

Abstract
Somatic mutation and gene expression dysregulation are considered two major tumorigenesis factors. While independent investigations of either factor pervade, studies of associations between somatic mutations and gene expression changes have been sporadic and nonsystematic. Utilizing genomic data collected from 11 315 subjects of 33 distinct cancer types, we constructed MutEx, a pan-cancer integrative genomic database. This database records the relationships among gene expression, somatic mutation and survival data for cancer patients. MutEx can be used to swiftly explore the relationship between these genomic/clinic features within and across cancer types and, more importantly, search for corroborating evidence for hypothesis inception. Our database also incorporated Gene Ontology and several pathway databases to enhance functional annotation, and elastic net and a gene expression composite score to aid in survival analysis. To demonstrate the usability of MutEx, we provide several application examples, including top somatic mutations associated with the most extensive expression dysregulation in breast cancer, differential mutational burden downstream of DNA mismatch repair gene mutations and composite gene expression score-based survival difference in breast cancer. MutEx can be accessed at http://www.innovebioinfo.com/Databases/Mutationdb_About.php.
Keywords: somatic mutation, gene expression, survival
Introduction
Cancer is a complex and highly heterogeneous disease [1]. Most common cancer types have been categorized into distinct subtypes based on either phenotype or genomic characteristic. To fully understand the complete landscape of cancer, often multiple types of genomic data were collected for large cancer consortium studies, with The Cancer Genome Atlas (TCGA) being a typical example. There are several resources utilizing large cancer consortium data resource for comprehensive cancer analysis, such as Human Protein Atlas [2], Broad Institute’s Firehose, Oncomine, etc. These resources offer the abilities to browse gene expression profile of multiple cancers and provide basic differential and survival analysis based on gene expression. However, these resources have some severe limitations. For example, Human Protein Atlas’s search function is limited to one gene only. Firehose stores correlational analysis results, but it is without any functional interpretation and visualization. Oncomine contains the largest collection of cancer data sets. However, many of its premium functions are not free. Inspired by these previously constructed resources, we aim to develop a free novel pan-cancer resource, which provides unique analysis functions not presented in the previous resources.
Clever integrative analyses among these data often reveal additional functional and regulatory aspects of cancers. A common example of integrative analysis between germline variants and gene expression is expression quantitative loci (eQTLs), in which the relationship between single-nucleotide polymorphism and gene expression is being modeled with linear regression. These eQTLs were often found to be regulating specific functions through the regulation of gene expression [3, 4]. Using a similar concept, we can also measure the association between somatic mutation and gene expression by dividing cancer subjects into mutant and wild-type groups based on a gene set’s mutation status. Using this strategy, we developed MutEx, a gateway to varied integrations of the curated data on mutation-associated gene expression dysregulation in cancer. With a gene-centric perspective and a scope of 33 cancer types, MutEx provides sophisticated query of somatic mutations and their associations with gene expression dysregulation, thereby providing additional insights into tumorigenesis mechanisms of cancers. With concrete examples, we demonstrate the effectiveness and unique features of MutEx.
Method
Based on TCGA genomic and clinical data, we developed MutEx, a tool for assessing somatic mutations and gene expression associated with cancer outcomes. MutEx is built upon the large pool of genomic data of 11 315 patients from 33 cancer types. Data retrieval was via Genomic Data Commons Data Portal with R package TCGAbiolinks (version 2.8.4). More than 10 billion computations were completed to integrate the diverse types of cancer data into a format permissible for exploring hypotheses surrounding mutations and dysregulations. The overall concept of MutEx is depicted in Figure 1.
Figure 1.
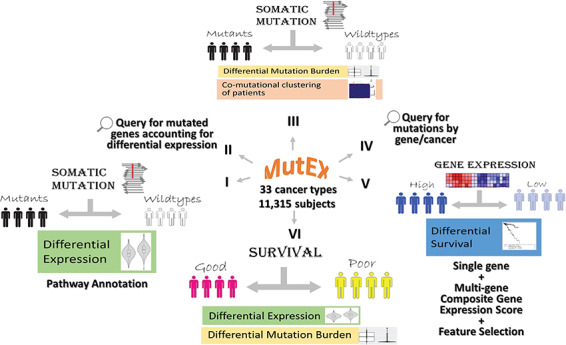
The six functionalities of MutEx. The infrastructure of each functionality consists of partitioning cancer patients by a certain genomic feature and then investigating into other genomic features.
