

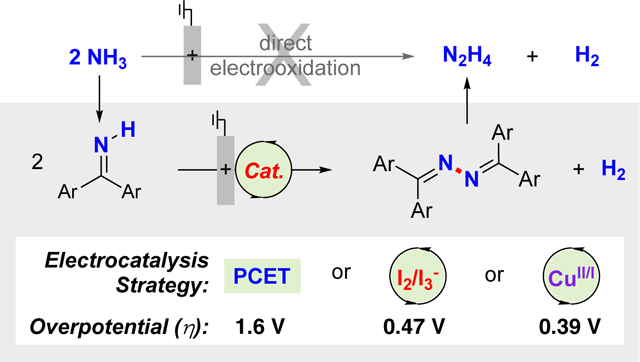
Abstract
Hydrazine is an important industrial chemical and fuel that has attracted considerable attention for use in liquid fuel cells. Ideally, hydrazine could be prepared via direct oxidative coupling of ammonia, but thermodynamic and kinetic factors limit the viability of this approach. The present study evaluates three different electrochemical strategies for the oxidative homocoupling of benzophenone imine, a readily accessible ammonia surrogate. Hydrolysis of the resulting benzophenone azine affords hydrazine and benzophenone, with the latter amenable to recycling. The three different electrochemical N–N coupling methods include (1) a proton-coupled electron-transfer process promoted by a phosphate base, (2) an iodine-mediated reaction involving intermediate N–I bond formation, and (3) a copper-catalyzed N–N coupling process. Analysis of the thermodynamic efficiencies for these electrochemical imine-to-azine oxidation reactions reveals low overpotentials (η) for the copper and iodine mediated processes (390 and 470 mV, respectively), but a much higher value for the proton-coupled pathway (η ~ 1.6 V). A similar approach is used to assess molecular electrocatalytic methods for electrochemical oxidation of ammonia to dinitrogen.
Graphical Abstract
Introduction
Nitrogen-based fuels represent compelling carbon-free alternatives to conventional fossil fuels for energy production. Ammonia (NH3) is an especially appealing energy carrier owing to its high energy density and established commercial sources of production and distribution networks.1–4 These considerations have led to the growing interest in electrocatalytic strategies for ammonia oxidation to dinitrogen (N2) in an effort to support improved performance of ammonia-based fuel cells.5–7 Hydrazine is a complementary fuel that has even higher energy density than ammonia,8 and it exhibits excellent performance in chemical and electrochemical power generation.9,10 While these merits are partially offset by its toxicity and safety hazards, hydrazine-based fuel cells employing non-precious metal electrocatalysts have already been demonstrated in electric vehicles.11,12 The appeal of hydrazine would be further improved if it could be synthesized efficiently on an as-needed basis. The (electro)catalytic oxidative coupling of ammonia represents an ideal target, but suitable methods do not exist. Dehydrogenative N–N coupling of ammonia into hydrazine is very unfavorable (ΔG° = +44 kcal/mol13,14,15) and would require the use of a strong oxidant or a suitable electrode potential to promote the reaction. But, any catalyst capable of promoting ammonia-to-hydrazine conversion will inevitably promote the highly favorable dehydrogenation of hydrazine to dinitrogen (ΔG° = −36 kcal/mol13,15,16). A complementary thermodynamic perspective is provided by the standard potentials for these reactions in aqueous solution (Figure 1A).17,18 The values show that dehydrogenation of hydrazine is 1.3–1.5 V more favorable than N–N coupling of ammonia to hydrazine, with the specific value depending on the protonation states of the different species at different pH. Consistent with these insights, molecular catalysts that have been reported for electrochemical ammonia oxidation in recent years generate dinitrogen as the N–N coupled product, including those proposed to proceed via hydrazine intermediates.19–26
Figure 1. (Electrochemical) Oxidative N–N bond formation.

A: Thermodynamic potentials of N2/N2H4/NH3 interconversion in aqueous media; B: The Hayashi process for hydrazine synthesis from ammonia; C: Our electrochemical approaches for formal hydrazine synthesis from ammonia via catalytic N–N bond formation.
The considerations outlined above demonstrate that alternative strategies are needed for electrochemical synthesis of hydrazine, and relevant insights come from industrial chemistry. Some of the most important industrial processes employ ketones as recyclable ‘protecting groups’,27 wherein condensation of ammonia with a ketone affords a primary imine as an ammonia surrogate with only one N–H bond. Oxidative coupling of the imine affords the N–N-coupled ketazine, and subsequent hydrolysis affords hydrazine and regenerates the ketone. These precedents suggested that an analogous electrochemical method could be developed, leading to a selective N–N coupling method that avoids overoxidation of hydrazine. The Hayashi process,28,29,30 which employs benzophenone as the ketone protecting group and has been used to generate hydrazine via the three-step sequence shown in Figure 1B, provided the platform for the present investigation of electrocatalytic N–N bond formation. The focal point of this process is the copper-catalyzed aerobic oxidative N–N coupling of benzophenone imine to afford the corresponding azine. The condensation and hydrolysis steps have also been thoroughly studied.31 Herein, we demonstrate three different approaches to achieve electrochemical oxidative N–N coupling of benzophenone imine (Figure 1C): (1) a proton-coupled electron-transfer process promoted by a phosphate base, (2) an iodine-mediated reaction involving intermediate N–I bond formation, and (3) a copper-catalyzed N–N coupling process, resembling the key sequence involved in the Hayashi process. To our knowledge, these results represent the first formal electrocatalytic synthesis of hydrazine from ammonia. The electrochemical data for each of these processes are then analyzed to determine thermodynamic efficiency for the different approaches, and the effective overpotentials are compared to other molecular catalyst systems that have been reported for electrochemical oxidation of ammonia.
Results and Discussion
Electrolysis Methods for Oxidative Coupling of Benzophenone Imine.
4,4’-Difluorobenzophenone imine (1) was selected as the substrate for the oxidative coupling studies as it allows straightforward analysis of the reactions by 19F NMR spectroscopy. In a preliminary test, the bulk electrolysis of 1 was evaluated in acetonitrile under constant current conditions, with nBu4NPF6 as supporting electrolyte, graphite rod as working electrode (anode) and platinum wire as counter electrode (cathode). Only trace amounts of the desired N–N-coupled azine product 2 was observed, together with a mixture of unidentified side products. It was expected that 1 could undergo proton-coupled oxidation at the anode to generate an N-centered radical (Figure 2A), but the substrate imine is the only species capable of serving as the proton acceptor under the initial conditions. The poor product yield and high anode potential observed under these conditions (cf. Figure 2B) prompted us to test various Brønsted bases in an effort to facilitate proton-coupled electron transfer (PCET) at the electrode. The bases included 2,4,6-collidine, 2,6-lutidine, carboxylates and phosphate (Figure 2C). No product was detected with carboxylates as the base, while moderate-to-low product yields were observed with 2,4,6-collidine (35%) and 2,6-lutidine (10%). Significantly improved results were obtained with dibutyl phosphate, [MeBu3N][OP(O)(OBu)2], as the base, and optimization of the reaction conditions led to an 84% yield of the desired azine product (Figure 2C, entry 7; see Table S1 of the Supporting Information for full optimization data). The beneficial effect of the phosphate base is evident in the cyclic voltammograms (CVs), which show a significant decrease in the anode potential when the imine and phosphate base are combined. The onset potential drops from 1.2 V (vs Fc+/Fc) with the imine alone to ~ 0.8 V upon addition of dibutyl phosphate (Figure 2B).
