

Abstract
Chagas heart disease is an inflammatory cardiomyopathy that develops in approximately one-third of people infected with the protozoan parasite Trypanosoma cruzi. One way T. cruzi is transmitted to people is through contact with infected kissing bugs, which are found in much of the Western Hemisphere, including in vast areas of the United States. The epidemiology of T. cruzi and Chagas heart disease and the varied mechanisms leading to myocyte destruction, mononuclear cell infiltration, fibrosis, and edema in the heart have been extensively studied by hundreds of scientists for more than 100 years. Despite this wealth of knowledge, it is still impossible to predict what will happen in an individual infected with T. cruzi because of the tremendous variability in clonal parasite virulence and human susceptibility to infection and the lack of definitive molecular predictors of outcome from either side of the host–parasite equation. Further, while several distinct mechanisms of pathogenesis have been studied in isolation, it is certain that multiple coincident mechanisms combine to determine the ultimate outcome. For these reasons, Chagas disease is best considered a collection of related but distinct illnesses. This review highlights the pathology and pathogenesis of the most common adverse sequela of T. cruzi infection—Chagas heart disease—and concludes with a discussion of key unanswered questions and a view to the future.
Keywords: Chagas, heart, trypanosome, myocarditis, cardiomyopathy, pathogenesis
INTRODUCTION
Chagas disease, also known as American trypanosomiasis, is caused by infection with the protozoan parasite Trypanosoma cruzi (1). T. cruzi is endemic to vast areas of the Western Hemisphere, from Argentina to the United States. T. cruzi is a zoonosis, naturally transferred among domestic and wild animals and humans by triatomine insects called reduviid bugs, or kissing bugs (2). In addition to insect transmission, which accounts for approximately 70% of infections, congenital transmission is responsible for 26% of cases, and a small number of people (<1% of each of the following categories) are infected by transfusion or transplantation from a T. cruzi–infected donor, laboratory accidents, or consumption of liquids or food contaminated with T. cruzi (Figure 1) (3). T. cruzi–infected individuals are found throughout the world due to the migration of infected individuals from areas of high Chagas disease endemicity (such as South and Central America) to areas with relatively low insect transmission (the United States) to no known insect transmission (the rest of the world) (4). Chagas disease currently affects 6–8 million people and is responsible for approximately 12,000 deaths per year (5). There are 28,000 new cases of T. cruzi infection per year. All of these numbers are much lower now than several decades ago due to increased public knowledge of the disease, blood screening to reduce transfusion-acquired disease, and insecticide spraying programs aimed at reducing transmission.
Figure 1.
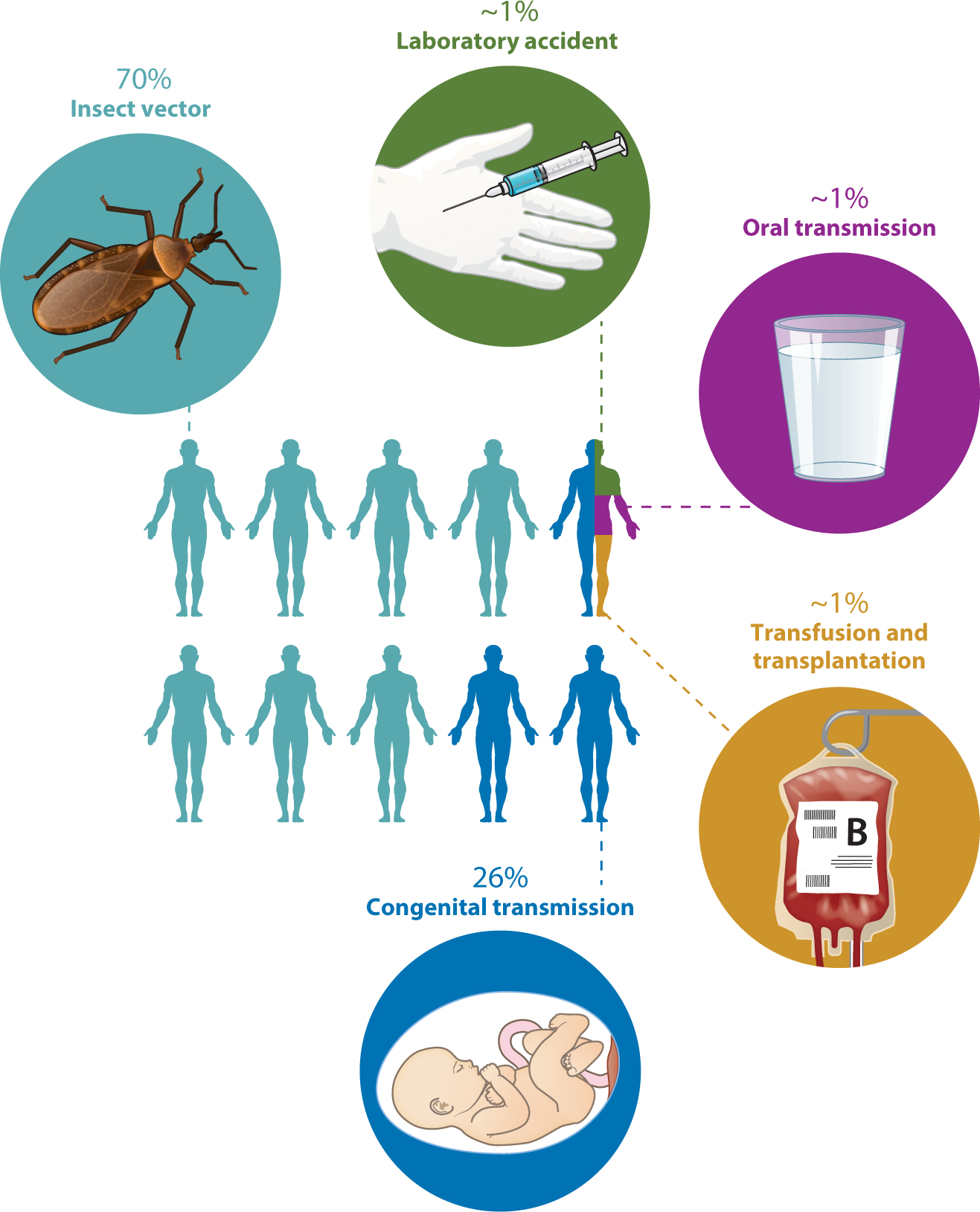
Modes of transmission of Trypanosoma cruzi. Approximately 70% of infections occur when infected reduviid bugs feed on or otherwise come into contact with people. The bug’s feces contain infective metacyclic trypomastigotes, which can enter the body through any break in the skin, for example the bite wound, or through any mucous membrane. Approximately 26% of infections are due to maternal–fetal transmission during pregnancy. A few infections occur as a result of laboratory accidents, the consumption of contaminated food or beverages, or blood transfusion or the transplantation of tissue from an infected donor. Oral transmission can occur when fresh juice is prepared by crushing plants such as sugar cane. If there are infected reduviids in the plants, trypomastigotes end up in the juice when the plants (and bugs) are crushed. Transmission by blood transfusion was a major problem before blood suppliers in many countries, including the United States, began screening for T. cruzi.
Benznidazole and nifurtimox are nitroimidazole compounds used to treat T. cruzi infection. They are reasonably effective in both acute and chronic disease (6), although treating chronically infected patients with cardiomyopathy did not affect progression to heart failure (7). A study by Morillo et al. (7) revealed a number of limitations in our ability to diagnose, accurately assess, and predict clinical outcome in T. cruzi infection and did not address the important question of whether treating the vast majority of infected individuals, who have no heart disease, might reduce the likelihood of them developing cardiomyopathy (8). There is currently no vaccine for T. cruzi infection, although many are hopeful that a vaccine will be developed in the coming years (9).
T. cruzi has a complex life cycle involving two hosts, the reduviid bug and virtually any vertebrate, and three distinct morphological and functional developmental stages: epimastigotes, trypomastigotes, and amastigotes. The epimastigote form replicates in the midgut of the reduviid bug and develops into nonreplicative metacyclic trypomastigotes in the hindgut. T. cruzi is transmitted to humans when the bite wound from a reduviid bug or a mucosal surface, such as the conjunctiva, is contaminated with parasites present in the triatomine excreta (10). After transmission, the bloodstream trypomastigote form of T. cruzi is able to enter a variety of host cells where it differentiates into the replicative amastigote form and multiplies in the cytoplasm. Eventually, parasitized cells rupture, releasing trypomastigotes, which may infect adjacent cells, be disseminated through the blood and infect cells at other locations in the body, or be taken up by another bug during a subsequent blood meal.
The clinical course of Chagas disease is commonly considered to have acute, indeterminate, and chronic phases (10, 11). If symptoms develop during acute infection, they are usually mild and nonspecific and often mistaken for a viral illness unless there is an obvious sign of infection, such as inflammation at the site of the bug bite (chagoma) or periorbital edema developing after conjunctival infection (Romaña’s sign). Approximately 1% of acutely infected people develop lymphadenopathy, hepatosplenomegaly, myocarditis, pericardial effusion, and heart failure or meningoencephalitis. Parasites are usually detectable in the blood for 2 to 4 months and thereafter are detectable by polymerase chain reaction (PCR) or indirectly by the presence of T. cruzi–specific antibodies. The infected person then enters the indeterminate phase, which is essentially a years-to-decades-long phase of low-level infection with no organ dysfunction and no symptoms of infection. The vast majority of people remain in the indeterminate phase for life without any clinical symptoms, but with a positive serology (Figure 2). Historically, it has not been possible to accurately predict which indeterminate-phase individuals will progress to a chronic disease state; however, a type of circulating plasma microRNA, referred to as microRNA-208a, has been studied as a potential biomarker for predicting the risk of Chagas disease progression (12).
Figure 2.
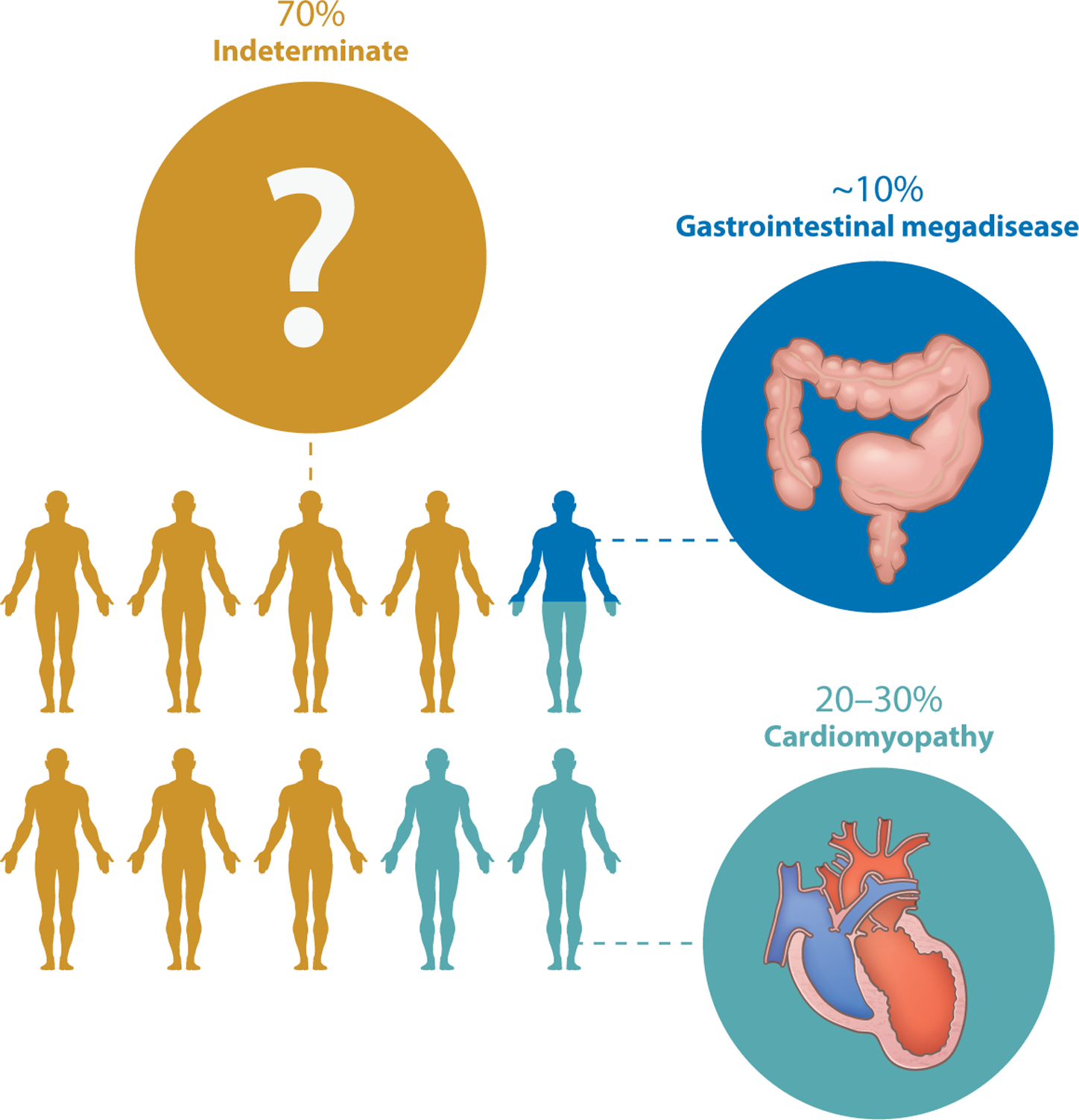
Outcomes of chronic Trypanosoma cruzi infection. Approximately 70% of T. cruzi–infected people live normal lives without developing any adverse sequelae of infection. Approximately 10% develop megaesophagus or megacolon, and 20–30% develop cardiomyopathy. A tiny number of people die during acute infection from fatal cardiac dysrhythmias (not shown), and a few others develop additional sequelae involving other systems.
Approximately 30% of infected individuals eventually progress from the indeterminate to the chronic (clinical) phase of Chagas disease. In these individuals, the indeterminate phase may involve years of undetected accumulation of diffuse myocardial damage prior to clinical detection. Cardiomyopathy is the most important clinical manifestation of chronic Chagas disease because of the frequency with which it develops (in 20–30% of infected individuals), and its severity, morbidity, and mortality. It is a complex disease that includes a wide spectrum of manifestations, ranging from minor myocardium involvement to left ventricular systolic dysfunction, dilated cardiomyopathy, arrhythmias, thromboembolic events, and terminal cardiac failure (13). In addition to cardiac involvement, Chagas disease may also affect the gastrointestinal tract and nervous system. Gastrointestinal symptoms in chronic Chagas patients are most often a result of damage to either excitatory or inhibitory enteric innervation by T. cruzi parasites (14). Although the most common gastrointestinal manifestations are megaesophagus and megacolon, essentially any part of the gastrointestinal tract can be involved, from the salivary glands to the rectum (14). Gastrointestinal Chagas disease (~10%) is most frequently observed in individuals infected south of the equator, in the region extending from central Brazil down through the Southern Cone countries of South America (15). The geographic distribution of this form of the disease is thought to result from subspecific variation among regional T. cruzi populations (15).
