

Abstract
The change in glycosylation of serum proteins is often associated with the development of various diseases and thus can be used for diagnosis. In this study, a liquid chromatography-tandem mass spectrometry-based method is used for accurate structural analysis and quantification of site-specific glycoforms of serum α-1-antitrypsin (A1AT) in early-stage HCC and cirrhosis patients. Serum protein A1AT was purified from patient sera by immunoprecipitation with anti-A1AT antibody conjugated agarose beads, and the isolated A1AT protein was digested and analyzed by LC-MS/MS. Two tandem mass spectrometry strategies are integrated in this study: a nontargeted stepped HCD strategy for structural analysis of A1AT glycopeptides and a targeted parallel reaction monitoring (PRM) strategy for quantification of site-specific glycoforms of A1AT in HCC and cirrhosis patient sera. Accordingly, pGlyco2.0 software was used for glycopeptide identification, and Skyline software was used for glycoform quantification using the Y1 ion (peptide+GlcNAc) in MS/MS spectra. Ten site-specific glycopeptides of A1AT were identified with stepped HCD-MS/MS in patient samples, 7 of which were further quantified using HCD-PRM-MS among patient samples. We found that our strategy was able to distinguish isomers of glycopeptides where several isomers showed distinctly different patterns between cirrhosis and HCC patients. We also found that the ratio of different charge states (2+/3+) of one glycopeptide of A1AT can significantly discriminate early-stage HCC from cirrhosis with the area under the receiver operating characteristic curve AUC of 0.9. Further analysis showed that the difference may be related to the sialic acid/galactose linkage of the glycan motif.
Graphical Abstract
The change in glycosylation of serum proteins is often associated with the development of various diseases and thus may be potential markers for early detection, diagnosis, and therapeutic monitoring.1,2 The clinically applied α-fetoprotein (AFP-L3) in serum is the core-fucosylated form of AFP with higher specificity for detection of HCC, and carbohydrate antigen 19-9 (CA19-9), the sialyl lewis A structure on glycoproteins, is the only available biomarker for pancreatic cancer.1 Many of the serum proteins are synthesized by the liver, which are involved in the complement system, in acute phase response, or are protease and protease inhibitors.3 One of these important serum proteins is α-1-antitrypsin (A1AT).
More than 80% of A1AT is synthesized and secreted by hepatocytes in the liver. A1AT has antitrypsin activity and protects elastin in the alveoli of the lung by binding to elastase, which is produced by white blood cells during infection and can otherwise attack elastin.4 A1AT is a relatively high abundance protein in the blood, with circulating levels of 0.9–1.75 mg/mL; upon infection or inflammation, the concentration of A1AT can reach up to 4-fold.4 There are three N-glycosylation sites in A1AT, i.e., Asn70, Asn107, and Asn271. The aberrant glycosylation of A1AT has been found in previous studies to be indicative for nonalcoholic steatohepatitis,5 hepatocellular carcinoma (HCC),6–8 and lung cancer.9 However, structural analysis and quantification of site-specific glycosylation remain challenging.
Current studies on intact glycopeptides focus on qualitative comparisons, which involve the development of glycopeptide enrichment methods (i.e., ZIC-HILIC beads,10 MAX beads,11,12 and cotton tips13), MS/MS collision methods (i.e., EThcD,14 CID/HCD,15 and stepped HCD16), and software for automatic glycopeptide data interpretation (i.e., GPQuest,12 Byonic,17 I-GPA,15 LaCyTools,18 and pGlyco16). These studies used nontargeted approaches, some of which have explored relative quantification of intact glycopeptides based either on precursor intensity of glycopeptides using the softwares of Byologic,19 I-GPA,15 pQuant,20 and LaCyTools,18 or on isobaric tags in MS/MS using GPQuest software.12,21
In comparison, targeted quantitation strategies, such as parallel reaction monitoring (PRM),22 have not been well explored for intact glycopeptide analysis due to the complexity of glycopeptides composed of both peptide sequence and heterogeneous glycan structures. In targeted proteomics, PRM uses parent ion → daughter transitions to quantify peptides and performs MS/MS scan on targeted precursors only, resulting in higher sensitivity and better reproducibility than the precursor level quantification.22 Skyline software is designed for automated processing of raw data generated by PRM strategy.23 Most recently, there have been some attempts to use PRM for targeted glycopeptide quantitation. Yuan and co-workers combined collision-induced dissociation (CID) with LC-MS PRM for quantitative characterization of intact glycopeptides, where CID serves as a “soft” fragmentation to extract informative Y ions for quantitative comparisons.24 Kim and co-workers used CID with LC-MS PRM for quantitative analysis of truncated glycopeptides.25 Conventional CID generates mostly glycan fragments, and the fragment information is not enough for peptide identification. Therefore, the conventional CID method can be used for quantitative analysis when the sample does not contain many glycopeptides and the interesting glycopeptide has been identified with high confidence using HCD or EThcD, etc., or it has been identified at peptide and glycan level separately. It would be simpler to use the same collision method for both nontargeted profiling and for targeted quantification, where the MS/MS spectra from the two approaches are the same. The workflow developed by this study employed stepped HCD which not only generates both glycan and peptide fragments but also has a higher scan speed than EThcD. This workflow can be applied to a more complex sample.
In this study, we integrated two tandem mass spectrometry strategies to study intact glycopeptides of A1AT. First, we used a nontargeted stepped HCD strategy for structural analysis of A1AT glycopeptides; then we developed a novel LC-HCD-PRM-MS method which combines stepped HCD with LC-MS PRM to quantitate the changes of targeted intact glycopeptides between disease states. With the application of the LC-HCD-PRM-MS method, we are able to identify site-specific glycosylation changes of A1AT in early-stage HCC compared with cirrhosis and to quantify possible changes that may be potential new markers for HCC. In addition, a data processing pipeline has also been established by conjugating pGlyco16 and Skyline23 to identify and quantify intact glycopeptides automatically, which can be readily applied to any other unknown glycoprotein and even complex glycoprotein mixtures. Notably, this novel LC-HCD-PRM-MS method shows the ability to discriminate glycopeptide isoforms.
