

Abstract
The exotoxins toxin A (TcdA) and toxin B (TcdB) are produced by the bacterial pathogen Clostridium difficile and are responsible for the pathology associated with C. difficile infection (CDI). The antitoxin antibodies actoxumab and bezlotoxumab bind to and neutralize TcdA and TcdB, respectively. Bezlotoxumab was recently approved by the FDA for reducing the recurrence of CDI. We have previously shown that a single molecule of bezlotoxumab binds to two distinct epitopes within the TcdB combined repetitive oligopeptide (CROP) domain, preventing toxin binding to host cells. In this study, we characterize the binding of actoxumab to TcdA and examine its mechanism of toxin neutralization. Using a combination of approaches including a number of biophysical techniques, we show that there are two distinct actoxumab binding sites within the CROP domain of TcdA centered on identical amino acid sequences at residues 2162–2189 and 2410–2437. Actoxumab binding caused the aggregation of TcdA especially at higher antibody:toxin concentration ratios. Actoxumab prevented the association of TcdA with target cells demonstrating that actoxumab neutralizes toxin activity by inhibiting the first step of the intoxication cascade. This mechanism of neutralization is similar to that observed with bezlotoxumab and TcdB. Comparisons of the putative TcdA epitope sequences across several C. difficile ribotypes and homologous repeat sequences within TcdA suggest a structural basis for observed differences in actoxumab binding and/or neutralization potency. These data provide a mechanistic basis for the protective effects of the antibody in vitro and in vivo, including in various preclinical models of CDI.
Keywords: Clostridium difficile infection, monoclonal antibody, toxin neutralization, epitope mapping, TcdA
Introduction
Clostridium difficile is a Gram-positive, spore-forming bacterium that infects the gastrointestinal tract of both humans and animals. In humans C. difficile infection (CDI) can cause mild symptoms such as a low-grade fever, watery stools, and minor abdominal cramping, as well as more severe symptoms such as bloody diarrhea, pseudomembrane colitis, toxic megacolon, and death [1]. Individuals whose normal gut flora has been compromised by treatment with antibiotics are most at risk for CDI. Over the past few decades, the incidence of CDI has increased throughout the developed world and is now a major health concern. Most often transmitted in a healthcare facility setting, C. difficile has become the most commonly reported pathogen in hospitals in the United States [2] and causes over 14,000 deaths per year†. Currently, CDI is treated with standard of care antibiotics vancomycin, metronidazole, and fidaxomicin. Despite the high efficacy of these agents in treating an initial episode of CDI, 25 to 30% of patients will suffer a recurrence within 3 months [3], with subsequent recurrences occurring at an even higher rate. Thus, there is a great need to develop novel therapies that will reduce the risk of recurrence.
The symptoms of CDI are primarily caused by the exotoxins toxin A (TcdA) and toxin B (TcdB), which are produced by the bacterium during the infection [4–7]. TcdA and TcdB are structurally similar proteins, each having four separate domains: an amino-terminal glucosyltransferase domain (GTD), internal autoprotease and translocation domains, and a combined repetitive oligopeptide (CROP) domain at the carboxy-terminus. The CROP domains of TcdA and TcdB are composed of multiple short repeats (SRs; 32 in TcdA and 20 in TcdB) interspersed with a smaller number of long repeats (LRs; 7 in TcdA and 4 in TcdB) and have been presumed to play a role in receptor binding [8]. Both toxins bind to intestinal epithelial cells, and possibly other mucosal cells, and are internalized through receptor-mediated endocytosis [9]. The low pH environment of the endosome triggers a conformational change in the protein, resulting in the translocation of the GTD across the endosomal membrane and into the cytoplasm [10–12]. The autoprotease domain then cleaves the GTD [13], allowing it to diffuse through the cytoplasm and inactivate small GTPases of the Ras superfamily (particularly the Rho subfamily but also Rap and Ras) through covalent glucosylation [14,15], resulting in actin depolymerization, inflammatory cytokine production, and cell death [16–18].
While much is known about the trafficking of TcdA and TcdB and their mechanisms of action once internalized into target cells, exactly how the toxins bind to cells and through which receptors is less clear. Because different cell types show different levels of susceptibility to each toxin, it is believed that TcdA and TcdB bind to different receptors. Truncated versions of TcdA and TcdB lacking the CROP domain are still capable of intoxicating cells, albeit with lower potency than intact toxins, showing that regions outside the CROP domain are also involved in receptor binding [19,20]. Recently, poliovirus receptor-like protein 3, chondroitin sulfate proteoglycan 4, and members of the Wnt receptor frizzled family have been identified as putative cellular receptors for TcdB [21–23]. The TcdB CROP domain appears to be not necessary for binding to poliovirus receptor-like protein 3 or frizzled family protein members. While the potential receptors for TcdB identified thus far are membrane proteins, the receptor for TcdA is thought to be a cell surface carbohydrate [24]. The LRs in the CROP domain may serve as receptor binding sites, since a crystal structure of a C-terminal fragment of the TcdA CROP domain in complex with α-Gal-(1,3)-β-Gal-(1,4)-β-GlcNAcO(CH2)8CO2CH3 shows binding of the carbohydrate to residues located around the LR regions [25].
The need for more effective treatments for CDI has led to the development of alternative non-antibiotic therapies. Foremost among these are the two fully human monoclonal antibodies actoxumab and bezlotoxumab, which target TcdA and TcdB, respectively. The combination of actoxumab and bezlotoxumab is highly protective in primary and recurrent animal models of CDI [26–29]. In clinical trials, bezlotoxumab alone or in combination with actoxumab significantly reduced the rate of CDI recurrence when co-administered with standard of care antibiotics [30]. We have previously shown that bezlotoxumab binds to two separate but homologous epitopes centered on LR1 and LR2 within the TcdB CROP domain and prevents toxin binding to host cells [31]. In this study, we characterized the binding of actoxumab to TcdA and show that the antibody binds at two distinct binding sites centered on two of the seven LRs in the TcdA CROP domain and that similar to bezlotoxumab, the antibody neutralizes toxin activity by preventing binding to host cells.
Results
Mechanism of TcdA neutralization by actoxumab
The CROP domain of TcdA has historically been considered to be the primary receptor binding domain of the toxin [32]. Specifically, pockets within the LR regions of the CROP domain have been implicated in mediating the binding of TcdA to carbohydrate moieties on the host cell surface [25]. Since actoxumab is known to bind to the CROP domain of TcdA [26], we assessed the possibility that the mechanism of neutralization of TcdA by actoxumab involves the blockade of TcdA binding to host cells, similar to the mechanism of TcdB neutralization by bezlotoxumab [31]. We first measured the binding of fluorescently labeled TcdA (TcdA-Atto488) to HT29 cells by flow cytometry in the presence and absence of actoxumab. As shown in Fig. 1a, mean fluorescence intensity (MFI) increases in a concentration-dependent manner in the presence of TcdA-Atto488, showing that TcdA binds to cells under these conditions. Preincubation of toxin with actoxumab or Fab fragments of actoxumab prevented the binding of TcdA to cells, with MFI values reduced to baseline levels. Actoxumab-Fab binds to TcdA with a Kd of 1900 ± 300 pM and fully neutralizes TcdA in a cell death assay, albeit at a higher EC50 when compared to full-length actoxumab (Supplementary Data Table S1 and Fig. S1). Conversely, bezlotoxumab did not affect MFI, showing that the blockade of TcdA binding is specific to actoxumab (Fig. 1 b). Similar results were obtained with Vero cells (data not shown). We also assessed the ability of actoxumab to prevent the binding of unlabeled TcdA by Western blot of isolated Vero cell membranes following incubation with TcdA in the presence or absence of actoxumab or bezlotoxumab. Consistent with the flow cytometry results, actoxumab blocked the binding of TcdA to Vero cells, while bezlotoxumab had no effect (Fig. 1c).
Fig. 1.
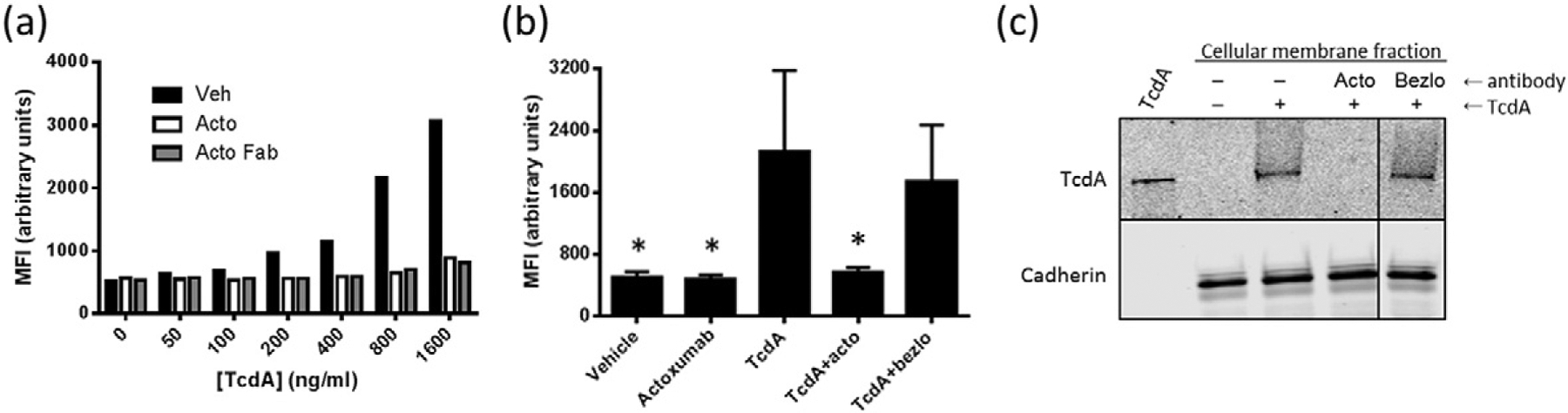
Actoxumab prevents the binding of TcdA to HT29 and Vero cells. (a) Flow cytometry analysis of HT29 cells preincubated with increasing concentrations of TcdA-Atto488 at 4 °C in the presence or absence of vehicle, actoxumab (200 μg/ml), or actoxumab-Fab (200 μg/ml). Following incubation, MFI was measured with excitation and emission wavelengths of 488 nm and 530 nm, respectively. A representative experiment is shown. (b) Flow cytometry analysis of HT29 cells preincubated with 800 ng/ml TcdA-Atto488 at 4 °C in the presence or absence of vehicle, actoxumab, or bezlotoxumab. MFIs were calculated as per panel (a). Values are means ± standard deviation of two independent experiments. acto = actoxumab; bezlo = bezlotoxumab. *p < 0.05 compared to TcdA alone by paired two-tailed t-test. (c) Western blot of membranes isolated from Vero cells following incubation with 1 μg/ml TcdA in the presence of vehicle, actoxumab, or bezlotoxumab (200 μg/ml). The top panel shows TcdA and the bottom panel shows cadherin (loading control).
