with Quantitative Colorimetric Peptide Assay (Thermo Scientific) before TMT-labeling to make sure the amount of peptides in each channel for TMT labeling is equal. TMT10plex amino reactive reagents (0.8 mg per vial, Thermo Scientific) were suspended in 41 μL of anhydrous acetonitrile respectively and suitable for the labeling of 100 μg peptide soups in each channel. According to the amount of the peptide mixtures, equal amounts of the peptide mixtures (∼90 μg) were labeling with three sets of the TMT10 labeling kit. Meanwhile, a peptide mixture consisted of aliquots of each sample was also labeled with TMT10 and spiked into each set of quantitation experiments as internal standards for protein normalization. Reactions were allowed to proceed at room temperature for 1 h, and then quenched by an additional 8 μL of 5% hydroxylamine for 15 min. The TMT-labeled samples were pooled at a 1:1:1:1:1:1:1:1:1:1 ratio. The mixture was vacuum centrifuged to near dryness and desalted on a MonoSpin™ C18 (GL Science).
Offline high pH-reversed phase fractionation
The desalted TMT-labeled peptides were resuspended in 300 ul 10% buffer C (150 mM ammonium hydroxide, pH 10). Offline fractionations were performed using Agilent 1260 HPLC with a 4.6 mm × 250 mm BEH C18, 3.5 μm column (Waters) by a 58 min gradient from 5% to 80% buffer B (100% acetonitrile) with buffer A (100% water) at a flow rate of 0.5 mL/min while buffer C was running constantly at 10% of total flow rate during the process. Fractions were collected over 100 min at 2 min intervals after the start of the gradient in 1.5 mL Eppendorf tubes. The peptide mixture was fractionated into a total of 50 fractions which were consolidated into 18 vials. Samples were subsequently desalted and vacuum centrifuged to near dryness. The desalted peptides were reconstituted in 0.1% formic acid for downstream LC-MS/MS analysis.
LC-MS/MS analysis of peptides
For proteome study, all the 18 fractions for each TMT experiment were analyzed by a home-made 40 cm-long pulled-tip analytical column (75 μm ID x 360 μm OD, ReproSil-Pur C18-AQ 1.9 μm resin, Dr. Maisch GmbH), the column was then placed in-line with an Easy-nLC 1200 nano HPLC (Thermo Scientific) for mass spectrometry analysis. The analytical column temperature was set at 55°C during the experiments. The mobile phase and elution gradient used for peptide separation were as follows: 0.1% formic acid in water as buffer A and 0.1% formic acid in 80% acetonitrile as buffer B, 0-1 min, 5%–10% B; 1-96 min, 10%–40% B; 96-104 min, 40%–60% B, 104-105 min, 60%–100% B, 105-120 min, 100% B. The flow rate was set at 300 nL/min. Data-dependent tandem mass spectrometry (MS/MS) analysis was performed with a Q Exactive Orbitrap mass spectrometer (Thermo Scientific). Peptides eluted from the LC column were directly electrosprayed into the mass spectrometer with the application of a distal 2.2-kV spray voltage. A cycle of one full-scan MS spectrum (m/z 300-1800) was acquired followed by top 20 MS/MS events (first mass fixed at 100 m/z scan range), sequentially generated on the first to the twentieth most intense ions selected from the full MS spectrum at a 35% normalized collision energy. Full scan resolution was set to 70,000 with automated gain control (AGC) target of 3e6. MS/MS scan resolution was set to 35,000 with an isolation window of 1.8 m/z and AGC target of 1e5. The number of micro scans was one for both MS and MS/MS scans and the maximum ion injection time was 50 and 100 ms, respectively. The dynamic exclusion settings used were as follows: charge exclusion, 1 and > 8; exclude isotopes, on; and exclusion duration, 30 s.
Data Analysis
The acquired MS/MS data were analyzed against a UniProtKB human (database released on Sept. 30, 2018) by Maxquant V1.6.10.43 using the default setting. Tolerance of precursor mass and fragment mass were set to ± 20ppm. The main search peptides tolerance was setting at 4.5ppm according to the feature of the instrument in the study. Carbamidomethylation of cysteine (+57.021 Da) and acetylation of protein N terminus were set as static modifications. The quantification search type chooses the reporter ion ms2 using TMT 10plex method. Oxidation of methionine (+15.995 Da) was set as variable modification. Trypsin was defined as cleavage enzyme, and the maximum missing cleavage was set at 2. All identified proteins had an FDR ≤ 1%, which was calculated at the peptide level. The unique peptides were selected for protein quantitation.
