

Abstract
Variants in genes which encode for polycystin-1 and polycystin-2 cause most forms of autosomal dominant polycystic disease (ADPKD). Despite our strong understanding of the genetic determinants of ADPKD, we do not understand the structural features which govern the function of polycystins at the molecular level, nor do we understand the impact of most disease-causing variants on the conformational state of these proteins. These questions have remained elusive because polycystins localize to several organelle membranes, including the primary cilia. Primary cilia are microtubule based organelles which function as cellular antennae. Polycystin-2 and related polycystin-2L1 are members of the transient receptor potential (TRP) ion channel family, and form distinct ion channels in the primary cilia of disparate cell types which can be directly measured. Polycystin-1 has both ion channel and adhesion G-protein coupled receptor (GPCR) features—but its role in forming a channel complex or as a channel subunit chaperone is undetermined. Nonetheless, recent polycystin structural determination by cryo-EM has provided a molecular template to understand their biophysical regulation and the impact of disease-causing variants. We will review these advances and discuss hypotheses regarding the regulation of polycystin channel opening by their structural domains within the context of the primary cilia.
Graphical Abstract
Challenges of studying polycystin channel function in ADPKD basic research
ADPKD impacts an estimated 12.5 million people worldwide, for whom current treatment consists of blood pressure regulation, dialysis, and kidney transplant1,2. ADPKD is the most common heritable form of cystic kidney disease, causing adult-onset focal cyst development which results from mutations in polycystin genes PKD1 or PKD2, which encode for polycystin-1 and polycystin-2 proteins, respectively3–6. While considered a dominant monogenic disease, the prevailing “two-hit model” states that ADPKD is recessive at the cellular level and that cysts develop from cells after acquiring a second somatic mutation to deactivate the remaining normal allele7–10. Previous work from several labs has demonstrated that haploinsufficiency or a complete loss of polycystin expression causes a cystic kidney phenotype in mice8,11–13. These in vivo results suggest that most ADPKD-causing variants inactivate polycystins in the collecting duct, but their mechanistic impact has not been directly assessed—do they impair trafficking or disrupt function? This question has eluded researchers for more than 20 years because polycystins traffic and function in the primary cilia. Primary cilia are tiny (<5 μm long) and privileged compartments, meaning that few protein types localize to this cellular organelle. Both aspects present significant technical challenges to researchers who study polycystins in this organelle. Contrary to initial reports14,15, co-expression of polycystin-1 and/or polycystin-2 does not produce ionic currents on the plasma membrane of native collecting duct cells or in heterologous systems16–19. Like all ion channels, much of the polycystin-2 protein localizes to the ER when it is overexpressed. Previous work using cytoplasmic Ca2+ fluorescence reporters supports the hypothesis that polycystin-2 can function as an ER Ca2+ release channel20–22, perhaps in addition to its duties in the primary cilia membrane. The function of polycystins within the context of the ER have been recently reviewed in this series23. Here, its function mimicks the Ca2+ response from bona fide ER ion channels— the inositol trisphosphate receptor (InsP3R) and ryanodine receptor (RyR)— thus it is difficult to determine the intrinsic function of polycystin-2 in the context of the ER. Previous reports measuring ER polycystin-2 channels in bilayers have created confusion regarding the basic properties of the channel (ion selectivity and conductance), which has raised doubts whether the same channel was being assayed (reviewed)24–26. Using this method, only a few ADPKD-causing missense variants have been described but we learned nothing about polycystin-2 biophysics or assembly20. Thus, our understanding of ADPKD pathogenesis has been hampered by the lack of tools to directly and reliably measure polycystin-2 function. However, new tools in the single particle cryogenic electron microscopy (cryo-EM) and direct cilia electrophysiology methods are being deployed to address this knowledge gap16,27,28. Outlined here is our current understanding of the mechanistic regulation of polycystin ion channels by their structural domains within the context of their native organelle— the primary cilia.
A cilia-centric hypothesis of ADPKD progression
ADPKD is a “ciliopathy”—a disorder affecting primary cilia proteins29. Primary cilia are singular, non-motile, hair-like structures which extend from the apical side of cells and are present in all organs of the human body. The role of the primary cilium in the biology of the cell is not well defined. Yet, its importance is highlighted by the number of cilia-related diseases such as Nephronophthesis (NPHS), Senior–Løken syndrome (SLS), Bardet-Biedl (BBS), Joubert (JS) and Alstrome syndrome (AS), which curiously share cystic kidneys and other renal defects as a common phenotype30. However the penetrance, onset during life and type of kidney cysts observed in non-ADPKD ciliopathies can be variable31. While typically tubulointerstitial cysts and normal size kidneys are most often observed in these ciliopathies, in some cases patients can show ADPKD-like enlarged kidneys. Typically, ADPKD symptoms do not become apparent before adulthood, whereas the aforementioned ciliopathies are present at birth or develop in early childhood. However, there are exceptions— a small proportion (2–5%) of ADPKD patients develop cystic kidneys in adolescence32. Many ciliopathy proteins form functional networks with other related and unrelated cilia effectors which has elucidated their interdependent regulation. While nearly all ciliopathies are associated with renal cysts, the penetrance of this comorbidity is variable depending on the disease. Thus, the cystic kidney phenotype associated with ADPKD and other ciliopathies likely converge on signaling pathways, but does not require a direct interaction between polycystins and other effected ciliopathy-related proteins themselves. However, an exception is the PKHD1 gene variants that encode for fibrocystin and are associated with childhood onset autosomal recessive polycystic kidney disease (ARPKD)33. Fibrocystin encodes for a single-pass transmembrane with large extracellular structure which may function as an adhesion molecule responsible for maintaining the collecting duct-lumen architecture of epithelium. Here, polycystin-2 directly associates with fibrocystin and is anterograde trafficked in the primary cilia via the kinesin motor protein family member (KIF3A)34,35. As expected, mutations the KIF3A-related intraflagellar transport protein kinesin IFT88 gene causes polycystic kidney disease in mice. These findings reinforce the hypothesis that dysregulation of cilia signaling is a common initiator of the cystogeneisis in the kidney, as it is observed in ADPKD and with variable penetrance in other rare ciliopathies.