Actoxumab binding in the CROP domain of TcdA
Actoxumab binds within the CROP domain of TcdA and does not cross-react with TcdB [26]. The number of binding sites and precise location(s) of the actoxumab epitopes within the CROP domain, however, are not known. To begin to address this, we expressed and purified several overlapping protein fragments of the CROP domain (Fig. 2a) and measured the binding of actoxumab to these fragments by Western blotting. As shown in Fig. 2b, actoxumab binds to full-length TcdA and to each of the TcdA CROP constructs tested, A0, A2, A3, and A4, but not to TcdB or to proteins B2 and B3, which are fragments of the TcdB CROP domain [31]. We also measured the binding of actoxumab to TcdA and constructs A1 and A4 by surface plasmon resonance (SPR). Binding kinetics analyses show that actoxumab binds to the TcdA CROP proteins with affinities comparable to full-length TcdA, with dissociation constants (Kds) of 610 ± 90 pM, 220 ± 110 pM, and 800 ± 490 pM for TcdA, A1, and A4, respectively (Table 1). As expected, actoxumab did not show any measurable binding to TcdB by SPR (Table 1 and Supplementary Data Fig. S2). Taken together, the Western blot and SPR data demonstrate that actoxumab binds to two or more sites within the TcdA CROP domain, with one or more in the N-terminal half (residues 1832–2233; protein A2) and one or more in the C-terminal half (residues 2234–2710; protein A4).
Fig. 2.
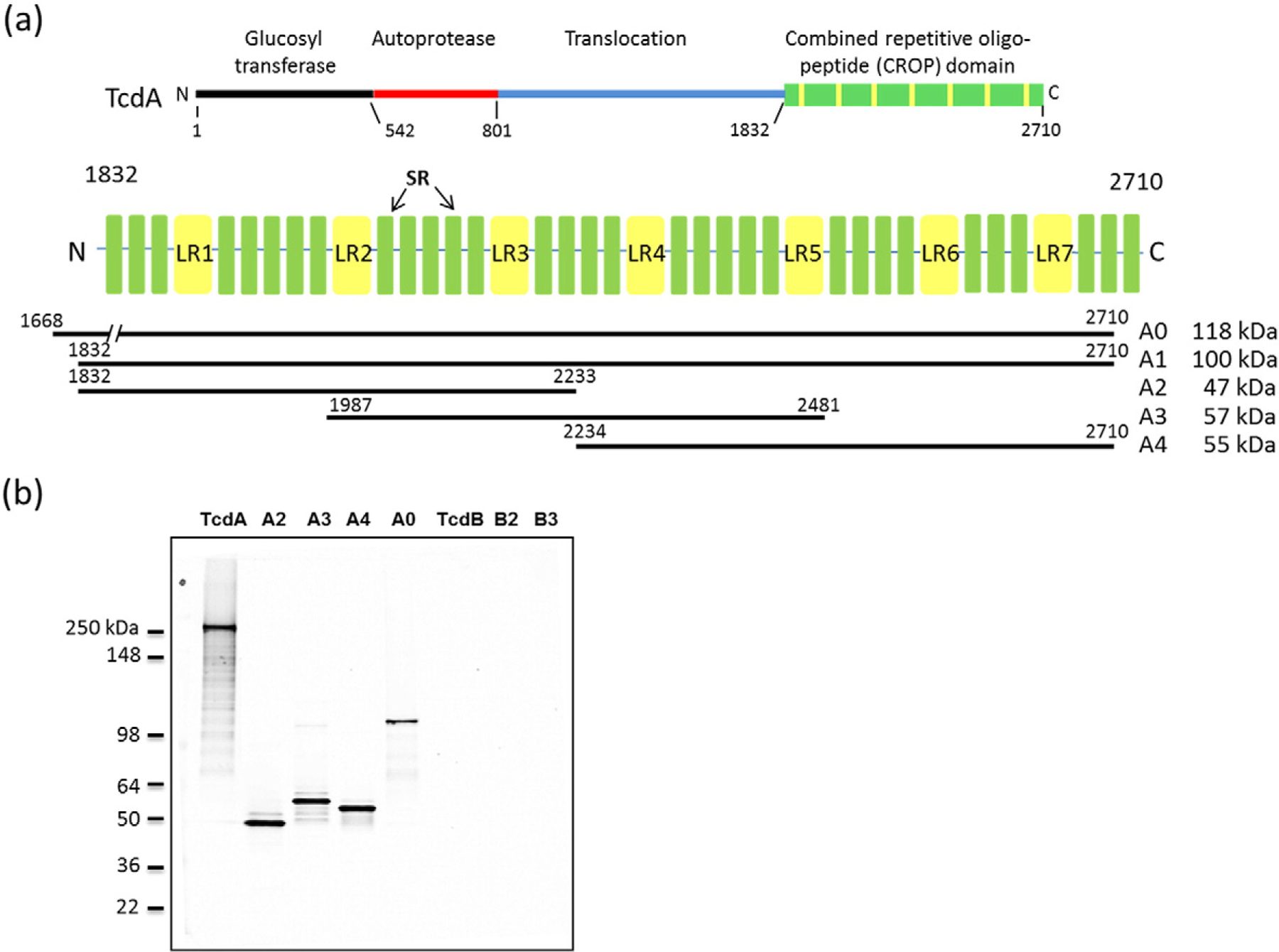
TcdA constructs used in this study and binding of actoxumab by Western blotting. (a) Domain organization of TcdA showing the CROP domain with LRs and SRs highlighted. The CROP fragment constructs (A0, A1, A2, A3, A4, and A5) used in this study are also shown. (b) Western blot of TcdA and TcdB holotoxins and of constructs A0, A2, A3, A4, B2, and B3.
Table 1.
Association and dissociation rates and affinities of actoxumab binding to purified TcdA and its fragments as determined by SPR
| Toxin/Kd (pM)a,b | kon (M−1 s−1)a,b | koff (sec−1)a,b | |
|---|---|---|---|
| TcdA | 1.87 × 105 ± 0.06 × 105 | 1.13 × 10−4 ± 0.13 × 10−4 | 610 ± 90 |
| A1 | 5.92 × 105 ± 1.65 × 105 | 1.22 × 10−4 ± 2.01 × 10−4 | 220 ± 110 |
| A4 | 3.19 × 105 ± 1.76 × 105 | 2.12 × 10−4 × 0.16 × 10−4 | 800 ± 490 |
| TcdB | Not measurable | Not measurable | Not measurable |
The data presented are average ± standard deviation from two independent experiments.
koff = dissociation binding constant; kon = association binding constant; Kd = equilibrium dissociation constant.
We next assessed the stoichiometry of actoxumab binding to TcdA and A1 protein by size-exclusion chromatography coupled with multiangle laser light scattering (SEC-MALLS). Immunocomplex formation was carried out at varying antibody:antigen molar ratios. When incubated at antibody:toxin molar ratios of 1:1 and 5:1, actoxumab and TcdA formed complexes with an average molecular weight of 2141 kDa and 2090 kDa, respectively (Fig. 3a and Table 2), corresponding approximately to the theoretical molecular weight of four molecules of actoxumab bound to five molecules of TcdA. At antibody:A1 ratios of 1:5 and 1:1, actoxumab bound to A1 with stoichiometries of 1:2 and 2:1, respectively (Fig. 3b and Table 2). At higher antibody:A1 M ratios, extensive actoxumab-peptide aggregation was detected, consistent with the results observed with full-length TcdA shown above. Crosslinking of TcdA and A1 by actoxumab and the formation of large immunocomplexes are consistent with the notion that multiple actoxumab binding sites exist within the TcdA CROP domain but do not confirm the number of epitopes. To eliminate issues arising from crosslinking of toxin and thereby more accurately measure the number of actoxumab binding sites in TcdA, we assessed immunocomplex formation with purified (monovalent) Fab fragments of actoxumab. At low Fab:TcdA ratios (1:1 and 1:2), actoxumab-Fab bound to TcdA as a 1:1 complex of 324 kDa (Fig. 3c and Table 2), but at a higher Fab:TcdA ratio of 5:1, the immunocomplex formed consisted of two Fab molecules bound to a single TcdA molecule (~390 kDa). Increasing the molar ratio to 10:1 did not further increase the size of the immunocomplex, consistent with the presence of no more than two binding sites for actoxumab within TcdA.
Fig. 3.