Merging of datasets and statistical analysis
The interpretation of three sets of proteomic results (protein Groups.txt) from MaxQuant was done in the Perseus software environment (version 1.6.2.3) and R4.0.0. Briefly, the datasets were merged by matching the protein accession numbers of each dataset using in-house developed Python scripts (comparepg.py). The protein abundances were normalized by the TMT-labeled internal standard samples which consisted of an aliquot of each sample and were spiked into the 3 sets of TMT labeling experiments. Protein features with less than 3 quantitative values in each condition (H&MS or MS&SS) were removed from the dataset to make sure each quantitative protein in the list have at least 3 values for H cases, MS cases, and SS cases during the proteomics analysis. The remaining missing quantitative values were replaced by values imputed using multivariate imputation algorithms from the MICE R package.63 Data were further normalized across the TMT 10plex sets using the median normalization method.64 All the normalized reporter ion abundances of biological repeats were averaged for fold-change calculation. For proteome analysis, P-value was calculated by empirical Bayes moderated t tests and then adjusted by Benjamini and Hochberg method, using the limma package57 built for R environment. Proteins were required to have a |log2 (fold change) | ≥ 0.58 and adj. P-value cut-off of 0.01 to be considered differentially expressed.58 Gene ontology (GO) analysis was performed using ClusterProfiler v3.14.3.60
Detection of SARS-CoV-2 intra-viral PPIs by yeast-two-hybrid
28 SAR-CoV-2 genes were cloned into both pGBTK7 (bait) and pACT2 (prey) vectors. The reporter Y2HGold yeast strain (Clontech) was co-transformed with different bait-prey combinations as indicated in Figure S1 in accordance with instructions for Matchmaker GAL4 Two-Hybrid System 3 (Clontech). The transformed yeast was then screened in 4-dropout plates for the protein-protein interaction.
RNA extraction and quantitative RT-PCR
Total RNA was isolated from cells with the RNeasy Mini Kit (QIAGEN, #74106) and treated with RNase-free DNase according to the manufacturer’s protocol. All gene transcripts were quantified by quantitative PCR using qScript™ One-Step qRT-PCR Kit (Quanta Biosciences, #95057-050) on CFX96 real-time PCR system (Bio-Rad). Primer sequences for qPCR were as follows: IL-8 forward: TCTTGCACAAATATTTGATGC, IL-8 reverse: CCACTGTGCCTTGGTTTC; GAPDH forward: GAGTCAACGGATTTGGTCGT, GAPDH reverse: TTGATTTTGGAGGGATCTCG; TNFα forward: CTCCAGGCGGTGCTTGTTC, TNFα reverse: GGCTACAGGCTTGTCACTCG; NKRF forward: GTAAACATGCAGCTGCCGAC, NKRF reverse: CGTGCACACGGGATTTGAAG; IL-1β forward: CTCGCCAGTGAAATGATGGCT; IL-1β revere: GTCGGAGATTCGTAGCTGGAT.
siRNA gene silencing
Small interfering RNA (siRNA) targeting human NKRF (#A10001) and negative control siRNAs were purchased from GenePharma. siRNAs were delivered to the A549 cells using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, #13778030) according to the manufacturer’s instructions. After 48 h of transfection, cells were verified for target gene knock-down by quantitative RT-PCR.
Phylogenetic and genetic analysis of SARS-CoV-2 sequences
160 SARS-CoV-2 isolates with complete genome sequences from 18 different countries collected from December 23, 2019 to February 29, 2020 were used for molecular evolutionary analysis in this study. Multiple alignments were performed by Clustal W and a phylogenetic tree was constructed by Neighbor-Joining statistical method based on the Maximum Composite Likelihood model with 500 replicates of bootstrap testing using MEGA.55
Quantification and Statistical Analysis
For AP-MS analysis (Figure 2), proteins with spectral counts no more than 8 and fold change no more than 5 were excluded as background. MiST algorithm was used to calculate the statistical significance of the dataset.34 To screening the high-confidence interactions, the MiST score setting at 0.8 was used to filter the interacting host targets. The ribosome proteins were also removed from the list due to their high background for protein purification. The interactome network was generated by Cytoscape software (https://cytoscape.org/).