A critical step in MutEx calculations is to divide cancer patients based on the mutation status of certain genes. To this end, the TCGA-annotated mutation terms were simplified into 12 categories and then further categorized into synonymous and nonsynonymous mutations. Unless explicitly stated, the results described below were based on nonsynonymous mutations only. For MutEx users, however, both options (all mutations and nonsynonymous mutations) are offered to divide cancer samples.
RNA-Seq data (HTSeq-counts) [5] were used for differential expression analyses through edgeR (version 3.22.3) [6], which involved tumor samples with both mutation and gene expression data available. Within each cancer, gene differential expression was examined between mutant and wild-type tumor samples. A mutated gene is considered if it is mutated in at least three patients or accounts for 5% or more of all tumor samples. Genes with a median counts per million of 0 across all samples were not interrogated for differential expression. Differential expression detected with a <0.05 false discovery rate-adjusted P-value was considered significant. All statistical analyses were performed in R 3.5.1. The original differential expression results consisted of more than 50 GB of data. To conserve space and improve performance, we only incorporated statistically significant results into the MutEx output.
For survival analyses where patients can be partitioned by the expression status of multiple genes, we implemented three strategies: (1) Single-gene approach: where univariable Kaplan–Meier analysis is performed to compare the survival of two groups of patients dichotomized by a single gene’s expression according to the user-defined cutoff (such as median). (2) Multigene approach: where MutEx fits survival data with a multivariable Cox regression model with all input genes and then uses the inferred coefficient of each gene as the individual weight in the following formula to compute a composite gene expression score (CGES):
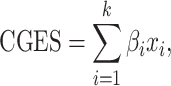 |
(1) |
where βi indicates the coefficients of genes, xi refers to the relative expression of the corresponding gene and k is the number of input genes used in calculating CGES. This composite score is then used to conduct the Kaplan–Meier survival curves. To avoid overfitting, we suggest limiting the number of input variables (genes) up to approximately one-tenth of the total number of survival events. This method is based on the concept that genes can form network and pathway; instead of assuming one gene can exert survival effect, we assume additive survival effect of a set of genes. (3) Dimensionality reduction approach (lasso [7] or elastic net) with Cox regression to select a subset of genes: this method allows the user to input a large gene set up to the entire transcriptome and find the optimal subset of genes that are associated with survival. Postselection inference is performed using the R package selectiveInference (version 1.2.4), which computes P-values and selection intervals that properly account for the inherent selection carried out by the procedure.
Whenever applicable, we offer hierarchical clustering through the R package heatmap3 [8]. Gene Ontology enrichment analyses, with respect to biological process, cellular component and molecular function aspects, are enabled by the R packages RDAVIDWebService and WebGestaltR. Pathway analyses with respect to KEGG and Wikipathway are mediated by WebGestaltR.
Results
Interface
The interface of MutEx was developed in a combination of PHP, HTML and Javascript. The backbone database was MySQL with custom R scripts. A detailed tutorial page is available on the MutEx webpage.
Functionalities
MutEx allows users to tap into the vast resources of available cancer patient genomic and survival data and provides different levels of analyses for mutation-associated expression dysregulation. MutEx stores the precomputed associations between somatic mutation and gene expression dysregulation and exploits the data through six major functionalities: (I) Identify differentially expressed genes between opposite mutation status (mutant vs. wild type) for a specified set of genes. (II) Conversely, identify potential causative genes whose mutations are associated with differential expression in the set of interested genes. (III) Explore the mutation profiles of cancer patients by comparing mutational burdens or examining co-mutational patterns. (IV) Search for mutated genes by genomic location or mutation type. (V) Perform survival analysis of cancer patients based on the collective expression of a set of genes with feature selection options and post feature selection inference evaluation. (VI) Examine expression difference or mutational burden difference between subjects with good and poor survival. The results are displayed in an HTML page through dynamic searchable tables and companion analytic graphs if applicable. All tables and high-resolution figures are available for download.
Functionality I of MutEx affords the ability to query our massive analysis results of somatic mutation-associated differential expression. The goal is to identify the differentially expressed genes contingent upon the mutation status of the input gene or genes. These genes could be from a specific pathway or of a particular interest. A single gene’s mutation might only disrupt the function of a pathway slightly, while mutations in all genes in the pathway might produce a more detrimental effect on the pathway collectively. For convenient references, MutEx provides 379 pathway gene lists retrieved from Pathway Commons V10. In addition, two existing clinical gene sets, Foundation One (336 genes) and ThermoFisher’s Ion AmpliSeq Comprehensive Cancer Panel (409 genes), were also preloaded for the user to select. The user may also upload his or her own list of genes of interest for analysis.