Figure 2. Base-Promoted Electrochemical Dehydrogenative Homocoupling of 1.
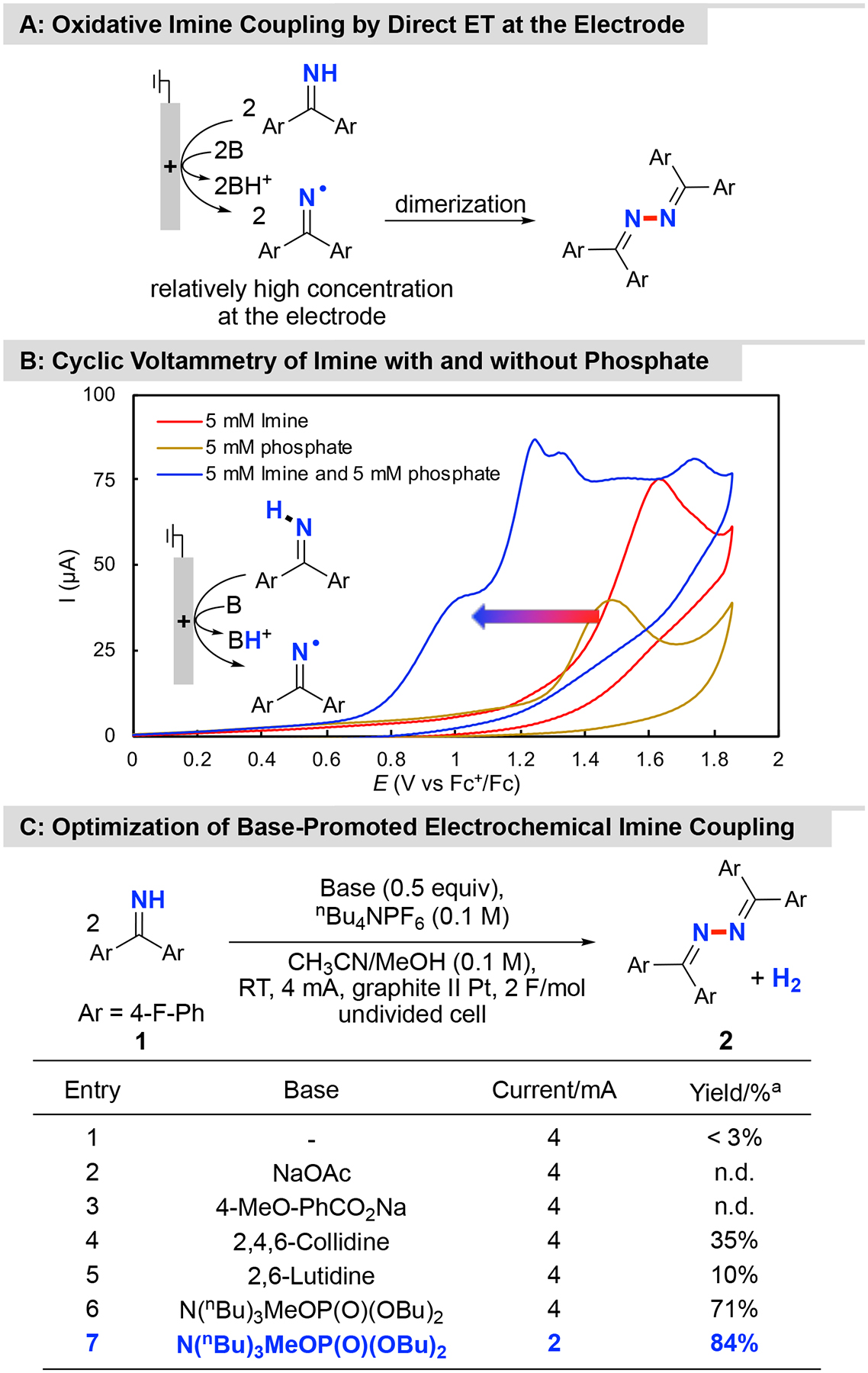
A: Oxidative imine coupling by direct electrolysis; B: Optimization of a base-promoted electrochemical imine coupling. aYields were determined by 19F NMR analysis with α,α,α-trifluorotoluene as internal standard.
Historical precedent for N–N coupling of iminyl reagents promoted by stoichiometric iodine32–34 raised the possibility of an electrochemical process for azine synthesis from benzophenone imine, using iodine as an electrocatalytic mediator.35 CV studies of iodide showed two distinct redox features, corresponding to the I− → I3− and I3− → I2 redox processes.36 Addition of 1 to this mixture led to a distinct increase in current at the second oxidation feature, suggesting that 1 reacts with iodine (but not I3−) on the CV time scale. For example, the reaction of 1 with I2 is expected to generate an N-iodo imine species,37 and the resulting iodide by-product can undergo oxidation at the electrode and lead to the observed current increase (Figure 3B). Initial attempts to perform bulk electrolysis with tetrabutylammonium iodide (TBAI) as the mediator in acetonitrile resulted in only trace product formation (Figure 3C, entry 1; see Table S2 of the Supporting Information for full optimization data). Inclusion of 8 equiv of MeOH under the conditions, however, increased the yield to 38% (Figure 3C, entry 2). The use of trifluoroethanol (TFE) or hexafluoroisopropanol (HFIP) instead of methanol led to inferior results (33% and 19% yield, respectively (Figure 3C, entries 3–4). Methanol and other alcohols are postulated to serve as proton donors that facilitate H2 formation at the cathode, and the resulting alkoxide co-products may then diffuse into the bulk solution where they can serve as bases to promote formation of the N-iodo imine and azine formation (cf. “B” in Figure 3A).38 Bromide and chloride-based mediators showed no formation of the desired azine product, even while leading to significant conversion of starting material (Figure 3C, entry 6–7). KI exhibited even better performance than TBAI, and use of 10 mol % KI with 12 equiv of MeOH led to an 86% yield of the desired azine (Figure 3C, entry 10).