For statistics of proteome analysis (Figures 3A–3F and S4A–S4F), The acquired MS/MS data were analyzed against a UniProtKB human (database released on Sept. 30, 2018) by Maxquant V1.6.10.43 using the default setting. The interpretation of three sets of proteomic results (protein Groups.txt) from MaxQuant was done in the Perseus software environment (version 1.6.2.3) and R4.0.0. P-value was calculated by empirical Bayes moderated t tests and then adjusted by Benjamini and Hochberg method, using the limma package57 built for R environment. Proteins were required to have a |log2 (fold change) | ≥ 0.58 and adj. P-value cut-off of 0.01 to be considered differentially expressed.58
All data were expressed as mean ± SEM when appropriate. For parametric analysis, the F test was used to determine the equality of variances between the groups compared; statistical significance across two groups was tested by Student’s t test (Figures 3K, S4J, and S4K); one-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test was used to determine statistically significant differences between multiple groups (Figures S3G, S3H, S3J, S3L, and S4H). P-values of less than 0.05 were considered significant.
Acknowledgments
This work was supported by grants from the National Key Research and Development Project of China (2018YFA0900802), the National Natural Science Foundation of China (31770176 and 31500667), the National Science and Technology Major Project (2020ZX09201001), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, the Shanghai Science and Technology Commission (2017QA1403200 and 20YF1442500), the Shanghai Municipal Health Commission (2018YQ40 and 201940179), the Shanghai Municipal Key Clinical Specialty (shslczdzk01102), the Innovative Research Team of High-level Local Universities in Shanghai, the Interdisciplinary Program of Shanghai Jiao Tong University (YG2020YQ14), the SII Challenge Fund for COVID-19 Research, the Chinese Academy of Sciences Large Research Infrastructure of Maintenance and Remolding Project (DSS-WXGZ-2020-0001), and the Chinese Academy of Sciences Key Technology Talent Program.
Author Contributions
Q.L., T.Z., and C.P. conceived of the research, designed the study, and wrote the manuscript. M.G. isolated and lysed PBMCs in denaturing buffer. Z.L., J.L., C.L., X.W., and X.Y. performed the experiments and analyzed the data. X.T., P.W., and Y.Y. performed the LC-MS/MS analysis. Z.X. helped with the visual representation of the data and edited the manuscript. All of the authors commented on the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: July 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.medj.2020.07.002.
Supplemental Information
SARS-CoV-2-Host Interactome List, Related to Figure 2
COVID-19 Patient Information for Quantitative Proteomic Analysis, Related to Figure 3
PBMC Proteomics, Related to Figure 3
Mutations of SARS-CoV-2 during Evolution, Related to Figure 3
References
- 1.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson M.E., Chen L.H. Travelers Give Wings to Novel Coronavirus (2019-nCoV) J. Travel Med. 2020;27 doi: 10.1093/jtm/taaa015. taaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Editorial Emerging understandings of 2019-nCoV. Lancet. 2020;395:311. doi: 10.1016/S0140-6736(20)30186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guan W., Ni Z., Hu Y., Liang W., Ou C., He J., Liu L., Shan H., Lei C., Hui D.S., et al. Clinical characteristics of 2019 novel coronavirus infection in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ong E.Z., Chan Y.F.Z., Leong W.Y., Lee N.M.Y., Kalimuddin S., Mohideen S.M.H., Chan K.S., Tan A.T., Bertoletti A., Ooi E.E., et al. A Dynamic Immune Response Shapes COVID-19 Progression. Cell Host Microbe. 2020;27:879–882.e2. doi: 10.1016/j.chom.2020.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vabret N., Britton G.J., Gruber C., Hegde S., Kim J., Kuksin M., Levantovsky R., Malle L., Moreira A., Park M.D., et al. Immunology of COVID-19: current state of the science. Immunity. 