THE PATHOLOGY OF CHAGAS HEART DISEASE
The pathology of Chagas disease is complex, heterogeneous, and dependent on many variables, with parasite and host determinants of immunity and inflammation having critical roles (16–18). The heart is the organ most frequently affected in chronic disease (19). Current thinking is that the inflammation in the heart develops over years from indolent, low-grade processes that depend, at least in part, on the few parasites that persist in the heart. Intertwined cellular and molecular mechanisms contribute to degenerative, destructive, inflammatory, and reparative responses, which are detailed throughout the remainder of this review. It is the sum of these processes that culminates in the varied outcomes of infection: from no disease whatsoever to cardiac damage and remodeling that can ultimately lead to heart failure and related clinical sequelae, such as stroke (17).
The Pathology of Acute Chagas Heart Disease
The acute phase of Chagas heart disease occurs within the first few weeks of parasite inoculation, and it is usually of weeks to a few months in duration (11). In the mammalian host, T. cruzi under-goes initial rounds of replication near the site of inoculation, with released trypomastigotes able to infect a wide range of cell types throughout the body. Essentially, any nucleated cell can be infected by T. cruzi (20). In the heart, these include myocytes, endothelial cells, neurons, fibroblasts, and adipocytes (21). The parasite shows particular tropism for striated cardiac myofibers, which may explain why the heart is the organ most commonly involved in chronic disease. The mechanism underlying this potential tropism has not been conclusively determined, but it might be related to the highly developed plasma membrane repair mechanisms that are operative in these cells, which facilitate parasite entry (22). In the rare cases (<1%) in which clinically apparent myocarditis develops during the acute phase, the myocardial inflammation is similar to that of other forms of myocarditis. Depending on the particular combination of T. cruzi strain and host genetic and immunologic variants, fulminant intracardiac replication may occur, with the accumulation of large numbers of intracellular amastigote forms that can be seen easily by light microscopy and that appear as a cyst in the tissue. Since each amastigote cyst is actually the parasitized cytoplasm of a single cell, it is a false cyst or pseudocyst.
Upon microscopic examination, T. cruzi–infected myocardial fibers show evidence of damage, characterized by vacuolization, myocytolysis, and myofibrillar degeneration. These changes are invariably associated with intense mononuclear cell infiltration, which is initially composed of macrophages and neutrophils and then becomes more mixed, with the addition of lymphocytes, plasma cells, eosinophils, and mast cells (23). The infiltrating lymphocytes have broad antigen specificity, including to parasite, host, and nonspecific antigens. Other factors contribute to and exacerbate the inflammatory response, including the local production of cytokines, chemokines, and their receptors; the upregulation of adhesion molecules; complement activation; platelet aggregation and adhesion; and antibody production and opsonization. Interstitial edema, hemorrhage, and vasculitis may be present. Within weeks, as the host adaptive immune response develops, the number of parasitized cells diminishes dramatically and the inflammatory cell response wanes, usually with resolution of myocardial tissue to a near-normal state. Residual abnormalities of acute myocarditis include scarring (interstitial fibrosis) and myofiber hypertrophy, with a minimal, or low-grade, ongoing inflammatory reaction. The gross features of the acute phase of Chagas cardiomyopathy range from minimal change to dilatation of the atria and ventricles that gives a globular architecture in severe cases. As mentioned above, acute infection is typically subclinical, although infection may be suspected if there is a chagoma or Romaña’s sign, when there has been a laboratory accident, or transfusion of blood or transplantation of tissue from a T. cruzi–infected donor to a recipient who develops symptoms of systemic infection.
The Pathology of Chronic Chagas Heart Disease
Most people with chronic infection live their lives with no sequelae of infection (Figure 2). Many were infected as children and are unaware of being infected. However, approximately 10% of these individuals develop megaesophagus or megacolon, both of which are fascinating illnesses that result from defects in the enteric nervous system (24) and are not discussed further in this review. Slowly developing inflammatory changes and scarring of the myocardium ultimately lead to changes in gross cardiac morphology in approximately 20–30% of infected individuals (25). Hearts are typically globally enlarged, with marked alteration in size and shape (Figure 3). Chamber dilatation predominates over hypertrophy, resulting in the globular appearance. The conus arteriosis may be prominent or bulging, and distinct delineation of the right and left ventricles gives a cor bifidum appearance (26). The epicardium may have dense fibroinflammatory plaques that align along the coronary arteries, sometimes in what is described as a rosary pattern. In about half of those who develop chronic disease, the heart shows an unusual thinning of the ventricular wall—usually near the apex of the left ventricle, but sometimes near the right—that bulges in the form of an aneurysm (Figure 3).
Figure 3.
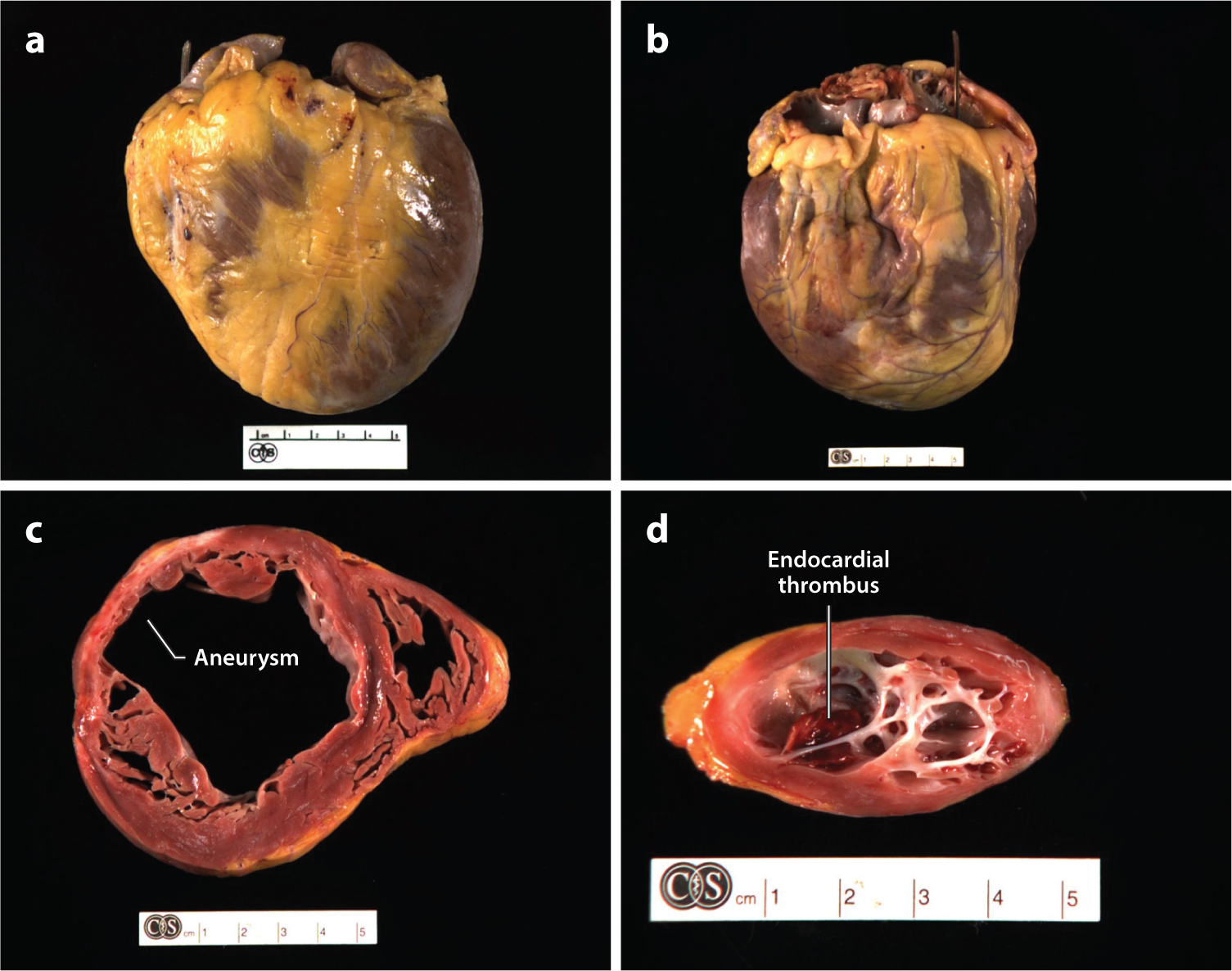
Gross pathology of Chagas heart disease. Examples of explanted hearts from patients with chronic Chagas cardiomyopathy highlight the characteristic features of (a,b) cardiomegaly, multichamber dilatation, and epicardial scarring. Cross-sections through (c) the ventricles and (d ) left apex reveal patchy myocardial, endocardial, and epicardial fibrosis. Aneurysms are frequently present in different stages of development and are visible in the 10 o’clock position in panel c and viewed from above in panel d. In the aneurysm in panel d, the thinning of the ventricular wall can be appreciated, and an apical endocardial thrombus (deep red ) is visible in the aneurysm cavity. All explants are from Cedars-Sinai Medical Center in Los Angeles.
Microscopic evaluation of these areas shows dramatic atrophy, with the loss of myocardial fibers and their replacement by dense, fibrous scar tissue and fat between the endocardium and epicardium (Figure 4). Inflammation is generally lacking in these areas, and there are no parasites. Endocardial thrombi are frequently present in the aneurysm, showing varying degrees of organization (Figure 3d). Mural thrombi can be found elsewhere in the heart as well. Large areas of dense white scar tissue are commonly identified throughout the myocardium. Coronary arteries may be dilated and have minimal evidence of atherosclerotic changes.
Figure 4.
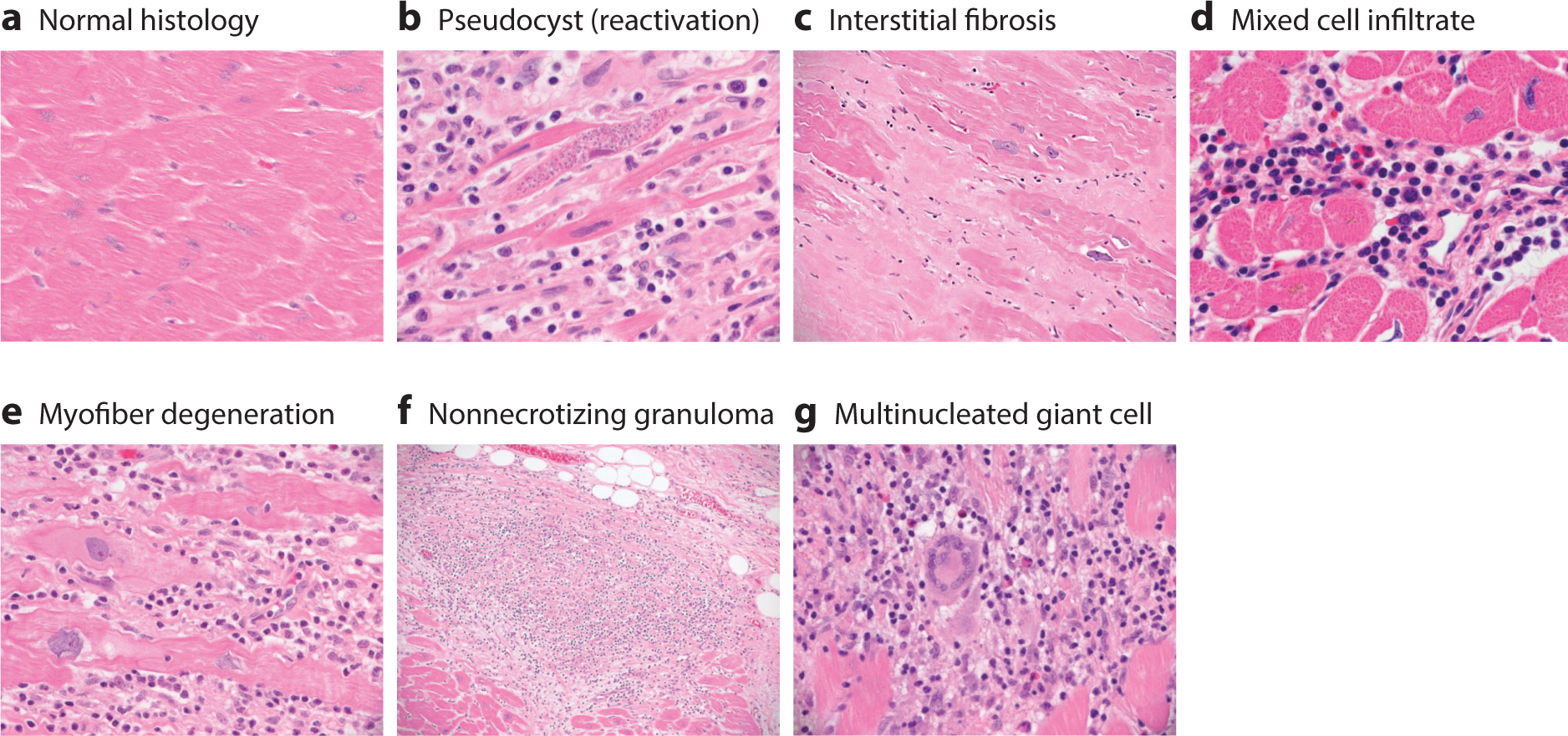
Microscopic pathology of Chagas heart disease. Essentially, every possible histologic picture of Trypanosoma cruzi infection can be observed by examining the hearts of patients with acute, chronic, or chronic reactivation (e.g., upon immunosuppression) disease. (a) Normal cardiac histology. (b) Heart section with mononuclear cell infiltration, myofibrillar degeneration, fibrosis, and an amastigote pseudocyst, commonly seen in reactivation disease. (c) Massive interstitial fibrosis replacing depleted myocytes. (d ) A mixed cell infiltrate comprising histiocytes, lymphocytes, eosinophils, and mast cells. (e) In addition to the chronic inflammation described in the earlier panels, myocardial fibers show evidence of degeneration, with expanded basophilic cytoplasm, corrugated cell membranes, and nuclei with degenerative features, including open and clumped chromatin and irregular nuclear borders. ( f ) Nonnecrotizing granulomas are occasionally observed. ( g) Multinucleated giant cells are commonly found. We emphasize that T. cruzi amastigotes are rarely observed, except in the case of reactivation (b).