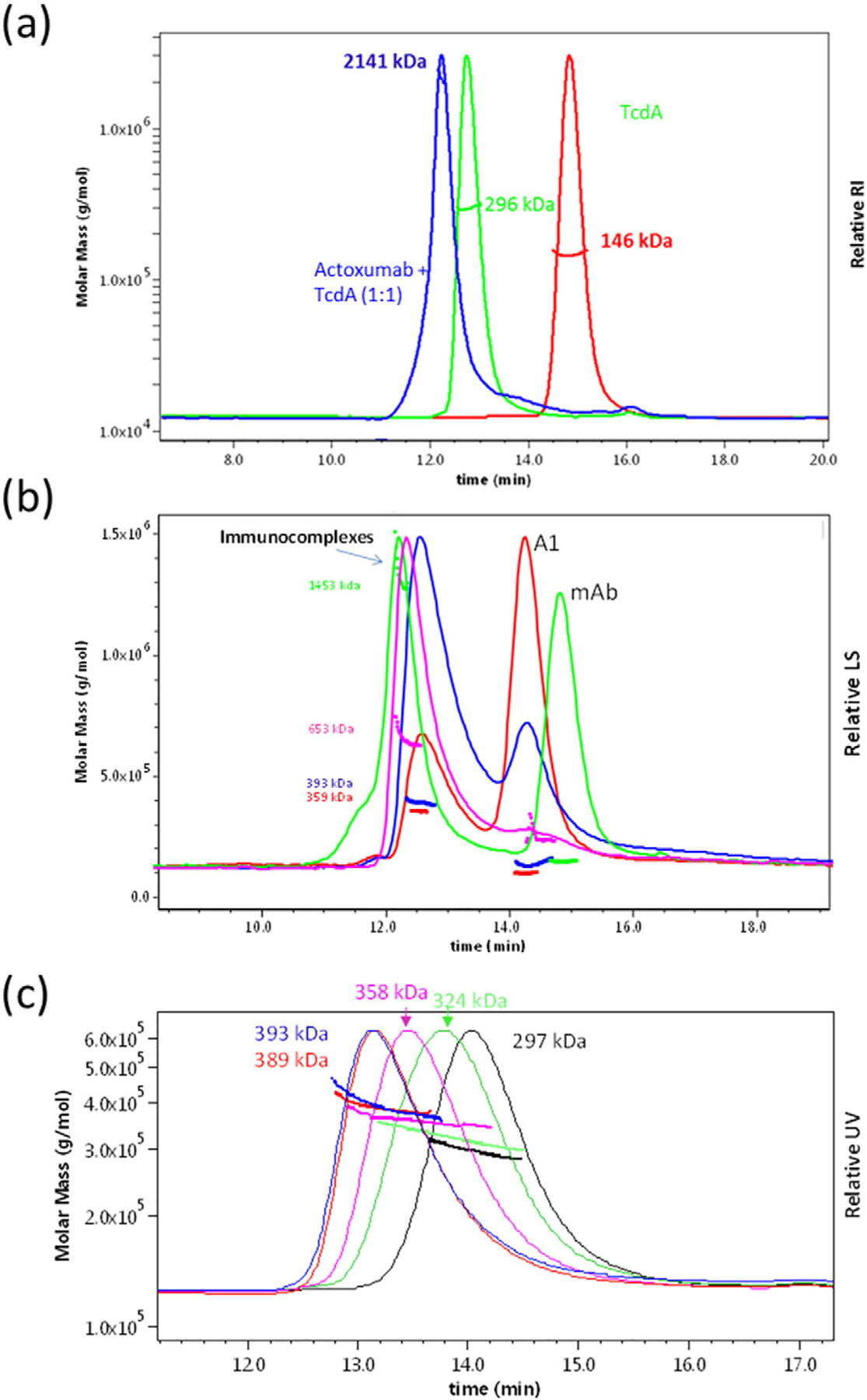
SEC-MALLS analysis of actoxumab/TcdA immunocomplexes. Light scattering and molecular mass distributions of immunocomplexes formed at various antibody:antigen molar ratios as a function of elution time. (a) Chromatograms of TcdA (green), actoxumab (red), and of actoxumab/TcdA immunocomplexes formed at 1:1 actoxumab:TcdA molar ratio (blue). (b) Chromatograms of actoxumab/A1 immunocomplexes formed at 1:5 (red), 1:1 (blue), 2:1 (pink), and 5:1 (green) actoxumab:A1 molar ratios. (c) Chromatograms of actoxumab-Fab alone (red) and of actoxumab-Fab/TcdA immunocomplexes formed at 1:5 (green), 1:1 (pink), 5:1 (dark red), and 10:1 (blue) Fab:TcdA molar ratios.
Table 2.
Summary of immunocomplexes formed between Actoxumab and TcdA and their fragments as determined by SEC-MALLS
| Antibody | Antigen | Molar ratio (Antibody:antigen) | MW of complex (kDa) | Putative stoichiometry (Antibody:antigen) |
|---|---|---|---|---|
| Actoxumab | – | – | 146 | 1:0 |
| Fab | – | – | 47 | 1:0 |
| – | TcdA | – | 296 | 0:1 |
| – | A1 | – | 100 | 0:1 |
| Actoxumab | TcdA | 1:1 | 2141 | 5:4 |
| 5:1 | 2090 | 5:4 | ||
| Actoxumab | A1 | 1:5 | 359 | 1:2 |
| 1:1 | 394 | 2:1 | ||
| 2:1 | 653 | 3:2 or 4:1 | ||
| 5:1 | 1453 | 6:5, 7:4, or 8:3 | ||
| Fab | TcdA | 1:5 | 324 | 1:1 |
| 1:1 | 358 | 1:1 or 2:1 | ||
| 5:1 | 389 | 2:1 | ||
| 10:1 | 393 | 2:1 |
Electron microscopic visualization of Fab fragments of actoxumab in complex with TcdA
To confirm the number of actoxumab binding sites within TcdA, we used negative-stain electron microscopy (EM), a technique that was previously used to resolve the structural organization of the functional domains of TcdA [33]. Within the EM structure, the glucosyltransferase and autoprotease domains appear as a “pincher-like” head domain, with the translocation and CROP domains extending from the head as short and long tails, respectively [33,34]. Since actoxumab caused the extensive aggregation of TcdA even at a 1:1 molar ratio (consistent with the SEC-MALLS data shown above; data not shown), Fab fragments of actoxumab were again used in place of full-length antibody for these experiments. Reference-free alignment of Fab:TcdA complexed at a 3:1 molar ratio produced multiple class averages with one Fab bound near the center of the CROP domain, with the retention of a stable orientation of the CROP domain relative to the body of the toxin (Fig. 4a). Using a relatively large number of classes in the alignment allowed the capture of scarcer bound species and revealed the occupation of a second binding site closer to the C terminus and on a different face of the CROP domain, extending in the opposite direction (Fig. 4b and d). Binding at the second site appeared to interfere with the stable conformation of the CROP region, resulting in particle averages with indistinct toxin images (Fig. 4b). The loss of resolution of the main body of the toxin in the two-site class average image suggests a higher degree of flexibility in the toxin when both sites are occupied. All particles from this class have both Fab binding sites occupied (Fig. 4c). Even at a 3:1 molar ratio, the most prevalent species had Fab clearly bound only to the first (i.e., N-terminal) binding site, with no particles identified having only the second (i.e., C-terminal) binding site occupied. These results suggest that the N-terminal site is the higher affinity site and support the presence of two spatially distinct binding sites for actoxumab, both within the CROP domain of TcdA.
Fig. 4.

Negative-stain EM images of actoxumab-Fab bound to TcdA. Representative class averages of actoxumab-Fab bound to TcdA holotoxin (6996 total particles, in dataset). (a) Individual classes for a single-site Fab (204 particles) or (b) two-site Fabs (40 particles) bound to the TcdA CROP domain at Fab:toxin ratio of 3:1. Side length of each panel is 52.7 nm (scale bar represents 10 nm). (c) Schematic representation of TcdA with arrows showing regions where Fab fragments bind (model based on EM data from Ref. [33]). (d) Gallery of selected particles from the two-site class (top), with schematic (below) outlining individual components (TcdA white, Fab red).
HDX-MS analysis of the TcdA CROP domain with and without bound actoxumab
Negative-stain EM confirmed two actoxumab binding sites within the TcdA CROP domain. To more precisely define the location of actoxumab epitopes, we utilized hydrogen deuterium exchange-mass spectrometry (HDX-MS), a method that compares the rate and levels of deuterium exchange in a protein that has been incubated in D2O in the presence and absence of a binding partner. In this case, we measured deuterium incorporation in protein A1 (corresponding to the entire CROP domain of TcdA) in the presence and absence of actoxumab. HDX-MS analysis of A1 shows that a 28-aa fragment (GVFKGPNGFEYFAPANTDANNIEGQAIL) is significantly protected from deuteration (17%) in the presence of actoxumab (Fig. 5a and b), suggesting that the antibody binds to this region. This exact sequence is located at residues 2162–2189 (within LR3) and again at residues 2410–2437 (within LR5). Based on our EM results showing the presence of one antibody binding site near the middle of the CROP domain and a second site for actoxumab closer to the C-terminal end, we conclude that the antibody interacts with both regions. Several other smaller regions showed lower levels of protection in the presence of actoxumab, suggesting either that actoxumab interacts weakly with these regions or that these areas are affected by localized conformational changes that occur upon antibody binding.
Fig.5.