We identified most of the glycosylations of A1AT at the glycopeptide level which corresponded to those found at the glycan level previously.26 We chose 7 of the identified glycopeptides, which were observed in most patient samples, for quantification analysis with parallel reaction monitoring (PRM) among patient samples. Our results indicate that the PRM strategy could distinguish isomers of A1AT glycopeptides, some of which showed distinctly different patterns between cirrhosis and HCC patients. We further evaluated the ratio of different charge states (2+/3+) of glycopeptides in A1AT for distinguishing early HCC from cirrhosis patients.
■ MATERIALS AND METHODS
Purification of A1AT from Patient Serum with Antibody-Based Affinity Beads.
Clinical samples were provided by the Division of Gastroenterology, University of Michigan, Ann Arbor, MI, according to IRB approval, including 10 cirrhosis samples and 10 early-stage HCC samples, all of which were HCV related. The clinical features of patients with HCC and cirrhosis are summarized in Table 1. The HCC group contains primary HCC samples at early stages (TNM staging system): stage I (n = 3) and stage II (n = 7). Serum samples were aliquoted and stored at −80 °C before further usage. Serum protein A1AT was immunoprecipitated from 20 μL of patient serum using the Pierce cross-link immunoprecipitation kit according to the manufacturer’s instruction. Briefly, serum was thawed and diluted with 250 μL of phosphate-buffered saline (PBS), followed by IgG depletion using Protein A/G agarose beads (Pierce Scientific, Rockford, µL). A 10 μg amount of monoclonal antihuman A1AT antibody (Abcam, #ab116604) was covalently bound to Protein A/G plus agarose beads by the cross-linking reagent, disuccinimidyl suberate (DSS). The antibody-conjugated beads were then incubated with IgG-depleted serum at 4 °C overnight. After 4 washes with PBS, A1AT was eluted with 60 μL of elution buffer, followed by desalination using Zeba desalting spin columns (Pierce Scientific, Rockford, IL). The purified serum A1AT was then dried in a SpeedVac concentrator (Thermo fisher Scientific, San Jose, CA) and subjected to double-enzymatic digestion with Glu-C and chymotrypsin.
Table 1.
Summary of Patient Sample Characteristics
| disease dignosis | HCC | cirrhosis |
|---|---|---|
| number | 10 | 10 |
| etiology | HCV | HCV |
| gender (M/F) | 7/3 | 9/1 |
| age (mean ± SD) | 62.9 ± 12 | 56.1 ± 9 |
| AFP level (median, ng/mL) | 10.4 | 2.0 |
| MELD score | 10.0 ± 4.6 | NA |
| TNM stage (I/II) | 3/7 | NA |
| BCLC stage | A | NA |
Enzymatic Digestion of Protein into Peptides.
The purified serum A1AT was dissolved in 50 mM NH4HCO3, reduced with 20 mM DTT, and alkylated with 50 mM IAA, followed by dilution three times with 50 mM NH4HCO3. Glu-C (Promega, Madison, WI) was then added to the sample (1:30, enzyme to protein) and incubated at 37 °C overnight. After quenching the reaction at 95 °C for 10 min, chrymotrypsin (Promega, Madison, WI) was added and incubated at 37 °C for 12 h. The digests were dried down using a SpeedVac and reconstituted with 10 μL of 0.1% TFA. ZipTip C18 desalting was then performed as described previously.27
LC-mS Identification and Quantification of Glyco-peptides.
A C18 capillary column (100 μm × 15 cm; 3 μm particles, 200 Å) (Thermo fisher Scientific, San Jose, CA) was used for LC separation, and gradient elution was performed using an Ultimate 3000 nanoLC system (ThermoFisher Scientific, San Jose, CA) with a flow rate of 350 nL/min. The mobile phase A was 2% acetonitrile with 0.1% formic acid in water, and mobile phase B was 2% water with 0.1% formic acid in acetonitrile. The analytical gradient was 70 min long, where after 15 min of balancing time, the composition of solvent B rose from 3% to 32% in 45 min, from 32% to 40% in 3 min, and from 40% to 90% in 2 min, followed by a washing and equilibration step where solvent B remained in 90% for 5 min and then returned to 3% in 0.1 min and was held for 15 min.
An Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) operated in positive ion mode was used for analysis. We used optimized MS setting for A1AT glycopeptide analysis, where the RF was set as 20%, electrospray voltage at 2300 V, and ion transfer tube at 150 °C, to minimize the in-source dissociation of glycopeptides (<3%).27 Stepped collision energy CE (19%–26%–33%) was used to increase the fragmentation of both glycan and peptide. Other conditions included an MS1 scan range of 800–2000 m/z, an Orbitrap resolution of 60K, max ion injection time of 100 ms, AGC target 4E5, an MS/MS scan range of 130–2000 m/z, an isolation window of 1.6 Da, an Orbitrap resolution of 30K, a max ion injection time of 250 ms, and an AGC target of 2E5. The difference between profiling and PRM analysis is that the latter performs MS/MS only on precursors shown on the preloaded target list.
Mass Spectrometry Data Analysis.
Glycopeptide Identification with pGlyco2.0.
The search engine pGlyco2.0 was used for glycoprotein analysis16 with the following(1) fixed modification: cysteine carbamidomethylation (+57.021 Da); (2) dynamic modification: methionine oxidation (+15.995 Da), acetylation (+42.011) at protein N-terminus; (3) one missed cleavage was allowed; (4) peptide ion tolerance = 15 ppm and fragment ion tolerance = 25 ppm; (5) built-in glycan database from human. All identified glycopeptides were manually checked by GlycoWorkbench Software developed by the EUROCarbDB.28 The nomenclature of glycans is used according to Essentials of Glycobiology,29 and the abbreviations are used according to the NIBRT GlycoBase.
Glycopeptide Quantification with Skyline.