Under Functionality I, MutEx offers a way to sort all mutated genes by the number of their affected differentially expressed genes and highlights user-specified input genes in the ranked list. This utility provides an empirical measure of the relative mutation caused gene dysregulation exerted by the query genes. Somatic mutations with greater importance may cause a higher number of differentially expressed genes. To test this, we examined the top 10 genes whose somatic mutations led to the most extensive expression dysregulation in breast invasive carcinoma (BRCA) (Table 1; Figure 2A). A majority of these genes are well-known cancer genes, such as TP53, PIK3CA, MAP3K1, etc. USH2A (6th), SPTA1 (7th) and KMT2C (10th) are relatively understudied compared to other common cancer genes. USH2A has been suggested as a tumor suppressor in hepatocellular carcinoma [9], yet lacking a direct link to breast cancer so far. Similarly, few studies focused on SPTA1’s and KMT2C’s roles in cancers. Only very recently, a study found that KMT2C mediates the estrogen dependence of breast cancer through regulation of estrogen receptor (ER) alpha enhancer function [10]. TTN and DMD were ranked in the 8th and 9th places, respectively. However, the enormous gene length must have contributed partly to the extent of TTN’s and DMD’s mutation impact on expression dysregulation, because a longer gene body tends to harbor more instances of mutations and mutation frequency was found positively correlated with the number of genes differentially expressed between the wild-type and mutant groups. A higher mutation frequency improves the sample size balance between wild type and mutation and increases the power to detect differential expression. To account for mutation rate, a scatter plot of mutation frequency versus number of differentially expressed genes is delineated for each cancer, in which user-specified input genes are highlighted (Figure 2B). Given the expected and observed correlation between mutation frequency and extent of exerted differential expression, researchers are advised to pay special attention to the bottom-right region of the scatterplot, which is populated with less popular cancer genes with low to moderate mutation frequency yet produce a significant effect on expression dysregulation.
Table 1.
Top 10 genes that cause the most gene expression dysregulation in breast cancer
| Gene | Differentially expressed genes | Mutated samples | Mutation rate |
|---|---|---|---|
| TP53 | 13104 | 333 | 30.33% |
| CDH1 | 9692 | 133 | 12.11% |
| PIK3CA | 8612 | 311 | 28.32% |
| GATA3 | 7949 | 131 | 11.93% |
| MAP3K1 | 5408 | 79 | 7.19% |
| USH2A | 2688 | 63 | 5.74% |
| SPTA1 | 2656 | 54 | 4.92% |
| TTN | 2248 | 189 | 17.21% |
| DMD | 1377 | 53 | 4.83% |
| KMT2C | 1177 | 90 | 8.20% |
Figure 2.
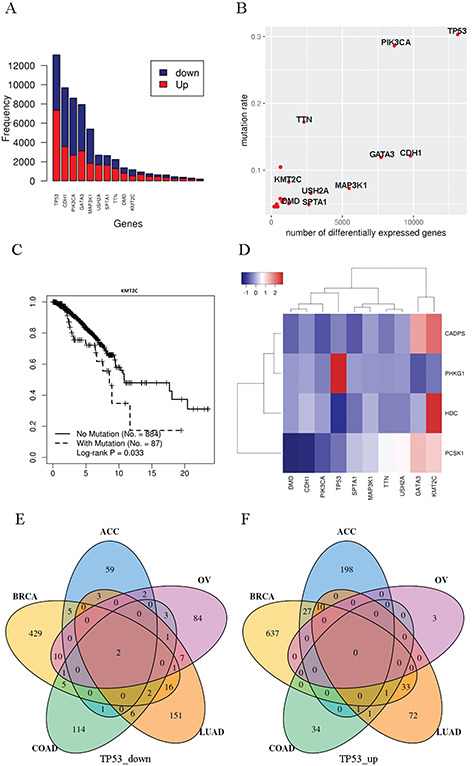
Example outputs of MutEx’s Functionality I. (A) Histogram that shows the number of genes differentially expressed in both directions by the top 10 genes in breast cancer. Known cancer genes such as TP53 and CDH1 are associated with the most gene expression dysregulation. (B) Scatter plot of the number of differentially expressed genes versus mutation rate. Because the number of differentially expressed genes is also dependent on the mutation rate, this scatter plot helps to visualize both measurements together. (C) Kaplan–Meier survival curve for KMT2C in breast cancer that shows significant survival difference in favor of wild-type KMT2C over mutants. KMT2C ranks number 10 in terms of the most expression dysregulation in breast cancer, and it is a relatively less studied gene in breast cancer. (D) Example clustering and heatmap using data from breast cancer. The columns are the top 10 breast cancer genes whose somatic mutations are associated with the most extensive gene expression dysregulation. The rows are the genes that are commonly dysregulated by these top 10 genes’ mutation status (fold change ≥ 2 and false discovery rate ≤ 0.05). The color in each cell denotes the scale of gene expression fold change between mutant versus wild type. (E) Venn diagram of downregulated genes based on TP53’s mutation status for five cancer types. (F) Venn diagram of upregulated genes based on TP53’s mutation status for five cancer types. The detailed gene list of each section of the Venn diagrams can be downloaded from the MutEx website.