In both the kidney collecting duct and in the embryonic node (covered in the next section), the function of the primary cilium is proposed to be either an antenna to amplify chemical signaling or a fluid flow sensor 36,37. The attenuated Ca2+ conductance caused by dysfunctional polycystin channels in the ciliary membrane are proposed to initiate a cystogenic signal in ADPKD38. In mouse models, the signaling from the cilia is strongly implicated in cyst formation. Complete genetic knockout of either polycystin-1 or polycystin-2 in mice results in embryonic lethality due to structural defects in the cardiovascular system, pancreas, and kidneys39–43. However, the allelic impact of variants on the cilia channel functions and Ca2+ levels has yet to be tested. The onset of kidney cyst development in adult mice following conditional inactivation of polycystin-1 or the intraflagellar transport protein kinesin, KIF3a (required for cilia formation), progresses well into adulthood, similar to the late onset of ADPKD in humans44–46. Mice with conditional repression of either PKD1 (Pax8rtTA; TetO-cre; PKD1fl/fl)46 or PKD2 (Pax8rtTA; TetO-cre; PKD2fl/fl)13 develop kidney cysts within 10 weeks after the start of induction, suggesting that expression of both genes is necessary to prohibit cyst development in mature mice. Recently, it was shown that the cystic phenotype found in mice deficient in PKD2 can be dose-dependently rescued by transgene expression of the channel47. Thus, these compelling findings implicate cilia-localized polycystin signaling in ADPKD progression and warrants investigation of their function directly from this organelle.
Aberrant ciliary calcium signaling as a unifying mechanism of ciliopathies
After twenty days in our mother’s womb, our cells establish three germ layers (ectoderm, mesoderm and endoderm) and subsequent organization of left-right axial structures that are essential to the orientation of our organs. This fundamental developmental process is initiated by a structure called the “embryonic node”— a stratified, pit-shaped cluster of motile and primary ciliated cells whose signaling coordinate an asymmetric expression gradient of Nodal and Hedgehog signaling pathways48–50. Beyond early development, primary cilia are found on all polarized cells in every organ of the human body. Here, they are proposed to have an antenna-like function–amplifying chemical signaling via molecular activation of cilia localized GPCRs and ion channels 51. The importance of the primary cilia in human health is highlighted by 35 congenital diseases called “ciliopathies”, which impact the development of organ systems such as the brain, heart and eye 52. Ciliopathies can either involve single or multiple organ systems with phenotypically variable and overlapping disease manifestations (Figure 1). Aberrant Ca2+ signaling caused by dysfunctional polycystin-2 ion channels in the collecting duct cilia is proposed to contribute to cystogenesis in ADPKD. The products of several gene variants responsible for ciliopathies are directly regulated by or are downstream of Ca2+ signaling effectors— such as CEP290 and NPHP4 that when mutated cause Joubert-related syndromes and nephronophthisis 53. Calcium also modulates cilia cargo trafficking by IFT88 (or KIF3a) and cilia formation by axonemal heavy chain DNAH7 via direct association with their EF-hand motifs. Here, variants in KIF3a and DNAH7 are associated with Retinitis Pigmentosa and impaired pulmonary function found in Kartagener’s Syndrome54,55. As discussed previously, other ciliopathies which affect organ development also exhibit either kidney cysts or kidney deformation comorbidities (Figure 1) 29. Thus, the implications of ciliary calcium may extend to other renal and non-renal ciliopathies, where aberrant cilia-to-cell Ca2+ transduction is a possible unifying signaling mechanism.
Figure 1. Ciliary calcium dysregulation as a mechanism connecting the shared PKD phenotype found in several congenital ciliopathies.
