

ABSTRACT
Introduction:
Biosimiliar, a copy of reference biological product, is making a buzz across the globe for its upper edge therapeutic usage. According to the market research report published by P&S Intelligence, biosimilars market is expected to generate $26.7 billion revenue by 2024, advancing at a CAGR of 29.6% during the forecast period. The first biosimilar to medicine Omnitrope, was approved in Europe by EMA (European Medicines Agency) in year 2006. Till date countries like US, China, Japan, India and many more have generated regulatory guidelines for biosimilars.
Aim:
Current study addresses the issues and challenges faced by Industry and regulators with their potential solutions and recommendations.
Materials and methods:
The questionnaire having 21 important questions/comments was given to participants after explaining the purpose of the study. The response in terms of responders V/s non-responders, agree V/s disagree, yes V/s no was recorded and analyzed by descriptive statistics.
Results and Discussion:
The study shows the limitation regarding the qualified personnel involved in biosimilars, as approx. 91% people believe that there is lack of expertise in this field. The same can be achieved through government initiatives for bridge courses which is also strongly felt by the major (83.6%) stakeholders participated in the study.
KEYWORDS: Biobetters, biologic medicine, biosimilar, global regulations, Omnitrope, recombinant deoxyribonucleic acid products, vaccine
INTRODUCTION
Biological products are defined as substances of biological origin that are assayed by biological tests and used in the prophylaxis, therapy, or diagnosis of human diseases (Reference: World Health Organization Technical Report Series [WHO TRS] 963). Various types of biological products include vaccine (a biological preparation that improves immunity to a particular disease, e.g., polio vaccine, Bacillus Calmette–Guérin (BCG), diphtheria-pertussis-tetanus (DPT), and hepatitis B), recombinant deoxyribonucleic acid (r-DNA)-based drugs (combination of two r-DNA strands artificially from two different sources to produce bio therapeutically active recombinant proteins, e.g., adalimumab and rituximab), blood, and blood products (obtained from pooled plasma of blood by fractionation, drawn from donors, e.g., coagulation factor VIII, human albumin, and prothrombin).[1,2] All vaccines and r-DNA products are new drugs under Rule 122-E and also vide G.S.R. 45(E) dated January 24, 2011.[3] The biosimiliars can be imported (Rule 122-A) and also can be manufactured (Rule 122-B) as per Drugs and Cosmetics Act 1940 & Rule 1945.[2]
Process of approval/testing before the products are released in the market is as follows: (1) Clinical trial application in Form 44 along with common technical document module[4] and other related documents is submitted to the Central Drugs Standard Control Organisation (CDSCO) on SUGAM portal[4] for evaluation. (2) After satisfactory review of the application, the proposal may be referred to subject expert committee (SEC) for evaluation and subsequently to the technical committee for further deliberation. (3) On receipt of recommendations from both the committees, the clinical trial permission is granted to the firm. In the case of new chemical entities (NCE), the proposal may be referred to investigational new drug committee followed by technical committee and Apex committee for recommendation and further action. (4) On the basis of evaluation, the firm may be granted the permission to go for further trials or the permission to import/manufacture and market the new drug; in addition, in the case of vaccines, a parallel review of Chemistry, Manufacturing and Controls (CMC) is performed by Central Drugs Laboratory (CDL), Kasauli and the permission to import/manufacture and market the vaccine is granted on receipt of no objection certificate from CDL, Kasauli.
The Guidelines on Similar Biologics was prepared by CDSCO and Department of Biotechnology (DBT) laid down the regulatory pathway for similar biologic claiming to be similar to an already-authorized reference biologic, which have come into effect from September 2012 [Table 1]. The revision of the 2012 DBT-CDSCO guidance document for the development and approval of Similar Biologics took place and the “Guidelines on Similar Biologics: Regulatory Requirements for Market Authorization in India 2016”[5,6] are now effective.
Table 1.