2020;52:910–941. doi: 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A., et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren L.-L., Wang Y.-M., Wu Z.-Q., Xiang Z.-C., Guo L., Xu T., Jiang Y.-Z., Xiong Y., Li Y.-J., Li X.-W., et al. Identification of a novel coronavirus causing severe pneumonia in human: a descriptive study. Chin. Med. J. 2020;133:1015–1024. doi: 10.1097/CM9.0000000000000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X., Tan Y., Ling Y., Lu G., Liu F., Yi Z., Jia X., Wu M., Shi B., Xu S., et al. Viral and host factors related to the clinical outcome of COVID-19. Nature. 2020;583:437–440. doi: 10.1038/s41586-020-2355-0. [DOI] [PubMed] [Google Scholar]
- 17.Németh T., Sperandio M., Mócsai A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020;19:253–275. doi: 10.1038/s41573-019-0054-z. [DOI] [PubMed] [Google Scholar]
- 18.Kolaczkowska E., Kubes P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 19.Wang C., Xie J., Fei X., Zhang H., Tan Y., Zhou L., Liu Z., Ren Y., Yuan L., Zhang Y., et al. Aveolar Macrophage Activation and Cytokine Storm in the Pathogenesis of Severe COVID-19. EBioMedicine. 2020:102833. doi: 10.1016/j.ebiom.2020.102833. 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim D., Lee J.-Y., Yang J.-S., Kim J.W., Kim V.N., Chang H. The Architecture of SARS-CoV-2 Transcriptome. Cell. 2020;181:914–921.e10. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu A., Peng Y., Huang B., Ding X., Wang X., Niu P., Meng J., Zhu Z., Zhang Z., Wang J., et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe. 2020;27:325–328. doi: 10.1016/j.chom.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Brunn A., Teepe C., Simpson J.C., Pepperkok R., Friedel C.C., Zimmer R., Roberts R., Baric R., Haas J. Analysis of intraviral protein-protein interactions of the SARS coronavirus ORFeome. PLOS ONE. 2007;2:e459. doi: 10.1371/journal.pone.0000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan J., Peng X., Gao Y., Li Z., Lu X., Chen Y., Ishaq M., Liu D., Dediego M.L., Enjuanes L., Guo D. Genome-wide analysis of protein-protein interactions and involvement of viral proteins in SARS-CoV replication. PLOS ONE. 2008;3:e3299. doi: 10.1371/journal.pone.0003299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hillen H.S., Kokic G., Farnung L., Dienemann C., Tegunov D., Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020 doi: 10.1038/s41586-020-2368-8. [DOI] [PubMed] [Google Scholar]
- 26.Wang Q., Wu J., Wang H., Gao Y., Liu Q., Mu A., Ji W., Yan L., Zhu Y., Zhu C., et al. Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell. 2020 doi: 10.1016/j.cell.2020.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decroly E., Debarnot C., Ferron F., Bouvet M., Coutard B., Imbert I., Gluais L., Papageorgiou N., Sharff A., Bricogne G., et al. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2′-O-methyltransferase nsp10/nsp16 complex. PLOS Pathog. 2011;7:e1002059. doi: 10.1371/journal.ppat.1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Y., Wu L., Shaw N., Gao Y., Wang J., Sun Y., Lou Z., Yan L., Zhang R., Rao Z. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. USA. 2015;112:9436–9441. doi: 10.1073/pnas.1508686112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y., Sun Y., Wu A., Xu S., Pan R., Zeng C., Jin X., Ge X., Shi Z., Ahola T., et al. Coronavirus nsp10/nsp16 Methyltransferase Can Be Targeted by nsp10-Derived Peptide In Vitro and In Vivo To Reduce Replication and Pathogenesis. J. Virol. 2015;89:8416–8427. doi: 10.1128/JVI.00948-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bojkova D., Klann K., Koch B., Widera M., Krause D., Ciesek S., Cinatl J., Münch C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature. 2020;583:469–472. doi: 10.1038/s41586-020-2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ni L., Ye F., Cheng M.-L., Feng Y., Deng Y.-Q., Zhao H., Wei P., Ge J., Gou M., Li X., et al. Detection of SARS-CoV-2-Specific Humoral and Cellular Immunity in COVID-19 Convalescent Individuals. Immunity. 2020;52:971–977.e3. doi: 10.1016/j.immuni.