Skyline software23 was used for quantification of selected glycopeptides, where the Y1 ion (peptide+GlcNAc), oxonium ions (138.055 and 204.087), and/or possible b/y ions were used for identification and the Y1 ion was used for quantification.25 Skyline analysis requires two types of settings, peptide settings and transition settings, to generate a spectral library which is applied to patient samples for quantification. In peptide settings, a background library was generated by uploading raw data, the fasta sequence, and a list of identified glycopeptides which were identified from the same raw data by pGlyco2.0. The list using *.ssl format (.txt based) includes the following information: file name of the raw data, scan number, charge, peptide sequence with [+glycan mass] at the modified Asn, and approximate retention time. The digestion method and filter for peptide length were set accordingly. All possible glycan side chains were added as structural modifications, so that Skyline can correlate the glycan mass indicated in the *.ssl with a glycan structure. In transition settings, ion types were set as b, y, and special ions where oxonium ion 138.055, 204.087 and Y1 ions were added, so that Skyline would automatically assign oxonium ions, Y1 ions, and other possible b, y ions as transitions. The ion match tolerance was set as 0.020 Da for parent ions and for transitions. A spectral library was then generated, where oxonium ions, Y1 ions, and the optimal b, y ions were selected to identify the glycopeptides. Skyline is then ready for importing the raw data of patients for quantitative analysis. Integration of all transitions was automatically calculated and could be exported as a report after manual checking. The Skyline analysis workflow is shown in Supporting Figure S1.
■ RESULTS AND DISCUSSION
The experimental workflow is shown in Figure S2. We used Glu-C/chymotrypsin to digest A1AT to produce shorter glycopeptides compared to trypsin digestion (Supporting Information Table S1). The glycosylation profile at the glycopeptide level of A1AT from patient samples showed that Asn107 has the most diversified glycosylation forms with biantennary and triantennary N-glycans with/without fucosylation compared to Asn271 and Asn70, which is consistent with our previous study.26 For statistical reasons, we chose 7 out of 10 glycopeptides of A1AT which were found in most samples for PRM analysis (Table 2), including glycopeptides (at site Asn107) with bi- and triantennary glycans and their fucosylated forms, glycopeptides (at site Asn70) with a biantennary glycan and its fucosylated form, and glycopeptides (at site Asn271) with a biantennary glycan at both charge 2+ and charge 3+ states. Oxonium ions (138.055 and 204.087) and Y1 (peptide+GlcNAc) of each corresponding glycopeptide were used for identification, and Y1 ion was used for the quantification of glycopeptides. The coefficient of variation (CV) for quantification using Y1 ion was compared with oxonium ions (138.055 and 204.085) and five other Y ions (Y2, pep+2GlcNAc; Y3, pep+2GlcNAc+Hex; Y4, pep+2GlcNAc+2Hex; Y5, pep+2GlcNAc+ 3Hex; Y6, pep+3GlcNAc+3Hex) in Supporting Table S2, where the Y1 ion showed the interday CV = 4.13%, lower than oxonium ions and comparable with that of summed Y ions. Y1 has the strongest signal intensity among all Y ions (Figure 1B) and thus is a better choice for quantification compared to the use of the summed Y ions. Also, the linearity using Y ions to quantitate the parent ion has been shown in a previous study.25 Therefore, we used the Y1 ion for quantification.
Table 2.
Glycopeptides of A1AT with Glu-C/Chymotrypsin Digestiona
| peptide | glycan | experimental m/z | charge (z) | theoretical m/z | Δppm | Y1 ion |
|---|---|---|---|---|---|---|
| with glycosylation site Asn70 | ||||||
| AHQSnSTNIF | A2G2S2(54200)* | 1108.4446 | 3+ | 1108.4363 | 7.5 | 1321.6060 |
| FA2G2S2(54201)* | 1157.1300 | 3+ | 1157.1233 | 6.7 | ||
| AHQSnSTNIFF | A2G2S2(54200) | 1157.4679 | 3+ | 1157.4589 | 7.8 | |
| FA2G2S2(54201) | 1206.1545 | 3+ | 1206.1449 | 8.0 | ||
| with glycosylation site Asn107 | ||||||
| nLTEIPE | A2G2S2(54200)* | 1007.4070 | 3+ | 1007.4003 | 6.7 | 1018.4965 |
| FA2G2S2(54201)* | 1056.0955 | 3+ | 1056.0863 | 8.7 | ||
| A3G3S3(65300)* | 1226.1520 | 3+ | 1226.1428 | 7.5 | ||
| A3FG3S3(65301)* | 1274.8389 | 3+ | 1274.8288 | 7.9 | ||
| with glycosylation site Asn271 | ||||||
| LGnATAIF | A2G2S2(54200)* | 1004.4170 | 3+ | 1004.4090 | 8.0 | 1009.5201 |
| A2G2S2(54200)* | 1506.1220 | 2+ | 1506.1135 | 5.6 | ||
| GnATAIF | A2G2S2(54200) | 966.7211 | 3+ | 966.7169 | 4.3 | |
aGlycopeptides selected for PRM quantification are labeled with an asterisk, and corresponding Y1 ions are shown.
Figure 1.