Various stakeholders involved in decision making of biosimilars
| S. no. | Stakeholders | Role |
|---|---|---|
| 1. | CDSCO | National Regulatory Authority in India that evaluates safety, efficacy, and quality of drugs in the country |
| 2. | Ministry of Environment | Issued Biosafety Guidelines under Environment Protection Act (EPA) |
| 3. | Review Committee on Genetic Manipulation (RCGM) under Department of Biotechnology (DBT), Ministry of Science and Technology | Is responsible for authorizing import/export for research and development and review of data up to preclinical evaluation. |
| 4. | Genetic Engineering Appraisal Committee (GEAC) under the Ministry of Environment and Forests (MoEF) | Is responsible for review and approval of activities involving large-scale use of genetically engineered organisms (LMOs) and products thereof in R&D, industrial production, environmental release, and field applications |
| 5. | State Licensing Authority | Works with CDSCO in each state and is responsible for issuance of license to manufacture r-DNA products in India. |
| 6. | Ministry of Health & Family Welfare’s | Constituted New Drug Advisory Committee (NDAC) on March 31, 2012 to advise Drugs Controller General (India) in matters related to approval of New Drugs including biopharmaceutical products and Clinical Trials. |
CDSCO = Central Drugs Standard Control Organisation, r-DNA = recombinant deoxyribonucleic acid
As per Biosimilar guidelines, Similar biologics can only be developed against an authorized reference biologic that has been approved using a complete data package in India. In case, Reference biologic is not authorized in India, it should have been licensed in any international council for harmonization countries. The reference biologic product can be imported for developing the Similar biologic for quality, preclinical, and clinical comparability. The dosage form, strength, and route of administration of the similar biologic should be the same as that of reference biologic. Any product can be considered as Similar Biologic only if it is proven to be similar using extensive quality characterization against reference biologic product. Quality-based consideration for Similar Biologics includes the following: analytical methods; product characterization, specifications, stability, quality comparability study by quality attributes, which are divided into two categories––critical quality attributes and key quality attributes. It is important to establish a formal Risk Management Plan to monitor and detect both known inherent safety concerns and potential unknown safety signals that may arise from the Similar Biologic.[7]
To further reduce the residual risk of the Similar Biologics, additional safety data may need to be collected after market approval through a predefined single-arm study of generally more than 200 evaluable patients and compared to historical data of the reference biologics. The study should be completed preferably within 2 years of market authorization/license. The primary aim of the post-marketing phase-IV study is safety and the secondary aim is efficacy and immunogenicity
Due to increase in competition among the pharmaceutical companies, new products that are cheaper are introduced in the market every now and then.[8] Biological products, which are less expensive and show similar function to the original product, called the biosimilars, are ruling the pharmaceutical market. Biosimilars show similar effect as the parent or original biological product but it is not necessarily made up of same active ingredient or chemicals. Once the patent of the original biological product or biologic expires, it is legal for other pharmaceutical companies to manufacture the drug.[9] Therefore, it is impossible for a biosimilars to be the exact identical of the original product and thus they cannot be compared to generic drugs.[10] Biosimilars are estimated to be the top upcoming product in the field of medicines. Biosimilar drugs promise treatment of diseases such as cancer, HIV/AIDS, rheumatoid arthritis, and inflammatory bowel diseases.
They are not only up to the mark in treating the patients but are also becoming the primary choice in the market due to their cheaper rate than the original product. As biosimilars are the replicas of the reference product, they require approvals along with successful clinical trials that assure the product’s quality, safety, and efficacy. Despite the several inherited advantages with the biosimilars, in India these are nascent phase because of several warming and warming challenges.[11,12] The following study had been undertaken to the issues and challenges faced by industry and regulators with their potential solutions and recommendations [Table 2].
Table 2.