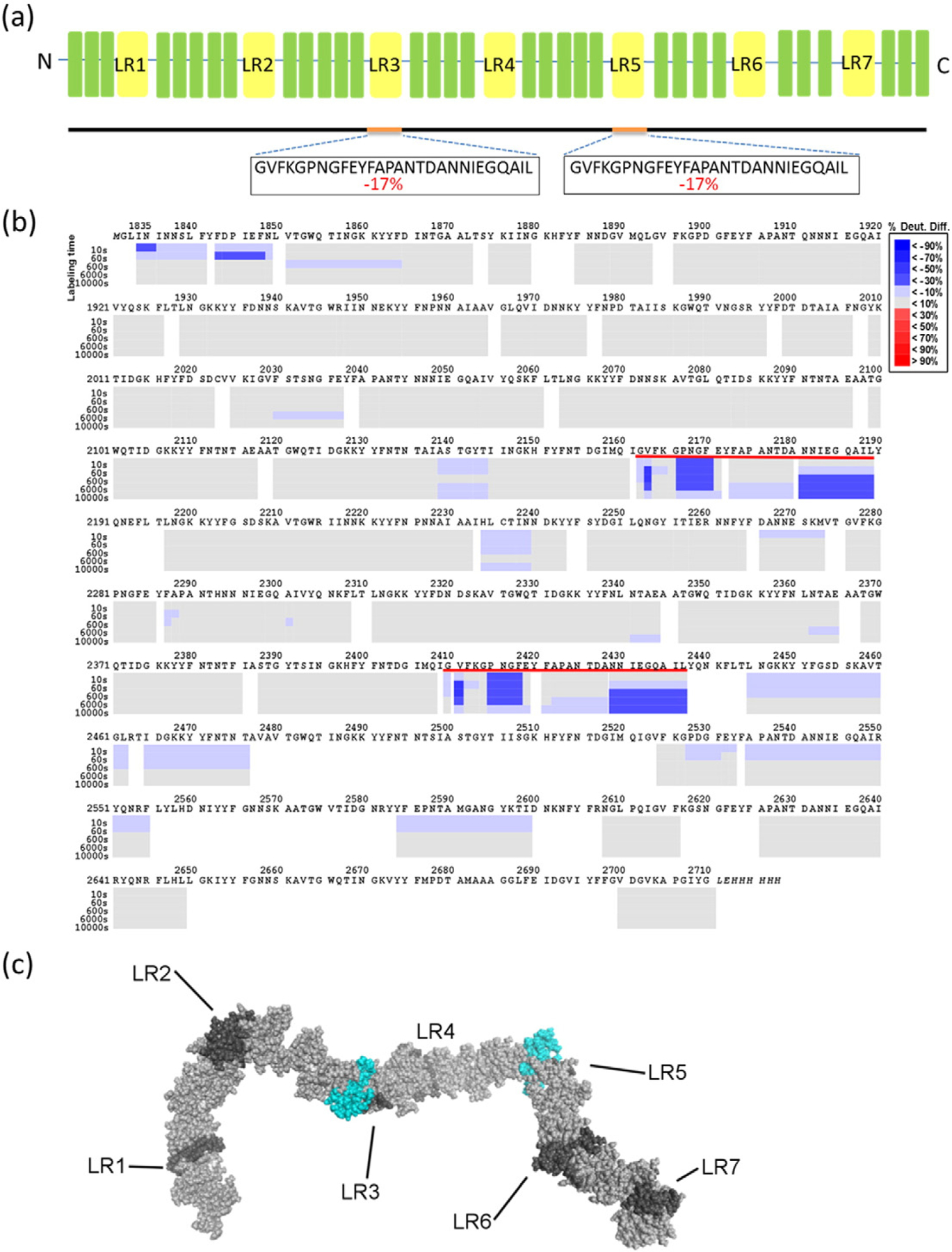
Identification of actoxumab epitopes by HDX-MS analysis. (a) Summary of HDX-MS analysis showing regions of TcdA CROP domain (construct A1) that have significantly lower deuterium incorporation in the presence of actoxumab, with % difference in deuteration level in the presence versus the absence of actoxumab indicated. (b) Peptide map showing regions of protein construct A1 containing lower (in blue) or higher (in red) deuterium incorporation following preincubation with actoxumab. Each gray box represents a distinct peptide identified by MS and is subdivided by incubation time point (10, 60, 600, 6000, and 10,000 s), as indicated. Sequences underlined in red are identical to each other and correspond to the putative epitopes of actoxumab. The numbering used is that of full-length TcdA. (c) Model of the TcdA CROP domain showing the putative actoxumab epitopes centered on LR3 and LR5. LRs are shown in dark gray, SRs in light gray, and regions protected from deuteration (putative actoxumab epitopes) in blue.
Comparison of actoxumab epitope sequences across homologous sequences within TcdA and across different strains of C. difficile
Our EM and HDX-MS data demonstrate that actoxumab binds to two distinct but identical regions centered at LR3 and LR5 of the TcdA CROP domain. As a consequence of its repetitive nature, five additional regions homologous to the two actoxumab epitopes are present within the TcdA CROP domain, to which actoxumab does not bind (LR1, LR2, LR4, LR6, and LR7). Furthermore, we have previously shown that actoxumab binds to and neutralizes TcdA of ribotype 027/toxinotype III and ribotype 078/toxinotype V with a lower affinity/potency compared to the control strain VPI 10463 (ribotype 087/toxinotype 0) [35]. In an effort to identify residues that are important for actoxumab binding, we compared the two identical epitopes of actoxumab (LR3 and LR5) within the VPI 10463 (Ribotype 087) TcdA, as determined by HDX-MS, to the homologous non-binding regions within TcdA and to the LR3 and LR5 epitopes in the TcdA sequences of other ribotypes. As shown in Fig. 6, amino acid differences between the actoxumab binding sites at LR3 and LR5 and these other homologous sequences tend to cluster at three distinct locations within the epitope, specifically between positions 4–7 and 18–19 and at position 28. Of these clusters, amino acids at positions 18 and 19 are unlikely to play an important role in actoxumab binding since LR5 of ribotype 012/-toxinotype I has substitutions at these positions, despite actoxumab having a similar neutralization potency against TcdA of this strain compared to toxinotype 0 strains [35]. On the other hand, substitution of asparagine to aspartate at position 7 of the epitope is likely deleterious to actoxumab binding since this substitution is present in both LR3 and LR5 of ribotype 078/toxinotype V TcdA and represents the only difference between this strain and ribotype 012/toxinotype I, despite a significantly lower potency of actoxumab against TcdA of 078 versus 012 [35]. Finally, substitutions of the leucine residue at position 28 for either valine or arginine are also likely involved in actoxumab binding since the epitope at LR4, to which actoxumab does not bind, has a Leu-to-Val substitution at this position and is otherwise identical to epitopes at LR3 and LR5 (except for substitutions at positions 18 and 19, which we argue above do not play a role).
Fig. 6.
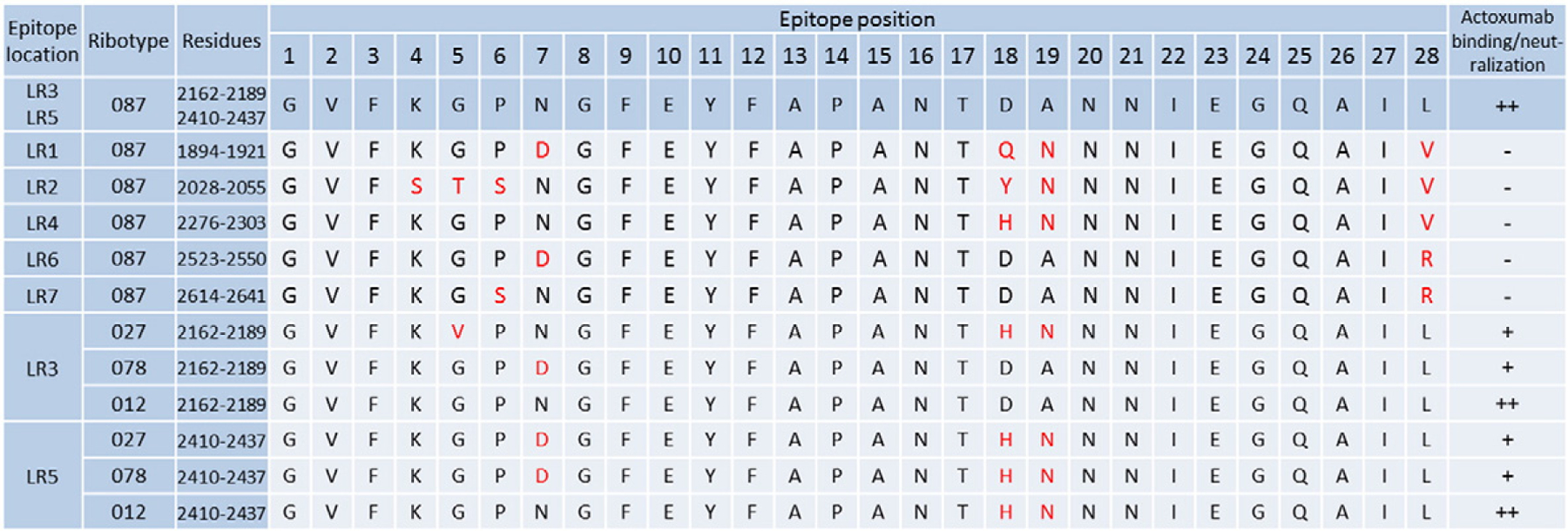
Alignment of actoxumab epitope sequences in TcdA. Residues within the putative actoxumab epitopes at LR3 and LR5 of ribotype 087 (VPI 10463) TcdA (as identified by HDX-MS) are shown by position (1–28) and compared to the homologous repeat sequences (LR1, LR2, LR4, LR6, and LR7) in the TcdA CROP domain and to the corresponding LR3 and LR5 epitopes within TcdA of ribotypes 027, 078, and 012. Residues that are different from those found at the same position in the actoxumab epitopes (LR 3 and 5) of ribotype 087 TcdA are marked in red. The ability of actoxumab to bind to individual homologous regions (as determined in this report) orto neutralize TcdA from various ribotypes (as demonstrated in Ref. [35]) is indicated (-, no binding; +, moderate neutralization; ++, high binding/neutralization).