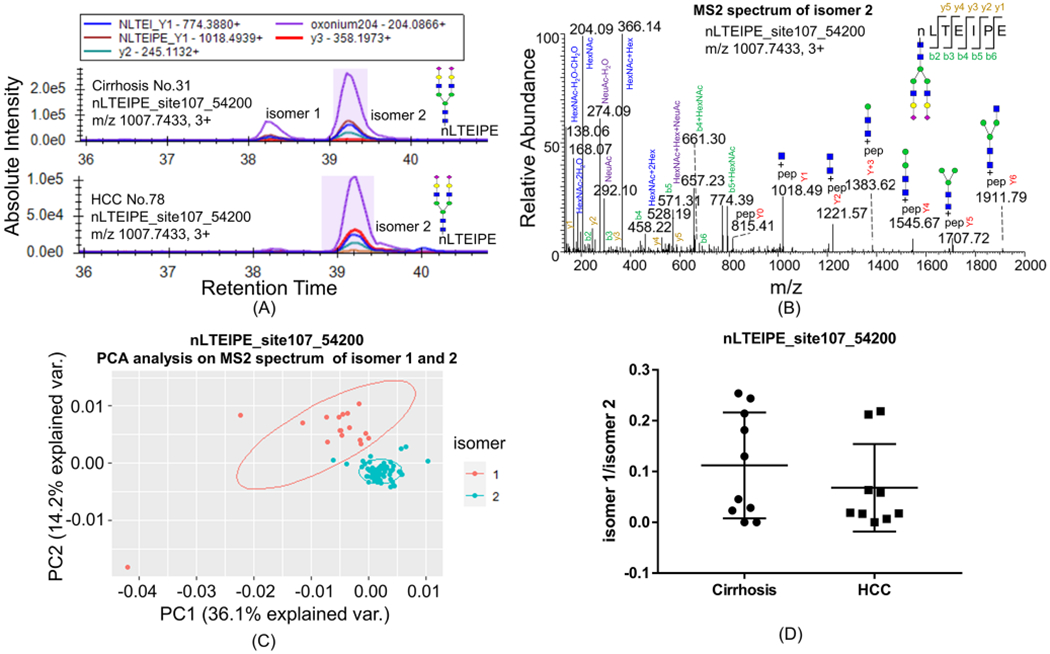
Isomers of glycopeptides nLTEIPE_54200 on a C18 column with the PRM method. (A) Most cirrhosis samples have both isomers 1 and 2 of nLTEIPE_54200 (top), whereas only a few early-stage HCC have both isomers (bottom). (B) MS2 spectrum of isomer 2. (C)PCA analysis of MS2 spectra of isomer 1 and isomer 2. MS2 spectra with ≥3 Y ions were included for analysis (total 77 scans); common fragments from all spectra (<20 ppm) were used as variables; peak intensity was converted to square root and normalized as a percentage of total summed intensity before PCA analysis. (D) Ratio of isomer1/isomer 2 in cirrhosis and early-stage HCC samples does not show a significant difference with Student’s t test.
Ten cirrhosis samples and 10 HCV-related early-stage HCC samples were used in this study. We found that some isomers of the glycopeptides were readily separated by the C18 column. Seven out of 10 cirrhosis samples were found to have both isomers 1 and 2 of nLTEIPE_54200 (indicating 5 Hex, 4 HexNAc, 2 NeuAc, 0 NeuGc, and 0 dHex) with Asn107 (Figure 1A, top), whereas 3 out of 9 early-stage HCC have both isomers (Figure 1A, bottom) (one sample did not show either isomer). The MS/MS spectra profiles of isomers 1 and 2 are similar, and that of isomer 2 is shown in Figure 1B. Principal component analysis (PCA) of the relative intensity of MS/MS fragments showed two distinct clusters of the two isomers (Figure 1C), indicating these two are true isomers. The alignment of the MS/MS fragments of isomer 1 and isomer 2 is shown in Figure S3. The abundance ratio of isomer 1/isomer 2 of nLTEIPE_54200 in all samples is plotted (Figure 1D). Although most HCC samples do not have isomer 1, the change was found not to be significant with respect to the Student’s t test. Ji and co-workers found that the isomers separated on a C18 column at high temperature is due to the difference of sialic linkages.30 These two isomers of nLTEIPE_54200 are probably also due to the different sialic linkages.
The two isomers of nLTEIPE_54201 (Supporting Figure S4A) and the four isomers of nLTEIPE_65300 were not well separated (Supporting Figure S4B). However, the two isomers of nLTEIPE_65301 were well separated, where again the two isomers of nLTEIPE_65301 were found in most cirrhosis samples (Figure 2A, top), and only one isomer was found in most early-stage HCC samples (Figure 2A, bottom). The abundance ratio of isomer1/isomer 2 of nLTEIPE _65301 in cirrhosis and early-stage HCC samples is shown in Figure 2B. In a comparison of Figure 1D and Figure 2B, we found that the pattern of the two plots are similar. We performed a correlation test of nLTEIPE_54201 isomer 1/2 and nLTEIPE_65301 isomer 1/2, where R2 turns out to be 0.9537 (Figure 2C). Therefore, the relative abundance of isomer 1/isomer 2 of glycopeptide nLTEIPE_54200 and nLTEIPE_65301 are very much correlated. The MS/MS spectrum of nLTEIPE_65301 contains oxonium ion of 512.1959, indicating this is an antennary fucosylation (Supporting Figure S5), consistent with our previous study where triantennary core fucosylation of this site was not detected.26 Addition of an antennary fucosylated branch (GlcNAc-galactose (fucose)-sialic acid) of nLTEIPE_65301 did not change the isomer types of nLTEIPE_54200. This indicates that nLTEIPE_65301 may share the same core structure of nLTEIPE_54200. A previous study has shown that most of the third antenna of nLTEIPE_65301 with an antennary fucosylation is α2,3-linked sialic acid, indicating this antennary fucosylation is specifically related to α2,3-linked sialic acid.31 The physiological addition of the antennary fucosylation on the third antenna may be α2,3-linkage specific. Also, the antennary fucosylation of nLTEIPE_65300 on the α2,3-linkage branch does not seem to be indicative for HCC.
Figure 2.
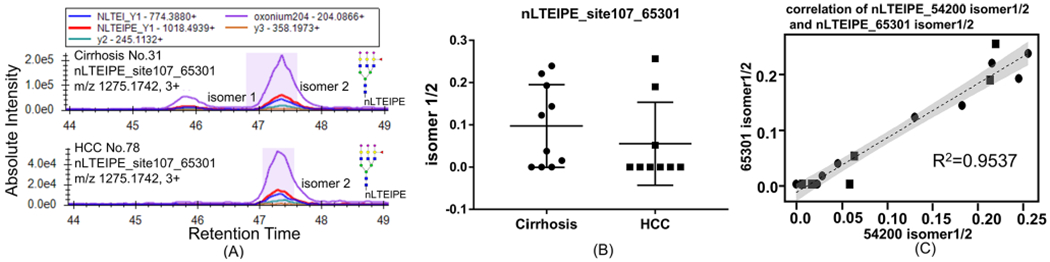
Isomers of glycopeptides nLTEIPE_65301 on a C18 column with the PRM method. (A) Two isomers of nLTEIPE_65301 were found in most cirrhosis samples (top), and only one isomer was found in most early-stage HCC samples (bottom). (B) Abundance ratio of isomer1/isomer 2 of nLTEIPE _65301 in cirrhosis and early-stage HCC samples. (C) Correlation between nLTEIPE_54201 isomer1/2 and nLTEIPE_65301 isomer 1/2. Cirrhosis sample indicated by a circle, and HCC sample indicated by a square.