Figure 3. Iodide-Mediated Electrochemical Dehydrogenative Homocoupling of 1.

A: A hypothesis for iodide-mediated imine coupling; B: Cyclic voltammetry of iodide w/wo imine, conditions: 2.5 mM nBu4NI in CH3CN (10 mL) with KPF6 (0.1 M) as supporting electrolyte, with glassy carbon as working electrode (~ 7.0 mm2) and a platinum wire (1.0 cm, spiral wire) as counter electrode, scan rate = 20 mV/s; C: Optimization of an iodide-mediated electrochemical imine coupling. aYields were determined by 19F NMR analysis with α,α,α-trifluorotoluene as internal standard; bConsumed charge is 2.2 F/mol; cWith 12 equiv of MeOH as additive.
The Hayashi process28,29 and other recent reports39,40 demonstrate that copper catalysts mediate the oxidative homocoupling of imines under aerobic conditions. Mechanistic studies have shown that CuII promotes efficient N–N coupling even in the absence of O2 (stoichiometrically), raising the possibility that Cu salts could serve as effective mediators for electrochemical N–N coupling (Figure 4A). CV analysis of [Cu(CH3CN)4]PF6 in the presence and absence of imine substrate 1 revealed that the presence of 1 substantially shifted the CuII/I redox potential. Moreover, the redox features became irreversible together with a current increase upon addition of 1, implicating the possibility of electrocatalytic turnover (Figure 4B). Pyridine is an effective ligand in aerobic Cu-catalyzed N–N coupling reactions,39,40 and also proved to be beneficial to bulk electrolysis reactions by ensuring that the imine substrate is not the sole ligand available for the Cu catalyst. Electrolysis of 1 in the presence of [Cu(CH3CN)4]PF6 and pyridine led to a low yield of the azine product (15%, Figure 4C, entry 1). Evaluation of various anionic Brønsted bases (e.g., K3PO4, K2CO3, or NaHCO3), however, revealed that substantially improved results could be obtained with bicarbonate (86%, Figure 4C, entry 4; see Table S3 of the Supporting Information for details).
Figure 4. Copper-Mediated Electrochemical Dehydrogenative Homocoupling of 1.
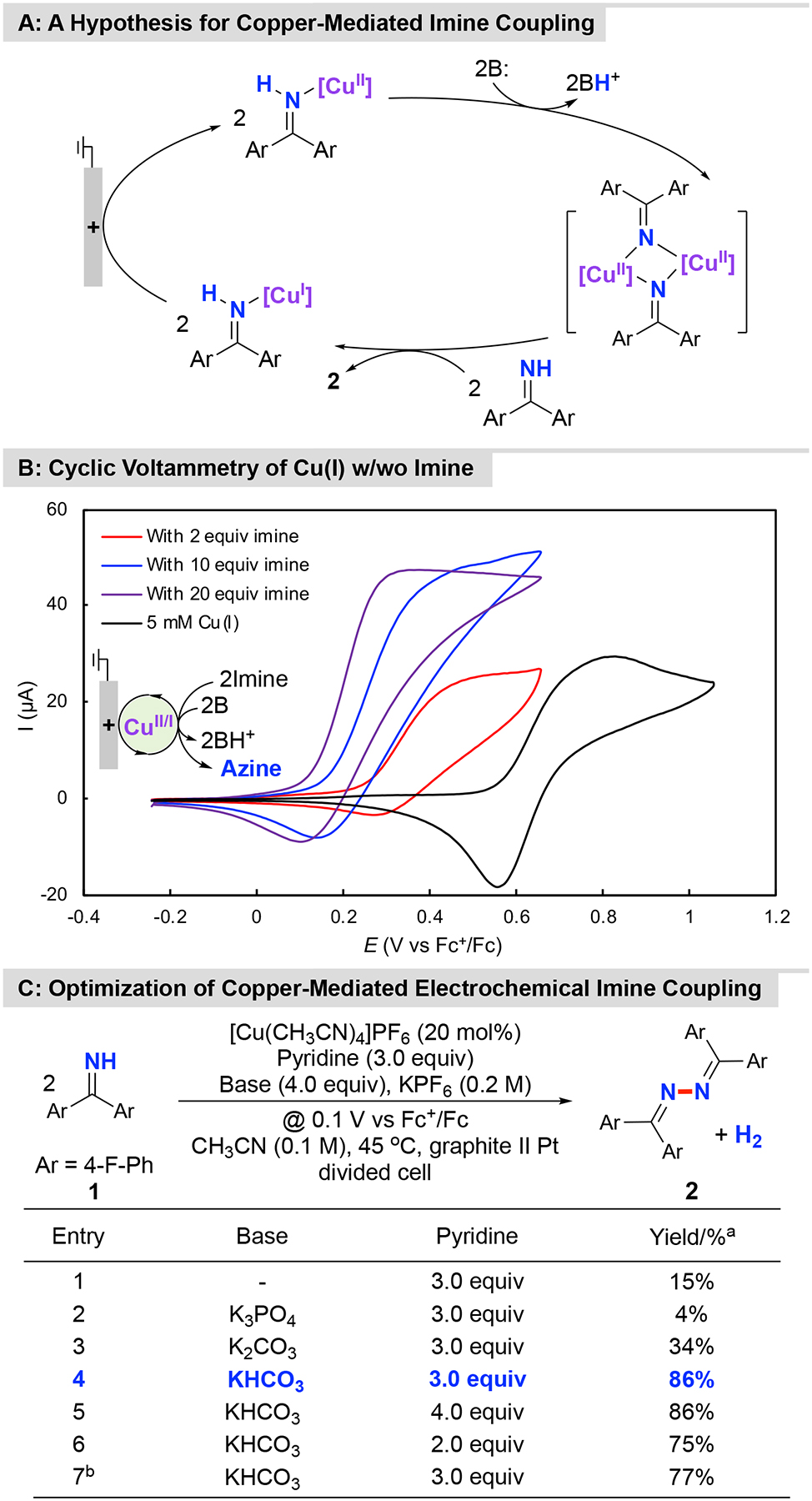
A: A hypothesis for copper-mediated imine coupling; B: Cyclic voltammetry of [Cu(CH3CN)4]PF6 w/wo imine, conditions: 5 mM [Cu(CH3CN)4]PF6 in CH3CN (10 mL) with nBu4NPF6 (0.1 M) as supporting electrolyte, with glassy carbon as working electrode (~ 7.0 mm2) and a platinum wire (1.0 cm, spiral wire) as counter electrode, scan rate = 20 mV/s; C: Optimization of a copper-mediated electrochemical imine coupling. aYields were determined by 19F NMR analysis with α,α,α-trifluorotoluene as internal standard; bUse of [Cu(CH3CN)4]PF6 (10 mol%) as mediator.