Perspective about biosimilar
| S. no. | Questions | Option 1 | Option 2 |
|---|---|---|---|
| 1. | Are you aware about biosimilar? | Yes | No |
| 2. | Are patients are aware about use of biosimilars in cancer? | Yes | No |
| 3. | Does patient survival rate increased by use of biosimilars? | Yes | No |
| 4. | Are biosimilars easily assessable and affordable? | Yes | No |
| 5. | Biosimilars better option than original drugs? | Agree | Disagree |
| 6. | Have entry of biosimilars affected the price pattern of reference drugs? | Yes | No |
| 7. | Is it must for a pharmacist to notify about the prescribes that he or she intends to substitute for a prescribed product? | Yes | No |
| 8. | Can we see biosimilars replacing original drugs when they lose patents? | Yes | No |
| 9. | Are you aware about difference between a biosimilar and generic? | Yes | No |
| 10. | The latest regulation on biosimilars were issued in 2012 | True | False |
| 11. | Does regulation on biosimilars needs to be updated? | Yes | No |
| 12. | Does current regulation on biosimilars comply with needs of pharmaceutical industry and patient safety both? | Yes | No |
| 13. | Does biosimilars have some serious adverse effects? | Yes | No |
| 14. | Is biosimilars entry into the market a boon? | True | False |
| 15. | Whether industry is potentially beneficial for biosimilars, exclusive of its five years in the market. | Yes | No |
| 16. | Whether current regulation supports the industry. | Yes | No |
| 17. | Does India have clear cut guidelines for naming the biosimilar. | Yes | No |
| 18. | As per the mission of current government, the single-window system should be followed. | Yes | No |
| 19. | Is multiple data generation is essential in case of biosimilar. | Yes | No |
| 20. | As per guidelines, the biosimilar should be compared with approved products or not. | Agree | Disagree |
| 21. | The comparison should be made with innovator products or the approved products. | Yes | No |
| 22. | Personal are involved in biosimilar are well trained and qualified or not. | Yes | No |
| 23. | Is India good in export of biosimilar. | Agree | Degree |
| 24. | Bridge courses to empower to manpower involved in Biosimilar are utmost necessary. | Yes | No |
| 25. | Does Govt. of India includes biosimilar in its popular scheme in Ayushman Bharat | Yes | No |
MATERIAL AND METHODS
It was a cross-sectional questionnaire-based study carried out among various stakeholders including researchers, regulators, and industry people. Two hundred and seventy-five stakeholders participated in this study. The questionnaire was designed and tested among small groups for conducting a pilot study. Modified questionnaire was given to all participants. The questionnaire having 21 important questions/comments was given to participants after explaining the purpose of the study. Any doubts regarding questionnaire were clarified by the investigators. 30min was given for filling the questionnaire. The response in terms of responders vs. nonresponders, agree vs. disagree, and yes vs. no was recorded and analyzed by descriptive statistics using Microsoft Excel.
RESULTS AND DISCUSSION
The European Medicine Agency (EMA)[12] was the first regulatory body to publish guidelines on specific regulatory pathway for the approval of biosimilars in 2005.[10] These guidelines were titled as “Guidelines on similar biological medicinal products” and with time have they have been updated and improvised according to the requirements. However in India, guidelines for biosimilars have been published in 2012 as given in Table 3.
Table 3.
| Countries | US | UK | India | Canada | Singapore | Japan |
|---|---|---|---|---|---|---|
| Regulatory Authority | FDA | EMA | CDSCO and DBT | Health Canada | HSA | PMDA |
| Terminology | Follow-on biologics | Biosimilars | Similar biologics | Subsequent entry biologics | Biosimilars | Follow-on biologics |
| Year issued | 2010 | 2004 | 2012 | 2010 | 2009 | 2009 |
| Approved biosimilars | 11 | 54 | 30 | 10 | – | 18 |
| Clinical trial/ additional clinical | Mandatory/ mandatory | Mandatory/optional | Mandatory/optional | Mandatory/ optional | Mandatory/mandatory | Optional/optional |
| Major steps | Analytical studies, PK, PD, clinical trials, animal studies, immunogenicity studies, and additional studies | Analytical studies, clinical trials, animal studies, PK, and PD | Analytical studies, animal trials, PK, PD, studies, clinical trials. | Analytical studies, clinical trials, animal studies, PK, and PD | Analytical studies, PK, PD, clinical trials, animal studies, immunogenicity studies, additional studies, and pharmacovigilance. | Analytical studies, clinical trials, animal studies, PK, and PD |
| Clinical data | 50-week and 102- week study for PK and PD. 54-week study for efficacy and safety. Post- marketing study. | PK and PD studies (1–2 Cts depending on the indications); efficacy and safety studies (1–2 Cts depending on the indications) | Phase I: intravenous dose study in 84 healthy individuals for 12 weeks. Phase III: to compare efficacy and safety, 189 patients, and unhealthy. | 50-week and 102- week study for PK and PD. 54-week study for efficacy and safety. | – | For PK and PD studies:104 patients, (include Cts); for efficacy and safety: 602 patients, 30-week data. |
FDA = Food and Drug Administration, EMA = European Medicines Agency, CDSCO = Central Drugs Standard Control Organisation, DBT = Department of Biotechnology, PK = Pharmacokinetics, PD = Pharmacodynamics, HSA = Health Sciences Authority, PMDA = Pharmaceuticals and Medical Devices Agency
Approximately 27.3% people are aware of the biosimilars [Figure 1]. In addition, the potential beneficiaries, that is, patients, also have poor awareness (18.2%). A large section of people (72.7%) need proper awareness [Figures 2 and 3]; the same can be achieved through awareness program.