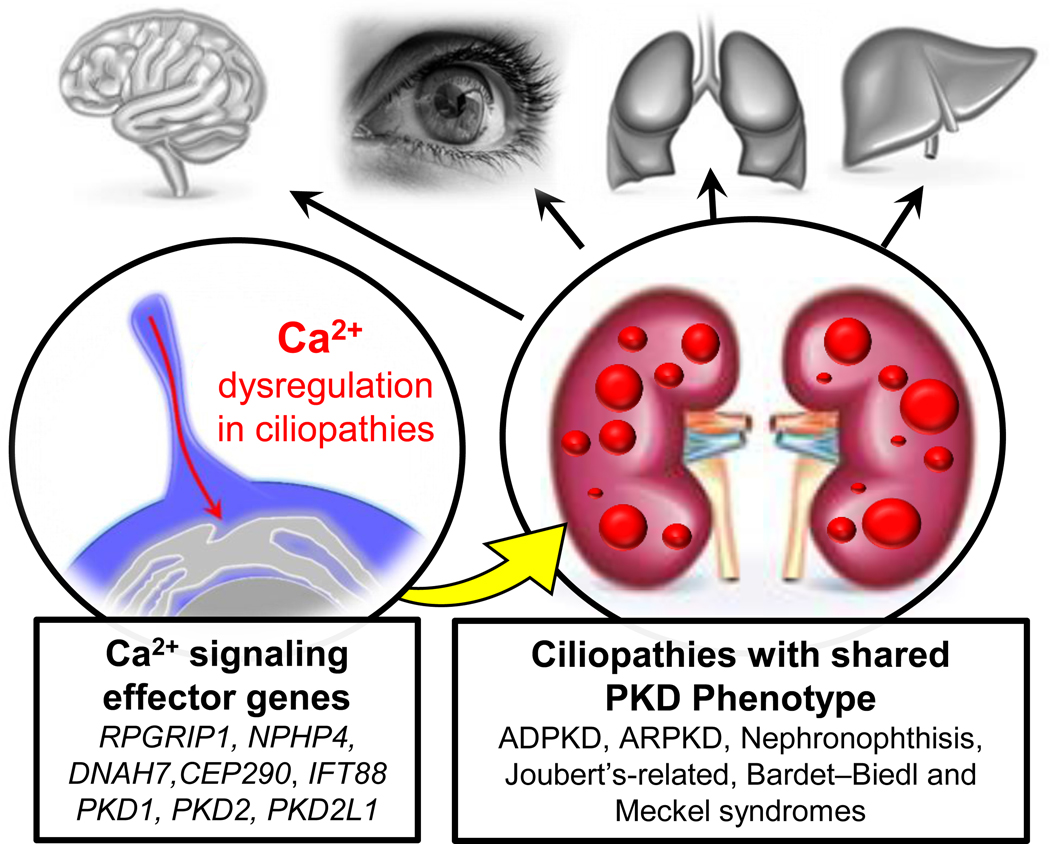
Left, examples of calcium effector genes which encode for cilia-localized proteins. Right, the associated ciliopathies which primarily impact specific organs (in grey) but share PKD as common co-morbidity.
Polycystins in primary cilia of divergent tissues
Polycystin-2 (PKD2) and polycystin-2L1 (PKD2L1) have six transmembrane helices and are members of the polycystin subfamily of transient receptor potential ion channels (Figure 2A, B) 56. Although they are 67% identical by amino acid identity, mutations in PKD2L1 are not associated with renal cysts in the human population or in animal models. The reason for this is that although polycystin-2 and polycystin-2L1 independently form ion channels in primary cilia, they do so in non-overlapping cell types. Two groups have independently identified polycystin-2 as an essential ion channel subunit by directly measuring its activity from the primary cilia membrane of kidney collecting duct cells27,57. In other cells types, polycystin-2L1 forms a cilia ion channel subunit in embryonic fibroblasts and retina pigmented epithelia16. Unlike polycystin-2, the function of polycystin-2L1 channels are not exclusive to the cilia membrane and can function as plasma membranes channel in cerebrospinal fluid-contacting neurons (CSF-cNs) and when heterologously expressed in host cells57,58. Both polycystin-2 and polycystin-2L1 are large conductance ion channel subunits which open at positive membrane potentials, creating an outwardly rectifying current when measured from cilia. However, the voltage dependence of these channels shifts to negative potentials when internal Ca2+ concentrations exceeds 1 μM (Figure 3A–C). Since the resting membrane potential of cilia is −17 mV, the voltage dependent gating of polycystins becomes physiologically relevant when ciliary Ca2+ is elevated (Figure 3D)27,57. While both channels are potentiated by elevated ciliary Ca2+, polycystin-2L1 channels are more sensitive to calcium-dependent desensitization— a process that irreversibly inactivates the channel59. Desensitization of polycystin-2L1 is induced when the cilia calcium accumulates, and outwardly moving Ca2+ occupies and blocks the selectivity filter. Surprisingly, the C-terminal EF-hands and coiled-coil domains of polycystin-2L1 do not contribute to its potentiation or desensitization, but the equivalent effects have not been tested in polycystin-259. Nonetheless, polycystin channel sensitization is hypothesized to amplify the Ca2+ signal within the cilia and subsequent desensitization acts as a form of negative feedback— turning off Ca2+ conductance and rectifying the signal towards the cell body from the ciliary compartment. Both features in polycystin-2 and polycystin-2L1 are expected to have a significance impact on the local Ca2+ levels and the downstream signaling from the cilia organelle into the cellular compartment. Thus, understanding the calcium-dependent regulatory mechanisms employed by these channels is important for our understanding of ADPKD and other cilia diseases.
Figure 2. Topology and structure of hetero and homomeric polycystins.