The reason that isomers were readily observed in our glycopeptide MS spectra could be due to the short peptide sequences and the noncharged glycopeptide C-terminus generated by double Glu-C/chymotrypsin digestion. The most commonly used tryptic digestion would generate longer glycopeptides of A1AT and would generate glycopeptides with the C-terminal positively charged R and K, which may reduce the linkage difference of glycans, especially the negatively charged sialic acid. Quantification of glycopeptides with the PRM strategy is essential where without the PRM strategy we may only have identification at the elution time of one isomer and would quantify that isomer based only on the precursor ion (Figure S6).
Most interestingly, we found that the abundance ratio of two charge states of LGnATAIF_54200 with Asn271, i.e., 2+ versus 3+, significantly increased in early HCC compared to cirrhosis samples. The XIC of daughter ions of LGnATAIF_54200 2+ and 3+ is shown in Figure 3A (top) and 3A (bottom), respectively. The MS1 spectrum of parent ions with the two charge states (top) and the MS2 spectrum of the charge 2+ form (bottom) are shown in Figure 3B. The ratio of LGnATAIF_54200 2+/3+ in cirrhosis and early-stage HCC is plotted in Figure 3C (p = 0.0003). The receiver operating characteristic curve (ROC) analysis is shown in Figure 3D, where the area under the receiver operating characteristic curve (AUC) of LGnATAIF_54200 2+/3+ alone is 0.900 and a combined analysis with AFP is 0.944, whereas AFP alone provides an AUC of 0.839. The same pattern holds for MS1 level quantification data shown in Supporting Figure S7. The XIC of parent ions in two different charge states are similar (Figure S7A); the box plot of 2+/3+ from MS1 level quantification in cirrhosis and HCC is shown in Figure S7B, and the ROC analysis of MS1 level quantification is shown in Figure S7C, from which we found that the ratio of 2+/3+ is also higher in HCC samples at MS1 level quantification but with lower AUC(0.84) compared with the above PRM-based quantification (AUC = 0.90).
Figure 3.
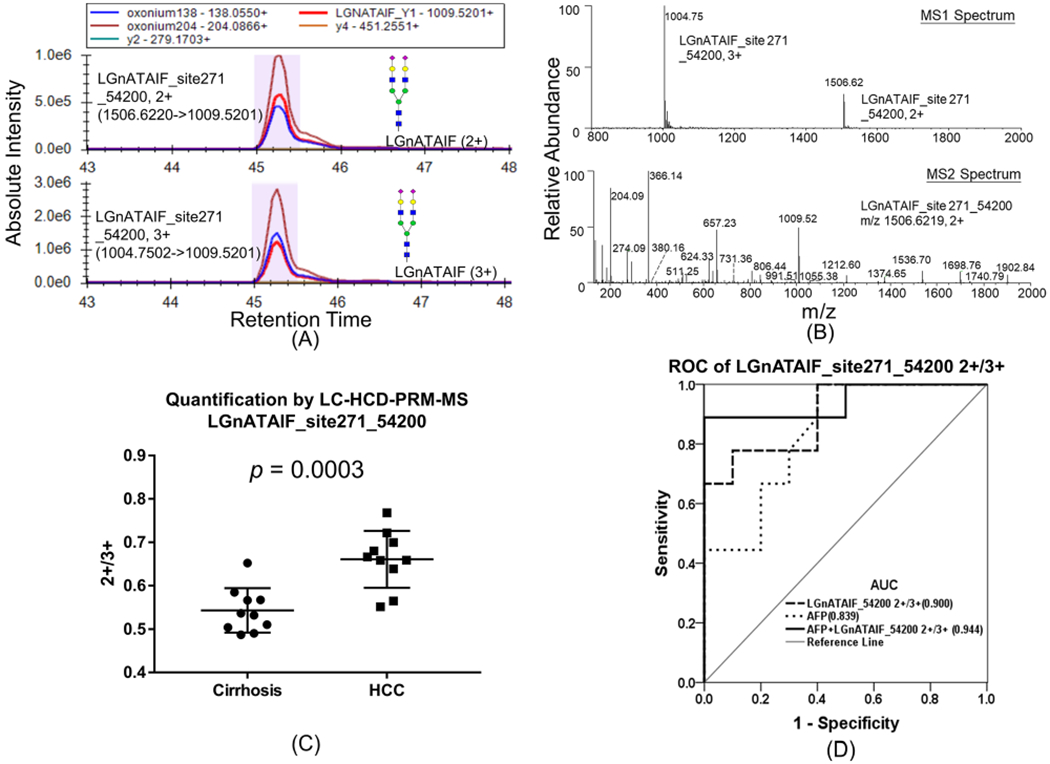
(A) PRM quantification of glycopeptide LGnATAIF_54200 2+ (top) and LGnATAIF_54200 3+ (bottom). (B) MS1 spectrum of parent ions with two charge states (top), and MS2 spectrum of charge 2+ state (bottom). (C) Relative abundance of glycopeptide LGnATAIF_54200 2+/3+ in cirrhosis and HCC patients. (D) ROC analysis of LGnATAIF_54200 2+/3+ in patients.