Figure 1.
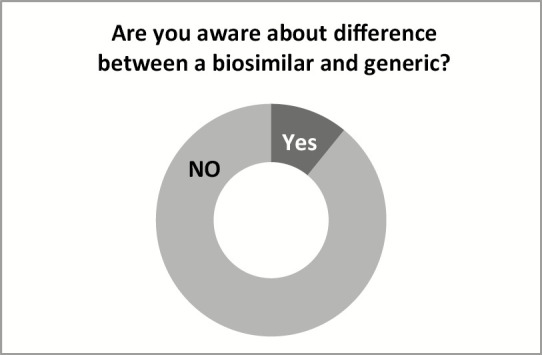
Awareness of biosimiliars Vs generics among the stakeholders and patients
Figure 2.
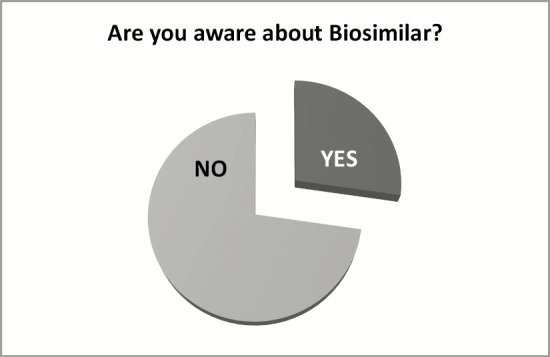
Awareness of biosimilars among the stakeholders and patients
Figure 3.
Stakeholders’ perception for biosimilars entry into India
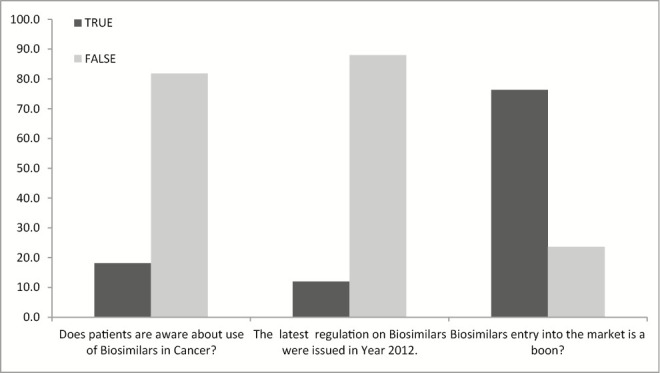
India has also introduced regulations as per majority of countries including USA, UK, Japan, Singapore, and Canada, later in 2012. The majority of stakeholders (76.4%) in opinion that entry of biosimilar is a welcome step from government and same can be considered as a boon for the patients undergoing life-threatening diseases [Figure 4].
Figure 4.
General perception of stakeholders for biosimilars
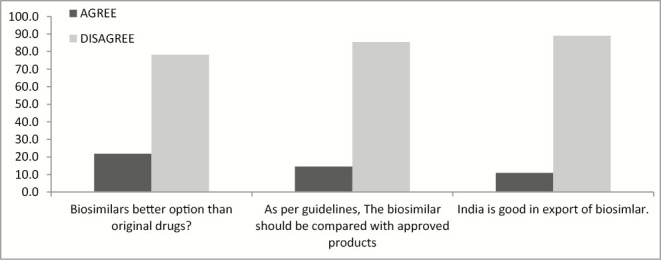
There is no doubt that biosimilar enhances the life span of patients (agreed by 69.1%). However, 83.6% stakeholders disagree that it can be better option from innovator product [Figure 5].
Figure 5.

Responses of various stakeholders against important issues of biosimilars
The study shows the limitation regarding the qualified personnel involved in biosimilars, as approximately 91% people believe that there is lack of expertise in this field. The same can be achieved through government initiatives for bridge courses, which is also strongly felt by the major (83.6%) stakeholders who participated in the study. Another important concern raised by participants was the affordability and pricing. There is no proper regulation for the pricing and market access. Also biosimilars are compared with approved product rather than innovator products that affect the quality. However, industry has further limitation of timeline of the product access to the market. Considering the data generation as per need of regulators is uphill task for industry. Industry also raises the concern for single-window approval system of biosimilars. As government giving export incentives in varied products, industry is advocating similar incentive to support the production and export of biosimilars.