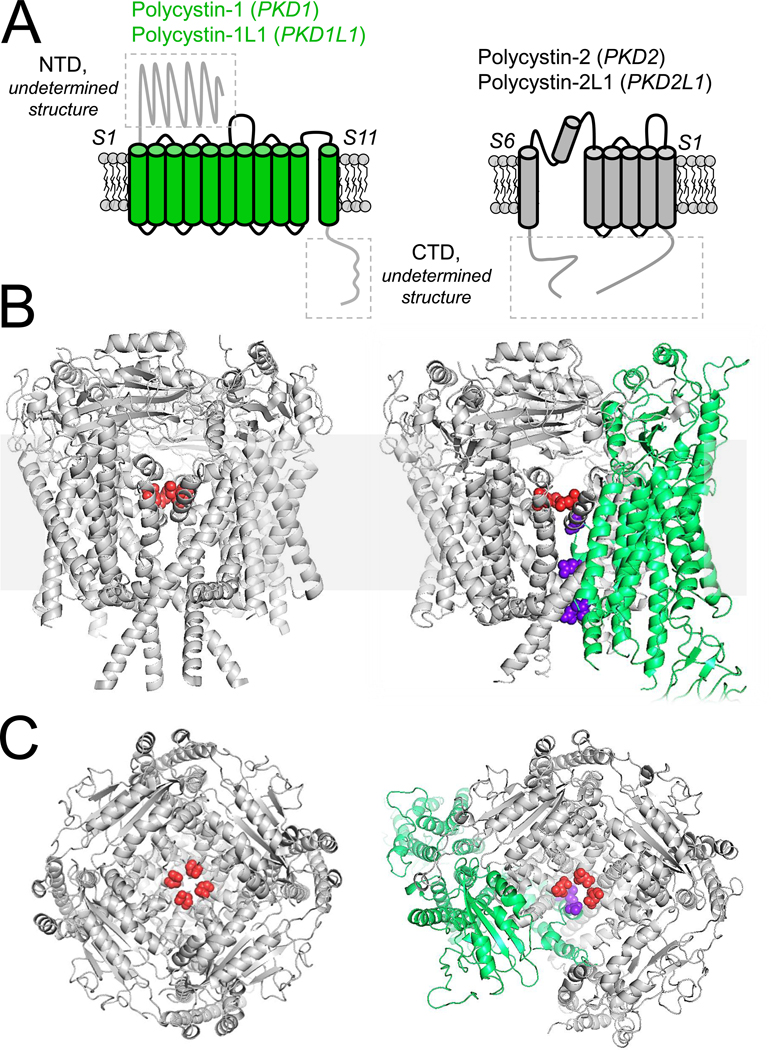
A) Left, the topology of the eleven transmembrane proteins, polycystin-1 and polycystin-1L1 (green). Right, the topology of the six transmembrane polycystin-2 (PKD2) and polycystin-2L1 (PKD2L1) TRP channels. The boxed areas of the proteins have not been structurally solved. B) Transmembrane and C) extracellular structural views of the homomeric polycystin-2 channel (PDB: 5K47, Grieben et al 2017)72 and heteromeric polycystin-1 + polycystin-2 complex (PDB: 6A70, Su et al 2019)67. The polycystin-1 positively charged pore residues are colored purple, whereas the negatively charged polycystin-2 selectivity filter residues are colored red.
Figure 3. Calcium-dependent potentiation of polycystin-2 channels in the primary cilia.
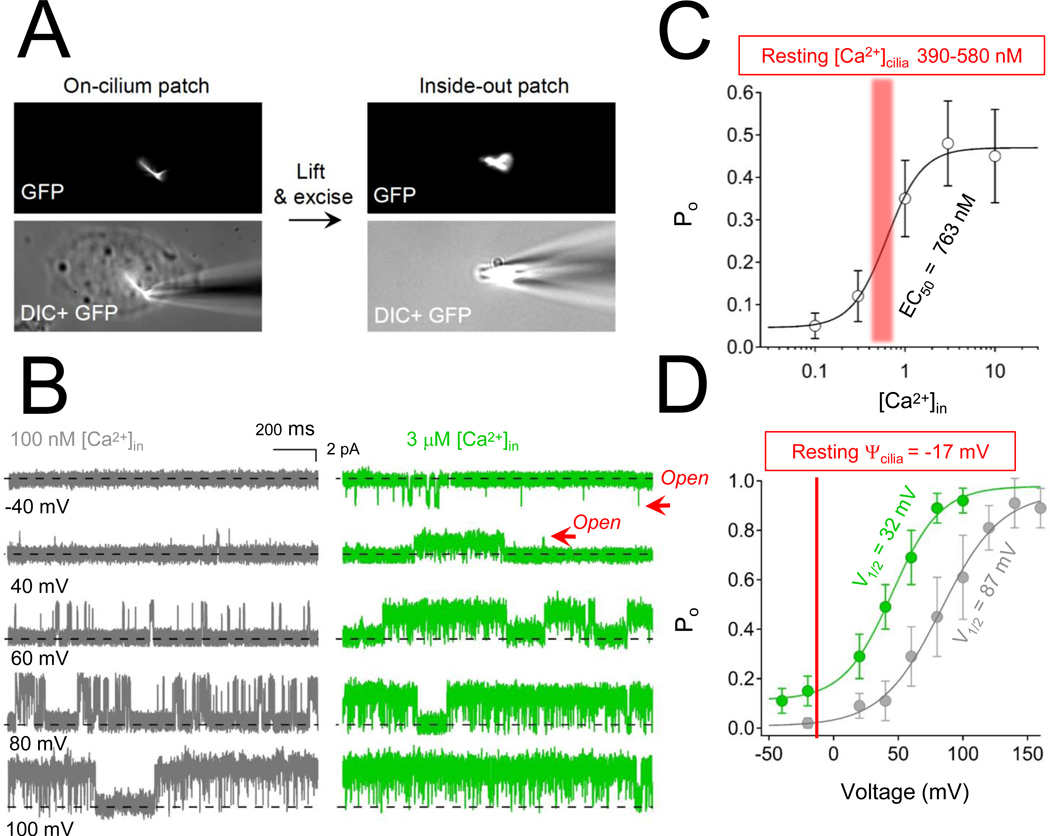
A) Images of the inside out cilia patch configuration. B) Here, the inner membrane of the cilia faces the external solution, so that calcium concentrations can be adjusted while measuring singe channel activity while holding the membrane potential at different voltages. C) Polycystin-2 open probability (Po) and intraciliary calcium concentration relationship. The resting ciliary calcium concentration (red range) has been measured at 390 nM and 580 nM for RPE and IMCD cells, respectively106,107. D) The voltage dependence of polycystin-2 channel opening captured at low and high intra-ciliary calcium. The resting membrane potential of the cilia is shown in red, indicating that most of the polycytin-2 channels are closed when intraciliary calcium is low.