Analysis of Overpotentials for the Different Electrochemical N–N Coupling Processes
The above results highlight three independent strategies for electrochemical oxidative N–N coupling of benzophenone imine, and good yields of the azine were obtained in each case. Nonetheless, each of the reactions was optimized independently to maximize the yield, and the different conditions and experimental parameters make it difficult to compare their relative performance. One important metric that arises from the potential utility of hydrazine as an energy carrier is the overpotential associated with each reaction, which corresponds to the difference between the anode potential needed to support electrochemical N–N coupling and the thermodynamic potential for the reaction under the different reaction conditions.
Several recent reports have outlined an effective protocol for evaluating overpotentials for electrochemical reactions in non-aqueous solutions.17,41,42 The foundation of this approach is determination of the H+/H2 open-circuit potential (OCP) under catalytic reaction conditions that employ buffered electrolytes. (Acid/base buffering is essential to establish a stable thermodynamic reference.) The H+/H2 OCP may be used together with (i) the standard aqueous cell potentials for the reactions of interest and (ii) the free energies associated with the transfer of molecules from water to organic solvent to obtain thermodynamic potentials in the organic medium. Miller and coworkers recently employed this approach to obtain the free energies and thermodynamic potentials for the equilibria of N2 with NH3 and with N2H4 in acetonitrile (Scheme 1A and 1B).17 These equations may be used to derive the equilibrium potential for NH3 and N2H4 (Scheme 1C), and a similar approach was used to obtain thermodynamic potentials for N2/hydrazinium/ammonium equilibria, relevant to the analysis below (cf. Scheme S2).43 The involvement of protons in each of these reactions lead to a Nernstian dependence of the thermodynamic potential on the pKa of the acid.
Scheme 1.
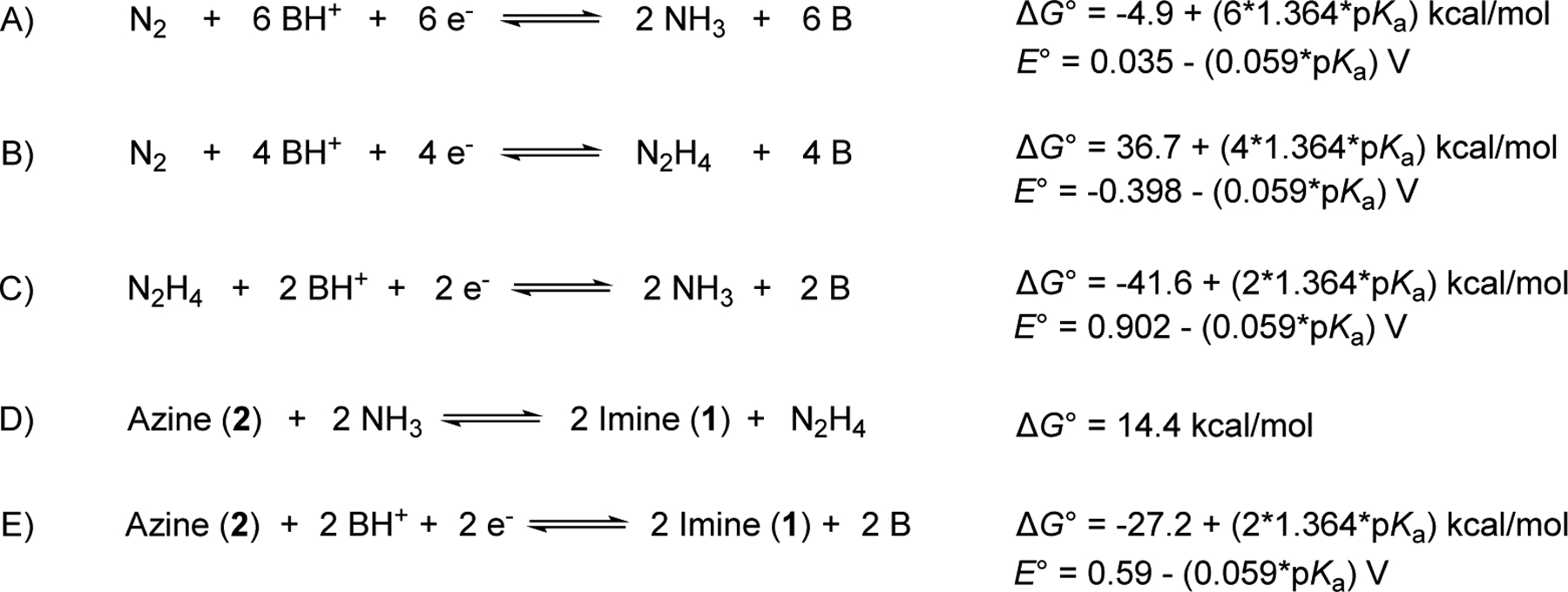
Thermodynamics of Interconversion of N2/N2H4/NH3 and Azine/Imine in MeCN
Full analysis for the catalytic reactions presented above required determination of the thermodynamic potentials for the 2e−/2H+ oxidative coupling of imine 1 to azine 2 and the free energy for azine ammonolysis. The latter was estimated from DFT calculations: ΔG°ammonolysis = 14.4 kcal/mol (Scheme 1D; B3LYP, 6–311+G(2d,p) basis set, see section 8 of the Supporting Information for details). Then, the sum of the reactions in Scheme 1C and 1D yields the azine/imine equilibrium free energy and thermodynamic potential (Scheme 1E).
The thermodynamic data for the azine/imine equilibrium and the previously reported data17 enabled creation of a Pourbaix-type diagram that correlates thermodynamic potentials of the relevant redox reactions with the electrolyte pKa (Figure 5). Each of the lines exhibits a change in slope where the protonation state of the substrate and/or product results in a change in the e−/H+ ratio for the overall reaction. For example, the azine/imine potential, which lies between the N2H4/NH3 and N2/NH3 redox couples, exhibits a change in slope at the pKa of the iminium ion (pKa = 13.3).
Figure 5. Pourbaix-type diagram for azine (2)/benzophenone imine (1), N2/NH3/N2H4 species, and redox potentials for the three electrochemical processes for azine synthesis.