The classic pathologic features of the chronic phase of Chagas cardiomyopathy include low-grade myocarditis accompanied by myocytolysis, myofiber hypertrophy, and interstitial fibrosis (26). In contrast to the acute phase, only focal areas of inflammation are generally found in the hearts of chronic Chagas patients. Inflammatory cell infiltrates are composed primarily of T cells and macrophages, with a few eosinophils, plasma cells, and mast cells (Figure 4). Unlike during the acute phase, parasites are rarely observed by conventional microscopic analysis of cardiac biopsy samples or random sections of explants or autopsy specimens (27). Myofiber degeneration is focal and associated with areas of inflammation. The scarring and fibrosis related to the chronic phase are prominent features that can be focal or diffuse, and they involve almost any area of the myocardium, although some older resources describe a ventricular predominance over an atrial predominance, a predominance of left over right, and a predominance of subepicardial myocardium over mural myocardium. Fibrosis exhibits a somewhat unique pattern of distribution, often surrounding and enveloping individual myocardial fibers, a feature that loosely separates it from the patterns of fibrosis noted in idiopathic dilated cardiomyopathy (28, 29). Rarely, nonnecrotizing granulomas may be present with multinucleated giant cells. Vasculitis is not a characteristic feature of chronic Chagas heart disease (30). With time, myofiber hypertrophy may become prominent, characterized by significant variation in myofiber and nuclear size and shape and increased perinuclear deposits of lipofuscin. Small blood vessels may show abnormalities, which include luminal narrowing, a feature that potentially supports a hypothesis of local injury related to ischemia (31, 32).
These chronic inflammatory changes with evidence of remodeling are found throughout the myocardium, including in regions harboring conducting fibers and nodes (33). Andrade et al. (34) also described a range of microscopic inflammatory changes, including scleroatrophy, fibrosis, and vascular ectasia in the atrioventricular conducting system, proposing a direct causal relationship to the clinical arrhythmic abnormalities in the study’s cohort. The destruction of parasympathetic nerves in the epicardial–mediastinal region has been documented, potentially suggesting a causative mechanism for arrhythmias and sudden death (35, 36). It has been suggested that direct damage by parasitism, inflammation, and antineuronal autoimmune reactions may all contribute to parasympathetic nerve damage, although this clinical feature is not specific to Chagas patients (32). Further, although this mechanism is consistent with autopsy reports showing significant denervation in some Chagas patients who died suddenly, this theory has not been rigorously tested, and recently the role of parasympathetic impairment in Chagas pathogenesis has been questioned more generally (32).
The chronic phase of the disease likely represents, at least in part, a slowly progressive, cyclical reaction to the presence of intramyocardial parasites, which results in a smoldering, ongoing inflammatory reaction, myocardial cell necrosis and dropout, and fibrosis that develop over years to decades. This theory has been advanced by studies in which low numbers of T. cruzi organisms, fragments, or antigens can be demonstrated in many chronic cases by immunohistochemical and PCR assays (37), which have greater sensitivity than conventional microscopy. This is supported by observations in human and animal studies that indicate trypanocidal therapies frequently do not completely clear T. cruzi, potentially leaving a stimulus for ongoing inflammation (38). However, foci of inflammation often have no geographic or temporal relationship to the presence of T. cruzi, suggesting that other mechanisms might contribute to ongoing inflammatory cascades and subsequent clinical decompensation. Finally, recent results using highly luminescent parasites and detailed molecular analyses show that myocarditis can develop in the complete absence of cardiac parasitism (39).
MECHANISMS OF THE PATHOGENESIS OF CHAGAS HEART DISEASE
Many of the mechanisms of the pathogenesis of Chagas heart disease have been described and defined in detail in humans and experimental animals (40). Most proposed mechanisms require the presence of T. cruzi somewhere in the host, whether in the heart or elsewhere, to produce and/or propagate cardiac pathology. Indeed, even cardiac autoimmunity, which develops in some people and animals infected with T. cruzi, resolves upon removal of the parasite (41). It is essentially impossible to study any individual mechanism of pathogenesis in an infected person or animal in isolation because other mechanisms may also be operative and can either accentuate or moderate the mechanism under study. For example, T regulatory (Treg) responses that develop to downregulate the autoimmunity that can develop in some individuals may also suppress parasite-specific immunity and permit T. cruzi to persist and replicate in the tissue. However, because most investigations have been designed to explore only one mechanism, we present them separately below, with the understanding that the final outcome of T. cruzi infection is an integration of many distinct processes rather than resulting from only one.
The absence or paucity of parasites in the hearts of those who succumb to Chagas heart disease has been a conspicuous feature of the disease since it was first described. The natural course of disease development in humans is largely unknown since (i) most individuals are unaware of being infected; (ii) those who develop Chagas disease typically do so many years after infection; and (iii) those who are acutely infected are often treated. The recent development of luminescent parasite systems now permits longitudinal and noninvasive assessment of host parasitism in experimental animals, and it is hoped that these models will be valuable both for assessing the mechanisms of pathogenesis and for developing new and more effective therapies (39, 42, 43).
Host Responses to T. cruzi Infection
Although the term host response encompasses complexity and early innate immune responses, nonimmune cellular responses to parasite products, cell invasion and intracellular replication, and adaptive immunity, all of which integrate to give an outcome of infection, the early response deserves separate consideration. Dendritic cells, macrophages, natural killer cells, B cells, CD4+ T cells, and CD8+ T cells, as well as protective and inflammatory factors—including reactive oxygen species (ROS), bioactive lipids, cytokines, and antibodies—contribute both to controlling parasite levels and to promoting tissue inflammation. Additionally, complement deposition on bloodstream trypomastigotes may be important, yet its role is not straightforward (44). While it has been shown that complement deposition enhances host cell invasion by T. cruzi (45), T. cruzi trypomastigotes are known to be resistant to complement-mediated lysis (46). This is an excellent example of the many ways that T. cruzi has evolved to exploit host molecules and processes that are harmful to other pathogens to advance its missions.
Disease outcome is determined by a highly complex interplay between the virulence of the T. cruzi strain and the genetic susceptibility of the individual, with both parasite virulence and host susceptibility varying with the parasite–host combination. That is, one parasite strain may be more virulent for one host than for another, and one host may be more susceptible to one parasite strain than to another. Further, this diversity in the species T. cruzi also extends to diagnosis and drug susceptibility (47). Innate and adaptive immune responses may contribute to heart damage and increase the risk of heart failure through inducing sustained inflammation, fibrosis, and oxidative stress injury, leading to the disruption of myofibrils, myocyte necrosis, microvascular dysfunction, autonomic dysfunction, and cardiac hypertrophy and fibrosis (Figure 5). Therefore, the outcome of infection results from a culmination of many processes. In the end, the ultimate goal of T. cruzi is the same as for any other pathogen: to replicate and persist in its human host until it can be transmitted to another host. For T. cruzi, this means circulating in the blood at levels sufficient to be picked up by a reduviid bug during a blood meal and transmitted to another animal or human. An autopsy-based study employing histology, immunohistochemistry, and PCR determined that there are vanishingly small numbers of T. cruzi in chronic infection, and the heart is the only solid organ with detectable parasites (and, then, these are detected only by PCR and only in 57% of cases) (48). Of course, this paucity of parasites in the heart has been a notable feature of Chagas heart disease for more than 100 years, and it is important to bear this in mind as we explore the pathogenesis of this important illness that afflicts millions throughout the world and especially in the Western Hemisphere (49).
Figure 5.
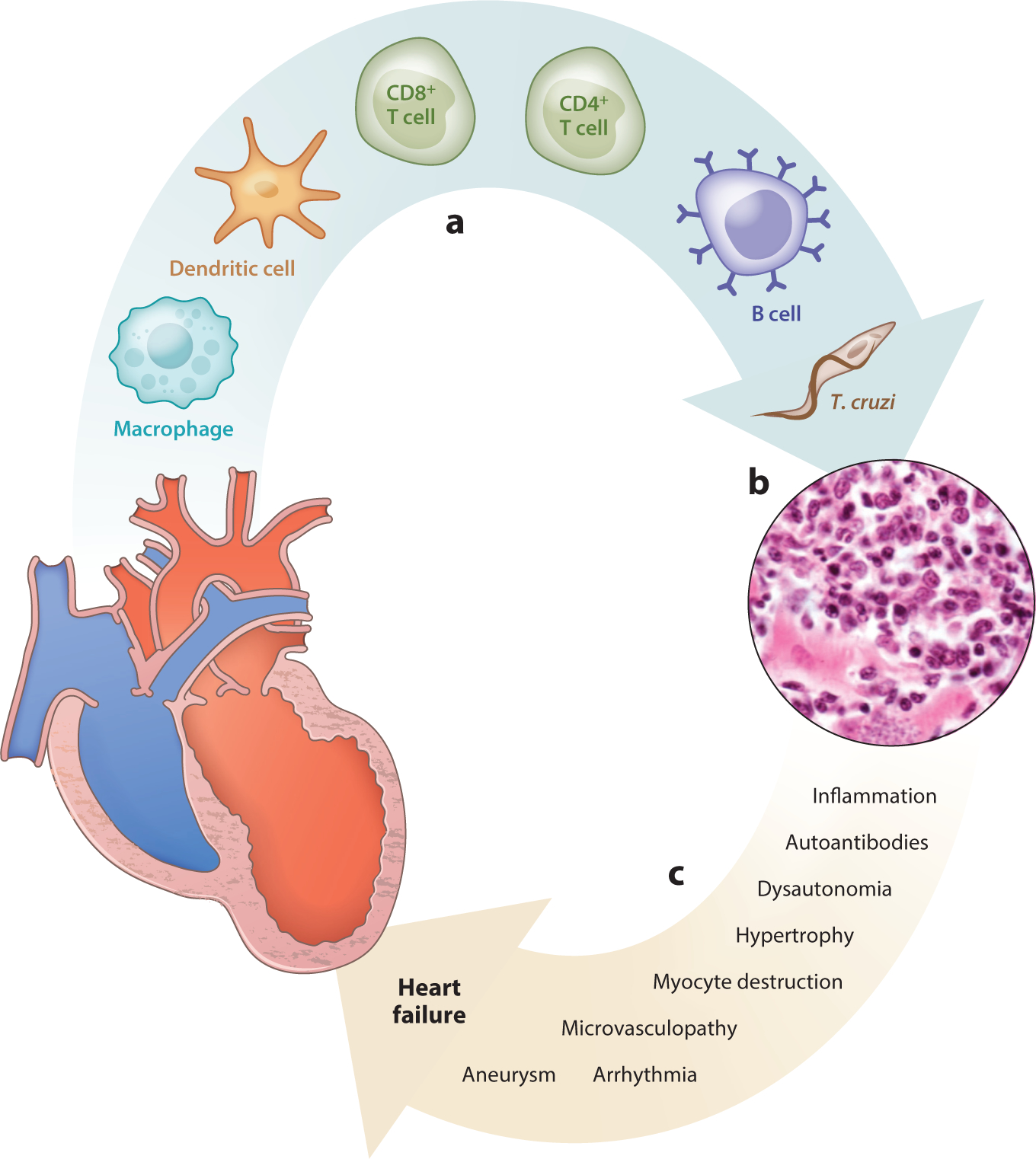
Pathogenesis of Chagas heart disease. (a) A variety of cells, including Trypanosoma cruzi parasites, infiltrate the myocardium, which develops (b) histopathology characterized by mononuclear cell infiltration, edema, myocyte destruction, and fibrosis. Parasites are typically not observed in chronic infection. (c) Mechanisms are listed that may lead to progressive cardiac dysfunction that progresses to heart failure during T. cruzi infection.
Innate Immune and Nonimmune Mechanisms of Chagas Pathogenesis
There have been many fine reviews of the innate immune mechanisms in T. cruzi infection (50), including those relating to Toll-like receptors (51) and inflammasomes and Nod-like receptors (52–54). In this review, we focus on the areas of innate immunity and nonimmune mechanisms that have not been emphasized in previous reviews.
Macrophages and oxidative stress.
In addition to producing cytokines and chemokines, activated macrophages exert cytotoxic effects against microbes by producing ROS and reactive nitrogen species. NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (NOX2) is a multimeric complex that utilizes NADPH as substrate and reduces O2 to produce superoxide (O2•−) that is then further dismutated into the stable and diffusible pro-oxidant H2O2. In infected mice, blocking NOX2 ROS arrests the activation and proliferation of splenic phagocytes and the production of inflammatory cytokines [e.g., interleukin (IL)-1, IL-6, interferon (IFN)-γ, tumor necrosis factor (TNF)-α] (55). This was confirmed by the finding that p47phox−/− mice exhibit increased susceptibility to T. cruzi and succumb to infection (56).
Likewise, inducible nitric oxide synthase utilizes l-arginine and O2 for the synthesis of l-citrulline and nitric oxide (NO) in a complex oxidoreductase reaction (57). The reaction of NO with O2•− produces peroxynitrite, which is a strong cytotoxic oxidant that kills T. cruzi in macrophages (58, 59). However, the extent of the ROS and NO responses in human and mouse macrophages infected with T. cruzi is significantly lower than that observed in lipopolysaccharide–IFN-γ-induced proinflammatory macrophages (60). Indeed, trypanosomes have evolved an elaborate antioxidant system to scavenge ROS NO from macrophages (61, 62). Parasite isolates overexpressing cytoplasmic and mitochondrial tryparedoxin peroxidases are able to infect and multiply more efficiently in macrophages (59, 63) and, thus, utilize macrophages to disseminate to different tissues.
In nonimmune cells such as cardiac myocytes, T. cruzi infection induces ROS production through mitochondria (64). Mitochondrial (mt) ROS signals nuclear factor κB (NF-κB)-dependent cytokine gene expression (64, 65) and causes mtDNA damage (e.g., 8-hydroxyguanine lesions) that signals activation of the DNA repair enzyme polyadenosine ribose polymerase 1 (PARP1). PARP1-dependent posttranslational modification of RelA (p65)-interacting nuclear proteins facilitates the assembly of the NF-κB transcription complex and boosts cytokine gene expression (64, 65). Further, Sirtuin 1 (SIRT1), a highly conserved member of the family of NAD+-dependent Sir2 histone deacetylases, competes with PARP1 for its NAD+ substrate and integrates mitochondrial metabolism and inflammation. Treating mice with the SIRT1 agonist SRT1720 has no effect on T. cruzi burden, but it suppresses NF-κB-dependent transcription and reduces oxidative and inflammatory stress in infected cells and mice (66). These studies point to the possibility that ROS–PARP1/PAR–RelA contributes to inflammatory pathology in T. cruzi infection and that the pathology might be reduced through SIRT1 activation.
Macrophages and chronic inflammation.