Polycystin-1 (PKD1), and polycystin-1L1 (PKD1L1) are eleven transmembrane helix proteins with both channel-like and adhesion G-protein coupled receptor (aGPCR) features (Figure 2A, C). The first five transmembrane segments (S1–5) resemble the adhesion class of GPCRs—containing a proteolysis site (GPS) that is part of a large (>3000 amino acids) extracellular domain called the autoproteolysis-inducing (GAIN) domain which was first identified in CIRL/latrophilin proteins3. Latrophilins are Gαq11-coupled and are essential components in development 60. Human variants in aGPCR loci are associated with disorders of the nervous and cardiovascular systems, and neoplasias of all major tissues61. While it is speculated that latrophilins function as mechanosensitive metabotropic GPCRs, the physiological stimuli for these receptors is unclear. It is reported that the C-terminus of polycystin-1 binds to heterotrimeric G-proteins, and signals through regulators of G-protein signaling (RGS) GTPase activity62. However, there are no reports of the related PKD1L1 functioning as a GPCR. Based on biochemical and immunolocalization studies using exogenous overexpression methods, polycystin-1 localizes to the primary cilia of the collecting duct63. In addition, polycystin-1 may function at adhesion junctions, where it may modulate junctional turnover64. However, detecting polycystin-1 in native tissue is challenging because of its low expression level and because most commercially available antibodies are not specific for the polycystin epitope. Cleavage of polycystin-1 at the GPS site has been implicated in its liberation from the ER, but results from separate studies suggest that its localization to the cilia is not dependent on this post-translational processing 63,65. The ciliary pool of polycystin-1 appears to bypass Brefeldin A-sensitive golgi compartment, where it rapidly repopulates the cilia membrane66. While neither polycystin-1 or polycystin-1L1 form ion channels on their own, they can form complexes with and modulate the function of polycystin-2 and polycystin-2L1, respectively9,16,67. Interdependence between polycystin-1 and polycystin-2 in ciliary trafficking is widely reported68,69. Yet, other reports have demonstrated that polycystin-2 traffics to the cilia through a recognition motif found in its N-terminus (RVxP motif) and without expression of polycystin-157,70. In the embryonic node, calcium transients induced by morphogens that establish right-left symmetry are dependent on the expression of polycystin-2 and polycystin-1L171. In embryonic fibroblasts and retina pigmented epithelial cells, the number of polycystin-2L1 channels and ion conductance type is modulated by the co-expression of polycystin-1L116. Surprisingly, polycystin-1 and polycystin-1L1 was dispensable for the collecting duct primary cilia channel, as current levels were unaltered when PKD1L1 or PKD1 was genetically attenuated57. Thus, while there is no doubt that polycystin-1 is a central determinant in ADPKD, its contribution as an ion channel subunit or as channel-chaperone is an open question. However, the polycystin-1 + polycystin-2 channel complex might become active when occupied by an undetermined ligand or stimulus (Figure 2C), or its putative ion channel activity might be modulated by initiating an autoregulatory step involving association or disassociation of polycystin-1 with its N-terminus.
Overview of the molecular architecture of ciliary polycystin-2 channels and heteromeric complexes
The first and high-resolution (3Å) homotetrameric polycystin-2 structure was solved using cryo-EM17. The channel was captured in the closed state, and subsequent lower resolution structures were also reported for polycystin-2 and polycystin-2L172–74. Each subunit of polycystin-2 and polycystin-2L1 contain six transmembrane helical segments (S1-S6) and four distinct domains: voltage sensor domain (VSD), pore domain (PD), extracellular TOP domain and unresolved cilioplasmic domain (CTD) which is comprised of the N- and C-termini75. Aside from members of the polycystin-1 family (PKD1, PKD1L1), polycystin-2 is proposed to form heteromeric complexes with other members of the TRP class of ion channels. Using heterologous overexpression methods recording currents from plasma membranes— TRPV4 and TRPC1 are independently proposed to form heteromeric channels with polycystin-276,77. However, the currents attributed to heteromeric channels incorporating these TRP subunits were not supported by siRNA knockdown results where currents were recorded directly from the primary cilia. The best evidence for a ciliary polycystin-2 heteromer comes from the Kleene group, where the ciliary polycystin-2 conductance was attenuated by ablating TRPM3 expression using the CRISPR/Cas9 genome editing method in immortalized kidney collecting duct cells27,78. However, it is not apparent how the structure of the polycystin-2 and TRPM3 would oligomerize—as TRPM3 is missing a TOP domain and coil-coil motifs, which are essential for ciliary trafficking and stability of the assembled channel. Furthermore, it is important to note that the disorders associated with variants of TRPM3 (intellectual disability), TRPV4 (skeletal dysplasia) and TRPC1 (cardiomyopathies) are not classified as ciliopathies and polycystic kidney disease is not a comorbidity in these conditions. As mentioned previously, polycystin-1 and polycystin-2 can form heteromeric complexes, and the structural features of the oligomer is briefly covered in the upcoming section. However, the function of polycystin-1 as a cilia ion channel or channel-subunit-chaperone remains unclear. Outlined in proceeding sections are key features of the structural domains that comprise the homomeric form of polycystin-2 and when it complexes with polycystin-1.