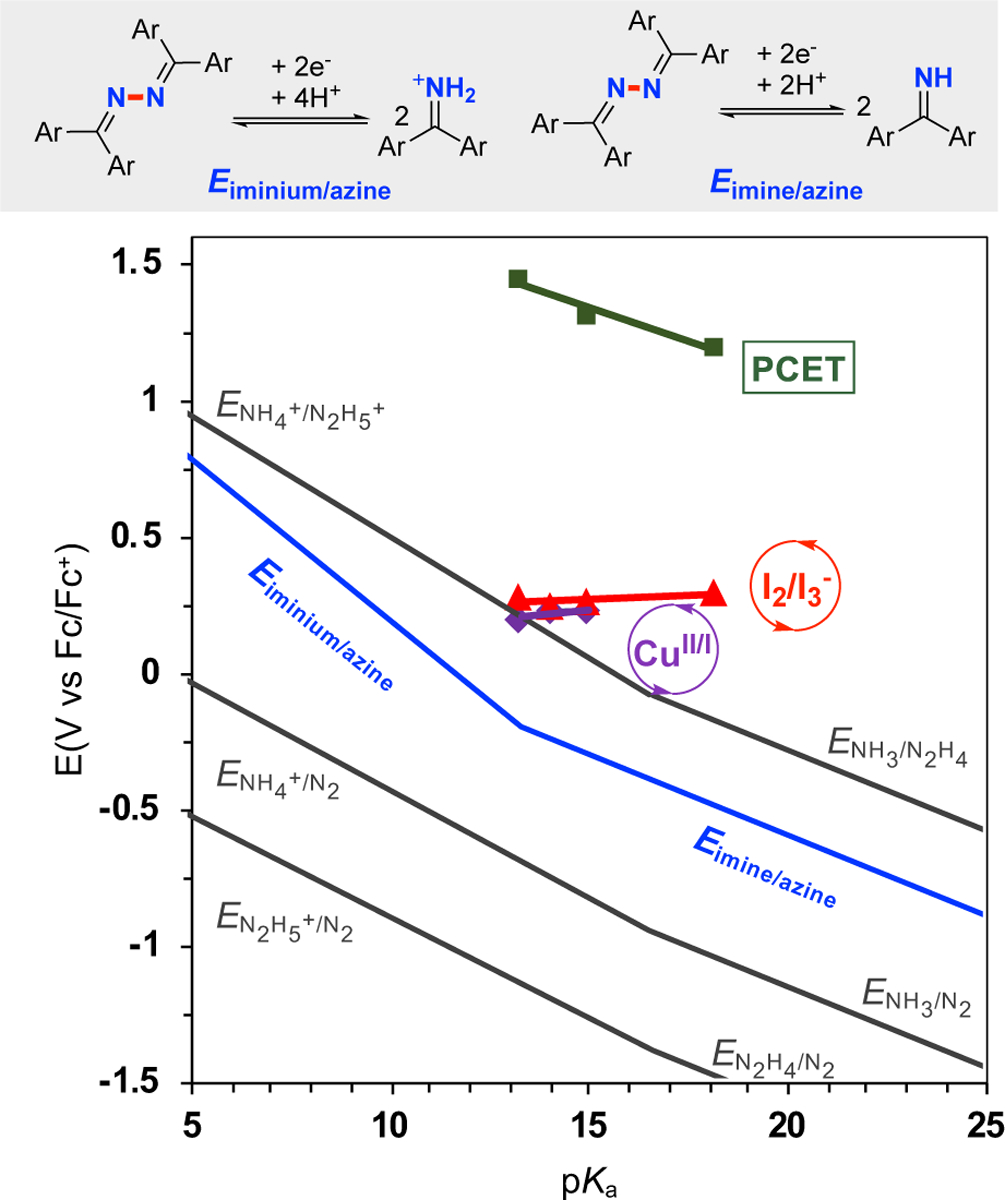
Potentials for processes (gray lines) involving N2 can be found in ref. 17. The blue line is the thermodynamics of azine 2 and imine 1 equilibrium. Green squares correspond to the PCET potentials with different buffer solutions: 5 mM 1 in CH3CN (10 mL) with nBu4NPF6 (0.1 M). Red triangles correspond to I 2/I3− potentials with different buffer solutions: 5 mM nBu4NI in CH3CN (10 mL) with 50 mM 1 and nBu4NPF6 (0.1 M). Purple diamonds correspond to the relevant CuII/I potentials with different buffer solutions: 5 mM [Cu(CH3CN)4]PF6 in CH3CN (10 mL) with 100 mM 1 and nBu4NPF6 (0.1 M). All the CV studies were performed with glassy carbon as working electrode (~ 7.0 mm2) and a platinum wire (1.0 cm, spiral wire) as counter electrode, scan rate = 20 mV/s.
This diagram enables straightforward assessment of the thermodynamic efficiency of the three electrochemical imine coupling methods presented above. CV studies were performed in buffered electrolytes with different pKa values at room temperature (i.e., somewhat different conditions from those shown in Figures 2–4), and E1/2 values were obtained from anodic waves (see Tables S4–S6 of the Supporting Information). The redox potentials observed for PCET-mediated N–N coupling exhibits an approximately Nernstian slope of 50 mV/pKa unit, consistent with a one electron-one proton transfer pathway (Figure 5, green squares). The applied potential is >1.5 V above the thermodynamic azine/imine potential, indicating a very large overpotential for this pathway. The I2/I3− and CuII/I redox couples are unaffected by the electrolyte pKa because their potential-determining steps do not involve protons. In addition, these processes occur at potentials much lower than that of the PCET process (Figure 5, red triangles and purple diamonds, respectively). For example, at a pKa of 13.3, corresponding to the pKa of the iminium species 1H+,44 the overpotentials for each of the three reactions are 1.6 V (PCET, Figure 2), 0.47 V (I2/I3−, Figure 3) and 0.39 V (CuII/I, Figure 3), respectively.
The high overpotential associated with the PCET pathway is attributed to generation of a high-energy iminyl radical prior to N–N coupling. Although the presence of a Brønsted base supports a PCET pathway, allowing the process to proceed at a lower potential than in the absence of added base, the applied potential is still much higher than the reactions catalyzed by I2 and CuII. The mechanisms with I2 and CuII avoid high-energy intermediates by bonding/coordination of iminyl intermediates to iodine and copper.32,36,40
The data in Figure 5 show that the potentials for the I2/I3− and CuII/I processes approach the thermodynamic potential for oxidation of NH4+ to N2H5+ at pKa = 13.3. This observation suggests the possibility of electrochemical oxidation of NH4+ with these catalysts; however, I2 reacts with ammonia to generate the explosive compound NI3,45 while CuII forms Cu/ammine complexes with much lower redox potentials that will be incapable of generating N2H4 or N2H5+.46 Moreover, the direct formation of hydrazine as a product will likely result in rapid oxidation of hydrazine to N2, which is favoured by >1.25 V (cf. Figure 1A). These considerations highlight the strategic benefit of using benzophenone imine as an ammonia surrogate as it supports formation of a single N–N bond and allows the process to proceed at a potential that preserves much of the energy stored in hydrazine.