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong Y., Liu Y., Cao L., Wang D., Guo M., Jiang A., Guo D., Hu W., Yang J., Tang Z., et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020;9:761–770. doi: 10.1080/22221751.2020.1747363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Z., Ren L., Zhang L., Zhong J., Xiao Y., Jia Z., Guo L., Yang J., Wang C., Jiang S., et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe. 2020;27:883–890.e2. doi: 10.1016/j.chom.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verschueren E., Von Dollen J., Cimermancic P., Gulbahce N., Sali A., Krogan N.J. Scoring Large-Scale Affinity Purification Mass Spectrometry Datasets with MiST. Curr. Protoc. Bioinformatics. 2015;49:8.19.1–8.19.16. doi: 10.1002/0471250953.bi0819s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scholte F.E.M., Tas A., Albulescu I.C., Žusinaite E., Merits A., Snijder E.J., van Hemert M.J. Stress granule components G3BP1 and G3BP2 play a proviral role early in Chikungunya virus replication. J. Virol. 2015;89:4457–4469. doi: 10.1128/JVI.03612-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faria P.A., Chakraborty P., Levay A., Barber G.N., Ezelle H.J., Enninga J., Arana C., van Deursen J., Fontoura B.M.A. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol. Cell. 2005;17:93–102. doi: 10.1016/j.molcel.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 38.Fitzgerald K.A., McWhirter S.M., Faia K.L., Rowe D.C., Latz E., Golenbock D.T., Coyle A.J., Liao S.-M., Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 39.Kondo T., Watanabe M., Hatakeyama S. TRIM59 interacts with ECSIT and negatively regulates NF-κB and IRF-3/7-mediated signal pathways. Biochem. Biophys. Res. Commun. 2012;422:501–507. doi: 10.1016/j.bbrc.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 40.Nourbakhsh M., Kälble S., Dörrie A., Hauser H., Resch K., Kracht M. The NF-κ b repressing factor is involved in basal repression and interleukin (IL)-1-induced activation of IL-8 transcription by binding to a conserved NF-κ b-flanking sequence element. J. Biol. Chem. 2001;276:4501–4508. doi: 10.1074/jbc.M007532200. [DOI] [PubMed] [Google Scholar]
- 41.Huang K.-H., Wang C.-H., Lin C.-H., Kuo H.-P. NF-κB repressing factor downregulates basal expression and mycobacterium tuberculosis induced IP-10 and IL-8 synthesis via interference with NF-κB in monocytes. J. Biomed. Sci. 2014;21:71. doi: 10.1186/s12929-014-0071-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pomerantz J.L., Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999;18:6694–6704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaix A., Lopez S., Voisset E., Gros L., Dubreuil P., De Sepulveda P. Mechanisms of STAT protein activation by oncogenic KIT mutants in neoplastic mast cells. J. Biol. Chem. 2011;286:5956–5966. doi: 10.1074/jbc.M110.182642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amelot A.A., Tagzirt M., Ducouret G., Kuen R.L., Le Bonniec B.F. Platelet factor 4 (CXCL4) seals blood clots by altering the structure of fibrin. J. Biol. Chem. 2007;282:710–720. doi: 10.1074/jbc.M606650200. [DOI] [PubMed] [Google Scholar]
- 45.Gupta S.K., Hassel T., Singh J.P. A potent inhibitor of endothelial cell proliferation is generated by proteolytic cleavage of the chemokine platelet factor 4. Proc. Natl. Acad. Sci. USA. 1995;92:7799–7803. doi: 10.1073/pnas.92.17.7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kunicki T.J., Williams S.A., Salomon D.R., Harrison P., Crisler P., Nakagawa P., Mondala T.S., Head S.R., Nugent D.J. Genetics of platelet reactivity in normal, healthy individuals. J. Thromb. Haemost. 2009;7:2116–2122. doi: 10.1111/j.1538-7836.2009.03610.x. [DOI] [PubMed] [Google Scholar]
- 47.Elisen M.G., von dem Borne P.A., Bouma B.N., Meijers J.C. Protein C inhibitor acts as a procoagulant by inhibiting the thrombomodulin-induced activation of protein C in human plasma. Blood. 1998;91:1542–1547. [PubMed] [Google Scholar]