Discussion
Actoxumab and bezlotoxumab bind to TcdA and TcdB, respectively, neutralizing the activity of the toxins both in vitro and in vivo [26–30,35–37]. Because of their potential therapeutic use for the prevention of recurrent CDI and their possible use as tools to gain further insight into toxin structure and function, there is much interest in determining the nature of the interactions of the antibodies with their respective toxin. The interaction of bezlotoxumab with TcdB and the mechanism of neutralization of the antibody have previously been characterized. An X-ray crystal structure of the antibody in complex with the amino-terminal half of the TcdB CROP domain was obtained, showing that bezlotoxumab binds to two homologous but distinct epitopes that overlap with LR1 and LR2 [31]. When bound to bezlotoxumab, TcdB cannot bind to the surface of host cells and is effectively neutralized. In contrast to bezlotoxumab, little is known about the interaction of actoxumab with its target toxin TcdA, other than the fact that the antibody binds to the CROP domain of TcdA [26]. In this study, we characterized the binding of actoxumab to TcdA and identified its mechanism of neutralization. Our results show that there are two binding sites for actoxumab within the TcdA CROP domain located at or around identical regions within LR3 (residues 2162–2189; epitope 1) and LR5 (residues 2410–2437; epitope 2; Figs. 5 and 6). This is in agreement with a study by Davies et al. [38], which demonstrated the binding of their version of actoxumab (CDA1) to both an N-terminal and a C-terminal half of the TcdA CROP domain. Although the actoxumab epitopes appear to have identical sequences based on HDX-MS, our EM studies suggest that actoxumab has a higher affinity for epitope 1 at LR3, as it is preferentially bound, whereas epitope 2 at LR5 is sparsely occupied at the same Fab concentration. It is possible that epitope 1 is more accessible to the antibody due to its positioning relative to the rest of the toxin and is therefore preferentially bound by actoxumab. Indeed, EM images suggest that epitope 2 is located in proximity to the presumed translocation domain of TcdA, which may impact the accessibility of actoxumab to bind to this epitope (Fig. 4). In contrast to toxin particles with only epitope 1 occupied, particles with epitope 1 and 2 bound by actoxumab-Fab show changes in the orientation of the CROP domain relative to the rest of the toxin that result in particle averages with indistinct images for the main body of the toxin (compare Fig. 4a and b). Alternatively, binding at epitope 1 may bring about intramolecular conformational changes in TcdA that permit binding to epitope 2.
While both actoxumab and bezlotoxumab have two epitopes within the CROP domain of their respective toxins, their relative locations are vastly different. The bezlotoxumab epitopes are in close proximity to each other and can be bound by one molecule of bezlotoxumab with each Fab region of the antibody binding to one epitope [31]. In the case of actoxumab, the two epitopes lie on different faces of the TcdA CROP domain, precluding the binding of both epitopes by a single molecule of actoxumab (Fig. 5c). Consistent with this, SEC-MALLS analysis of actoxumab/TcdA or actoxumab/A1 combinations showed the formation of large immunocomplexes at high actoxumab:TcdA molar ratios (Fig. 3 and Table 2), indicating that a single molecule of actoxumab can bind to two distinct molecules of TcdA, thereby leading to crosslinking of multiple toxin molecules. Furthermore, there were no immunocomplexes observed consisting of a single molecule of TcdA or A1 bound to one actoxumab molecule even at low antibody:antigen molar ratios (Fig. 3 and Table 2), supporting the notion that one antibody molecule cannot simultaneously bind two sites within the same TcdA molecule. This is in contrast to observations with bezlotoxumab, whose Fab regions bind simultaneously to two homologous regions within the CROP domain of a single TcdB molecule, precluding the formation of higher-order immunocomplexes. This observation may have therapeutic implications since larger immunocomplexes are cleared faster than smaller ones in vivo [39–41].
Comparison of the amino acid sequences of the actoxumab epitopes across the 027, 078, and 012 ribotypes and across homologous repeat sequences within the TcdA CROP domain that do not bind actoxumab (Fig. 6) revealed potential amino acid substitutions within these regions that negatively affect actoxumab binding and neutralization potency. The substitutions that are predicted to have the most significant impact cluster at epitope positions 4–7 (KGPN) and a leucine at epitope position 28. Interestingly, these epitope positions overlap with the regions having the highest level of protection from deuteration as measured in our HDX-MS analysis (Fig. 5b). As shown in Fig. 7, we mapped the actoxumab epitope 2 at LR5 to our TcdA model to examine this area more closely. The two regions mentioned above (residues 4–7 and residue 28) are in close proximity to each other in the 3D structure, suggesting that they could constitute a binding site for actoxumab. Amino acid changes in the KGPN loop region or mutations in the leucine may either disrupt the direct binding interactions with the antibody or disturb the stability of the toxin at this site, thereby impacting antibody binding. Further experiments will be needed to confirm this.
Fig. 7.
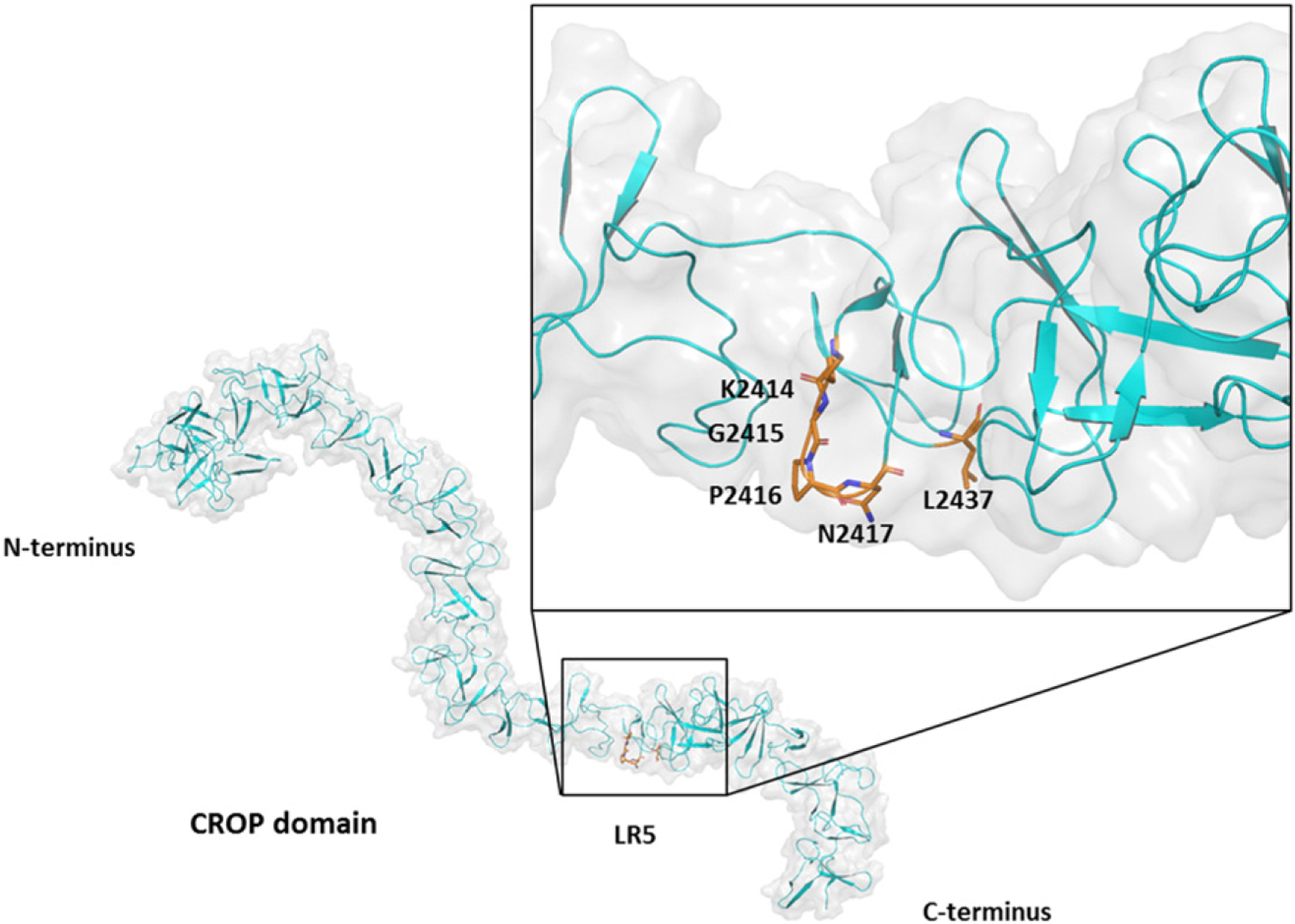
Proposed model of actoxumab epitope 2 at LR 5. Epitope positions 4–7 (KGPN) and 28 (L) are highlighted to show conformation.