CONCLUSIONS AND RECOMMENDATIONS
Biosimilars show immense hope in the field of medicine because of their potential use and low cost than their original biologics. Although there are many countries that have not approved the use of biosimilars, there are few countries that have issued regulatory guidelines for biosimilars but hesitate to accept in using them. Their nonacceptance is due to very low knowledge of the biosimilars. This issue can be resolved by initiating new schemes that could educate patients and distributors about the efficacy and safety of the biosimilar. Different countries have issued different set of guidelines with no common regulation worldwide. Countries should work toward setting a new common set of guidelines for the easy flow of trade. This would allow a uniform harmonization of a biosimilar and their easy acceptance in different countries, which would further help in fast treatment of a disease and easy availability and accessibility of the medicine to the patients.
Following needed to be taken into consideration for improving perceptions: enhance the perspective usage of biosimilar through promulgation and implementation of rules and regulation for all the stakeholders; educate patients and professionals’ about biosimilars; make better marketing and pricing policies to ensure benefits to the manufactures and patients; and assess patients’ indifferent geographical locations through patent studies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1. [Last accessed on 2019 Sept 1]. Available from: https://www.globenewswire.com/news-release/2019/04/15/1803693/0/en/Biosimilars-Market-Size-to-Reach-26-7-Billion-by-2024-P-S-Intelligence.html .
- 2. [Last accessed on 2019 Sept 10]. Available from: https://cdsco.gov.in/opencms/opencms/en/Notifications/Gazette-Notifications/ .
- 3. [Last accessed on 2020 March 7]. Available from: http://www.dbtindia.gov.in/sites/default/files/uploadfiles/Guidelines_on_Similar_Biologics%2C2016.pdf .
- 4. [Last accessed on 2019 Sept 14]. Available from: https://cdsco.gov.in/opencms/export/sites/CDSCO_WEB/Pdf-documents/SUGAM_user_manual.pdf .
- 5. [Last accessed on 2019 Sept 14]. Available from: http://nib.gov.in/NIB-DBT2016.pdf .
- 6.Drugs and Cosmetics Act 1940 & Rules. [Last accessed on 2020 Mar 7]. Available from: https://cliniexperts.com/draft-guidelines-on-similar-biologics-2016/ .
- 7. [Last accessed on 2019 Sept 14]. Available from: https://www.birac.nic.in/webcontent/Guidelines_on_Similar_Biologics_06_10_2017.pdf .
- 8.Richard O, Dolinar MD, Michael S. Reilly, Esq. The future of biological therapy: a pathway forward for biosimilars Generics and Biosimilars Initiative Journal (GaBI Journal) 2013;2:36–40. [Google Scholar]
- 9.Canadian Society of Intestinal Research (GI Society) [Last accessed on 2019 Sept 14]. Available from: https://www.badgut.org/information-centre/a-z-digestive-topics/biosimilars-pamphlet/ .
- 10.Gupta SK, Chaudhari PS, Nath R. Opportunities and challenges in biosimilar development. [Last accessed on 2018 May 17]. Available from: https://bioprocessintl.com/manufacturing/biosimilars/opportunities-challenges-biosimilar-development/ .
- 11.DrugPatentWatch. [Last accessed on 2019 Sept 10]. Top 5 challenges faced by biosimilars. Available from: https://www.drugpatentwatch.com/blog/top-5-challenges-faced-biosimilars/ .
- 12.GaBi. [Last accessed on 2019 Sept 10]. Biosimilars approved in Europe. Available from: http://www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe .
- 13.Mahdis D. Regulatory approvals of biosimilars in Canada. Biotechnol Focus. [Last accessed on 2020 Mar 7]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/biosimilar-biologic-drugs.html .
- 14.The Pharma Innovation. Current scenario of biosimilars. Pharm Innovation J. 2018;7:188–93. [Google Scholar]
- 15.Kumar R, Sigala S, Bertini Malgarini R, Pimpinella G, Pani L, Pecorelli S, et al. Biosimilars: regulatory status and implications across the world. J Pharmacovigilance. 2015;S3 Available from: https://www.omicsonline.org/open-access/biosimilars-regulatory-status-and-implications-across-the-world-2329-6887-S3-002.php?aid=61505. page 1-14. [Google Scholar]
- 16.Krishna A, Mody R, Malhorta H. Global regulatory landscape of biosimilars: established market perspectives. Biosimilars. 2015;5:19–32. [Google Scholar]