Yet, the charge ratio may change under different experimental conditions where it is not practical to directly apply the charge ratio for early HCC diagnosis. We thus sought to uncover the mechanism of the charge ratio change. The structure level difference (such as glycan linkages) which leads to the charge ratio difference may be used as a biomarker. We then studied the difference in MS/MS spectra between LGnATAIF_54200 2+ and 3+ glycopeptides. The common daughter ion fragments of the two glycopeptides with 2+ or 3+ were aligned (Figure 4A) based on the fragment intensity. The MS2 spectrum of LGnATAIF_54200 3+ is shown in Figure 4B. Most daughter ion fragments in charge 2+ and charge 3+ forms were linearly correlated, except four fragments, m/z 366.1385, 657.2346, and 1901.8370, were much higher in the charge 2+ form and 274.0917 was much higher in the charge 3+ form. Fragments m/z 366.1385 and 657.2346 are the oxonium ions of GlcNAc-galactose and GlcNAc-galactosesialic acid, respectively,32 and m/z 1901.8370 is the Y6 ion (peptide+2GlcNAc-3mannose-GlcNAc). Fragment m/z 274.0917 is the oxonium ion of sialic acid.32 The three fragments m/z 366.1385, 657.2346, and 1901.8370 were more abundant in the charge 2+ form, indicating that linkage X is easier to break in charge 2+, whereas the fragment 274.0917 was more abundant in charge 3+ form, indicating that linkage Y (indicated in Figure 4C) is easier to break in the charge 3+ form, and the extra H+ in the charge 3+ form may closely cluster around the sialic acid. The fact that the ratio of LGnATAIF_54200 2+/3+ significantly increased in early-stage HCC relative to cirrhosis samples indicates that LGnATAIF_54200 may have a glycan linkage difference beyond the Y6 fragment in HCC samples which weakly attracts H+ ions compared to that in cirrhosis samples.
Figure 4.
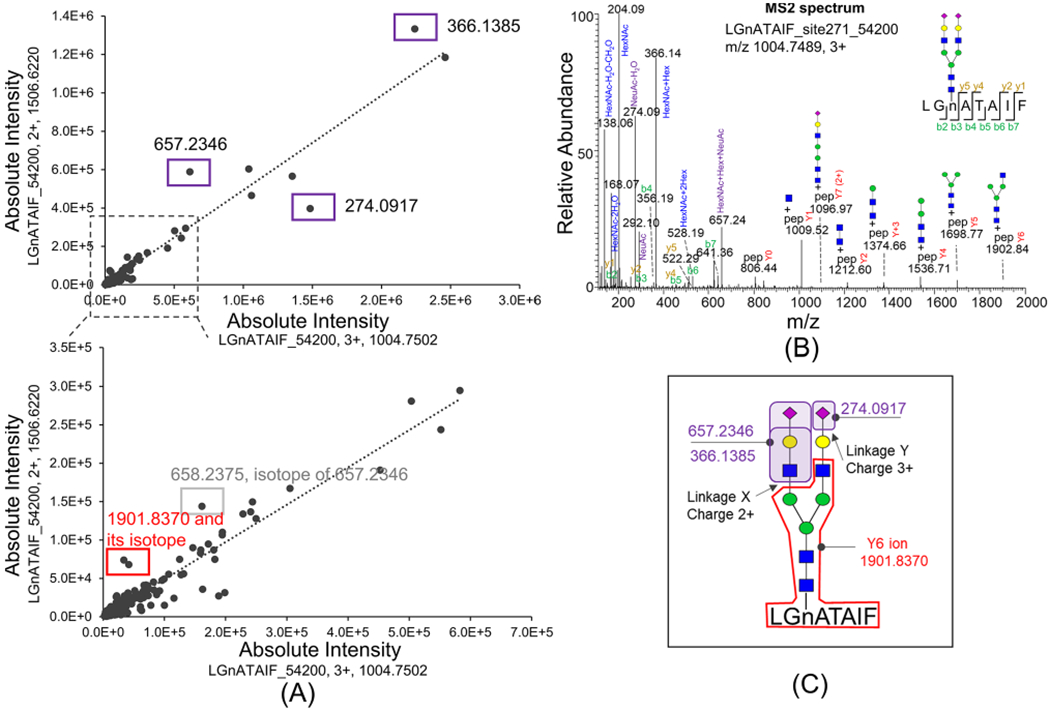
MS2 spectral comparison of glycopeptide LGnATAIF_54200 with charge 2+ and charge 3+. (A) Common fragments in the LGnATAIF_54200 2+ MS/MS spectrum versus those in its 3+ form (<20 ppm) are aligned. Significantly different fragments are labeled in boxes: 366.1385, 657.2346, and 274.0917 are oxonium ions of glycans; 1901.8370 is the Y6 ion. (B) MS2 spectrum of LGnATAIF_54200 with charge 3+. (C) LGnATAIF_54200 with charge 2+ state favors fragmenting at linkage X, and with charge 3+ state it favors fragmenting at linkage Y.
The glycan linkage microheterogeneity in LGnATAIF_54200 with Asn271 beyond the Y6 fragment could be due to galactose linkage or sialac acid linkage. The glycopeptide mixture of commercial A1T digested by trypsin was further treated with different exoglycosidases to differentiate the galactose and sialic acid linkages of glycans on Asn271 (Figure S8). The XIC of glycopeptides after digestion are shown in Supporting Figure S9. Biantennary glycan on YLGnATAIFFLPDEGK at site Asn271 has about 15% β1,6-linked galactose, about 81.6% β1,4-linked galactose about 91.6% α2,6-linked sialic acid, and about 8.4% α2,3-linked sialic acid (Figure S8). The A1AT glycopeptide LGnATAIF_54200 2+/3+ in early-stage HCC samples is significantly higher than that in cirrhosis samples, which may possibly be due to its aberrant galactose and sialic linkages at glycosylation site Asn271 in HCC samples.
The change of sialic acid linkage has been shown on prostate-specific antigen (PSA) for aggressive prostate cancer33 and on blood glycans of type II diabetes.34 The blood aberrant glycan profile is found to be indicative for fibrosis, cirrhosis, and HCC by Glycomics assays from Helena-Biosciences, which is based on electrophoretic N-glycan separation with UV detection. Core fucosylated AFP showed an improved diagnosis value compared to AFP for early diagnosis of HCC, but the galactose/sialic acid linkage differences have not been considered. In a recent study, the PRM strategy has been applied to analysis of the fucosylation level of AFP glycopeptide VnFTEIQK_54000 (AUC = 0.889) and vitronectin glycopeptide nGSLFAFR_65000 (AUC = 0.792) in HCC compared with cirrhosis samples.25 In our results, the fucosylation level of neither A1AT glycopeptides showed a significant difference (Supporting Figure S10), but the galactose/sialic acid linkage change of A1AT may serve as a new marker for early-stage HCC.