We show here that actoxumab neutralizes TcdA by preventing the binding of the toxin to host cells. Given that the host receptor(s) for TcdA are thought to be cell surface carbohydrates and that the actoxumab epitopes overlap with two potential carbohydrate binding sites at LR3 and LR5, it is conceivable that antibody binding directly blocks these receptor binding sites, thereby preventing surface attachment to host cells. However, there are seven putative carbohydrate binding sites within the TcdA CROP domain, and only two of these are bound by actoxumab, theoretically leaving five sites free to interact with carbohydrate receptors even in the context of bound actoxumab. Despite this, actoxumab fully neutralizes TcdA in a number of cellular assays, including TcdA-induced cell death in Vero cells [35]. One explanation may be that toxin binding to host cells is the result of an additive effect of multiple binding sites, with each site contributing to the overall affinity of the toxin for the cell surface. Supporting this theory, multiple antibodies or antibody combinations targeting the CROP domain have been reported to neutralize TcdA, with the most potent ones binding to two or more locations at or near putative receptor binding sites [38,42–46]. Blocking at least two binding sites with actoxumab at LR3 and LR5 may be sufficient to lower the affinity of the toxin for host cells and prevent toxin binding to the host cell surface. Additionally, different regions of the CROP domain have been reported to have different roles in toxin binding and internalization and to potentially recognize distinct host cell receptors. For example, Huang et al. carried out studies with TcdA CROP fragments corresponding to the N-terminal (F1), middle (F2), and C-terminal (F3) regions of the domain [47]. While all three fragments were able to bind Vero cells, only F3, which contained LRs 5–7, was internalized by the cell following binding, and F1 (LRs 1–3) appeared to contain a binding site that was unique from the other two fragments. Actoxumab binding to LR3 and LR5 may therefore block binding sites for two different receptors, making it a more potent neutralizing antibody. Alternatively, it is possible that the binding of actoxumab to LR3 and LR5 causes steric hindrance, preventing other LRs from binding to cell surface receptors. A final possibility is that crosslinking of TcdA by actoxumab effectively decreases the concentration of free TcdA available for cell surface binding, although this is unlikely since monovalent Fab fragments of actoxumab block binding (Fig. 1a) and neutralize TcdA (Fig. S1) despite not being able to crosslink TcdA (Fig. 3 and Table 2).
While the CROP domain is thought to play an important role in toxin binding to the cell surface, a truncated version of TcdA lacking the CROP domain is still cytotoxic at high concentrations, suggesting that an alternative receptor binding site exists outside of the CROP domain [19]. Our EM studies suggest that Fab binding to TcdA at both epitopes results in structural changes within TcdA, with loss of resolution of the N-terminal domains of the toxin (consisting of the glucosyltransferase, autoprotease, and translocation domains; see Ref. [34]). Therefore, in addition to directly occluding receptor binding sites within the CROP domain, actoxumab binding to TcdA may also prevent binding at an alternative site outside of the CROP domain by causing unfavorable conformational changes at this site.
Our characterization of actoxumab binding to TcdA improves our understanding of the mechanism of TcdA neutralization, highlighting the roles of receptor binding sites both within and outside the CROP domain in the binding of TcdA to the host cell surface. Identification of the actoxumab epitopes may also allow us to better predict the potency of actoxumab against TcdA of existing and emerging clinically important strains of C. difficile.
Materials and Methods
Cell lines, antibodies, and purified toxins
Vero and HT29 cells were purchased from the American Type Culture Collection and grown at 37 °C in 5% CO2. Vero cells were maintained in Eagle’s minimal essential medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 U/ml streptomycin. HT29 cells were maintained in McCoy’s 5A Modified Medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 0.75% sodium bicarbonate, 100 U/mL penicillin, and 100 U/mL streptomycin. Actoxumab and bezlotoxumab were generated in humanized mice as previously described [26]. Purified native TcdA and TcdB from ribotype 087 were purchased from The Native Antigen Company and used in all experiments as indicated except for the negative-stain eM studies. For these studies, recombinant TcdA was expressed in Bacillus megaterium and purified as previously described [33].
Expression and purification of TcdA constructs
The DNAs encoding different TcdA fragments derived from the CROP domain of TcdA from C. difficile ribotype 087 (strain VP110463) were cloned into the pET30a vector (Novagen-EMD Millipore), which encodes a C- terminal His-tag. The following constructs were cloned (see Fig. 2): A0 (residues 1668 to 2710; contains the full-length CROP domain with additional 165 aa on the N-terminal end), A1 (residues 1831 to 2710; the full-length CROP domain), A2 (residues 1832 to 2233), A3 (residues 1987 to 2481), A4 (residues 2234 to 2710), and A5 (residues 2387 to 2710). All TcdA constructs were expressed in Escherichia coli strain BLR(DE3) (Novagen-EMD Millipore) grown in Terrific Broth supplemented with 50 μg/ml kanamycin in a 10-L batch under IPTG induction conditions (performed at Blue Sky Biotech). Cells were harvested by centrifugation for 15 min at 6000g. For A2 and A3 constructs, cell pellets were resuspended in IMAC Buffer A (containing 50 mM NaCl, 5 mM imidazole, 20 mM Tris, 125 mM Brij-35, 10 mM Triton X-100, 5 mM Tween 20 at pH 7.5, and protease inhibitor cocktail) and lysed by high-pressure homogenization. The lysates were clarified by centrifuging for 60 min at 4 °C at 10000g. The supernatants containing the soluble crude constructs were loaded onto a Ni2+ Sepharose 6 Fast Flow resin column (GE Healthcare), and the columns were washed with IMAC Buffer A. Constructs were eluted using a linear imidazole gradient of 0% to 100% IMAC Buffer B (IMAC Buffer A with 1 M imidazole). Fractions containing constructs were analyzed by SDS-PAGE. Prominent construct-containing fractions were pooled and dialyzed overnight against 50 mM Hepes with 150 mM NaCl at pH 7.5. For A0, A1, A4, and A5 constructs, cell pellets were lysed and centrifuged as described above. However, since the constructs in this case were not soluble, the supernatants were discarded and the pellets consisting of construct-containing inclusion bodies were solubilized in Refold buffer A (containing 20 mM Tris-HCland8 M urea at pH 7.5) and mixed overnight at room temperature (RT). The solutions containing dissolved denatured constructs were clarified by centrifuging for 60 min at RT at 10000g and loaded onto a Ni2+ Sepharose 6 Fast Flow resin column. The columns were washed with Refold Buffer A containing 8 M urea (pH 7.5), and constructs were eluted using a step imidazole gradient in Refold Buffer A. Prominent fractions containing constructs were pooled, and construct refolding was achieved by step dialysis into buffers containing reduced amounts of urea. For the final prep, soluble (refolded) constructs were dialyzed overnight against 50 mM Hepes with 150 mM NaCl at pH 7.5. Concentrations of constructs were determined by the Bradford method using standard methodology.
Western blotting
TcdA toxin and purified TcdA constructs from C. difficile ribotype 087 were dissolved in Laemmli sample buffer, incubated for 5 min at 95 °C, and separated by SdS-PAGE on a 4% to 12% gradient polyacrylamide gel. Then, 200 ng of protein was loaded per well. Gels were transferred to nitrocellulose for Western blotting according to standard methodology. The nitrocellulose membrane containing transferred protein was blocked in Odyssey blocking buffer (LI-COR) followed by incubation with actoxumab for 1 h at RT. After washing, the nitrocellulose membrane was incubated with a goat anti-human IgG antibody coupled to IRDye® 800CW (LI-COR) for 30 min at RT. After additional washing, bands were visualized using the Odyssey Imaging System (LI-COR). For Western blots of Vero cell membrane proteins, nitrocellulose membranes were also incubated with rabbit anti-cadherin polyclonal antiserum (Cell Signaling) using the same procedure as described above with donkey anti-rabbit IgG antibody coupled to IRDye® 800CW (LI-COR) used as the secondary antibody to ensure that equivalent amounts of protein were loaded into each lane.
Generation of actoxumab Fab fragments
Fab fragments from actoxumab were generated using the Pierce Fab Preparation Kit (ThermoScien-tific) according to the manufacturer’s instructions. Antibody was incubated with immobilized papain resin at 37 °C. After 5 h, undigested antibody and Fc fragments were removed by passage through a protein A column. Fab fragments were collected in the flow through and purified by gel-filtration chromatography. Fab fragment identity was confirmed by N-terminal sequencing.
SPR
Binding of actoxumab to TcdA from C. difficile ribotype 087 and their fragments was assessed by SPR using a ProteOn XPR36 instrument as described by Orth et al. [31]. Actoxumab was immobilized to the sensor chip surface using an antibody-capture method. Briefly, a ProteOn GLC Sensor chip was docked to the system, and after standard cleaning according to the manufacturer’s recommendations, a mixture of 1 × 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride with N-hydroxysulfosuccinimide was injected over the chip to activate the chip surface. A 25 μg/mL solution of goat anti-human IgG F(ab’)2 in ProteOn immobilization buffer (10 mM sodium acetate at pH 5.5) was injected over 1.5–5 min. Then, 1 M ethanolamine-HCl was injected over 5 min to block any unoccupied reactive sites on the surface of the chip. Finally, 5 μg/mL actoxumab in ProteOn running buffer (containing phosphate-buffered saline (PBS) and 0.005% Tween-20 at pH 7.4) was injected over 1.5–5 min. TcdA and fragments were diluted at various concentrations in ProteOn running buffer and injected in horizontal orientation for 4 min (flow rate of 25 μL/min). Association and dissociation were measured overtime as changes in the refractive index. Data were analyzed using the ProteOn instrument software to determine the association and dissociation binding constants (kon and koff) and equilibrium dissociation constant (Kd). The data presented are average ±standard deviation from two independent experiments.