The voltage sensor domain
The VSD consists of a bundle of transmembrane segments (S1-S4) that are is sandwiched between the intracellular CTD and extracellular TOP domain. In most TRP channels, this part of the protein is called the voltage sensor-like domain (VSDL), as it is not clear if it is regulated by membrane potential. However, the VSD S4 of polycystin-2 and polycystin-2L1 contain two positively charged residues called “gating charges”. These residues are conserved in voltage-gated ion channels at charge positions K3 and K4, These charges electrostatically “sense” changes in membrane potential, causing the S4 to move outwardly in the plane of the membrane when the cell is depolarized. This conformation change is coupled to opening of the PD, and allows ions to flow into the cell79. Based on work carried out in insect cells expressing PKD2L1, each VSD transfers at least one gating charge while forming state-dependent cation-π interactions during the activation process80. VSD gating charge delivery occurs prior to channel opening and is required to open the ion conducting pore of polycystin-2L1, regardless of polymodal stimuli (e.g. heat and membrane swelling)80. Since these interacting positions are conserved among voltage-gated ion channels, we propose that this gating mechanism was preserved during evolution when polycystins diverged from members of canonical voltage-gated channels (e.g. Cav, Nav, and Kv families). As discussed in the previous sections, elevation of internal calcium (see cilioplasmic domain section) shifts the voltage-dependent threshold of channel opening to negative membrane potentials, analogous to the effects observed in epithelial sodium channels measured in the plasma membrane. Thus although polycystin-2 and polycystin-2L1 channels exhibit voltage dependence, their opening at physiological membrane potentials is positively modulated by the level of intraciliary calcium.
The pore domain
The PD consists of S5 and S6 and form the central ion permeation pathway which houses the selectivity filter responsible for ion permeation through polycystin-2 (γK=139 pS, γNa=89 pS, γCa=4 pS)19 and polycystin-2L1 (γK= 160 pS, γNa= 137 pS, γCa= 34 pS)16,17. In both channels, the selectivity filter is scaffolded by two short alpha helices that were first observed in voltage-gated sodium channels and are believed to contribute to the electronegativity of the PD81. This feature is shared by members of the Nav, Cav, TRPA and TRPM families but is notably missing in Kir, Kv and TRPV channels, which only have a single pore helix. Both polycystin channels conduct multiple cations but differ in their selectivity for Ca2+. Polycystin-2L1 is more selective for Ca2+ than polycystin-2, and this functional difference is conferred by a second ring of aspartate residues (D525) which is not present in polycystin-259. Both polycystin-2 and polycystin-2L1 are blocked by trivalent ions (La3+, Gd3+), which occupy the selectivity filter aspartate as demonstrated in mutagenesis studies. Based on the cryo-EM structure of polycystin-2L1, the pore is proposed to have an upper and lower gate. The lower gate is found on the S6 and is proposed to be mobilized by uncoiling its secondary structure from a π-to-α helix82,83. The lower gate closely resembles the gate found in voltage-gated ion channels and involves a lateral displacement or “splaying” of the S6 through a series of hydrogen bonds formed with positions on the S4–S5 linker84. The opening of the lower gate was modeled using the Rosetta method while utilizing the polycystin-2 and polycystin-2L1 cryo-EM structures as templates. Here, membrane depolarization causes the S4 to move outwardly (~3 Å) and the S6 splays 11° to allow the passage of cations80. The function of the proposed upper gate is controversial and is found at sites within the selectivity filter: L521, D523 in polycystin-2L1, and L641, D643 in polycystin-2. The presence of an upper gate is also proposed for members of the vanilloid TRP channels (TRPV)85,86, and is supported by structural evidence and enhanced activation modes where ion selectivity is reduced by a proposed dilation of the channel pore87,88. However, recent work suggests that the selectivity filters in TRPV1–3 channels do not function as activation gates but changes in the selectivity filter upper gate might be coupled to rearrangements in the S6 lower gate89,90. It is unclear how motions from the polycystin VSD would be transferred to the upper gate, or if this site is most akin to an inactivation gate found in voltage-gated ion channels. Here, collapse of the selectivity filter has been widely described in Nav and Kv channels and is functionally linked to inactivation modes91–93. Thus, it is possible that the polycystin inactivation might be conferred by structural changes in the proposed upper gate.
The TOP domain
The TOP domains of polycystin-2 and polycystin-2L1 extend from the S1 and S2 helices and forms extracellular contacts with the S3-S4 loop of the VSD. The four TOP domains form homotypic contacts between each subunit in the assembled tetrameric channel. Although this protein fold is unique to polycystins, it is structurally similar to the extraluminal domain of the lysosomal mucolipin 1 TRP channels (TRPML1)94. Importantly, the TOP domain is the site of most (16/27) of the missense variants that cause ADPKD95. Similarly, many variants in the TRPML1 channel (MCOLN1 gene), which are associated with mucolipidosis type IV, also localize to its extraluminal domain96. Thus, these external sites in mucolipin and polycystin TRP channels strongly implicate their importance in disease progression and possibly channel function. The TOP domains of polycystin-2 and polycystin-2L1 are comprised of five beta strands with two intervening alpha helices. The five beta strands form two prominent hairpin turns, which we call Finger 1 and Finger 2 that serve as the interface between the homotypic contacts made between each polycystin subunit. Interestingly, the Finger intersubunit interface is where most of the disease-causing missense PKD2 variants localize within the TOP domain. In polycystin-2, Finger 1 projects from the β1-β2 sheet, which is stabilized by a hydrogen bond network involving Q323-T419-R325 and a cation-π interaction between R322 and F423. Thus, the multiple ADPKD-causing variant substitutions at positions R322 (R322Q and R322W) and R325 (R325Q and R325P) would likely destabilize the Finger 1 motif. C331 forms a disulfide bond (C331-C344), which likely constrains Finger 1’s association with its neighboring TOP domain. C331S is an ADPKD-causing variant whose hydroxyl side chain cannot form a disulfide bond with C344. Importantly, these polycystin-2 TOP domain interactions are conserved in polycystin-2L1, suggesting that these chemical bonds are important for both channel types. One hypothetical function of the TOP domain is to stabilize the channel’s tetrameric structure, and consequently, variants in this domain would alter its cilia membrane trafficking and formation of functional channels. An alternative hypothesis is that variants in the TOP domain may alter the voltage-dependence of channel gating since the TOP domain rests on the extracellular side of the VSD and would allosterically regulates the opening of the PD. This control of channel function by the polycystin TOP domain may be analogous to how proteinaceous toxins from spiders and scorpions interact with the VSDs of Kv and Nav ion channels97–99. These toxins trap their ion channel targets open and closed states and are voltage-dependent modifiers. Thus, both hypotheses for channel regulation by the TOP domain are plausible. The function of this domain remains undefined but its importance is underscored by its alterations in the ADPKD population.