The New York Heart Association (NYHA) functional classification of heart failure places patients in one of four categories according to the severity of their symptoms. Chagas patients with NYHA class III–IV display a proinflammatory transcrip-tomic and proteomic profile in peripheral blood mononuclear cells (67–70) and increased levels of TNF-α+ monocytes (71–73) in the circulation. In contrast, IL-10+ monocytes with an anti-inflammatory transcriptome are present in the peripheral blood of infected individuals classified as NYHA class I–II who have no-to-minimal left ventricular dysfunction. Several studies conducted during the 2010s suggested that chronic injurious processes generate cellular debris that signals a proinflammatory macrophage response in class III–IV patients (74). Cardiac proteins oxidized during T. cruzi infection are immunogenic, and immunity to these oxidized proteins can cause cardiac inflammation and decreased cardiac contractility. Preventing protein oxidation by treatment with the spin-trapping antioxidant α-phenyl-N-tert-butyl nitrone (PBN) prevents immunity to these oxidized proteins and maintains normal cardiac histology and cardiac contractility (75).
Mitochondrial dysfunction and oxidative stress.
T. cruzi invasion elicits massive calcium mobilization in the host cell (76), mitochondrial membrane potential transition (64, 65), and O2•− production in cardiac myocytes (77, 78). In vivo studies show that mitochondrial defects of the respiratory chain contribute to consistently high levels of mtROS and oxidative adducts in experimental animals and humans, and elevated mtROS is associated with a decline in host antioxidant capacity due to inhibition of the nuclear factor (erythroid 2)-like 2 (NFE2L2, also known as NRF2) antioxidant response element pathway and other mechanisms (77, 79–83). Enhancing antioxidant capacity by treatment with antioxidant PBN or vitamin A or overexpression of the Mn isoform of superoxide dismutase (known as MnSOD) can prevent oxidative stress and preserve mitochondrial and left ventricular function (82, 84–86). Likewise, treatment with sildenafil, an inhibitor of phosphodiesterase 5, provides cardioprotection through preservation of cGMP-dependent protein kinase activity and antioxidant–oxidant balance in chronically infected mice (87). These studies conclusively establish the pathogenic importance of mtROS and chronic oxidative stress in Chagas disease. The benefits of ROS scavengers in suppressing hypertrophic gene expression were also noted in experimental studies (77, 80), while treatment of T. cruzi–infected rodents with the trypanocidal drug benznidazole suppresses the classical mediators of inflammatory ROS (e.g., NOX2 and myeloperoxidase) but not the hypertrophic response. These observations imply that ROS of mitochondrial origin contributes to cardiac remodeling in Chagas disease.
It was not understood until recently why the antioxidant response is not triggered in the presence of continued oxidative stress in the T. cruzi–infected myocardium. NFE2L2 is a transcription factor that regulates the expression of antioxidant proteins. NFE2L2 activation is decreased in cardiac myocytes and the myocardium of mice infected with T. cruzi. Importantly, inhibiting T. cruzi–induced mtROS by overexpression of MnSOD in cardiac myocytes preserves NFE2L2 transcriptional activity and antioxidant–oxidant balance, and MnSODtg mice overexpressing MnSOD also preserve normal cardiac structure and function (85). These findings provide evidence that mtROS inhibition of the NFE2L2/antioxidant responsive element (or ARE) pathway constitutes a key mechanism in stimulating the expression of genes promoting fibrosis and the evolution of chronic Chagas cardiomyopathy. Several drugs that stimulate the NFE2L2 pathway are being evaluated for treating diseases that are caused by oxidative stress, and these may also be helpful as adjuvant therapy in Chagas disease. In summary, T. cruzi–induced ROS production by mitochondria and inflammatory cells is crucial for clearing parasites, yet it may lead to prolonged oxidative stress, resulting in pathologic changes within the myocardium in Chagas patients.
Lipid mediators.
Lipid derivatives of arachidonic acid, including leukotrienes (LTB4) and cys-teinyl lymphotoxins (LTC4, LTD4, and LTE4), also contribute to reducing cardiac pathology in Chagas disease by modulating cytokine production, macrophage function, and NO-dependent killing of T. cruzi (86). Many inflammatory mediators are stored in specialized organelles known as lipid bodies, which increase in number inside macrophages in response to T. cruzi infection (88). LTB4 synthesis is mainly directed by dendritic cells, granulocytes, macrophages, and mast cells through the production of the enzyme 5-lipoxygenase (5-LO). 5-LO deficiency in mice permits an increase in peak parasitemia during T. cruzi infection. However, these mice also survive longer upon infection and have lower cardiac parasitosis, inflammation, and fibrosis (89, 90). This is yet another example of the complexity of innate immunity in balancing parasite control and tissue pathology. 5-LO also induces the synthesis of lipoxin A4 (LXA4), which induces the anti-inflammatory suppressor of cytokine signaling 2 (SOCS2). In T. cruzi–infected mice, SOCS2 deficiency decreases cardiac inflammation, but not parasitosis, and also decreases myocyte dysfunction and cardiac hypertrophy (91). This is accompanied by a reduction in levels of IL-12, IFN-γ, TNF-α, and other proinflammatory cytokines. SOCS2 deficiency also leads to an increase in LXA4 expression and the number of Tregs and increased macrophage sensitivity to IFN-γ, which could explain the ability to clear parasites despite a reduction in proinflammatory mediators (91). LXA4 may function through both SOCS2-dependent and SOCS2-independent mechanisms, and its effects may be augmented by other lipid mediators, such as thromboxane A4 (TXA4). By suppressing the proliferation of intracellular T. cruzi and thereby enhancing survival in acute infection, TXA4 may promote a state of low-level chronic infection. Additional studies also suggest that the induction of prothrombotic lipids, such as TXA, increases parasitemia and cardiovascular damage (92). Clearly, bioactive lipids are important modulators of T. cruzi infection and of the cardiac pathology resulting from chronic infection, and thus pharmacologic modulation of these lipids may have therapeutic benefit.
Additional factors.
All of the innate immune mechanisms mentioned above vary depending on the strain of T. cruzi involved and the genetic background of the host, both in humans (93) and experimental animals (94). In addition to innate and adaptive immunity, other mechanisms—including microvasculopathy, autonomic neuropathy, and kallikrein–kinin dysregulation—may also influence outcomes (40, 95–97).
Adipocytes appear to serve as an important reservoir for T. cruzi as well as to contribute to the development of a chronic inflammatory state that promotes cardiac damage in chronically infected individuals (21). The detection of T. cruzi DNA in the adipose tissue of elderly Chagas patients combined with experimental observation of T. cruzi infection in human and mouse adipocytes is consistent with a role for parasite persistence in Chagas pathogenesis, and it is suggestive of a role for adipose tissue in maintaining and reactivating chronic infection. Due to the prominent role of adipose tissue in energy metabolism, insulin sensitivity, and inflammation, it has been suggested that T. cruzi infection may be linked to other adipocyte-associated diseases, such as diabetes and obesity (98). Chagas cardiomyopathy may be further affected by the activation of the kallikrein–kinin system, leading to a proinflammatory response that is characterized by mast cell expansion, vasodilation, increased vascular permeability, and cardiac edema and is accompanied by increased myocardial parasitism (99). Evidence that cardiac dysautonomia is a significant component of Chagas cardiomyopathy includes reports of impaired parasympathetic heart rate regulation, tachycardia, and other functional abnormalities linked to cardiac neuropathy, as well as a notable reduction of parasympathetic innervation in the myocardium observed upon autopsy of individuals who had chronic Chagas disease (35, 36). Reversible perfusion defects and reductions in the poststress left ventricle ejection fraction have also been observed in infected individuals in both the indeterminate and chronic phases of Chagas heart disease. However, the subtleness and variability of denervation and the lack of a clear relationship between dysautonomia and death in Chagas patients raises questions about the role of neuropathy in Chagas pathogenesis. Due to the heterogeneity in disease progression, the precise contribution of each of these factors is likely to vary considerably among patients.
Adaptive Immunity to T. cruzi Infection
Innate immunity to T. cruzi infection is important to prevent unchecked intracellular parasite proliferation through all of the mechanisms described above. These innate immunity responses bide time until antigen-specific adaptive immunity develops, which is ultimately responsible for clearing infected cells.
Cell-mediated immunity.
By the early 1990s, much work had already been done to delineate the importance of lymphocyte subsets in immunity to T. cruzi (100–102). It is now known that essentially every lymphocyte subset is important in immunity to T. cruzi. Infection elicits a pre-dominately T helper 1 (Th1)–type immune response, both in the myocardium and peripherally. This response is characterized by the expression of the Th1 cell transcription factor Tbet and the production of Th1 cytokines (i.e., IFN-γ, IL-2, IL-12, and TNF-α), which are accompanied by suppressed levels of transcription factors and cytokines associated with Th2 (i.e., GATA-3, IL-4, and IL-10) and Th17 responses (i.e., IL-17) (103–107). Among chronically infected individuals, those with heart disease produce higher levels of IFN-γ, TNF-α, and IL-6 and lower levels of IL-4 and IL-10 than those in the indeterminate phase (108). This promotes Th1 differentiation and proliferation in CD4+ T cells, leading to activation of antiparasite responses by CD8+ T cells and macrophages (109). Effector CD8+ T cells are crucial for recognizing T. cruzi–infected cells and controlling infection (110). Reduced CD8+ T cell responses are associated with more severe clinical disease, and treatments that improve CD8+ T cell responses improve clinical outcome, presumably through cytotoxicity that clears infected cells (111, 112).
Within the CD4+ T cell compartment, both Th1 and Th17 immunity are protective against T. cruzi infection, yet they may also contribute to deleterious inflammation and increase the severity of cardiac lesions (104, 113–115). The complexity of Th1 and Th17 responses to T. cruzi infection is highlighted by the observation that selective downregulation of Th1 cells and Th17 cells by parasite trans-sialidases or the expression of other immunomodulatory factors promotes both host survival and low-level parasite persistence by reducing both protective and harmful effects (113, 116). Levels of IFN-γ, IL-2, and IL-10 may predict the risks of arrhythmia and sudden death in Chagas patients, with elevated IL-2 most strongly correlated with a high risk of death (117). However, the frequency of activated T cells is inversely related to disease severity, and chronic infection may lead to clonal exhaustion and reduced numbers of memory T cells (118–121). This effect is greater for CD8+ T cells than for CD4+ T cells.
The presence of active myocarditis during the chronic phase of disease increases the chance of progression to heart failure (122). Chagas myocarditis typically is characterized by diffuse foci of myofibril disruption, fibrosis, few if any intracellular parasites, and mononuclear cell infiltration (123, 124). The inflammatory infiltrate is composed primarily of lymphocytes and macrophages, with smaller numbers of natural killer cells, dendritic cells, and granulocytes. Among infiltrating lymphocytes, parasite-specific CD8+ T cells are thought to predominate among other populations of CD4+ and CD8+ T cells.
In addition to directly damaging cardiac myocytes and disrupting myofibrils, infiltrating lymphocytes and other inflammatory mediators produced in response to the primary infection may trigger autoimmune responses. Polyantigenic autoimmunity involving T cells has been widely reported both in humans and experimental animals infected with T. cruzi, yet the significance of autoimmunity in the pathogenesis of human Chagas heart disease is not known, despite there being hundreds of published articles characterizing the autoimmunity in detail. Pathogenic autoimmunity involving CD45+, CD8γδ+, CD8α, and other autoreactive T lymphocytes has been demonstrated both in chicken and mouse models of experimental Chagas disease (125, 126), but a clear link between cell-mediated autoimmunity and myocardial inflammation in humans has not been demonstrated. The prevalence, significance, and mechanisms of the development of autoimmunity in human Chagas heart disease are major unresolved questions in the field (127).
It is advantageous to both the host and parasite to moderate T cell immunity during T. cruzi infection: a response that promotes parasite persistence to enable the parasite to transfer to new hosts but has no-to-moderate morbidity and no premature mortality serves the needs of both partners in the symbiotic relationship. CD4+CD25+Foxp3+ Treg cells modulate proinflammatory immune responses by downregulating prolonged activation of CD4+ T cells and CD8+ T cells. Peripheral Treg cell populations are reduced during acute T. cruzi infection, and they expand during chronic infection (128, 129). This may promote the expansion of effector and Th cells during acute infection when parasitemia is highest, and it may later control tissue damage resulting from exposure to the persistent inflammatory state that develops during chronic disease. This may also facilitate the persistence of low-level infection in patients with indeterminate or chronic Chagas disease. In experimental Chagas disease, the depletion of Tregs during acute infection increases the levels of inflammatory mediators, myocarditis, and tissue parasitosis, leading to earlier mortality (130, 131). This effect may depend on several factors, including the method of Treg depletion. Other studies have attributed a range of effects to the loss of Treg function during T. cruzi infection, from no significant impact to a notable increase in the expression of inflammatory mediators that is accompanied by reduced parasitemia and cardiac pathology (130, 132, 133). These pathologic changes are associated with an elevated Th1 response early in the course of infection, followed by a downregulation of Th1 cells and an increase in Th17 cells during later phases of infection (132). Genetic polymorphisms associated with lower expression of transforming growth factor (TGF)-β—a regulatory cytokine produced by Treg cells that promotes the differentiation of naive T cells into Th17 cells—are also associated with a lower susceptibility to developing cardiomyopathy (134). This suggests that Tregs help control cardiac inflammation by modulating the proinflammatory Th1 and Th17 cell responses required for parasite clearance, yet they also contribute to immune-mediated cardiac damage. Various methods for promoting Treg cell functionality have been suggested as potential therapeutic approaches. These include the adoptive transfer of Treg cells, intravenous immunoglobulin therapy, administration of low-dose IL-2–IL-2 antibody complex, administration of sphingosine 1–phosphate receptor 1 agonists, and vitamin D supplementation (135). The efficacies of these interventions are yet to be tested.
T. cruzi may indirectly influence T cell responses through interactions with dendritic cells. Exposure to low-virulence strains of T. cruzi triggers upregulation of major histocompatibility complex (MHC) class I and II on dendritic cells, leading to increased production of IL-6, IL-12, and TNF-α, which promote the activation and proliferation of Th cells and effector T cells (136). This protective immunity results in increased clearance of T. cruzi. In contrast, high levels of virulence are achieved by some parasite strains that are capable of manipulating dendritic cells to downregulate the expression of MHC class I and II molecules, while increasing the expression of IL-10 and TGF-β (136). This promotes the differentiation of naive T cells into Treg cells, leading to a suppressed immune response that promotes parasite replication. Although TGF-β promotes the differentiation of T cells into Tregs, somewhat paradoxically, it has the potential to induce inflammation and fibrosis through other mechanisms. Due to its function in activating growth factors and directing tissue repair and remodeling, high levels of TGF-β have been independently linked to the induction of inflammation, fibrosis, and cardiac hypertrophy, and it is associated with a higher incidence of heart failure and death in patients with chronic Chagas disease (137, 138). However, lower serum TGF-β together with elevated TNF-α are associated with decreased left ventricular function (139). Thus, TGF-β can have different effects depending on other factors.