Comparison with Other Molecular Electrocatalysts for Oxidative N–N Coupling
As noted in the Introduction, a number of transition metal complexes have been identified as catalysts for ammonia oxidation to N2.21–26 Meyer and coworkers demonstrated stoichiometric oxidation of Ru-polypyridyl ammine complexes into metal-imido and/or metal nitride species that lead to N–N coupling and release of N2.19,20 This work provided the basis for the first molecular electrocatalysts for NH3 oxidation to N2. In 2019, Hamann and Smith showed that the mononuclear Ru complex [(tpy)(dmabpy)RuII(NH3)](PF6)2 (tpy = 2,2’:6’,2”-terpyridine; dmabpy = 4,4’-bis(dimethylamino)-2,2’-bipyridine) is an effective electrocatalyst in THF.21 Independently, Nishibayashi and Sakata demonstrated catalytic NH3 oxidation with a mononuclear Ru complex (bpy-dicarboxylate)RuL2 (bpy-dicarboxylate = 2,2’-bipyridyl-6,6’-dicarboxylate, L = isoquinoline) in acetonitrile.24 Although the majority of studies were conducted with a triarylaminium-based oxidant, CV studies provided evidence for electrocatalysis. More recently, Warren26 and Peters23 demonstrated iron-based catalysts for electrochemical ammonia oxidation in acetonitrile. Peters and co-workers used an FeII(TPA) complex, [FeII(TPA)(NH3)2](OTf)2, [TPA = tris(2-pyridylmethyl)amine], as the catalyst, while Warren and coworkers showed that ferrocene (Fc) is an effective electrocatalyst. Each of these catalytic reactions showed exquisite formation of N2 as the N–N coupling product, consistent with the much higher reactivity of N2H4 (and N2H2), relative to NH3, if they are formed as intermediates.
Analysis of the overpotential has not been rigorously addressed in these reactions, in part, due to the lack of buffered reaction conditions24,26 or the absence of a suitable thermodynamic references.21 Nonetheless, reasonable estimates may be obtained by employing an analysis similar to that described above. In order to estimate the overpotential for the Ru catalyst system reported by Hamann and Smith, the H+/H2 OCP was measured in THF under the reported conditions (0.20 M NH4PF6, 0.34 M NH3 in THF; see section 9 in the Supporting Information for details).21 The resulting value, −0.86 V vs. Fc+/Fc, was then used to determine the thermodynamic N2/NH3 potential (−0.81 V vs. Fc+/Fc), and the difference between this value and the applied electrolysis potential (0.20 V vs. Fc+/Fc) reveals an effective overpotential of ~ 1.0 V. The other three catalyst systems were used in acetonitrile, allowing literature data17 to be used to estimate the N2/NH3 potential; however, only the Fe(TPA)-catalyzed reaction was conducted in buffered electrolyte (i.e., with both NH4+ and NH3 present).23 In this case, the catalytic onset potential of 0.70 V vs. Fc+/Fc may be compared to N2/NH3 potential of −0.94 V in acetonitrile at the NH4+/NH3 electrolyte pKa of 16.5 (cf. Figure 5) to obtain an estimated overpotential of ~1.6 V. The analysis is more complicated for the other two reactions due to the lack of buffered electrolyte.24,26 Nevertheless, overpotentials of 0.94 and 1.1 V vs. Fc+/Fc are estimated for the ammonia oxidation reactions catalyzed by ferrocene and (bpy-dicarboxylate)RuL2 by assuming an electrolyte pKa of 16.5.
In spite of the approximate nature of the overpotential estimates for the ammonia oxidation catalysts, the values represent relevant benchmarks for consideration in future studies,47 and they also provide relevant points for comparison with the imine oxidative coupling methods described here. The PCET-based N–N coupling method mediated by a Brønsted base exhibits an overpotential of 1.6 V, which is as large or larger than that observed for the ammonia oxidation reactions. In contrast, the I2 and Cu-catalyzed methods feature a comparatively low overpotential, presumably reflecting their ability to stabilize reactive intermediates (i.e., an iminyl radical). Future progress toward lower overpotential ammonia oxidation will presumably arise from application of similar concepts, for example, identifying catalysts that can stabilize relatively high-energy intermediates such as hydrazine- and/or azine-derived species.
The I2 and Cu-catalyzed methods for oxidative N–N coupling of imines operate at potentials 1.03 V and 0.95 V, respectively, above the thermodynamic N2/NH3 potentials. The similarity between these values and the overpotentials associated with the ammonia oxidation catalysts in Table 1 is probably not a coincidence and may be rationalized by the need for each of these processes to access a similarly reactive species capable of forming an N–N bond. The lower overpotential in the present system reflects its ability to stop at a high energy azine product without cascading further to the lower energy N2 product.
Table 1.
Overpotential Comparison of Molecular Electrocatalysts for N–N Coupling.
| Electrocatalyst/Mediator | Solvent | E° (N2/NH3) (V)a@/pKa=16.5 | CV/Electrolysis Potential (V)a | ƞ(V) | Ref. |
|---|---|---|---|---|---|
| [(trpy)(dmabpy)RuII (NH3)](PF6)2 | TDF | −0.81 | 0.20 | 1.0 | 21 |
| [(bpy-dicarboxylate)RuL2] | CH3CN | −0.94 | 0.20 | 1.1 | 24 |
| Ferrocene | CH3CN | −0.94 | 0 | 0.94 | 26 |
| [FeII (TPA)(NH3)2](OTf)2 | CH3CN | −0.94 | 0.7 | 1.6 | 23 |
| Electrocatalyst/Mediator | Solvent |
E° (Azine/Imine) (V)a @pKa=13.3 |
CV/Electrolysis Potential (V)a | ƞ(V) | Ref. |
| Base/PCETb | CH3CN | −0.19 | 1.45 | 1.6 | this work |
| Iodide | CH3CN | −0.19 | 0.28 | 0.47 | |
| [Cu(CH3CN)PF6 | CH3CN | −0.19 | 0.20 | 0.39 | |
Potentials are reported vs. Fc+/Fc.
At pKa = 13.3, the imine substrate is the Brønsted base that promotes the PCET initiated N–N coupling reaction.
Conclusion
In this study, we have demonstrated three complementary methods to achieve electrochemical N–N coupling of benzophenone imine. Use of the imine as an ammonia surrogate ensures that the reaction stops at formation of a single N–N bond, avoiding the kinetically facile and thermodynamically favorable oxidation of hydrazine to dinitrogen that would occur in reactions that use ammonia as a feedstock. By leveraging known processes in the chemical industry, these results establish a formal strategy for electrochemical synthesis of hydrazine from ammonia. Analysis of the overpotentials for the three different processes shows that the Cu-catalyzed reaction exhibits good thermodynamic efficiency, establishing an important foundation for development of improved catalysts and potential consideration of methods for practical implementation of the concepts described herein.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award #DE-FG02-05ER15690. FW was partially supported by a postdoctoral fellowship from SIOC. Spectroscopic instrumentation was partially supported by a generous gift from Paul J. and Margaret M. Bender, the NIH (1S10 OD020022-1) and the NSF (CHE-1048642). The computational cluster at UW-Madison is funded by the NSF (CHE-0840494).