Additional optional analyses offered at the query interface include survival analysis (mutant versus wild type), pathway/Gene Ontology enrichment, hierarchical clustering and cross-cancer comparison. The survival analysis is another intuitive approach to evaluating mutation impact: Kaplan–Meier survival curves are constructed, and log-rank test is performed to compare the survival pattern between two groups of patients separated by the mutation status in one gene or a gene set. When a set of genes is used, the mutant status of a patient is determined by mutations in any of the genes in the set. KMT2C, a less well-known cancer gene, which is ranked in the 10th place in BRCA in terms of extent of consequential dysregulation, manifested a significant survival difference favoring the wild-type group (P = 0.03; Figure 2C). Enrichment of pathways and Gene Ontology terms are sought among the differentially expressed genes contingent upon each user-specified mutated gene. A hierarchical clustering heatmap is produced based on the expression fold changes of the common set of differentially expressed genes associated with the input genes (Figure 2D). All analyses above are conducted within individual cancer types. The last analysis, cross-cancer comparison, returns the intersection scenario of the upregulated/downregulated genes across multiple (≤5) cancer types, which is conducted for each mutated genes individually. This utility helps to investigate whether a gene’s somatic mutations exert ubiquitous effect across different cancer types. We tested TP53 on five cancer types adrenocortical carcinoma (ACC), BRCA, colon adenocarcinoma (COAD), lung adenocarcinoma (LUAD), ovarian serous cystadenocarcinoma (OV) to demonstrate this feature (Figure 2E, F). Remarkably, despite the nearly universal involvement of TP53 in cancer progression, only two genes, EDA2R and SPATA18, were found to be downregulated in all five cancer types tested. EDA2R was shown to be a p53 target [11], and SPATA18 encodes a p53-inducible protein. Thus, these results hint at important tissue-specific mechanisms that result in different changes in gene expression in different cancer types harboring TP53 mutations.
To further explore the heuristic potential of Functionality I, we asked if MutEx could provide more insight into the role of the BRCA1-associated protein 1 (BAP1) in cancer. BAP1 is a pleiotropic tumor suppressor, which may mediate its effects through chromatin regulation, transcription modulation, ubiquitin–proteasome system and the DNA damage response pathway [12]. Somatic mutations in BAP1 were identified in various cancers, but signal transduction pathways with its involvement remain to be fully characterized. Using Functionality I, we found nonsynonymous BAP1 mutations in CHOL (15%), KIRC (6%), LIHC (4.7%), MESO (25%), UCEC (5.3%) and UVM (27.5%). Sorting through genes affected by BAP1 mutations, we found that RNF43, a RING-type E3 ligase involved in downregulation of canonical Wnt/β-catenin signaling [13], is ~2.6- to 6-fold downregulated in the KIRC, MESO and UVM specimens, with BAP1 mutations suggesting that BAP1 inactivation has suppressive effects on RNF43 expression in at least four unrelated cancers (Table 2). Indeed, as inactivating RNF43 mutations were reported in various cancers, suggesting that RNF43 may function as a tumor suppressor [14–16], our analysis using MutEx provides the first suggestion of the stimulatory effect of BAP1 on Wnt/β-catenin signaling, which needs experimental validation.
Table 2.