Impaired peripheral T cell responses observed during acute T. cruzi infection may also be caused by parasite-induced inhibition of IL-2 receptor expression or by temporary clonal exhaustion of parasite-specific T cell subsets following an earlier vigorous response (140). In addition to the downregulation of IL-2 receptor expression, incubation of human peripheral blood mononuclear cells with T. cruzi trypomastigotes inhibits the expression of CD3, CD4, and CD8 by Th cells and cytotoxic lymphocytes, leading to reduced cell proliferation (141). A notable reduction in the frequency of T cells expressing the Vβ5 T cell receptor has been observed in patients with acute but not chronic Chagas disease (142). Robust expansion and activation of peripheral CD4+ and CD8+ T cells specific both for parasite and host antigens occur in patients with chronic Chagas disease.
Humoral immunity.
Polyclonal activation of B cells and hypergammaglobulinemia are characteristic of acute Chagas disease. The majority of antibodies produced by splenic B cells in the early phases of infection are not specific for parasite antigens, but parasite-specific antibodies may be detected within 3 weeks of infection (143). The classification of early and late acute infection and the indeterminate phase can be made based on predictable changes in antibody specificities and the ratio of lytic to nonlytic antibodies (144). Antibody quantification may also be a useful indicator of treatment efficacy in children; however, serologic tests may not be robust indicators of parasitologic cure in adults due to the prolonged persistence of circulating T. cruzi–specific antibodies (145). Promisingly, during the past year, a specific antibody was identified that may serve as an effective biomarker for monitoring parasite persistence in chronically infected people (146). A protective role for lytic antibodies is suggested by the association of increased levels of lytic immunoglobulin G with the indeterminate phase in patients, as well as with adults who have undetectable parasitemia after 10 years (147, 148). Although T. cruzi trypomastigotes are resistant to complement-mediated lysis, anti-gal antibodies may lyse trypomastigotes directly (149), and antibody opsonization may promote uptake and clearance by reticuloendothelial macrophages (150).
As is true of several of the immune and nonimmune mechanisms discussed above, humoral immunity may have deleterious as well as beneficial effects during T. cruzi infection. At least one parasite antigen acts as a B cell mitogen, triggering polyclonal lymphocyte activation (151). This activation may reduce the efficiency of parasite clearance by impeding the development of a robust, parasite-specific response, leading to increased cardiac pathology (152, 153). T. cruzi infection also triggers the production of an array of potentially pathogenic autoantibodies. Antibodies from chronically infected people may bind to cardiac myocytes and exert damage via antibody-dependent cytotoxicity, leading to the release of creatine phosphokinase (154). This may also explain why elevated levels of serum cardiac troponin I are observed both in T. cruzi–infected mice and uninfected mice immunized with T. cruzi antigens (155). Antibodies to a number of self-antigens are produced during T. cruzi infection; targets include cardiac antigens such as actin, myosin binding protein C, myosin, and tropomyosin, as well as muscarinic and adrenergic receptors, galectin 1, and laminin (155–158). High levels of antibodies that cross-react with cardiac myosin (anti-B13) and β-adrenergic receptors (anti-β1AR and anti-p2β) are linked with disease severity in patients with chronic Chagas disease, being most abundant in those experiencing dilated cardiomyopathy and systolic heart failure (159). Autoantibodies may also contribute to pathologic changes in the heart by disrupting cardiac conduction. However, some studies suggest that autoantibody titers do not correlate with disease severity and that cardiac damage resulting from T. cruzi–induced autoimmunity may not be antibody mediated at all (156, 160, 161).
Although the presence of humoral autoimmunity in T. cruzi–infected individuals is both widespread and widely acknowledged, questions remain regarding the genesis of this autoimmunity and its importance in pathogenesis. As in some other infection-induced, organ-specific autoimmune diseases, autoimmunity may develop as a result of pathogen–host molecular mimicry, bystander activation after tissue damage, or a combination of other immunologic factors that lead to a breach of self-tolerance (40, 162, 163). The observation of robust autoimmunity in T. cruzi–infected individuals in the indeterminate phase without signs or symptoms of disease supports the argument that autoimmunity is irrelevant to pathogenesis (164). Alternatively, the persistence of T. cruzi in anatomic sites such as adipose tissue (21) or the large intestine (39, 42) may be sufficient to promote heart disease pathogenesis in the absence of cardiac parasitosis (20, 165, 166). Indeed, the development of cardiac inflammation and fibrosis whether or not parasites are found in the heart illustrates the importance of a parasite reservoir somewhere in the body (43). Removing this reservoir from experimental animals leads to complete resolution of myocarditis (41), further supporting the importance of parasite persistence—even outside the heart—in the pathogenesis of Chagas heart disease.
CONCLUDING REMARKS
In this article we have attempted to review the gross and microscopic pathology and major mechanisms of the pathogenesis of Chagas heart disease, with an emphasis on innate immune, adaptive immune, and nonimmune processes. We refer the reader to reviews of other relevant topics, for example autoimmunity (124, 127, 167, 168) and the heterogeneity and geographic distribution of the species T. cruzi (169). In the current literature, it is clear that the variable and largely unpredictable outcomes of T. cruzi infection result from a complex interplay of host–parasite interactions. These include protective host responses—such as immune cell infiltration of infected tissues and the release of inflammatory cytokines, ROS, and lipid mediators—and an array of parasite-specific immune responses, as well as the pathogenic effects of infection, such as myocyte destruction, inflammation, autoimmunity, and pathogenic oxidative stress and adipocyte modulation (Figure 6). To treat Chagas disease most effectively, one must determine how to tip this delicate balance in favor of the host.
Figure 6.
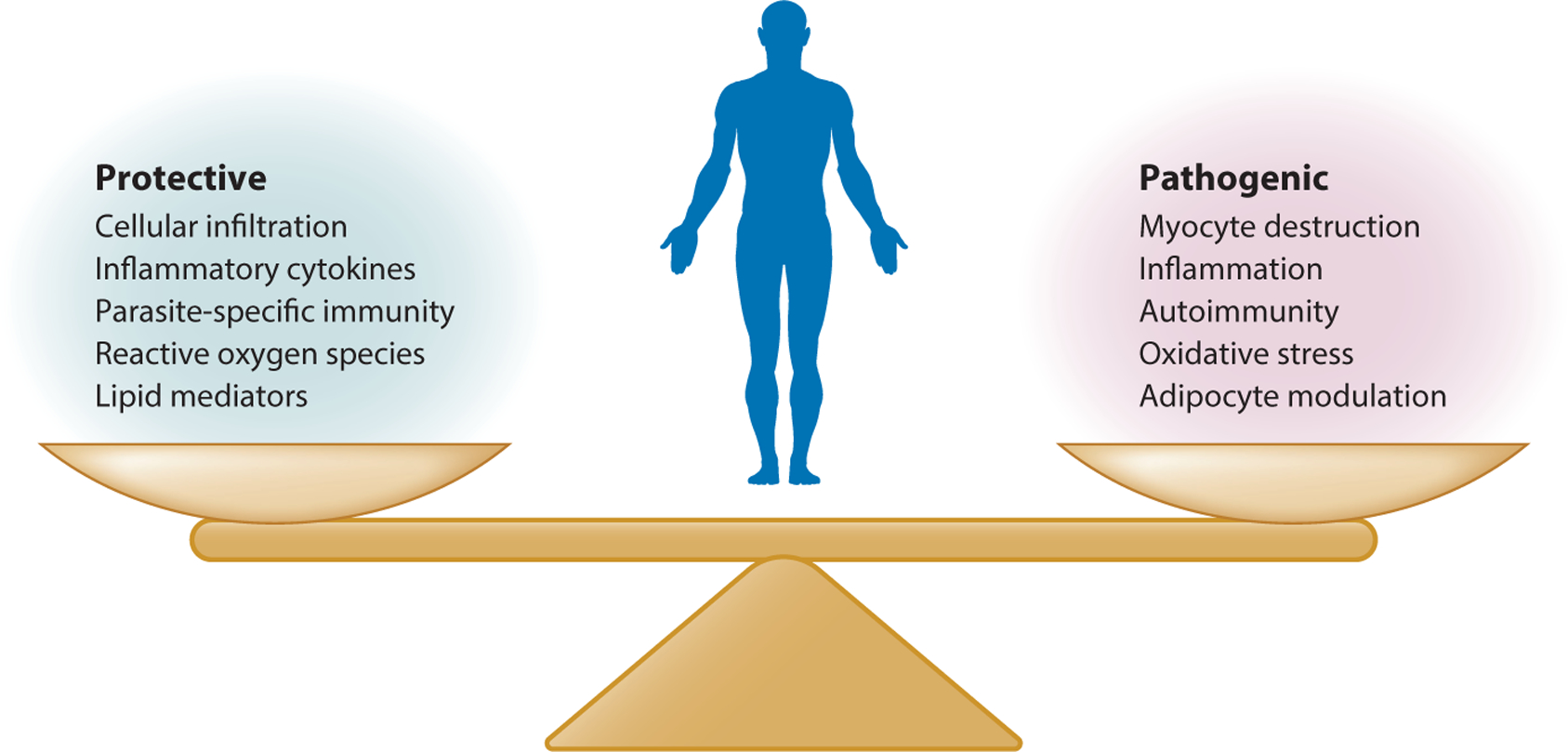
Protective and pathogenic responses to Trypanosoma cruzi infection. The various protective and pathogenic responses are shown. Importantly, many protective processes can also have deleterious effects, which is a general challenge in mounting innate and adaptive responses to pathogens.
Can We Prevent the Progression of Chagas Heart Disease?
The pathology of Chagas heart disease is fascinating. Why are cardiac apical aneurysms so common in Chagas but not in other cardiac infections? Why is cardiac pathology focal and not diffuse? How do tissue inflammation, edema, and fibrosis progress over time in an individual, and why do some lesions heal while others develop? How might the removal of T. cruzi (especially from reservoir sites) through drug treatment prevent progression from the indeterminate to the chronic phase? There was much excitement about testing the efficacy of drug therapy on chronic patients with cardiomyopathy. The Benznidazole Evaluation for Interrupting Trypanosomiasis (BENEFIT) Trial tested the safety of benznidazole and its efficacy in improving the clinical outcomes of individuals with cardiomyopathy (170). Unfortunately, although benznidazole treatment reduced parasitemia, it did not significantly prevent the progression of cardiac disease during 5 years of follow-up. What was not tested in this trial was whether drug treatment has benefit for those in the indeterminate phase by preventing progression to the chronic phase. Indeed, this is the treatment philosophy of many Chagas physicians, even in light of the BENEFIT Trial results.
Chagas in the United States
Another aspect of Chagas disease that has not been emphasized in general reviews such as this one is the importance of this disease in the United States. It is estimated that 300,000 persons living in the United States are chronically infected with T. cruzi (171), with the majority of these being people from South and Central America who were infected before migrating. This includes 30,000 people in Los Angeles alone (172). However, reduviid bugs are found throughout vast areas of the country, with more than half being infected with T. cruzi (173). Insect transmission of T. cruzi occurs in the United States as well. Although the first documented case of autochthonous transmission of T. cruzi was reported in 1955 (174), a person with Chagas megacolon who lived 1,200 years ago was discovered mummified in a Texas rock shelter in 2011 (175). Large percentages of wild animals, as well as nonhuman primates used in research (176) and working dogs along the Texas–Mexico border (177), have also been infected through insect transmission. A 2012 study of US blood donors estimated the prevalence of insect transmission as 1 in 354,000 eligible donors (178). A second, pilot study conducted prospectively in Houston blood donors identified individuals who were likely to have been exposed near their home, while hunting or camping, or through congenital transmission (179). A major question about autochthonous transmission relates to the pathogenic potential of the United States isolates. Many of them belong to a set of T. cruzi strains (TcI) known to cause chronic myocarditis (169). All of the pathology images included in Figures 3 and 4 were obtained at Cedars-Sinai Medical Center in Los Angeles (180).
Final Thoughts on Pathogenesis
Essentially every host response to T. cruzi infection that aims to kill or suppress the parasite may also harm the host. When several mechanisms are operative simultaneously, for example oxidative stress and adaptive immunity, they may be synergistic or antagonistic. Regulatory responses involved in restoring the self-tolerance that may be broken during infection may dampen parasite-specific immunity and permit T. cruzi to replicate and disseminate. In the end, balance is best for both host and parasite. This balance likely occurs in the vast majority of infected people, who remain in the indeterminate phase of infection and eventually die of causes other than Chagas disease. A healthy balance of parasite suppression and modulated immunity keeps tissue pathology to a minimum, but allows parasites to circulate and be transferred to other hosts (181). In this regard, the death of an infected individual during the acute phase of infection is a tragic accident from the parasite’s perspective. Key questions remain about the pathogenesis of Chagas disease and Chagas heart disease in particular: What determines the tissue distribution of T. cruzi over time? What are the mechanisms of transendothelial migration (182) and tissue tropism? What are the genetic, environmental, and physiologic factors that determine the outcome of infection in each individual? These are likely numerous, and the importance and complexity of this issue cannot be overstated. How prevalent is autochthonous infection in the United States, and what are the outcomes of those infected with natural T. cruzi isolates? In the end, Chagas disease is a collection of related diseases having different degrees of severity. Much work remains before we can accurately determine whether an infected person will progress from the indeterminate phase to the chronic phase and how to intervene to prevent this progression.