SEC-MALLS
Actoxumab was mixed with TcdA at molar ratios of 1:1 or 5:1 (Ab:toxin) or with construct A1 at molar ratios of 1:1, 2:1, 5:1, or 1:5. Actoxumab-Fab was mixed with TcdA at molar ratios (Fab:toxin) of 1:1, 2:1, 5:1, or 10:1. The mixtures were first incubated at RT for 45 min and then for 2 h at 4 °C in PBS. The final concentrations of actoxumab or actoxumab-Fab were kept constant at 0.17 mg/mL and 0.19 mg/mL, respectively. SEC-MALLS was performed using the Dawn Heleos-II 18 angle light scattering detector and Optilab T-rEX refractive index detector (Wyatt Technology). Samples were separated on a YMC-pack Diol 200 column (300 × 8 mm, 5 pm) on an Agilent 1200 system at RT with a mobile phase of PBS, a flow rate of 0.5 mL/min, and a running time of 30 min. Absorption of UV light at 280 nm and 214 nm was recorded. Light scattering and refractive index signals were collected and molecular masses of detected components calculated using the ASTRA VI software (Wyatt Technology).
Negative-stain EM
Purified TcdA (100 nM) was incubated with actoxumab (1:1 M ratio) or actoxumab-Fab at an optimized molar ratio (1:3 M excess Fab) for 30 min at RT in 20 mM Tris and 100 mM NaCl (pH 8.0). Reactions were diluted fivefold in the same buffer to give a 20 nM toxin (6 ng/μl) final concentration, and samples were immediately placed onto carbon-coated, glow-discharged copper grids. Grids were negative-stained with 0.75% uranyl formate for imaging. Initial toxin-mAb complexes were imaged on a Morgagni transmission electron microscope at 44-K magnification. Images of the negative-stained toxin-Fab complexes were collected at 62-K magnification on a Tecnai F20 (FEI) transmission electron microscope equipped with a Gatan 4 K × 4 K CCD camera. Images were converted to mixed raster content format and binned by a factor of 2, giving final images with 3.5 Å/pixel. Individual particles of the complexes (6996 total particles) were manually selected in Boxer [48] and windowed with a 150-pixel (52.7 nm) side length. Reference-free two-dimensional class and alignments (100 classes in total) were carried out using SPIDER [49].
HDX-MS
Protein construct A1 was incubated with actoxumab at a final concentration of 10pmol/μl and 5 pmol/μl, respectively. An antigen-only control sample was made with PBS (pH 7.4) in place of actoxumab. After incubation for 1 h at RT, 2.5 μl was aliquoted for each labeling time point and replicate. The deuterium labeling, quenching, and digestion were done by a LEAPTec H/D-X system. We mixed 25 μl of deuterium oxide buffer (10 mM NaH2PO4 and 75 mM NaCl in D2O) with the aliquoted samples and incubated it for 10, 60, 600, 6000, or 10,000 s. We then added 30 μl of chilled quenching buffer (8 M Urea and 100 mM TCEP), and the sample was incubated at 2 °C for 3 min. Then, 50 μl of the mixture was then injected into a chilled digestion and column chamber at 2 °C. The samples were flowed over a combined pepsin/protease XIII enzyme column (NovaBioAssays) at 100 μl/min (2% acetonitrile and 0.1% TFA) for 2 min and onto a trapping column (CSH C18 Vanguard column, Waters Corp.). The peptides were then separated over an analytical column (CSH C18, 1 ×50 mm, 1.7 μm; Waters Corp.) by HPLC (nanoAcquity; Waters Corp.) using a linear gradient from 2% to 36% Buffer B (Buffer A, 0.1% formic acid; and Buffer B, 0.1% formic acid in acetonitrile) for 10 min and analyzed by mass spectrometry (Orbitrap-Elite; Thermofisher Scientific). For labeled samples, full scan MS1 data were acquired from 350 to 2000 m/z in profile mode, 120,000 resolving power, and with 2 microscans. For peptide identification of unlabeled samples, the same full scan MS1 was done followed by 10 MS/MS scans in the ion trap. Each time point was done in triplicate and the run order randomized. Peptide identifications were done using the SEQUEST-HT algorithm in the Proteome Discoverer software (v1.4,Thermofisher Scientific). HDX data were analyzed using HDExaminer (Sierra Analytics).
Modeling of the TcdA CROP domain
The TcdA CROP domain was modeled using the amino acid sequence of C. difficile ribotype 087 (strain VPI 10463). Amino acid numbering and repeat units are based on that published by Ho et al. [50]. A known crystal structure of TcdA [Protein Data Bank (PDB) code 2QJ6; spanning amino acids 2391 to 2706] published by Albesa-Jove et al. [51] was used to build the model. The following intermediate models on the PDB code 2QJ6 template were made: A1, spanning amino acids 1832 to 1926; TcdA2, spanning amino acids 1945 to 2058; TcdA3, spanning amino acids 1894 to 1965; TcdA4, spanning amino acids 2079 to 2192; TcdA5, spanning amino acids 2028 to 2099; TcdA6, spanning amino acids 2193 to 2306; TcdA7, spanning amino acids 2276 to 2348; and TcdA8, spanning amino acids 2328 to 2440. They were assembled as TcdA9, spanning amino acids 1832 to 2706.
Fluorescent labeling of TcdA
TcdA was fluorescently labeled using the Lightning Link Atto488 Antibody Labeling kit (Novus Biosciences) according to the manufacturer’s instructions. Briefly, 50 μg of TcdA was resuspended in sterile ddH2O and incubated at RT for 30 min. LL-modifier buffer was added and mixed by pipetting. The toxin solution was then added to a vial containing lyophi-lized Lightning Link mix, pipetted up and down gently, and incubated at RT in the dark for 5 h. Following incubation, LL-quencher buffer was added and incubated at RT in the dark for 30 min. The labeled toxin (TcdA-Atto488) was immediately used in flow cytometry assays or stored at 4 °C for up to 4 days.
TcdA cell surface binding by flow cytometry
Various concentrations of TcdA-Atto488 were incubated with or without 200 μg/mL actoxumab or bezlotoxumab at RT for 60 min in the dark. Samples were then chilled on ice for a minimum of 25 min. Adherent HT29 cells were resuspended in cell medium following treatment with Accutase (Innovative Cell Technologies), chilled on ice, and washed once with cold Dulbecco’s PBS with calcium and magnesium (DPBS++) containing 1% bovine serum albumin (BSA). We incubated 3 × 105 cells with 100 μL of toxin/antibody mixture for 30 min on ice in the dark. Following incubation, 1 mL of ice cold DPBS++ with 1% BSA was added to each sample. Unbound toxin was removed by washing cells twice with ice cold DPBS++ with 1% BSA, by centrifuging for 5 min at 4 °C at 200g, and by removing the supernatant. Cells were finally resuspended in 500 μl cold DPBS++ with 1% BSA and subjected to flow cytometry on an LSRII instrument (BD Biosciences). We measured 10,000 events for each condition.
TcdA cell surface binding by Western blotting
TcdA (1 μg/ml) was incubated with or without 200 μg/mL actoxumab or bezlotoxumab in Vero cell culture medium for 30 min at 37 °C; these mixtures were then chilled on ice and added to plates of pre-chilled Vero cells. Plates were incubated for 30 min on ice to allow the binding of TcdA. Following incubation, plates were washed three times with cold DPBS++, and cells were harvested by scraping. Cell membranes were then isolated in the cold using the Mem-PER Plus Membrane Protein Extraction Kit (Thermo Scientific) according to the manufacturer’s instructions and solubilized in a total volume of 100 μL Solubilization Buffer with HALT protease/phosphatase inhibitors (Thermo Scientific). Following the addition of Laemmli sample buffer, samples were incubated for 5 min at 95 °C and resolved by SDS-PAGE on 4% to 12% polyacrylamide gels and processed for Western blotting as described above, utilizing either actoxumab or an anti-cadherin antibody (Cell Signaling Technology) as the primary antibody.
Supplementary Material
Abbreviations used:
- CDI
C. difficile infection(s)
- TcdA
toxin A
- TcdB
toxin B
- GTD
glucosyltransferase domain
- CROP
combined repetitive oligopeptide
- SR
short repeat
- LR
long repeat
- MFI
mean fluorescence intensity
- SPR
surface plasmon resonance
- SEC-MALLS
size-exclusion chromatography coupled with multiangle laser light scattering
- EM
electron microscopy
- HDX-MS
hydrogen deuterium exchange-mass spectrometry
- RT
room temperature
- PBS
phosphate-buffered saline
- PDB
Protein Data Bank
- DPBS++
Dulbecco’s PBS with calcium and magnesium
- BSA
bovine serum albumin
Footnotes
Appendix A. Supplementary Data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2017.02.010.