Effects of BAP1 mutations on RNF43 expression
| Gene | P | Fold change |
|---|---|---|
| KIRC | 1.00e−05 | −4.54 |
| MESO | 3.50e−02 | −3.15 |
| UVM | 1.00e−05 | −6.04 |
| LIHC | 6.50e−03 | −2.6 |
Functionality II of MutEx is conceptually the reverse of Functionality I, whereby the user can identify genes whose mutation status may account for the differential expression of input genes. This provides an inverse perspective on the relationship between somatic mutations and gene expression. For example, given CDKN1A in ovarian cancer, MutEx detects TP53 mutation status as the major determinant of CDKN1A expression dysregulation. The regulatory effect between TP53 mutations and expression of CDKN1A was previously reported [17]. All the directed links among mutated genes and the differentially expressed genes can be modeled into a network representation. Furthermore, Functionality II can also be used to identify genes whose mutation status does not cause gene expression dysregulation. For example, in skin cutaneous melanoma (SKCM), gene RGPD3’s mutation rate is 14.7%. However, only one gene was differentially expressed between RGPD3 mutants and wild types.
Functionality III of MutEx focuses on the exploration of mutation burden in pan-cancer TCGA data set. Mutational burden is quantified by the number of nonsynonymous mutations observed in each mutated gene and allows for comparisons of mutation burdens between two groups of patients separated by the mutation status of specified input genes. The mutational burden of a subject is defined as the number of nonsynonymous mutations in all genes excluding the gene set used to define mutant and wild type. The purpose is to assess whether mutations in the set of genes can affect overall mutational burden and survival (survival analysis can be performed in parallel to mutational burden analysis). To demonstrate this, we hypothesized that nonsynonymous mutations in DNA mismatch repair genes will result in higher mutational burden in other genes. Based on prior knowledge, we used MutEx to analyze mutation burden attributed to eight DNA mismatch repair genes (MLH1, MLH3, MSH3, MSH6, PMS1, PMS2 and PMS2L3) in all cancer types. Twenty-six of the 33 cancer types were eligible for this analysis with sufficient mutated samples (mutant ≥ 3) regarding those DNA mismatch repair genes. All 26 cancers showed higher mutational burden in the patients with mutant DNA mismatch repair genes compared to patients with wild-type genes, with 13 showing statistical significance (t-test P < 0.05) (Figure 3A). Besides mutational burden analysis, a co-mutation analysis is also provided, which performs hierarchical clustering on the mutational statuses of the input genes and allows users to examine whether the mutation status of the particular gene set can form distinct clusters among all patients.
Figure 3.
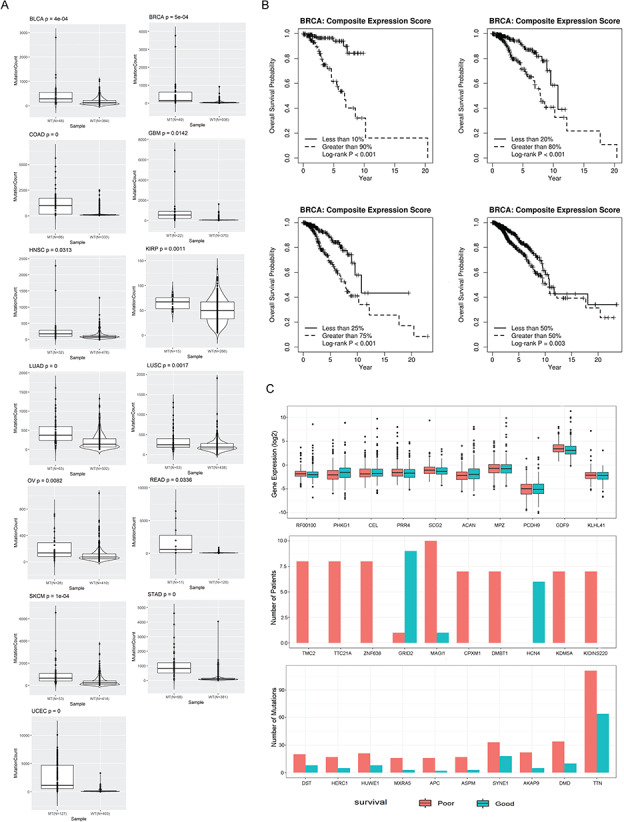
(A) Boxplot of the 13 cancers that showed significant (t-test P < 0.05) mutational burden difference between DNA mismatch repair gene mutant and wild types. (B) Example of Functionality V. Using all genes as input, elastic net Cox regression identified 17 genes that are associated with breast cancer survival. The 17 genes were further evaluated using selective inference package from R. A CGES was computed from these 17 genes, and significant associations between this score and survival time were found at different gene expression cutoff thresholds. (C) Example analysis results between good and poor survival groups (bottom 25% versus top 25%) in BRCA. Top: boxplot of top genes that are differentially expressed between good and poor survival groups. Middle: barplot that shows the top 10 genes ranked by Fisher’s exact test P values computed from the number of subjects with nonsynonymous somatic mutations between good and poor survival groups. Bottom: barplot that shows the top 10 genes ranked by raw nonsynonymous mutation count difference between good and poor survival groups.