ACKNOWLEDGMENTS
This work was supported in part by grants from the US National Institutes of Health (HL-075822 and AI-38022 to D.M.E. and AI-054578 and AI-136031 to N.J.G.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Brener Z 1973. Biology of Trypanosoma cruzi. Annu. Rev. Microbiol 27:347–82 [DOI] [PubMed] [Google Scholar]
- 2.WHO (World Health Organ.). 2002. Control of Chagas Disease: Second Report of the WHO Expert Committee. Geneva, Switzerland: WHO [Google Scholar]
- 3.Shikanai-Yasuda MA, Carvalho NB. 2012. Oral transmission of Chagas disease. Clin. Infect. Dis 54:845–52 [DOI] [PubMed] [Google Scholar]
- 4.Tanowitz HB, Weiss LM, Montgomery SP. 2011. Chagas disease has now gone global. PLOS Negl. Trop. Dis 5:e1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.PAHO (Pan Am. Health Organ.). 2018. Chagas disease portal. Pan Am. Health Organ http://www.paho.org/hq/index.php?option=com_topics&view=article&id=10&Itemid=40743&lang=en [Google Scholar]
- 6.Guedes PM, Silva GK, Gutierrez FR, Silva JS. 2011. Current status of Chagas disease chemotherapy. Expert Rev. Anti. Infect. Ther 9:609–20 [DOI] [PubMed] [Google Scholar]
- 7.Morillo CA, Marin-Neto JA, Avezum A, Sosa-Estani S, Rassi A Jr., et al. 2015. Randomized trial of benznidazole for chronic Chagas’ cardiomyopathy. N. Engl. J. Med 373:1295–306 [DOI] [PubMed] [Google Scholar]
- 8.Pecoul B, Batista C, Stobbaerts E, Ribeiro I, Vilasanjuan R, et al. 2016. The BENEFIT Trial: Where do we go from here? PLOS Negl. Trop. Dis 10:e0004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beaumier CM, Gillespie PM, Strych U, Hayward T, Hotez PJ, Bottazzi ME. 2016. Status of vaccine research and development of vaccines for Chagas disease. Vaccine 34:2996–3000 [DOI] [PubMed] [Google Scholar]
- 10.Tanowitz HB, Kirchhoff LV, Simon D, Morris SA, Weiss LM, Wittner M. 1992. Chagas’ disease. Clin. Microbiol. Rev 5:400–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benziger CP, do Carmo GAL, Ribeiro ALP. 2017. Chagas cardiomyopathy: clinical presentation and management in the Americas. Cardiol. Clin 35:31–47 [DOI] [PubMed] [Google Scholar]
- 12.Linhares-Lacerda L, Granato A, Gomes-Neto JF, Conde L, Freire-de-Lima L, et al. 2018. Circulating plasma microRNA-208a as potential biomarker of chronic indeterminate phase of Chagas disease. Front. Microbiol 9:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanowitz HB, Machado FS, Spray DC, Friedman JM, Weiss OS, et al. 2016. Developments in the management of Chagas cardiomyopathy. Expert Rev. Cardiovasc. Ther 13:1393–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuda NM, Miller SM, Evora PR. 2009. The chronic gastrointestinal manifestations of Chagas disease. Clinics 64:1219–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Ayala A, Perez-Molina JA, Norman F, Monge-Maillo B, Faro MV, Lopez-Velez R. 2011. Gastrointestinal Chagas disease in migrants to Spain: prevalence and methods for early diagnosis. Ann. Trop. Med. Parasitol 105:25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Machado FS, Dutra WO, Esper L, Gollob KJ, Teixeira MM, et al. 2012. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin. Immunopathol 34:753–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higuchi MDL. 1997. Chronic chagasic cardiopathy: the product of a turbulent host–parasite relationship. Rev. Inst. Med. Trop. Sao Paulo 39:53–60 [DOI] [PubMed] [Google Scholar]
- 18.Teixeira AR, Nascimento RJ, Sturm NR. 2006. Evolution and pathology in Chagas disease—a review. Mem. Inst. Oswaldo Cruz 101:463–91 [DOI] [PubMed] [Google Scholar]
- 19.Tanowitz HB, Machado FS, Jelicks LA, Shirani J, de Carvalho AC, et al. 2009. Perspectives on Trypanosoma cruzi–induced heart disease (Chagas disease). Prog. Cardiovasc. Dis 51:524–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis MD, Kelly JM. 2016. Putting infection dynamics at the heart of Chagas disease. Trends Parasitol. 32:899–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferreira AV, Segatto M, Menezes Z, Macedo AM, Gelape C, et al. 2011. Evidence for Trypanosoma cruzi in adipose tissue in human chronic Chagas disease. Microbes Infect. 13:1002–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandes MC, Andrews NW. 2012. Host cell invasion by Trypanosoma cruzi: a unique strategy that promotes persistence. FEMS Microbiol. Rev 36:734–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrade ZA. 1999. Immunopathology of Chagas disease. Mem. Inst. Oswaldo Cruz 94(Suppl. 1):71–80 [DOI] [PubMed] [Google Scholar]
- 24.Jabari S, de Oliveira EC, Brehmer A, da Silveira AB. 2014. Chagasic megacolon: enteric neurons and related structures. Histochem. Cell Biol 142:235–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alvarez JM, Fonseca R, Borges da Silva H, Marinho CR, Bortoluci KR, et al. 2014. Chagas disease: still many unsolved issues. Mediat. Inflamm 2014:912965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossi MA, Ramos SG, Bestetti RB. 2003. Chagas’ heart disease: clinical–pathological correlation. Front. Biosci 8:e94–109 [DOI] [PubMed] [Google Scholar]
- 27.Marcon GE, de Albuquerque DM, Batista AM, Andrade PD, Almeida EA, et al. 2011. Trypanosoma cruzi: parasite persistence in tissues in chronic chagasic Brazilian patients. Mem. Inst. Oswaldo Cruz 106:85–91 [DOI] [PubMed] [Google Scholar]
- 28.Rossi MA. 1991. The pattern of myocardial fibrosis in chronic Chagas’ heart disease. Int. J. Cardiol 30:335–40 [DOI] [PubMed] [Google Scholar]
- 29.Rossi MA. 1998. Fibrosis and inflammatory cells in human chronic chagasic myocarditis: scanning electron microscopy and immunohistochemical observations. Int. J. Cardiol 66:183–94 [DOI] [PubMed] [Google Scholar]
- 30.Roffe E, Marino AP, Weaver J, Wan W, de Araujo FF, et al. 2016. Trypanosoma cruzi causes paralyzing systemic necrotizing vasculitis driven by pathogen-specific type I immunity in mice. Infect. Immun 84:1123–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marin-Neto JA, Simoes MV, Rassi A Jr. 2013. Pathogenesis of chronic Chagas cardiomyopathy: the role of coronary microvascular derangements. Rev. Soc. Bras. Med. Trop 46:536–41 [DOI] [PubMed] [Google Scholar]
- 32.Marin-Neto JA, Cunha-Neto E, Maciel BC, Simoes MV. 2007. Pathogenesis of chronic Chagas heart disease. Circulation 115:1109–23 [DOI] [PubMed] [Google Scholar]
- 33.Mello de Oliveira JA, Meira Oliveira JS, Koberle F. 1972. Pathologic anatomy of the His-Tawara system and electrocardiographic abnormalities in chronic Chagas’ heart disease. Arq. Bras. Cardiol 25:17–25 [PubMed] [Google Scholar]
- 34.Andrade ZA, Andrade SG, Sadigursky M, Camara EJ. 1988. Pathology of complete atrioventricular block in chronic Chagas’ myocarditis. Rev. Soc. Bras. Med. Trop 21:7–13 [DOI] [PubMed] [Google Scholar]
- 35.Ramos SG, Matturri L, Rossi L, Rossi MA. 1996. Lesions of mediastinal paraganglia in chronic chagasic cardiomyopathy: cause of sudden death? Am. Heart J 131:417–20 [DOI] [PubMed] [Google Scholar]
- 36.Miranda CH, Figueiredo AB, Maciel BC, Marin-Neto JA, Simoes MV. 2011. Sustained ventricular tachycardia is associated with regional myocardial sympathetic denervation assessed with 123I-metaiodobenzylguanidine in chronic Chagas cardiomyopathy. J. Nucl. Med 52:504–10 [DOI] [PubMed] [Google Scholar]
- 37.Rassi A Jr., Marin JAN, Rassi A. 2017. Chronic Chagas cardiomyopathy: a review of the main pathogenic mechanisms and the efficacy of aetiological treatment following the BENznidazole Evaluation for Interrupting Trypanosomiasis (BENEFIT) Trial. Mem. Inst. Oswaldo Cruz 112:224–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gutierrez FR, Guedes PM, Gazzinelli RT, Silva JS. 2009. The role of parasite persistence in pathogenesis of Chagas heart disease. Parasite Immunol. 31:673–85 [DOI] [PubMed] [Google Scholar]
- 39.Lewis MD, Fortes Francisco A, Taylor MC, Burrell-Saward H, McLatchie AP, et al. 2014. Biolumines-cence imaging of chronic Trypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cell. Microbiol 16:1285–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonney KM, Engman DM. 2008. Chagas heart disease pathogenesis: one mechanism or many? Curr. Mol. Med 8:510–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hyland KV, Leon JS, Daniels MD, Giafis N, Woods LM, et al. 2007. Modulation of autoimmunity by treatment of an infectious disease. Infect. Immun 75:3641–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyland KV, Asfaw SH, Olson CL, Daniels MD, Engman DM. 2008. Bioluminescent imaging of Trypanosoma cruzi infection. Int. J. Parasitol 38:1391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis MD, Francisco AF, Taylor MC, Jayawardhana S, Kelly JM. 2016. Host and parasite genetics shape a link between Trypanosoma cruzi infection dynamics and chronic cardiomyopathy. Cell. Microbiol 18:1429–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lidani KCF, Bavia L, Ambrosio AR, de Messias-Reason IJ. 2017. The complement system: a prey of Trypanosoma cruzi. Front. Microbiol 8:607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rimoldi MT, Tenner AJ, Bobak DA, Joiner KA. 1989. Complement component C1q enhances invasion of human mononuclear phagocytes and fibroblasts by Trypanosoma cruzi trypomastigotes. J. Clin. Investig 84:1982–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joiner K, Sher A, Gaither T, Hammer C. 1986. Evasion of alternative complement pathway by Trypanosoma cruzi results from inefficient binding of factor B. PNAS 83:6593–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zingales B 2017. Trypanosoma cruzi genetic diversity: something new for something known about Chagas disease manifestations, serodiagnosis and drug sensitivity. Acta Trop. 184:38–52 [DOI] [PubMed] [Google Scholar]
- 48.Benvenuti LA, Roggerio A, Cavalcanti MM, Nishiya AS, Levi JE. 2017. An autopsy-based study of Trypanosoma cruzi persistence in organs of chronic chagasic patients and its relevance for transplantation. Transpl. Infect. Dis 19:e12783. [DOI] [PubMed] [Google Scholar]
- 49.Hotez PJ, Dumonteil E, Woc-Colburn L, Serpa JA, Bezek S, et al. 2012. Chagas disease: “The new HIV/AIDS of the Americas.” PLOS Negl. Trop. Dis 6:e1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kayama H, Takeda K. 2010. The innate immune response to Trypanosoma cruzi infection. Microbes Infect. 12:511–17 [DOI] [PubMed] [Google Scholar]
- 51.Campos MA, Gazzinelli RT. 2004. Trypanosoma cruzi and its components as exogenous mediators of inflammation recognized through Toll-like receptors. Mediat. Inflamm 13:139–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clay GM, Sutterwala FS, Wilson ME. 2014. NLR proteins and parasitic disease. Immunol. Res 59:142–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zamboni DS, Lima-Junior DS. 2015. Inflammasomes in host response to protozoan parasites. Immunol. Rev 265:156–71 [DOI] [PubMed] [Google Scholar]
- 54.Gurung P, Kanneganti TD. 2016. Immune responses against protozoan parasites: a focus on the emerging role of Nod-like receptors. Cell. Mol. Life Sci 73:3035–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dhiman M, Garg NJ. 2011. NADPH oxidase inhibition ameliorates Trypanosoma cruzi–induced myocarditis during Chagas disease. J. Pathol 225:583–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dhiman M, Garg NJ. 2014. P47phox−/− mice are compromised in expansion and activation of CD8+ T cells and susceptible to Trypanosoma cruzi infection. PLOS Pathog. 10:e1004516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bogdan C 2015. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. 36:161–78 [DOI] [PubMed] [Google Scholar]
- 58.Alvarez MN, Peluffo G, Piacenza L, Radi R. 2011. Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J. Biol. Chem 286:6627–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piacenza L, Peluffo G, Alvarez MN, Kelly JM, Wilkinson SR, Radi R. 2008. Peroxiredoxins play a major role in protecting Trypanosoma cruzi against macrophage- and endogenously-derived peroxynitrite. Biochem. J 410:359–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koo SJ, Chowdhury IH, Szczesny B, Wan X, Garg NJ. 2016. Macrophages promote oxidative metabolism to drive nitric oxide generation in response to Trypanosoma cruzi. Infect. Immun 84:3527–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Piacenza L, Zago MP, Peluffo G, Alvarez MN, Basombrio MA, Radi R. 2009. Enzymes of the antioxidant network as novel determiners of Trypanosoma cruzi virulence. Int. J. Parasitol 39:1455–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zago MP, Hosakote YM, Koo S-J, Dhiman M, Piñeyro MD, et al. 2016. TcI isolates of Trypanosoma cruzi exploit the antioxidant network for enhanced intracellular survival in macrophages and virulence in mice. Infect. Immun 84:1842–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pineyro MD, Parodi-Talice A, Arcari T, Robello C. 2008. Peroxiredoxins from Trypanosoma cruzi: virulence factors and drug targets for treatment of Chagas disease? Gene 408:45–50 [DOI] [PubMed] [Google Scholar]
- 64.Gupta S, Bhatia V, Wen J-J, Wu Y, Huang M-H, Garg NJ. 2009. Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic. Biol. Med 47:1414–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ba X, Gupta S, Davidson M, Garg NJ. 2010. Trypanosoma cruzi induces the reactive oxygen species–PARP-1–RelA pathway for up-regulation of cytokine expression in cardiomyocytes. J. Biol. Chem 285:11596–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wan X, Wen JJ, Koo SJ, Liang LY, Garg NJ. 2016. SIRT1–PGC1α–NFκB pathway of oxidative and inflammatory stress during Trypanosoma cruzi infection: benefits of SIRT1-targeted therapy in improving heart function in Chagas disease. PLOS Pathog. 12:e1005954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Keating SM, Deng X, Fernandes F, Cunha-Neto E, Ribeiro AL, et al. 2015. Inflammatory and cardiac biomarkers are differentially expressed in clinical stages of Chagas disease. Int. J. Cardiol 199:451–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cunha-Neto E, Teixeira PC, Fonseca SG, Bilate AM, Kalil J. 2011. Myocardial gene and protein expression profiles after autoimmune injury in Chagas’ disease cardiomyopathy. Autoimmun. Rev 10:163–65 [DOI] [PubMed] [Google Scholar]