- 48.Nishioka J., Ning M., Hayashi T., Suzuki K. Protein C inhibitor secreted from activated platelets efficiently inhibits activated protein C on phosphatidylethanolamine of platelet membrane and microvesicles. J. Biol. Chem. 1998;273:11281–11287. doi: 10.1074/jbc.273.18.11281. [DOI] [PubMed] [Google Scholar]
- 49.Vegh Z., Kew R.R., Gruber B.L., Ghebrehiwet B. Chemotaxis of human monocyte-derived dendritic cells to complement component C1q is mediated by the receptors gC1qR and cC1qR. Mol. Immunol. 2006;43:1402–1407. doi: 10.1016/j.molimm.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 50.Waggoner S.N., Hall C.H.T., Hahn Y.S. HCV core protein interaction with gC1q receptor inhibits Th1 differentiation of CD4+ T cells via suppression of dendritic cell IL-12 production. J. Leukoc. Biol. 2007;82:1407–1419. doi: 10.1189/jlb.0507268. [DOI] [PubMed] [Google Scholar]
- 51.Xu L., Xiao N., Liu F., Ren H., Gu J. Inhibition of RIG-I and MDA5-dependent antiviral response by gC1qR at mitochondria. Proc. Natl. Acad. Sci. USA. 2009;106:1530–1535. doi: 10.1073/pnas.0811029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delaney M.K., Liu J., Kim K., Shen B., Stojanovic-Terpo A., Zheng Y., Cho J., Du X. Agonist-induced platelet procoagulant activity requires shear and a Rac1-dependent signaling mechanism. Blood. 2014;124:1957–1967. doi: 10.1182/blood-2014-03-560821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fogarty H., Townsend L., Ni Cheallaigh C., Bergin C., Martin-Loeches I., Browne P., Bacon C.L., Gaule R., Gillett A., Byrne M., et al. COVID-19 Coagulopathy in Caucasian Patients. Br. J. Haematol. 2020;189:1044–1049. doi: 10.1111/bjh.16749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiong C., Jiang L., Chen Y., Jiang Q. Evolution and variation of 2019-novel coronavirus. bioRxiv. 2020 doi: 10.1101/2020.01.30.926477. [DOI] [Google Scholar]
- 55.Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bester J., Pretorius E. Effects of IL-1β, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci. Rep. 2016;6:32188. doi: 10.1038/srep32188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Connell J.D., Paulo J.A., O’Brien J.J., Gygi S.P. Proteome-Wide Evaluation of Two Common Protein Quantification Methods. J. Proteome Res. 2018;17:1934–1942. doi: 10.1021/acs.jproteome.8b00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang Q., Wei D., Chung B., Brulois K.F., Guo C., Dong S., Gao S.-J., Feng P., Liang C., Jung J.U. Novel Role of vBcl2 in the Virion Assembly of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2018 doi: 10.1128/JVI.00914-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma J., Chen T., Wu S., Yang C., Bai M., Shu K., Li K., Zhang G., Jin Z., He F., et al. iProX: an integrated proteome resource. Nucleic Acids Res. 2019;47(D1):D1211–D1217. doi: 10.1093/nar/gky869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Savaryn J.P., Toby T.K., Catherman A.D., Fellers R.T., LeDuc R.D., Thomas P.M., Friedewald J.J., Salomon D.R., Abecassis M.M., Kelleher N.L. Comparative top down proteomics of peripheral blood mononuclear cells from kidney transplant recipients with normal kidney biopsies or acute rejection. Proteomics. 2016;16:2048–2058. doi: 10.1002/pmic.201600008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Resche-Rigon M., White I.R. Multiple imputation by chained equations for systematically and sporadically missing multilevel data. Stat. Methods Med. Res. 2018;27:1634–1649. doi: 10.1177/0962280216666564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chawade A., Alexandersson E., Levander F. Normalyzer: a tool for rapid evaluation of normalization methods for omics data sets. J. Proteome Res. 2014;13:3114–3120. doi: 10.1021/pr401264n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SARS-CoV-2-Host Interactome List, Related to Figure 2
COVID-19 Patient Information for Quantitative Proteomic Analysis, Related to Figure 3
PBMC Proteomics, Related to Figure 3
Mutations of SARS-CoV-2 during Evolution, Related to Figure 3
Data Availability Statement
The datasets generated during this study were deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository61 with the dataset identifier IPX0002285000.