Functionality IV allows for quick and flexible searches of mutations by gene in one or multiple cancers. We implemented a query function for finding mutations based on a combination of patient characteristics, including cancer type, gene symbols, genomic position and mutation type. The results are displayed in a searchable and downloadable dynamic table.
Functionality V offers gene expression-based survival analysis. Existing TCGA data-based resource websites offer expression survival analysis by dividing patients into high- and low-expression groups based on one gene’s expression using the median. MutEx offers the option for using a user-defined cutoff to divide patients into two groups. Additionally, when a set of genes are specified as input genes, the user can choose to process them as a group. In such a scenario, a CGES is created and used to group the patients. The last MutEx option included in the Functionality V is the ability to perform feature selection using lasso or elastic net regularization. When this option is selected, MutEx will perform dimension reduction and select a subset of the input genes that together are associated with patient survival. The selected genes are then evaluated for postselection inference. Using BRCA as an example, the lasso method selected 17 potentially correlated genes that are altogether most associated with survival (Table 3). The CGES from these 17 genes showed a strong association with survival under multiple thresholds (Figure 3B).
Table 3.
Elastic net selected genes whose collective expressions are mostly associated with breast cancer survival
| Gene | Coefficient | P | 95% Confidence interval |
|---|---|---|---|
| ENPP4 | 1.07 | <0.0001 | [1.04–1.09] |
| STPG1 | 1.07 | <0.0001 | [1.05–1.10] |
| GCLC | 0.93 | 0.0002 | [1.04–1.11] |
| CFTR | 0.94 | 0.0002 | [1.04–1.09] |
| SCYL3 | 0.94 | 0.0004 | [1.04–1.10] |
| FGR | 0.94 | 0.0007 | [1.03–1.10] |
| SEMA3F | 0.95 | 0.0018 | [1.02–1.08] |
| CFH | 0.96 | 0.0141 | [1.01–1.08] |
| DPM1 | 1.03 | 0.017 | [1.01–1.05] |
| C1orf112 | 1.04 | 0.0263 | [1.01–1.07] |
| FUCA2 | 1.03 | 0.0358 | [1.00–1.06] |
| LAS1L | 0.96 | 0.0455 | [1.00–1.08] |
| TSPAN6 | 1.03 | 0.0702 | [1.00–1.06] |
| NIPAL3 | 0.97 | 0.0846 | [0.99–1.06] |
| TNMD | 0.97 | 0.0899 | [0.99–1.05] |
| NFYA | 0.97 | 0.101 | [0.99–1.05] |
| ANKIB1 | 1.02 | 0.243 | [0.97–1.05] |
Results generated from selectiveInference R package.
Functionality VI is designed to detect differentially expressed genes or genes with differential mutational burden between subjects of good and poor survival. Subjects are dichotomized into good and poor survival based on the real survival information provided by TCGA. Users are free to choose the survival dichotomizing percentage threshold. Using BRCA data as an example, we conducted a comparative analysis between the bottom and top 25% of patients and identified 1917 genes that were significantly differentially expressed between good and poor survival groups (Supplementary Table S1; Figure 3C, top). The most differentially expressed gene is RF00100, a noncoding RNA. Pathway analysis revealed multiple cancer-related pathways from the differentially expressed genes, such as TNF signaling pathway and NF-kappa B signaling pathway (Supplementary Table S2). Gene-based mutational burden analyses are carried out at both the sample and mutation count levels between the good and poor survival groups. At the sample level, mutational burden is computed as the number of samples with nonsynonymous mutations at each gene, whereas at the mutation count level, the mutational burden is computed as the total number of nonsynonymous mutations in each gene, where a gene could have multiple mutations. Analysis in BRCA identified 60 genes in which the poor survival group had significantly (Fisher’s exact test P < 0.05) more mutations than the good survival group (Supplementary Table S3; Figure 3C, middle). For example, mutations in the less-known cancer genes TMC2, TTC21A and ZNF638 were found to be more prevalent in poor survival groups, which may prompt additional follow-up studies. By mutation count, the poor survival group tends to have more mutations than the good survival group in most genes. This is probably caused by damage in DNA repair genes, which caused a larger genome-wide mutational burden. Large genes such as TTN and DMD may act as a sample region of the entire protein-coding regions; thus, they show the biggest difference for mutation count between good and poor survival groups (Figure 3C, bottom).