References
- [1].Rupnik M, Wilcox MH, Gerding DN, Clostridium difficile infection: new developments in epidemiology and pathogenesis, Nat. Rev. Microbiol 7 (2009) 526–536. [DOI] [PubMed] [Google Scholar]
- [2].Gerding DN, Lessa FC, The epidemiology of Clostridium difficile infection inside and outside health care institutions, Infect. Dis. Clin. N. Am 29 (2015) 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bassetti M, Villa G, Pecori D, Arzese A, Wilcox M, Epidemiology, diagnosis and treatment of Clostridium difficile infection, Expert Rev. Anti-Infect. Ther 10 (2012) 1405–1423. [DOI] [PubMed] [Google Scholar]
- [4].Carter GP, Rood JI, Lyras D, The role of toxin A and toxin B in the virulence of Clostridium difficile, Trends Microbiol. 20 (2012) 21–29. [DOI] [PubMed] [Google Scholar]
- [5].Shen A, Clostridium difficile toxins: mediators of inflammation, J. Innate Immun 4 (2012) 149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Smits WK, Lyras D, Lacy DB, Wilcox MH, Kuijper EJ, Clostridium difficile infection, Nat. Rev. Dis. Primers 2 (2016) 16,020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pruitt RN, Lacy DB, Toward a structural understanding of Clostridium difficile toxins A and B, Front. Cell. Infect. Microbiol 2 (2012) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].von Eichel-Streiber C, Sauerborn M, Clostridium difficile toxin A carries a C-terminal repetitive structure homologous to the carbohydrate binding region of streptococcal glycosyl-transferases, Gene 96 (1990) 107–113. [DOI] [PubMed] [Google Scholar]
- [9].Papatheodorou P, Zamboglou C, Genisyuerek S, Guttenberg G, Aktories K, Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis, PLoS One 5 (2010) e10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Qa’Dan M, Spyres LM, Ballard JD, pH-induced conformational changes in Clostridium difficile toxin B, Infect. Immun 68 (2000) 2470–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Barth H, Pfeifer G, Hofmann F, Maier E, Benz R, Aktories K, Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells, J. Biol. Chem 276 (2001) 10,670–10,676. [DOI] [PubMed] [Google Scholar]
- [12].Pfeifer G, Schirmer J, Leemhuis J, Busch C, Meyer DK, Aktories K, et al. , Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells, J. Biol. Chem 278 (2003) 44,535–44,541. [DOI] [PubMed] [Google Scholar]
- [13].Reineke J, Tenzer S, Rupnik M, Koschinski A, Hasselmayer O, Schrattenholz A, et al. , Autocatalytic cleavage of Clostridium difficile toxin B, Nature 446 (2007) 415–419. [DOI] [PubMed] [Google Scholar]
- [14].Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K, Glucosylation of Rho proteins by Clostridium difficile toxin B, Nature 375 (1995) 500–503. [DOI] [PubMed] [Google Scholar]
- [15].Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, Mann M, et al. , The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins, J. Biol. Chem 270 (1995) 13,932–13,936. [DOI] [PubMed] [Google Scholar]
- [16].Qa’Dan M, Ramsey M, Daniel J, Spyres LM, Safiejko- Mroczka B, Ortiz-Leduc W, et al. , Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells, Cell. Microbiol 4 (2002) 425–434. [DOI] [PubMed] [Google Scholar]
- [17].Savidge TC, Pan WH, Newman P, O’Brien M, Anton PM, Pothoulakis C, Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine, Gastroenterology 125 (2003) 413–420. [DOI] [PubMed] [Google Scholar]
- [18].Gerhard R, Nottrott S, Schoentaube J, Tatge H, Olling A, Just I, Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells, J. Med. Microbiol 57 (2008) 765–770. [DOI] [PubMed] [Google Scholar]
- [19].Olling A, Goy S, Hoffmann F, Tatge H, Just I, Gerhard R, The repetitive oligopeptide sequences modulate cytopathic potency but are not crucial for cellular uptake of Clostridium difficile toxin A, PLoS One 6 (2011) e17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schorch B, Song S, van Diemen FR, Bock HH, May P, Herz J, et al. , LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 6431–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yuan P, Zhang H, Cai C, Zhu S, Zhou Y, Yang X, et al. , Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B, Cell Res. 25 (2015) 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].LaFrance ME, Farrow MA, Chandrasekaran R, Sheng J, Rubin DH, Lacy DB, Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 7073–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tao L, Zhang J, Meraner P, Tovaglieri A, Wu X, Gerhard R, et al. , Frizzled proteins are colonic epithelial receptors for C. difficile toxin B, Nature 538 (2016) 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Krivan HC, Clark GF, Smith DF, Wilkins TD, Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal alpha 1–3Gal beta 1–4GlcNAc, Infect. Immun 53 (1986) 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Greco A, Ho JG, Lin SJ, Palcic MM, Rupnik M, Ng KK, Carbohydrate recognition by Clostridium difficile toxin A, Nat. Struct. Mol. Biol 13 (2006) 460–461. [DOI] [PubMed] [Google Scholar]
- [26].Babcock GJ, Broering TJ, Hernandez HJ, Mandell RB, Donahue K, Boatright N, et al. , Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters, Infect. Immun 74 (2006) 6339–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yang Z, Ramsey J, Hamza T, Zhang Y, Li S, Yfantis HG, et al. , Mechanisms of protection against Clostridium difficile infection by the monoclonal antitoxin antibodies actoxumab and bezlotoxumab, Infect. Immun 83 (2015) 822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang Z, Chen X, Hernandez LD, Lipari P, Flattery A, Chen SC, et al. , Toxin-mediated paracellular transport of antitoxin antibodies facilitates protection against Clostridium difficile infection, Infect. Immun 83 (2015) 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Steele J, Mukherjee J, Parry N, Tzipori S, Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease, J. Infect. Dis 207 (2013) 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, et al. , Treatment with monoclonal antibodies against Clostridium difficile toxins, N. Engl. J. Med 362 (2010) 197–205. [DOI] [PubMed] [Google Scholar]
- [31].Orth P, Xiao L, Hernandez LD, Reichert P, Sheth PR, Beaumont M, et al. , Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography, J. Biol. Chem 289 (2013) 18,008–18,021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gerhard R, Receptors and binding structures for Clostridium difficile toxins A and B, Curr. Top. Microbiol. Immunol (2016) (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- [33].Pruitt RN, Chambers MG, Ng KK, Ohi MD, Lacy DB, Structural organization of the functional domains of Clostridium difficile toxins A and B, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 13,467–13,472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chumbler NM, Rutherford SA, Zhang Z, Farrow MA, Lisher JP, Farquhar E, et al. , Crystal structure of Clostridium difficile toxin A, Nat. Microbiol 1 (2016) 15,002. [DOI] [PubMed] [Google Scholar]
- [35].Hernandez LD, Racine F, Xiao L, DiNunzio E, Hairston N, Sheth PR, et al. , Broad coverage of genetically diverse strains of Clostridium difficile by actoxumab and bezlotoxumab predicted by in vitro neutralization and epitope modeling, Antimicrob. Agents Chemother 59 (2015) 1052–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Warn P, Thommes P, Sattar A, Corbett D, Flattery A, Zhang Z, et al. , Disease progression and resolution in rodent models of Clostridium difficile infection and impact of antitoxin antibodies and vancomycin, Antimicrob. Agents Chemother 60 (2016) 6471–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Koon HW, Shih DQ, Hing TC, Yoo JH, Ho S, Chen X, et al. , Human monoclonal antibodies against Clostridium difficile toxins A and B inhibit inflammatory and histologic responses to the toxins in human colon and peripheral blood monocytes, Antimicrob. Agents Chemother 57 (2013) 3214–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Davies NL, Compson JE, Mackenzie B, O’Dowd VL, Oxbrow AK, Heads JT, et al. , A mixture of functionally oligoclonal humanized monoclonal antibodies that neutralize Clostridium difficile TcdA and TcdB with high levels of in vitro potency shows in vivo protection in a hamster infection model, Clin. Vaccine Immunol 20 (2013) 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yousaf N, Howard JC, Williams BD, Studies in cobra venom factor treated rats of antibody coated erythrocyte clearance by the spleen: differential influence of red blood cell antigen number on the inhibitory effects of immune complexes on Fc-dependent clearance, Clin. Exp. Immunol 66 (1986) 654–660. [PMC free article] [PubMed] [Google Scholar]
- [40].Davies KA, Hird V, Stewart S, Sivolapenko GB, Jose P, Epenetos AA, et al. , A study of in vivo immune complex formation and clearance in man, J. Immunol (Baltimore, Md: 1950) 144 (1990) 4613–4620. [PubMed] [Google Scholar]
- [41].Humphreys DP, Wilcox MH, Antibodies for treatment of Clostridium difficile infection, Clin. Vaccine Immunol 21 (2014) 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Anosova NG, Cole LE, Li L, Zhang J, Brown AM, Mundle S, et al. , A combination of three fully human toxin A- and toxin B-specific monoclonal antibodies protects against challenge with highly virulent epidemic strains of Clostridium difficile in the hamster model, Clin. Vaccine Immunol 22 (2015) 711–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Murase T, Eugenio L, Schorr M, Hussack G, Tanha J, Kitova EN, et al. , Structural basis for antibody recognition in the receptor-binding domains of toxins A and B from Clostridium difficile, J. Biol. Chem 289 (2014) 2331–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Frey SM, Wilkins TD, Localization of two epitopes recognized by monoclonal antibody PCG-4 on Clostridium difficile toxin A, Infect. Immun 60 (1992) 2488–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Marozsan AJ, Ma D, Nagashima KA, Kennedy BJ, Kang YK, Arrigale RR, et al. , Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies, J Infect Dis 206 (2012) 706–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Demarest SJ, Hariharan M, Elia M, Salbato J, Ping J, Bird C, et al. , Neutralization of Clostridium difficile toxin A using antibody combinations, MAbs 2 (2010) 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Huang JH, Shen ZQ, Lien SP, Hsiao KN, Leng CH, Chen CC, et al. , Biochemical and immunological characterization of truncated fragments of the receptor-binding domains of C. difficile toxin A, PLoS One 10 (2015) e0135045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ludtke SJ, Baldwin PR, Chiu W, EMAN: semiautomated software for high-resolution single-particle reconstructions, J. Struct. Biol 128 (1999) 82–97. [DOI] [PubMed] [Google Scholar]
- [49].Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, et al. , SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields, J. Struct. Biol 116 (1996) 190–199. [DOI] [PubMed] [Google Scholar]
- [50].Ho JG, Greco A, Rupnik M, Ng KK, Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A, Proc. Natl. Acad. Sci. U. S.A 102 (2005) 18,373–18,378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Albesa-Jove D, Bertrand T, Carpenter EP, Swain GV, Lim J, Zhang J, et al. , Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS, J. Mol. Biol 396 (2010) 1260–1270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.