Cilioplasmic domain
Based on the EM density maps used to solve the structures of polycystin-1 and polycystin-2, the N- and C-terminal ends of the peptide form a single structure on the cilioplasmic side of the channels called the CTD17,72,73. Although the CTD is poorly resolved, the domain engages the pore through the extended S6 helix, which reaches ~20 Å into the cilioplasm. Thus, structural changes in the CTD may control dilation of the lower gate through its interactions with the S6. Although the contiguous structure of the CTD has not been determined, the coiled-coil and EF-hand found in the CTD has been structurally resolved using crystallographic and nuclear magnetic resonance methods100–102. Here, the coiled-coil of polycystin-2 forms a trimeric complex, which is unexpected given that the transmembrane portions of polycystin-2 and polycystin-2L1 are tetrameric. However, this difference in stoichiometry might be satisfied in the heteromeric complex with polycystin-1 as a heterotetramer (see next section)103. The EF- hands found in polycystin-2 and polycystin-2L1 coordinate calcium, but with relatively low affinity (~20 μM) which would seem irrelevant at low cytoplasmic levels of calcium (90 nM){Yang, 2015 #440}. However, given that polycystins localize to cilia and ER, these calcium privileged environments undoubtedly expose the protein to higher calcium levels102. Additionally, there is conflicting information as to the necessity of the EF-hand and coiled-coil domains for channel formation and function of polycystin channels. C-terminal truncations of polycystin-2, which truncates the EF hand and coiled-coil, results in channels with decreased open probability when reconstituted from the ER into lipid bilayers 20. It is important to note that these results have not been validated from the ciliary pool of polycystins. By biochemical analysis, truncations also prohibit its association with polycystin-1 (see next section)9. However, several groups have captured high resolution structures of polycystins with and without the CTD, indicating that in vitro oligomerization of the channel is not dependent on this portion of the channel17,72,73. Similarly, the related polycystin-2L1 channel tetramerizes without its coiled coil and EF-hand by cryo-EM structural analysis74,82 and retains its functionality when heterologously expressed59. Thus, the impact of the C-terminal coiled coil and EF-hand for channel assembly, ciliary trafficking and function among the polycystins remains an open question.
The heteromeric polycystin-1 + polycystin-2 complex
The heteromeric structure polycystin-1 + polycystin-2 complex was determined by cryo-EM in a 1:3 ratio, respectively (Figure 2C)67. The central oligomerization site contains the electronegative carboxylate side chains contributed by three aspartate residue of the polycystin-2 subunits, but also four positively charged amine side chains contributed by arginine from a single polycystin-1 subunit. On its face, the amine side chains from the arginines would create a large electrostatic barrier to a permeating cation. It is important to note that these barrier charges are missing in polycystin-1L1, which is a bona fide channel subunit when it is complexed with polycystin-2 or polycystin-2L1. Here, co-expression of polycystin-1L1 attenuates the single channel conductance of polycystin-2L1 when measured from the primary cilia16. When co-expressed in oocytes, polycystin-1 increases calcium selectivity of polycystin-2, but only when drastic gain-of-function mutations are introduced at the gate of polycystin-2104. Thus, it is difficult to know if the polycystin-1 is functioning as part of a channel complex or these findings are apparent to what is normally a chaperone complex. As discussed early, it is possible that electrophysiologists have missed the heteromeric channel properties because the channel-complex only opens by an unidentified stimulus or ligand. Here, a stimulus may initiate a conformational change in the complex, tilting the S11 of polycystin-1 away from the channel or splay the pore forming helices of the polycystin-2. Thus, when moved out of the ion conducting pathway, the barrier charges may serve a channel function by bringing water into the pore vestibule, so that the partially dehydrated ions passing through the selectivity filter can be rehydrated upon entry. With limited understanding of its function as a putative ion channel subunit, it is difficult to speculate what the unidentified gating mechanism of polycystin-1 subunits may involve. Based on its extracellular location and its physiological localization to the unique lipid found in the cilia, it is hypothesized that polycystin-1 might be activated by ligands akin to aGPCRs or perhaps by lipid effectors. Interestingly, there is a gap in the structure of the complex located on one side of the intersubunit contact sites with polycystin-1. One possibility is that this site might be occupied by either the removed N-terminus of polycystin-1 (not present in the construct used to resolve the structure) or perhaps a third interacting molecule not expressed in the heterologous expression system. Thus, perhaps polycystin-1 and polycystin-2 are not the only members of this macromolecular complex. Enumerating structural components of the complex will direct research efforts in determining additional gene targets that are altered or modulate ADPKD progression. Determining these structural elements will also bring insight into the downstream effects of polycystin dysregulation and represent an intersection of the PKD phenotype found in other ciliopathies that affect other organ systems.