Discussion
It is interesting to note that our expression-based mutation effect analysis indicates TP53 mutations produced the greatest change in gene expression. This may be due to the fact that TP53 is a tumor suppressor gene that functions as a transcription factor, and it is the most frequently mutated gene in human cancers. Another interesting observation is that CDH1 mutations produce the 2nd largest effect on gene expression. CDH1 encodes cadherin 1 cell–cell adhesion protein, and the loss of function mutations in this gene contributes to cancer progression. It is not known why CDH1 mutations would produce stronger effects on gene expression dysregulation than mutations in a master signal transducer PIK3CA, which was more frequently mutated than CDH1. GATA3 is a transcription factor, and mutations in GATA3 produce the 4th largest effect on expression dysregulation. Mutations in KMT2C top out the 10th largest effects on expression dysregulation. Interestingly, mutations in KMT2C are observed in approximately 10% of breast cancer cases, and these mutations are associated with poor outcome. It is not known if these mutations could serve as an independent prognostic factor. Additional multivariate analysis is needed to discern if mutations in KMT2C could serve as an independent prognostic factor. Finally, the heatmap produced by mutation–expression clustering analysis of top 10 mutations with the greatest effect on expression dysregulation indicates PHKG1 is upregulated in cancer with TP53 mutations. PHKG1 encodes for the serine/threonine protein kinase catalytic subunit of a kinase complex that regulates glycogenolysis and metabolism. It was previously shown to be upregulated in HCT-116 p53−/− cells compared to HCT-116 p53+/+ cells [18]. PHKG1 is amplified in several cancer types [19]. However, our study is the first systematic analysis of TP53 mutations in tumor samples that show a potential effect on PHKG1 expression in tumor samples. Glycogenolysis is an important alternative energy source for cancer cells, and disruption of this pathway contributes to oxidative stress, induction of senescence and suppression of tumor growth in vivo [20]. Therefore, our observation that TP53 mutations are associated with the upregulation of PHKG1 provides biological insights into a potential role of TP53 mutations in the PHKG1 dysregulation and cancer cell metabolism.
While significant effort and considerations have been put into the development and implementation of MutEx, there are several notable limitations of MutEx. Because MutEx is based on empirical data, novel somatic mutations that are not included in the existing data sets cannot be analyzed in MutEx. However, novel somatic mutations not covered by existing data sets in cancer are likely to be rare. Furthermore, TCGA subjects were primarily Caucasian; thus, minority races are underrepresented. These limitations can be mitigated by incorporating additional data sets into MutEx in the future. MutEx presents the analysis results from a gene-centric point of view by aggregating individual loci to the gene level by searching for occurrence of at least one mutation within the gene body. An alternative strategy would be to partition the subjects into mutant and wild type for each mutation. However, this would result in very few patients in the mutant group for the majority of the cases, thus tremendously lowering the statistical power of the differential expression analysis. For this reason, we opted to use gene-level mutation status instead of individual mutation status. Our gene-level mutation classification approach has limitations in some cases. For example, mutations in TP53 can be classified into loss-of-function mutations and gain-of-function mutations, because particular mutations in TP53 result not only in loss of function but also in gain of oncogenic functions. Currently, all mutations in TP53 are considered as one property without distinguishing the gain-of-function or loss-of-function mutants. This limitation can be addressed in the future as the classification of gain-of-function TP53 mutations becomes more robust. Lastly, we suspect that mutated allele frequency (expressed in percentage of mutated sequence reads) may be associated with expression as well. However, such analyses will also be subject to small sample size issues. We will seek additional opportunities to investigate this hypothesis in future studies.
While the current iteration of development is complete, we plan to add several additional features to MutEx in the near future, including miRNA expression and methylation information. miRNA and other long noncoding RNAs (lncRNA) have been the focus of much functional research recently [21, 22]. Many studies have constructed disease prediction models using miRNA [23, 24] and lncRNA [25]. The algorithms used in these models can be applied to cancer setting to predict survival and treatment response.
Using several proof of concept examples, we demonstrated that MutEx is a valuable tool for evaluating gene-based cancer hypothesis. It is capable of finding novel cancer genes and estimating cancer mutation and gene expression association with survival outcomes. MutEx is constructed with component design, which simplifies the development of additional functionalities and the incorporation of additional data sets. Many of the existing features of MutEx were inspired by our collaborations with oncologists, and we plan for MutEx to undergo constant improvement to incorporate feedbacks from MutEx real-time users.