■ CONCLUDING REMARKS
In this study, we identified 10 glycopetides of serum A1AT on three glycosylation sites (Asn70, Asn107, and Asn271) in cirrhosis and early-stage HCC patients. Seven of these glycopeptides were selected for PRM quantification using a novel LC-HCD-PRM-MS method. The m/z list of the selected glycopeptides was used for parallel reaction monitoring MS analysis. The raw data were analyzed by Skyline software for quantitative comparison using parent ion → Y1 ion transition. Several glycopeptide isomers were readily separated on a C18 column, some of which showed different patterns between cirrhosis and HCC patients. One glycopeptide LGnATAIF_54200 showed a significantly different distribution of charge states in cirrhosis compared to HCC with an AUC of 0.9. Further glycan linkage analysis with exoglycosidases suggested that the different charge states in a glycopeptide may be related to the galactose/sialic acid linkage differences of the glycan motif.
Supplementary Material
Table S1, Glu-C/chymotrypsin for A1AT digestion compared with trypsin digestion; Table S2, reproducibility of the PRM quantification using various transition ions; Figure S1, Skyline analysis flowchart for quantification of glycopeptides; Figure S2, experimental workflow to identify glycopeptides and to quantify them by LC-HCD-PRM-MS; Figure S3, MS/MS spectra of the two isomers of nLTEIPE_54200 with Asn107; Figure S4, other isomers of nLTEIPE glycopeptide; Figure S5, MS/MS spectrum of nLEIPE_65301 with Asn107; Figure S6, the importance to quantitate glycopeptides with the PRM strategy; Figure S7, MS1 level analysis of LGnATAIF_54200 with Asn271 in charge 2+ and 3+ forms; Figure S8, exoglycosidase digestion on commercial A1AT standard from human serum for galactose and sialic acid linkage differentiation; Figure S9, extracted chromatogram of glycopeptides with various glycosidase digestion; Figure S10, fucosylation level of A1AT at Asn107 (nLTEIPE) and Asn70 (AHQSnSTNIF) among early-stage HCC and cirrhosis patients (PDF)
■ ACKNOWLEDGMENTS
This work was funded by the National Natural Science Foundation of China (81601828). We are thankful for software support from Dr. Zeng WF from the pGlyco group in the Institute of Computing Technology, CAS, Beijing, and software support from the MacCross lab at the University of Washington, Seattle, WA. We are thankful for R programming support from Ms. Liu FJ from China Pharmaceutical University, Nanjing. Also, we are thankful for support from the University Research Facility in Chemical and Environmental Analysis and the University Research Facility in Life Sciences of Hong Kong Polytechnic University. Partial support was also provided by the National Cancer Institute under grant nos. R01 CA160254 (D.M.L.), U01 CA225753 (D.M.L.), R50 CA221808 (J.Z.), and by the Hong Kong RGC CRF grants C5031-14E (Z.Y.), C4002-17G (Z.Y.).
■ ABBREVIATIONS:
- A1AT
α-1-antitrypsin
- HCC
hepatocellular carcinoma
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- PRM
parallel reaction monitoring
- DTT
dithiothreitol
- IAA
iodoacetamide
- TFA
trifluoroacetic acid
- ROC
receiver operating characteristic curve
- AUC
area under the receiver operating characteristic curve
- HCD
higher energy collision dissociation
- XIC
extracted ion chromatogram
- CE
collision energy
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c00420.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c00420
The authors declare no competing financial interest.
Contributor Information
Haidi Yin, The Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen, People’s Republic of China; Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong.
Jianhui Zhu, Department of Surgery, University of Michigan Medical Center, Ann Arbor, Michigan 48109, United States.
Mengmeng Wang, Department of Surgery, University of Michigan Medical Center, Ann Arbor, Michigan 48109, United States.
Zhong-Ping Yao, The Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen, People’s Republic of China; Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong.
David M. Lubman, Department of Surgery, University of Michigan Medical Center, Ann Arbor, Michigan 48109, United States.