- 69.Garg NJ, Soman KV, Zago MP, Koo SJ, Spratt H, et al. 2016. Changes in proteome profile of peripheral blood mononuclear cells in chronic Chagas disease. PLOS Negl. Trop. Dis 10:e0004490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferreira LR, Ferreira FM, Nakaya HI, Deng X, Cândido DD, et al. 2017. Blood gene signatures of Chagas disease cardiomyopathy with or without ventricular dysfunction. J. Infect. Dis 215:387–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Souza PE, Rocha MO, Menezes CA, Coelho JS, Chaves AC, et al. 2007. Trypanosoma cruzi infection induces differential modulation of costimulatory molecules and cytokines by monocytes and T cells from patients with indeterminate and cardiac Chagas’ disease. Infect. Immun 75:1886–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Souza PE, Rocha MO, Rocha-Vieira E, Menezes CA, Chaves AC, et al. 2004. Monocytes from patients with indeterminate and cardiac forms of Chagas’ disease display distinct phenotypic and functional characteristics associated with morbidity. Infect. Immun 72:5283–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Machado FS, Dutra WO, Esper L, Gollob KJ, Teixeira MM, et al. 2012. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin. Immunopathol 34:753–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chowdhury IH, Koo SJ, Gupta S, Liang LY, Bahar B, et al. 2017. Gene expression profiling and functional characterization of macrophages in response to circulatory microparticles produced during Trypanosoma cruzi infection and Chagas disease. J. Innate Immun 9:203–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dhiman M, Zago MP, Nunez S, Nunez-Burgio F, Garg NJ. 2012. Cardiac oxidized antigens are targets of immune recognition by antibodies and potential molecular determinants in Chagas disease pathogenesis. PLOS ONE 7:e28449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tardieux I, Nathanson MH, Andrews NW. 1994. Role in host cell invasion of Trypanosoma cruzi–induced cytosolic-free Ca2+ transients. J. Exp. Med 179:1017–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wen JJ, Garg NJ. 2008. Mitochondrial generation of reactive oxygen species is enhanced at the Qo site of the complex III in the myocardium of Trypanosoma cruzi–infected mice: beneficial effects of an antioxidant. J. Bioenerg. Biomembr 40:587–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wen J-J, Garg NJ. 2010. Mitochondrial complex III defects contribute to inefficient respiration and ATP synthesis in the myocardium of Trypanosoma cruzi–infected mice. Antioxid. Redox Signal 12:27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wen JJ, Dhiman M, Whorton EB, Garg NJ. 2008. Tissue-specific oxidative imbalance and mitochondrial dysfunction during Trypanosoma cruzi infection in mice. Microbes Infect. 10:1201–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perez-Fuentes R, Guegan JF, Barnabe C, Lopez-Colombo A, Salgado-Rosas H, et al. 2003. Severity of chronic Chagas disease is associated with cytokine/antioxidant imbalance in chronically infected individuals. Int. J. Parasitol 33:293–99 [DOI] [PubMed] [Google Scholar]
- 81.de Oliveira TB, Pedrosa RC, Filho DW. 2007. Oxidative stress in chronic cardiopathy associated with Chagas disease. Int. J. Cardiol 116:357–63 [DOI] [PubMed] [Google Scholar]
- 82.Wen J-J, Yachelini PC, Sembaj A, Manzur RE, Garg NJ. 2006. Increased oxidative stress is correlated with mitochondrial dysfunction in chagasic patients. Free Radic. Biol. Med 41:270–76 [DOI] [PubMed] [Google Scholar]
- 83.Wan X-X, Gupta S, Zago MP, Davidson MM, Dousset P, et al. 2012. Defects of mtDNA replication impaired the mitochondrial biogenesis during Trypanosoma cruzi infection in human cardiomyocytes and chagasic patients: the role of Nrf1/2 and antioxidant response. J. Am. Heart Assoc 1:e003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wen J-J, Gupta S, Guan Z, Dhiman M, Condon D, et al. 2010. Phenyl-α-tert-butyl-nitrone and ben-zonidazole treatment controlled the mitochondrial oxidative stress and evolution of cardiomyopathy in chronic chagasic rats. J. Am. Coll. Cardiol 55:2499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wen JJ, Porter C, Garg NJ. 2017. Inhibition of NFE2L2–ARE pathway by mitochondrial ROS contributes to development of cardiomyopathy and left ventricular dysfunction in Chagas disease. Antioxid. Redox Signal 27:550–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Panis C, Mazzuco TL, Costa CZ, Victorino VJ, Tatakihara VL, et al. 2011. Trypanosoma cruzi: effect of the absence of 5-lipoxygenase (5-LO)–derived leukotrienes on levels of cytokines, nitric oxide and iNOS expression in cardiac tissue in the acute phase of infection in mice. Exp. Parasitol 127:58–65 [DOI] [PubMed] [Google Scholar]
- 87.Wen JJ, Wan X, Thacker J, Garg NJ. 2016. Chemotherapeutic efficacy of phosphodiesterase inhibitors in chagasic cardiomyopathy. JACC Basic Transl. Sci 1:235–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.D’Avila H, Toledo DA, Melo RC. 2012. Lipid bodies: inflammatory organelles implicated in host–Trypanosoma cruzi interplay during innate immune responses. Mediat. Inflamm 2012:478601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Canavaci AM, Sorgi CA, Martins VP, Morais FR, de Sousa EV, et al. 2014. The acute phase of Trypanosoma cruzi infection is attenuated in 5-lipoxygenase-deficient mice. Mediat. Inflamm 2014:893634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pavanelli WR, Gutierrez FR, Mariano FS, Prado CM, Ferreira BR, et al. 2010. 5-lipoxygenase is a key determinant of acute myocardial inflammation and mortality during Trypanosoma cruzi infection. Microbes Infect. 12:587–97 [DOI] [PubMed] [Google Scholar]
- 91.Esper L, Roman-Campos D, Lara A, Brant F, Castro LL, et al. 2012. Role of SOCS2 in modulating heart damage and function in a murine model of acute Chagas disease. Am. J. Pathol 181:130–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tanowitz HB, Mukhopadhyay A, Ashton AW, Lisanti MP, Machado FS, et al. 2011. Microarray analysis of the mammalian thromboxane receptor–Trypanosoma cruzi interaction. Cell Cycle 10:1132–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vago AR, Andrade LO, Leite AA, d’Avila Reis D, Macedo AM, et al. 2000. Genetic characterization of Trypanosoma cruzi directly from tissues of patients with chronic Chagas disease: differential distribution of genetic types into diverse organs. Am. J. Pathol 156:1805–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.dos Santos DM, Talvani A, Guedes PM, Machado-Coelho GL, de Lana M, Bahia MT. 2009. Trypanosoma cruzi: Genetic diversity influences the profile of immunoglobulins during experimental infection. Exp. Parasitol 121:8–14 [DOI] [PubMed] [Google Scholar]
- 95.Scharfstein J, Andrade D. 2011. Infection-associated vasculopathy in experimental Chagas disease: pathogenic roles of endothelin and kinin pathways. Adv. Parasitol 76:101–27 [DOI] [PubMed] [Google Scholar]
- 96.Scharfstein J, Schmitz V, Svensjo E, Granato A, Monteiro AC. 2007. Kininogens coordinate adaptive immunity through the proteolytic release of bradykinin, an endogenous danger signal driving dendritic cell maturation. Scand. J. Immunol 66:128–36 [DOI] [PubMed] [Google Scholar]
- 97.Scharfstein J, Ramos PIP, Barral-Netto M. 2017. G protein–coupled kinin receptors and immunity against pathogens. Adv. Immunol 136:29–84 [DOI] [PubMed] [Google Scholar]
- 98.Tanowitz HB, Jelicks LA, Machado FS, Esper L, Qi X, et al. 2011. Adipose tissue, diabetes and Chagas disease. Adv. Parasitol 76:235–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nascimento CR, Andrade D, Carvalho-Pinto CE, Serra RR, Vellasco L, et al. 2017. Mast cell coupling to the kallikrein–kinin system fuels intracardiac parasitism and worsens heart pathology in experimental Chagas disease. Front. Immunol 8:840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tarleton RL. 1990. Depletion of CD8+ T cells increases susceptibility and reverses vaccine-induced immunity in mice infected with Trypanosoma cruzi. J. Immunol 144:717–24 [PubMed] [Google Scholar]
- 101.Tarleton RL, Sun J, Zhang L, Postan M. 1994. Depletion of T-cell subpopulations results in exacerbation of myocarditis and parasitism in experimental Chagas’ disease. Infect. Immun 62:1820–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tarleton RL, Koller BH, Latour A, Postan M. 1992. Susceptibility of β2-microglobulin-deficient mice to Trypanosoma cruzi infection. Nature 356:338–40 [DOI] [PubMed] [Google Scholar]
- 103.Sathler-Avelar R, Vitelli-Avelar DM, Eloi-Santos SM, Gontijo ED, Teixeira-Carvalho A, Martins-Filho OA. 2012. Blood leukocytes from benznidazole-treated indeterminate Chagas disease patients display an overall type-1-modulated cytokine profile upon short-term in vitro stimulation with Trypanosoma cruzi antigens. BMC Infect. Dis 12:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guedes PM, Gutierrez FR, Silva GK, Dellalibera-Joviliano R, Rodrigues GJ, et al. 2012. Deficient regulatory T cell activity and low frequency of IL-17-producing T cells correlate with the extent of cardiomyopathy in human Chagas’ disease. PLOS Negl. Trop. Dis 6:e1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guo S, Cobb D, Smeltz RB. 2009. T-bet inhibits the in vivo differentiation of parasite-specific CD4+ Th17 cells in a T cell–intrinsic manner. J. Immunol 182:6179–86 [DOI] [PubMed] [Google Scholar]
- 106.Cunha-Neto E, Kalil J. 2001. Heart-infiltrating and peripheral T cells in the pathogenesis of human Chagas’ disease cardiomyopathy. Autoimmunity 34:187–92 [DOI] [PubMed] [Google Scholar]
- 107.Dutra WO, Gollob KJ, Pinto-Dias JC, Gazzinelli G, Correa-Oliveira R, et al. 1997. Cytokine mRNA profile of peripheral blood mononuclear cells isolated from individuals with Trypanosoma cruzi chronic infection. Scand. J. Immunol 45:74–80 [DOI] [PubMed] [Google Scholar]
- 108.Gomes JA, Bahia-Oliveira LM, Rocha MO, Martins-Filho OA, Gazzinelli G, Correa-Oliveira R. 2003. Evidence that development of severe cardiomyopathy in human Chagas’ disease is due to a Th1-specific immune response. Infect. Immun 71:1185–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Da Matta Guedes PM, Gutierrez FR, Maia FL, Milanezi CM, Silva GK, et al. 2010. IL-17 produced during Trypanosoma cruzi infection plays a central role in regulating parasite-induced myocarditis. PLOS Negl. Trop. Dis 4:e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martin D, Tarleton R. 2004. Generation, specificity, and function of CD8+ T cells in Trypanosoma cruzi infection. Immunol. Rev 201:304–17 [DOI] [PubMed] [Google Scholar]
- 111.Mateus J, Perez-Anton E, Lasso P, Egui A, Roa N, et al. 2017. Antiparasitic treatment induces an improved CD8+ T cell response in chronic chagasic patients. J. Immunol 198:3170–80 [DOI] [PubMed] [Google Scholar]
- 112.Albareda MC, Laucella SA, Alvarez MG, Armenti AH, Bertochi G, et al. 2006. Trypanosoma cruzi modulates the profile of memory CD8+ T cells in chronic Chagas’ disease patients. Int. Immunol 18:465–71 [DOI] [PubMed] [Google Scholar]
- 113.Cai CW, Blase JR, Zhang X, Eickhoff CS, Hoft DF. 2016. Th17 cells are more protective than Th1 cells against the intracellular parasite Trypanosoma cruzi. PLOS Pathog. 12:e1005902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rocha Rodrigues DB, dos Reis MA, Romano A, Pereira SADL, Teixeira VDPA, et al. 2012. In situ expression of regulatory cytokines by heart inflammatory cells in Chagas’ disease patients with heart failure. Clin. Dev. Immunol 2012:361730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cunha-Neto E, Dzau VJ, Allen PD, Stamatiou D, Benvenutti L, et al. 2005. Cardiac gene expression profiling provides evidence for cytokinopathy as a molecular mechanism in Chagas’ disease cardiomyopathy. Am. J. Pathol 167:305–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ruiz Diaz P, Mucci J, Meira MA, Bogliotti Y, Musikant D, et al. 2015. Trypanosoma cruzi trans-sialidase prevents elicitation of Th1 cell response via interleukin 10 and downregulates Th1 effector cells. Infect. Immun 83:2099–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rodriguez-Angulo H, Marques J, Mendoza I, Villegas M, Mijares A, et al. 2017. Differential cytokine profiling in chagasic patients according to their arrhythmogenic-status. BMC Infect. Dis 17:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mateus J, Lasso P, Pavia P, Rosas F, Roa N, et al. 2015. Low frequency of circulating CD8+ T stem cell memory cells in chronic chagasic patients with severe forms of the disease. PLOS Negl. Trop. Dis 9:e3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Arguello RJ, Vigliano C, Cabeza-Meckert P, Viotti R, Garelli F, et al. 2014. Presence of antigen-experienced T cells with low grade of differentiation and proliferative potential in chronic Chagas disease myocarditis. PLOS Negl. Trop. Dis 8:e2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Giraldo NA, Bolanos NI, Cuellar A, Guzman F, Uribe AM, et al. 2011. Increased CD4+/CD8+ double-positive T cells in chronic chagasic patients. PLOS Negl. Trop. Dis 5:e1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dutra WO, Martins-Filho OA, Cancado JR, Pinto-Dias JC, Brener Z, et al. 1994. Activated T and B lymphocytes in peripheral blood of patients with Chagas’ disease. Int. Immunol 6:499–506 [DOI] [PubMed] [Google Scholar]
- 122.Higuchi ML, De Morais CF, Pereira Barreto AC, Lopes EA, Stolf N, et al. 1987. The role of active myocarditis in the development of heart failure in chronic Chagas’ disease: a study based on endomyocardial biopsies. Clin. Cardiol 10:665–70 [DOI] [PubMed] [Google Scholar]
- 123.Dos Santos Virgilio F, Pontes C, Dominguez MR, Ersching J, Rodrigues MM, Vasconcelos JR. 2014. CD8+ T cell-mediated immunity during Trypanosoma cruzi infection: a path for vaccine development? Mediat. Inflamm 2014:243786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cunha-Neto E, Kalil J. 1995. Autoimmunity in Chagas’ heart disease. Sao Paulo Med. J 113:757–66 [DOI] [PubMed] [Google Scholar]