Key Points
MutEx is a multifaceted cancer genomic feature database.
MutEx provides the evaluation of somatic mutation effect on gene expression dysregulation.
MutEx provides several additional functionalities such as mutational burden analysis and gene expression composite score-based survival analysis.
Supplementary Material
Acknowledgment
This study was supported by the Cancer Center Support Grant P30CA118100 from the National Cancer Institute.
Yan Guo is an associate professor in the Department of Internal Medicine, University of New Mexico. He is also the Director of Bioinformatics Shared Resources for University of New Mexico Comprehensive Cancer Center.
References
- 1. Melo FDE, Vermeulen L, Fessler E, et al. Cancer heterogeneity—a multifaceted view. EMBO Rep 2013;14:686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uhlen M, Zhang C, Lee S, et al. A pathology atlas of the human cancer transcriptome. Science 2017;357. [DOI] [PubMed] [Google Scholar]
- 3. Westra HJ, Franke L. From genome to function by studying eQTLs. Biochim Biophys Acta 1842;2014:1896–902. [DOI] [PubMed] [Google Scholar]
- 4. Li J, Wang L, Jiang T, et al. eSNPO: an eQTL-based SNP ontology and SNP functional enrichment analysis platform. Sci Rep 2016;6:30595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anders S, Pyl PT, Huber W. HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nikolayeva O, Robinson MD. edgeR for differential RNA-seq and ChIP-seq analysis: an application to stem cell biology. Methods Mol Biol 2014;1150:45–79. [DOI] [PubMed] [Google Scholar]
- 7. Wang HS, Li GD, Tsai CL. Regression coefficient and autoregressive order shrinkage and selection via the lasso. J R Stat Soc Series B Stat Methodol 2007;69:63–78. [Google Scholar]
- 8. Zhao S, Guo Y, Sheng Q, et al. Advanced heat map and clustering analysis using heatmap3. Biomed Res Int 2014;2014:986048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gala K, Li Q, Sinha A, et al. KMT2C mediates the estrogen dependence of breast cancer through regulation of ER alpha enhancer function. Oncogene 2018;37:4692–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanikawa C, Ri C, Kumar V, et al. Crosstalk of EDA-A2/XEDAR in the p53 signaling pathway. Mol Cancer Res 2010;8:855–63. [DOI] [PubMed] [Google Scholar]
- 12. Murali R, Wiesner T, Scolyer RA. Tumours associated with BAP1 mutations. Pathology 2013;45:116–26. [DOI] [PubMed] [Google Scholar]
- 13. Loregger A, Grandl M, Mejias-Luque R, et al. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated beta-catenin by sequestering TCF4 to the nuclear membrane. Sci Signal 2015;8:ra90. [DOI] [PubMed] [Google Scholar]
- 14. Liu L, Wong CC, Gong B, et al. Functional significance and therapeutic implication of ring-type E3 ligases in colorectal cancer. Oncogene 2018;37:148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giannakis M, Hodis E, Mu XJ, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet 2014;46:1264–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang XM, Hao HX, Growney JD, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 2013;110:12649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993;75:817–25. [DOI] [PubMed] [Google Scholar]
- 18. Daoud SS, Munson PJ, Reinhold W, et al. Impact of p53 knockout and topotecan treatment on gene expression profiles in human colon carcinoma cells: a pharmacogenomic study. Cancer Res 2003;63:2782–93. [PubMed] [Google Scholar]
- 19. Camus S, Quevedo C, Menendez S, et al. Identification of phosphorylase kinase as a novel therapeutic target through high-throughput screening for anti-angiogenesis compounds in zebrafish. Oncogene 2012;31:4333–42. [DOI] [PubMed] [Google Scholar]
- 20. Favaro E, Bensaad K, Chong MG, et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab 2012;16:751–64. [DOI] [PubMed] [Google Scholar]
- 21. Chen X, Yan CC, Zhang X, et al. Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform 2017;18:558–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen X, Xie D, Zhao Q, et al. MicroRNAs and complex diseases: from experimental results to computational models. Brief Bioinform 2019;20:515–39. [DOI] [PubMed] [Google Scholar]
- 23. Chen X, Wang L, Qu J, et al. Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics 2018;34:4256–65. [DOI] [PubMed] [Google Scholar]
- 24. Chen X, Yin J, Qu J, et al. MDHGI: matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput Biol 2018;14:e1006418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen X, Yan GY. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 2013;29:2617–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.