■ REFERENCES
- (1).Pinho SS; Reis CA Nat. Rev. Cancer 2015, 15 (9), 540–555. [DOI] [PubMed] [Google Scholar]
- (2).Stowell SR; Ju T; Cummings RD Annu. Rev. Pathol.: Mech. Dis 2015, 10, 473–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Putnam FW The Plasma Proteins: Structure, Function, and Genetic Control; Academic Press, 1975. [Google Scholar]
- (4).de Serres F; Blanco IJ Intern. Med 2014, 276 (4), 311–35. [DOI] [PubMed] [Google Scholar]
- (5).Ogawa K; Kobayashi T; Furukawa J.-i.; Hanamatsu H; Nakamura A; Suzuki K; Kawagishi N; Ohara M; Umemura M; Nakai M; Sho T; Suda G; Morikawa K; Baba M; Furuya K; Terashita K; Kobayashi T; Onodera M; Horimoto T; Shinada K; Tsunematsu S; Tsunematsu I; Meguro T; Mitsuhashi T; Hato M; Higashino K; Shinohara Y; Sakamoto N Sci. Rep 2020, 10 (1), 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ahn YH; Shin PM; Oh NR; Park GW; Kim H; Yoo JS J. Proteomics 2012, 75 (17), 5507–15. [DOI] [PubMed] [Google Scholar]
- (7).Ahn YH; Shin PM; Kim YS; Oh NR; Ji ES; Kim KH; Lee YJ; Kim SH; Yoo JS Analyst 2013, 138 (21), 6454–62. [DOI] [PubMed] [Google Scholar]
- (8).Zhu J; Warner E; Parikh ND; Lubman DM Mass Spectrom. Rev 2019, 38 (3), 265–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liang Y; Ma T; Thakur A; Yu H; Gao L; Shi P; Li X; Ren H; Jia L; Zhang S; Li Z; Chen M Glycobiology 2015, 25 (3), 331–40. [DOI] [PubMed] [Google Scholar]
- (10).Mysling S; Palmisano G; Hojrup P; Thaysen-Andersen M Anal. Chem 2010, 82 (13), 5598–609. [DOI] [PubMed] [Google Scholar]
- (11).Sun SS; Shah P; Eshghi ST; Yang WM; Trikannad N; Yang S; Chen LJ; Aiyetan P; Hoti N; Zhang Z; Chan DW; Zhang H Nat. Biotechnol 2016, 34 (1), 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cho KC; Chen L; Hu Y; Schnaubelt M; Zhang H ACS Chem. Biol 2019, 14 (1), 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Selman MH; Hemayatkar M; Deelder AM; Wuhrer M Anal. Chem 2011, 83 (7), 2492–9. [DOI] [PubMed] [Google Scholar]
- (14).Yu Q; Wang B; Chen Z; Urabe G; Glover MS; Shi X; Guo LW; Kent KC; Li LJ Am. Soc. Mass Spectrom 2017, 28 (9), 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Park GW; Kim JY; Hwang H; Lee JY; Ahn YH; Lee HK; Ji ES; Kim KH; Jeong HK; Yun KN; Kim YS; Ko JH; An HJ; Kim JH; Paik YK; Yoo JS Sci. Rep 2016, 6, 21175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Liu MQ; Zeng WF; Fang P; Cao WQ; Liu C; Yan GQ; Zhang Y; Peng C; Wu JQ; Zhang XJ; Tu HJ; Chi H; Sun RX; Cao Y; Dong MQ; Jiang BY; Huang JM; Shen HL; Wong CCL; He SM; Yang PY Nat. Commun 2017, 8 (1), 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bern M; Kil YJ; Becker C Curr. Protoc Bioinformatics 2012, 40, 13.20.1–13.20.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jansen BC; Falck D; de Haan N; Hipgrave Ederveen AL; Razdorov G; Lauc G; Wuhrer MJ Proteome Res. 2016, 15 (7), 2198–210. [DOI] [PubMed] [Google Scholar]
- (19).Zhu J; Chen Z; Zhang J; An M; Wu J; Yu Q; Skilton SJ; Bern M; Ilker Sen K; Li L; Lubman DM J. Proteome Res 2018, 18 (1), 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Qin H; Dong X; Mao J; Chen Y; Dong M; Wang L; Guo Z; Liang X; Ye MJ Proteome Res. 2019, 18 (9), 3439–3446. [DOI] [PubMed] [Google Scholar]
- (21).Sun S; Shah P; Eshghi ST; Yang W; Trikannad N; Yang S; Chen L; Aiyetan P; Hoti N; Zhang Z; Chan DW; Zhang H Nat. Biotechnol 2016, 34 (1), 84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Peterson AC; Russell JD; Bailey DJ; Westphall MS; Coon JJ Mol. Cell. Proteomics 2012, 11 (11), 1475–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26 (7), 966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yuan W; Benicky J; Wei R; Goldman R; Sanda MJ Proteome Res. 2018, 17 (8), 2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kim KH; Park GW; Jeong JE; Ji ES; An HJ; Kim JY; Yoo JS Anal. Bioanal. Chem 2019, 411 (14), 3009–3019. [DOI] [PubMed] [Google Scholar]
- (26).Yin H; Zhu J; Wu J; Tan Z; An M; Zhou S; Mechref Y; Lubman DM Electrophoresis 2016, 37 (20), 2624–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yin H; An M; So PK; Wong MY; Lubman DM; Yao Z Electrophoresis 2018, 39 (18), 2351–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ceroni A; Maass K; Geyer H; Geyer R; Dell A; Haslam SM J. Proteome Res 2008, 7 (4), 1650–9. [DOI] [PubMed] [Google Scholar]
- (29).In Essentials of Glycobiology, 2nd ed.; Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, Eds.; Cold Spring Harbor Laboratory Press: New York, 2008; p 107. [PubMed] [Google Scholar]
- (30).Ji ES; Lee HK; Park GW; Kim KH; Kim JY; Yoo JS J. Chromatogr. B: Anal. Technol. Biomed. Life Sci 2019, 1110-1111, 101–107. [DOI] [PubMed] [Google Scholar]
- (31).Kolarich D; Weber A; Turecek PL; Schwarz HP; Altmann F Proteomics 2006, 6 (11), 3369–80. [DOI] [PubMed] [Google Scholar]
- (32).Huddleston MJ; Bean MF; Carr SA Anal. Chem 1993, 65 (7), 877–84. [DOI] [PubMed] [Google Scholar]
- (33).Ferrer-Batalle M; Llop E; Ramirez M; Aleixandre RN; Saez M; Comet J; de Llorens R; Peracaula R Int. J. Mol. Sci 2017, 18 (4), 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Dotz V; Wuhrer M FEBS Lett. 2019, 593 (21), 2966–2976. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Glu-C/chymotrypsin for A1AT digestion compared with trypsin digestion; Table S2, reproducibility of the PRM quantification using various transition ions; Figure S1, Skyline analysis flowchart for quantification of glycopeptides; Figure S2, experimental workflow to identify glycopeptides and to quantify them by LC-HCD-PRM-MS; Figure S3, MS/MS spectra of the two isomers of nLTEIPE_54200 with Asn107; Figure S4, other isomers of nLTEIPE glycopeptide; Figure S5, MS/MS spectrum of nLEIPE_65301 with Asn107; Figure S6, the importance to quantitate glycopeptides with the PRM strategy; Figure S7, MS1 level analysis of LGnATAIF_54200 with Asn271 in charge 2+ and 3+ forms; Figure S8, exoglycosidase digestion on commercial A1AT standard from human serum for galactose and sialic acid linkage differentiation; Figure S9, extracted chromatogram of glycopeptides with various glycosidase digestion; Figure S10, fucosylation level of A1AT at Asn107 (nLTEIPE) and Asn70 (AHQSnSTNIF) among early-stage HCC and cirrhosis patients (PDF)