Footnotes
Supporting Information.
Complete experimental details, procedures for bulk electrolysis and CV data and analysis, and DFT calculation (PDF).
References
- 1.Erisman JW; Sutton MA; Galloway J; Klimont Z; Winiwarter W How a century of ammonia synthesis changed the world. Nat. Geosci 2008, 1, 636–639. [Google Scholar]
- 2.Lan R; Tao S Ammonia as a suitable fuel for fuel cells. Front. Energy Res 2014, 2, 35. [Google Scholar]
- 3.Afif A; Radenahmad N; Cheok Q; Shams S; Kim JH; Azad AK Ammonia-fed fuel cells: a comprehensive review. Renewable Sustainable Energy Rev. 2016, 60, 822–835. [Google Scholar]
- 4.Cheddie D Ammonia as a hydrogen source for fuel cells: A review In Hydrogen Energy–Challenges and Perspectives; Minić D, Ed.; INTECH: Rijeka, Croatia, 2012; pp 333–362. [Google Scholar]
- 5.Klerke A; Christensen CH; Nørskov JK; Vegge T Ammonia for hydrogen storage: challenges and opportunities. J. Mater. Chem 2008, 18, 2304–2310. [Google Scholar]
- 6.Adli NM; Zhang H; Mukherjee S; Wu G Review-ammonia oxidation electrocatalysis for hydrogen generation and fuel cells. J. Electrochem. Soc 2018, 165, J3130–J3147. [Google Scholar]
- 7.Schüth F; Palkovits R; Schlögl R; Su DS Ammonia as a possible element in an energy infrastructure: catalysts for ammonia decomposition. Energy Environ. Sci 2012, 5, 6278–6289. [Google Scholar]
- 8.Rothgery EF Hydrazine and its derivatives, In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons: New York, 2004; Vol. 13, pp 562–607. [Google Scholar]
- 9.Serov A; Kwak C Direct hydrazine fuel cells: A review. Appl. Catal. B 2010, 98, 1–9. [Google Scholar]
- 10.Sanabria-Chinchilla J; Asazawa K; Sakamoto T; Yamada K; Tanaka H; Strasser P Noble metal-free hydrazine fuel cell catalysts: EPOC effect in competing chemical and electrochemical reaction pathways. J. Am. Chem. Soc 2011, 133, 5425–5431. [DOI] [PubMed] [Google Scholar]
- 11.Asazawa K; Yamada K; Tanaka H; Oka A; Taniguchi M; Kobayashi T A platinum-free zero-carbon-emission easy fuelling direct hydrazine fuel cell for vehicles. Angew. Chem. Int. Ed 2007, 46, 8024–8027. [DOI] [PubMed] [Google Scholar]
- 12.Serov A; Padilla M; Roy AJ; Atanassov P; Sakamoto T; Asazawa K; Tanaka H Anode catalysts for direct hydrazine fuel cells: From laboratory test to an electric vehicle. Angew. Chem. Int. Ed 2014, 53, 10336–10339. [DOI] [PubMed] [Google Scholar]
- 13. For the condensed phase thermochemistry data of hydrazine, see:; https://webbook.nist.gov/cgi/cbook.cgi?ID=C302012&Units=SI&Mask=2#Thermo-Condensed (accessed April 19, 2020).
- 14. For the gas phase thermochemistry data of ammonia, see:; https://webbook.nist.gov/cgi/cbook.cgi?ID=C7664417&Units=SI&Mask=1#Thermo-Gas (accessed April 19, 2020).
- 15. For the gas phase thermochemistry data of hydrogen, see:; https://webbook.nist.gov/cgi/cbook.cgi?Name=hydrogen&Units=SI (accessed April 19, 2020).
- 16. For the gas phase thermochemistry data of nitrogen, see:; https://webbook.nist.gov/cgi/cbook.cgi?ID=C7727379&Units=SI&Mask=1#Thermo-Gas (accessed April 19, 2020).
- 17.Lindley BM; Appel AM; Krogh-Jespersen K; Mayer JM; Miller AJM Evaluating the thermodynamics of electrocatalytic N2 reduction in acetonitrile. ACS Energy Lett. 2016, 1, 698–704. [Google Scholar]
- 18. The redox potential for the N2/N2H5+ couple in Figure 1A(iii) (−0.21 V) deviates somewhat from the value reported in Table 1 of ref. 17 (−0.23 V); however, it matches that in the Supporting Information of the same publication. The redox potential −0.21 V is the correct value on the basis of standard heats of formation and entropies for the different reaction components.
- 19.Ishitani O; White PS; Meyer TJ Formation of dinitrogen by oxidation of [(bpy)2(NH3)RuORu(NH3)(bpy)2]4+. Inorg. Chem 1996, 35, 2167–2168. [DOI] [PubMed] [Google Scholar]
- 20.Ishitani O; Ando E; Meyer TJ Dinitrogen formation by oxidative intramolecular N–N coupling in cis,cis-[(bpy)2(NH3)RuORu(NH3)(bpy)2]4+. Inorg. Chem 2003, 42, 1707–1710. [DOI] [PubMed] [Google Scholar]
- 21.Habibzadeh F; Miller SL; Hamann TW; Smith III MR Homogeneous electrocatalytic oxidation of ammonia to N2 under mild conditions. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 2849–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhattacharya P; Heiden ZM; Chambers GM; Johnson SI; Bullock RM; Mock MT Catalytic ammonia oxidation to dinitrogen by hydrogen atom abstraction. Angew. Chem. Int. Ed 2019, 58, 11618–11624. [DOI] [PubMed] [Google Scholar]
- 23.Zott MD; Garrido-Barros P; Peters JC Electrocatalytic ammonia oxidation mediated by a polypyridyl iron catalyst. ACS Catal. 2019, 9, 10101–10108. [Google Scholar]
- 24.Nakajima K; Toda H; Sakata K; Nishibayashi Y Ruthenium-catalysed oxidative conversion of ammonia into dinitrogen. Nat. Chem 2019, 11, 702–709. [DOI] [PubMed] [Google Scholar]
- 25.Dunn PL; Johnson SI; Kaminsky W; Bullock RM Diversion of catalytic C–N bond formation to catalytic oxidation of NH3 through modification of the hydrogen atom abstractor. J. Am. Chem. Soc 2020, 142, 3361–3365. [DOI] [PubMed] [Google Scholar]
- 26.Boroujeni MR; Greene C; Bertke JA; Warren TH Chemical and electrocatalytic ammonia oxidation by ferrocene. ChemRxiv 2019, doi.org/10.26434/chemrxiv.9729635.v1. [Google Scholar]
- 27.Schirmann J-P; Bourdauducq P. Hydrazine. Ullmann’s encyclopedia of industrial chemistry; Wiley-VCH: Weinheim, Germany, 2002; pp 79–96. [Google Scholar]
- 28.Hayashi H Hydrazine synthesis by a catalytic oxidation process. Catal. Rev 1990, 32, 229–277. [Google Scholar]
- 29.Hayashi H; Kainoh A; Katayama M; Kawasaki K; Okazaki T Hydrazine production from ammonia via azine. Ind. Eng. Chem. Prod. Res. Dev 1976, 15, 299–303. [Google Scholar]
- 30. A commercial ketazine process uses methyl ethyl ketone and proceeds in a “one-pot” process involving oxidation of the corresponding imine with H2O2 to generate an oxaziridine, followed by reaction with ammonia to generate a hydrazone. The Hayashi process involves Cu-catalyzed N–N homocoupling of benzophenone imine. The latter process has not yet been commercialized, primarily reflecting challenges in product separation and benzophenone recycling. In principle, electrochemical N–N coupling could employ various primary imine precursors; however, imines with α-C–H bonds, such as that derived from methyl ethyl ketone, can tautomerize to enamines and undergo decomposition. This consideration prompted us to select benzophenone imine as the ammonia surrogate for the present study. Further context for these considerations is elaborated in ref. 28.