- 125.Teixeira AR, Nitz N, Bernal FM, Hecht MM. 2012. Parasite induced genetically driven autoimmune Chagas heart disease in the chicken model. J. Vis. Exp 65:e3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Girones N, Carrasco-Marin E, Cuervo H, Guerrero NA, Sanoja C, et al. 2007. Role of Trypanosoma cruzi autoreactive T cells in the generation of cardiac pathology. Ann. N. Y. Acad. Sci 1107:434–44 [DOI] [PubMed] [Google Scholar]
- 127.Bonney KM, Engman DM. 2015. Autoimmune pathogenesis of Chagas heart disease: looking back, looking ahead. Am. J. Pathol 185:1537–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vitelli-Avelar DM, Sathler-Avelar R, Massara RL, Borges JD, Lage PS, et al. 2006. Are increased frequency of macrophage-like and natural killer (NK) cells, together with high levels of NKT and CD4+CD25high T cells balancing activated CD8+ T cells, the key to control Chagas’ disease morbidity? Clin. Exp. Immunol 145:81–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vitelli-Avelar DM, Sathler-Avelar R, Dias JC, Pascoal VP, Teixeira-Carvalho A, et al. 2005. Chagasic patients with indeterminate clinical form of the disease have high frequencies of circulating CD3+CD16−CD56+ natural killer T cells and CD4+CD25High regulatory T lymphocytes. Scand. J. Immunol 62:297–308 [DOI] [PubMed] [Google Scholar]
- 130.Mariano FS, Gutierrez FR, Pavanelli WR, Milanezi CM, Cavassani KA, et al. 2008. The involvement of CD4+CD25+ T cells in the acute phase of Trypanosoma cruzi infection. Microbes Infect. 10:825–33 [DOI] [PubMed] [Google Scholar]
- 131.Sales PA Jr., Golgher D, Oliveira RV, Vieira V, Arantes RM, et al. 2008. The regulatory CD4+CD25+T cells have a limited role on pathogenesis of infection with Trypanosoma cruzi. Microbes Infect. 10:680–88 [DOI] [PubMed] [Google Scholar]
- 132.Bonney KM, Taylor JM, Thorp EB, Epting CL, Engman DM. 2015. Depletion of regulatory T cells decreases cardiac parasitosis and inflammation in experimental Chagas disease. Parasitol. Res 114:1167–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nihei J, Cardillo F, Dos Santos WL, Pontes-de-Carvalho L, Mengel J. 2014. Administration of a non-depleting anti-CD25 monoclonal antibody reduces disease severity in mice infected with Trypanosoma cruzi. Eur. J. Microbiol. Immunol 4:128–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Calzada JE, Beraun Y, Gonzalez CI, Martin J. 2009. Transforming growth factor beta 1 (TGFβ1) gene polymorphisms and Chagas disease susceptibility in Peruvian and Colombian patients. Cytokine 45:149–53 [DOI] [PubMed] [Google Scholar]
- 135.Altara R, Mallat Z, Booz GW, Zouein FA. 2016. The CXCL10/CXCR3 axis and cardiac inflammation: implications for immunotherapy to treat infectious and noninfectious diseases of the heart. J. Immunol. Res 2016:4396368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gil-Jaramillo N, Motta FN, Favali CB, Bastos IM, Santana JM. 2016. Dendritic cells: a double-edged sword in immune responses during Chagas disease. Front. Microbiol 7:1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Saraiva RM, Waghabi MC, Vilela MF, Madeira FS, Sperandio da Silva GM, et al. 2013. Predictive value of transforming growth factor-β1 in Chagas disease: towards a biomarker surrogate of clinical outcome. Trans. R. Soc. Trop. Med. Hyg 107:518–25 [DOI] [PubMed] [Google Scholar]
- 138.Araujo-Jorge TC, Waghabi MC, Bailly S, Feige JJ. 2012. The TGF-β pathway as an emerging target for Chagas disease therapy. Clin. Pharmacol. Ther 92:613–21 [DOI] [PubMed] [Google Scholar]
- 139.Curvo EO, Ferreira RR, Madeira FS, Alves GF, Chambela MC, et al. 2018. Correlation of transforming growth factor-β1 and tumour necrosis factor levels with left ventricular function in Chagas disease. Mem. Inst. Oswaldo Cruz 113:e170440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kierszenbaum F, Cuna WR, Beltz LA, Sztein MB. 1990. Trypanosomal immunosuppressive factor: a secretion product(s) of Trypanosoma cruzi that inhibits proliferation and IL-2 receptor expression by activated human peripheral blood mononuclear cells. J. Immunol 144:4000–4 [PubMed] [Google Scholar]
- 141.Sztein MB, Cuna WR, Kierszenbaum F. 1990. Trypanosoma cruzi inhibits the expression of CD3, CD4, CD8, and IL-2R by mitogen-activated helper and cytotoxic human lymphocytes. J. Immunol 144:3558–62 [PubMed] [Google Scholar]
- 142.Costa RP, Gollob KJ, Fonseca LL, Rocha MO, Chaves AC, et al. 2000. T-cell repertoire analysis in acute and chronic human Chagas’ disease: differential frequencies of Vβ5 expressing T cells. Scand. J. Immunol 51:511–19 [DOI] [PubMed] [Google Scholar]
- 143.Bermejo DA, Amezcua Vesely MC, Khan M, Acosta Rodriguez EV, Montes CL, et al. 2011. Trypanosoma cruzi infection induces a massive extrafollicular and follicular splenic B-cell response which is a high source of non-parasite-specific antibodies. Immunology 132:123–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Antas PR, Medrano-Mercado N, Torrico F, Ugarte-Fernandez R, Gomez F, et al. 1999. Early, intermediate, and late acute stages in Chagas’ disease: a study combining anti-galactose IgG, specific serodiagnosis, and polymerase chain reaction analysis. Am. J. Trop. Med. Hyg 61:308–14 [DOI] [PubMed] [Google Scholar]
- 145.Jackson Y, Chatelain E, Mauris A, Holst M, Miao Q, et al. 2013. Serological and parasitological response in chronic Chagas patients 3 years after nifurtimox treatment. BMC Infect. Dis 13:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zrein M, Granjon E, Gueyffier L, Caillaudeau J, Liehl P, et al. 2018. A novel antibody surrogate biomarker to monitor parasite persistence in Trypanosoma cruzi–infected patients. PLOS Negl. Trop. Dis 12:e0006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cordeiro FD, Martins-Filho OA, Da Costa Rocha MO, Adad SJ, Correa-Oliveira R, Romanha AJ. 2001. Anti-Trypanosoma cruzi immunoglobulin G1 can be a useful tool for diagnosis and prognosis of human Chagas’ disease. Clin. Diagn. Lab. Immunol 8:112–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Galvao LM, Nunes RM, Cancado JR, Brener Z, Krettli AU. 1993. Lytic antibody titre as a means of assessing cure after treatment of Chagas disease: a 10 year follow-up study. Trans. R. Soc. Trop. Med. Hyg 87:220–23 [DOI] [PubMed] [Google Scholar]
- 149.Gazzinelli RT, Pereira ME, Romanha A, Gazzinelli G, Brener Z. 1991. Direct lysis of Trypanosoma cruzi: a novel effector mechanism of protection mediated by human anti-gal antibodies. Parasite Immunol. 13:345–56 [DOI] [PubMed] [Google Scholar]
- 150.Scott MT, Moyes L. 1982. 75Se-methionine labelled Trypanosoma cruzi blood trypomastigotes: opsonization by chronic infection serum facilitates killing in spleen and liver. Clin. Exp. Immunol 48:754–57 [PMC free article] [PubMed] [Google Scholar]
- 151.Gao W, Wortis HH, Pereira MA. 2002. The Trypanosoma cruzi trans-sialidase is a T cell–independent B cell mitogen and an inducer of non-specific Ig secretion. Int. Immunol 14:299–308 [DOI] [PubMed] [Google Scholar]
- 152.Reina-San-Martin B, Degrave W, Rougeot C, Cosson A, Chamond N, et al. 2000. A B-cell mitogen from a pathogenic trypanosome is a eukaryotic proline racemase. Nat. Med 6:890–97 [DOI] [PubMed] [Google Scholar]
- 153.Bryan MA, Norris KA. 2010. Genetic immunization converts the Trypanosoma cruzi B-cell mitogen proline racemase to an effective immunogen. Infect. Immun 78:810–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Laguens RP, Meckert PC, Chambo JG. 1988. Antiheart antibody–dependent cytotoxicity in the sera from mice chronically infected with Trypanosoma cruzi. Infect. Immun 56:993–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bonney KM, Taylor JM, Daniels MD, Epting CL, Engman DM. 2011. Heat-killed Trypanosoma cruzi induces acute cardiac damage and polyantigenic autoimmunity. PLOS ONE 6:e14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Daliry A, Pereira IR, Pereira-Junior PP, Ramos IP, Vilar-Pereira G, et al. 2014. Levels of circulating anti-muscarinic and anti-adrenergic antibodies and their effect on cardiac arrhythmias and dysautonomia in murine models of Chagas disease. Parasitology 141:1769–78 [DOI] [PubMed] [Google Scholar]
- 157.Florea F, Bernards C, Caproni M, Kleindienst J, Hashimoto T, et al. 2014. Ex vivo pathogenicity of anti-laminin γ1 autoantibodies. Am. J. Pathol 184:494–506 [DOI] [PubMed] [Google Scholar]
- 158.Giordanengo L, Gea S, Barbieri G, Rabinovich GA. 2001. Anti-galectin-1 autoantibodies in human Trypanosoma cruzi infection: differential expression of this β-galactoside-binding protein in cardiac Chagas’ disease. Clin. Exp. Immunol 124:266–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rodeles LM, Vicco MH, Bontempi IA, Siano A, Tonarelli G, et al. 2016. Combined analysis of cross-reacting antibodies anti-β1AR and anti-B13 in advanced stages of Chagas heart disease. Trop. Med. Int. Health 21:1545–51 [DOI] [PubMed] [Google Scholar]
- 160.Bonney KM, Gifford KM, Taylor JM, Chen CI, Engman DM. 2013. Cardiac damage induced by immunization with heat-killed Trypanosoma cruzi is not antibody mediated. Parasite Immunol. 35:1–10 [DOI] [PubMed] [Google Scholar]
- 161.Talvani A, Rocha MO, Ribeiro AL, Borda E, Sterin-Borda L, Teixeira MM. 2006. Levels of anti-M2 and anti-β1 autoantibodies do not correlate with the degree of heart dysfunction in Chagas’ heart disease. Microbes Infect. 8:2459–64 [DOI] [PubMed] [Google Scholar]
- 162.Lv H, Lipes MA. 2012. Role of impaired central tolerance to α-myosin in inflammatory heart disease. Trends Cardiovasc. Med 22:113–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cunha-Neto E, Bilate AM, Hyland KV, Fonseca SG, Kalil J, Engman DM. 2006. Induction of cardiac autoimmunity in Chagas heart disease: a case for molecular mimicry. Autoimmunity 39:41–54 [DOI] [PubMed] [Google Scholar]
- 164.Machado FS, Tyler KM, Brant F, Esper L, Teixeira MM, Tanowitz HB. 2012. Pathogenesis of Chagas disease: time to move on. Front. Biosci 4:1743–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cruz JS, Santos-Miranda A, Sales-Junior PA, Monti-Rocha R, Campos PP, et al. 2016. Altered cardiomyocyte function and Trypanosoma cruzi persistence in Chagas disease. Am. J. Trop. Med. Hyg 94:1028–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nagajyothi F, Machado FS, Burleigh BA, Jelicks LA, Scherer PE, et al. 2012. Mechanisms of Trypanosoma cruzi persistence in Chagas disease. Cell. Microbiol 14:634–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Girones N, Cuervo H, Fresno M. 2005. Trypanosoma cruzi–induced molecular mimicry and Chagas’ disease. Curr. Top. Microbiol. Immunol 296:89–123 [DOI] [PubMed] [Google Scholar]
- 168.Teixeira AR, Hecht MM, Guimaro MC, Sousa AO, Nitz N. 2011. Pathogenesis of Chagas’ disease: parasite persistence and autoimmunity. Clin. Microbiol. Rev 24:592–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zingales B, Miles MA, Campbell DA, Tibayrenc M, Macedo AM, et al. 2012. The revised Trypanosoma cruzi subspecific nomenclature: rationale, epidemiological relevance and research applications. Infect. Genet. Evol 12:240–53 [DOI] [PubMed] [Google Scholar]
- 170.Morillo CA, Marin-Neto JA, Avezum A, Sosa-Estani S, Rassi A Jr., et al. 2015. Randomized trial of benznidazole for chronic Chagas’ cardiomyopathy. N. Engl. J. Med 373:1295–306 [DOI] [PubMed] [Google Scholar]
- 171.Bern C, Montgomery SP. 2009. An estimate of the burden of Chagas disease in the United States. Clin. Infect. Dis 49:e52–54 [DOI] [PubMed] [Google Scholar]
- 172.Meymandi SK, Forsyth CJ, Soverow J, Hernandez S, Sanchez D, et al. 2017. Prevalence of Chagas disease in the Latin American–born population of Los Angeles. Clin. Infect. Dis 64:1182–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Curtis-Robles R, Auckland LD, Snowden KF, Hamer GL, Hamer SA. 2018. Analysis of over 1500 triatomine vectors from across the US, predominantly Texas, for Trypanosoma cruzi infection and discrete typing units. Infect. Genet. Evol 58:171–80 [DOI] [PubMed] [Google Scholar]
- 174.Woody NC, Woody HB. 1955. American trypanosomiasis (Chagas’ disease): first indigenous case in the United States. JAMA 159:676–77 [DOI] [PubMed] [Google Scholar]
- 175.Barth E, Kundrotas L. 2011. Megacolon from Chagas disease in an ancient Texan. Gastroenterology 141:35–404 [DOI] [PubMed] [Google Scholar]
- 176.Zabalgoitia M, Ventura J, Anderson L, Carey KD, Williams JT, Vandeberg JL. 2003. Morphologic and functional characterization of chagasic heart disease in non-human primates. Am. J. Trop. Med. Hyg 68:248–52 [PubMed] [Google Scholar]
- 177.Meyers AC, Meinders M, Hamer SA. 2017. Widespread Trypanosoma cruzi infection in government working dogs along the Texas–Mexico border: discordant serology, parasite genotyping and associated vectors. PLOS Negl. Trop. Dis 11:e0005819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cantey PT, Stramer SL, Townsend RL, Kamel H, Ofafa K, et al. 2012. The United States Trypanosoma cruzi Infection Study: evidence for vector-borne transmission of the parasite that causes Chagas disease among United States blood donors. Transfusion 52:1922–30 [DOI] [PubMed] [Google Scholar]
- 179.Garcia MN, Aguilar D, Gorchakov R, Rossmann SN, Montgomery SP, et al. 2015. Evidence of autochthonous Chagas disease in southeastern Texas. Am. J. Trop. Med. Hyg 92:325–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kransdorf EP, Fishbein MC, Czer LS, Patel JK, Velleca A, et al. 2016. Pathology of chronic Chagas cardiomyopathy in the United States: a detailed review of 13 cardiectomy cases. Am. J. Clin. Pathol 146:191–98 [DOI] [PubMed] [Google Scholar]
- 181.Cardillo F, de Pinho RT, Antas PR, Mengel J. 2015. Immunity and immune modulation in Trypanosoma cruzi infection. Pathog. Dis 73:ftv082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Coates BM, Sullivan DP, Makanji MY, Du NY, Olson CL, et al. 2013. Endothelial transmigration by Trypanosoma cruzi. PLOS ONE 8:e81187. [DOI] [PMC free article] [PubMed] [Google Scholar]