- 31.Hayashi H; Somei J; Okazaki T Ammonia-hydrazine conversion processes XVI. Conversion of benzophenone azine into hydrazine catalyzed by sulphonic acids in a two-phase system. Appl. Catal 1988, 41, 213–224. [Google Scholar]
- 32.Morton AA; Stevens JR Condensations by sodium instead of by the Grignard reaction. II. Reaction with benzonitrile. Preparation of diphenylketazine. J. Am. Chem. Soc 1931, 53, 2769–2772. [Google Scholar]
- 33.Song L; Tian X; Lv Z; Li E; Wu J; Liu Y; Yu W; Chang J I2/KI-Mediated oxidative N–N bond formation for the synthesis of 1,5-fused 1,2,4-triazoles from N-aryl amidines. J. Org. Chem 2015, 80, 7219–7225. [DOI] [PubMed] [Google Scholar]
- 34.Guo Q; Lu Z Recent advances in nitrogen–nitrogen bond formation. Synthesis 2017, 49, 3835–3847. [Google Scholar]
- 35. For a recent review on iodide-mediated electroorganic synthesis, see:; Liu K; Song C; Lei A Recent advances in iodine mediated electrochemical oxidative cross-coupling. Org. Biomol. Chem 2018, 16, 2375–2387. [DOI] [PubMed] [Google Scholar]
- 36.Wang F; Stahl SS Merging photochemistry with electrochemistry: functional-group tolerant electrochemical amination of C(sp3)–H Bonds. Angew. Chem. Int. Ed 2019, 58, 6385–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bosnidou AE; Duhamel T; Muñiz K Detection of the elusive nitrogen-centered radicals from catalytic Hofmann–Löffler reactions. Eur. J. Org. Chem 2019, DOI: 10.1002/ejoc.201900497. [DOI] [Google Scholar]
- 38.Lennox AJJ; Goes SL; Webster MP; Koolman HF; Djuric SW; Stahl SS Electrochemical aminoxyl-mediated α-cyanation of secondary piperidines for pharmaceutical building block diversification. J. Am. Chem. Soc 2018, 140, 11227–11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laouiti A; Rammah MM; Rammah MB; Marrot J; Couty F; Evano G Copper-catalyzed oxidative alkynylation of diaryl imines with terminal alkynes: A facile synthesis of ynimines. Org. Lett 2012, 14, 6–9. [DOI] [PubMed] [Google Scholar]
- 40.Ryan MC; Kim Y-J; Gerken JB; Wang F; Aristov MM; Martinelli JR; Stahl SS Mechanistic insights into copper-catalyzed aerobic oxidative coupling of N–N bonds. Chem. Sci 2020, 11, 1170–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts JAS; Bullock RM Direct determination of equilibrium potentials for hydrogen oxidation/production by open circuit potential measurements in acetonitrile. Inorg. Chem 2013, 52, 3823–3835. [DOI] [PubMed] [Google Scholar]
- 42.Pegis ML; Roberts JAS; Wasylenko DJ; Mader EA; Appel AM; Mayer JM Standard reduction potentials for oxygen and carbon dioxide couples in acetonitrile and N,N-dimethylformamide. Inorg. Chem 2015, 54, 11883–11888. [DOI] [PubMed] [Google Scholar]
- 43. In principle, diprotonated hydrazine should also be included in the diagram in Figure 5; however, the pKa of N2H62+ in acetonitrile has not been reported. The aqueous pKa of N2H62+ is estimated to be in the range of −0.5 to 0.27, according to the following studies: a) Doherty, A. M. M.; Dadcliffe, M. D.; Stedman, G. Kinetics of oxidation of nitrogen compounds by cerium (IV). J. Chem. Soc., Dalton Trans. 1999, 3311–3316; b) Rich, L. R. Inorganic Reactions in Water; Springer-Verlag: Berlin, Heidelberg, 2007: Section 1.
- 44. If there is no stronger base, the benzophenone imine will serve as the Brønsted base. The iminium pKa value was obtained by 1H NMR spectroscopy using acids of known pKa close to that of the iminium (see Table S6 of the Supporting Information). Reference acid pKa values are from: Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I. A. Extension of the self-consistent spectrophotometric basicity scale in acetonitrile to a full span of 28 pKa units: unification of different basicity scales. J. Org. Chem. 2005, 70, 1019–1028. [DOI] [PubMed]
- 45.Watt GW; Foerster DR Reactions of iodine in liquid ammonia. J. Inorg. Nucl. Chem 1960, 13, 313–317. [Google Scholar]
- 46.Sun Z; Cao H; Venkatesan P; Jin W; Xiao Y; Sietsma J; Yang Y Electrochemistry during efficient copper recovery from complex electronic waste using ammonia based solutions. Front. Chem. Sci. Eng 2017, 11, 308–316. [Google Scholar]
- 47. The method for estimating overpotentials employed here is considerably more reliable than approximations based on adapting aqueous thermodynamic and/or reference potentials to reactions conducted in organic solvent. For example, use of the latter method led to an overpotential estimate approximately 400 mV lower than that in Table 1 for the (bpy-dicarboxylate)RuL2 system (cf. ref. 24).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.