Summary and future directions in ADPKD basic research
In this review we have discussed advances determining the homomeric channel structure of polycytsin-2 and the structure of the polycystin-1 + polycystin-2 complex. We have also discussed the structural features and functional work which determined how polycystin-2 and polycystin-2L1 channels select for ions, oligomerize and open though conformational changes in its functional domains. The features of homoterameric polycystin-2 and polycystin-2L1 channels can be generally summarized as a voltage-gated ion channel that are modified by intra-ciliary calcium, and perhaps other ligands. As discussed previously, there is evidence that polycystic kidney disease can be initiated by gain- and loss-of-function of polycystins in mouse models. Since cilia electrophysiology is capable of directly measuring changes in channel biophysics, future work determining the dysregulation of polycystin-2 caused by ADPKD-variants is likely forthcoming. However, the functionality of polycystin-1 remains largely undetermined, which presents a large gap in our understanding of ADPKD since PKD1 variants are most frequently affected (≈ 80%) in the patient population. Since ADPKD in patients with PKD2 variants usually present with less severe cysts and typically development of end stage renal disease (ESRD) 10–20 years later than those with PKD1 variants— there is a clear impetus to determine polycystin-1’s biological function independently or in conjunction with polycystin-2 105. While it is likely that ciliary signaling involving several pathways can initiate kidney cystogenesis and other pathologies, it is uncertain if the ciliary population is the only pool of polycystins that contributes to ADPKD progression. Future work should be directed at determining the biological relevance of the ER populations of polycystins and their impact when dysregulated by variants induction.
Abbreviations used:
- ADPKD
Autosomal dominant polycystic kidney disease
- PKD
polycystic kidney disease
- ER
endoplasmic reticulum
- Ca2+
free calcium ions
- InsP3R
inositol trisphosphate receptor
- RyR
ryanodine receptor
- Cryo-EM
cryogenic electron microscopy
- Kv
members of voltage-gated potassium channel family
- Nav
members of voltage-gated sodium channel family
- Cav
members of voltage-gated calcium channel family
- VSD
voltage sensor domain
- PD
pore domain
- CTD
internal cilioplasmic domain, a singular domain comprised of the C- and N-termini of polycystins
- TRPV
members of transient receptor potential channel, vanilloid family
- TRPML
lysosomal mucolipin TRP channels
REFERENCES
- 1.Harris PC & Rossetti S Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol 6, 197–206, doi: 10.1038/nrneph.2010.18 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chebib FT & Torres VE Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am J Kidney Dis 67, 792–810, doi: 10.1053/j.ajkd.2015.07.037 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hughes J et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 10, 151–160, doi: 10.1038/ng0695-151 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Brasier JL & Henske EP Loss of the polycystic kidney disease (PKD1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. J Clin Invest 99, 194–199, doi: 10.1172/JCI119147 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mochizuki T et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272, 1339–1342 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Grantham JJ Polycystic kidney disease: from the bedside to the gene and back. Curr Opin Nephrol Hypertens 10, 533–542 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Koptides M, Hadjimichael C, Koupepidou P, Pierides A & Constantinou Deltas C Germinal and somatic mutations in the PKD2 gene of renal cysts in autosomal dominant polycystic kidney disease. Human molecular genetics 8, 509–513 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Wu G et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell 93, 177–188 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Qian F et al. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet 16, 179–183, doi: 10.1038/ng0697-179 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Pei YA “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 7, 151–156 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Happe H & Peters DJ Translational research in ADPKD: lessons from animal models. Nat Rev Nephrol 10, 587–601, doi: 10.1038/nrneph.2014.137 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Wilson PD Mouse models of polycystic kidney disease. Curr Top Dev Biol 84, 311–350, doi: 10.1016/S0070-2153(08)00606-6 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Ma M, Tian X, Igarashi P, Pazour GJ & Somlo S Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 45, 1004–1012, doi: 10.1038/ng.2715 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanaoka K et al. Co-assembly of polycystin-1 and −2 produces unique cation-permeable currents. Nature 408, 990–994, doi: 10.1038/35050128 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Delmas P et al. Gating of the polycystin ion channel signaling complex in neurons and kidney cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 18, 740–742, doi: 10.1096/fj.03-0319fje (2004). [DOI] [PubMed] [Google Scholar]
- 16.DeCaen PG, Delling M, Vien TN & Clapham DE Direct recording and molecular identification of the calcium channel of primary cilia. Nature 504, 315–318, doi: 10.1038/nature12832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen PS et al. The Structure of the Polycystic Kidney Disease Channel PKD2 in Lipid Nanodiscs. Cell 167, 763–773 e711, doi: 10.1016/j.cell.2016.09.048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arif Pavel M et al. Function and regulation of TRPP2 ion channel revealed by a gain-of-function mutant. Proc Natl Acad Sci U S A 113, E2363–2372, doi: 10.1073/pnas.1517066113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X et al. Polycystin-2 is an essential ion channel subunit in the primary cilium of the renal collecting duct epithelium. Elife, doi: 10.1101/215814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koulen P et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol 4, 191–197, doi: 10.1038/ncb754 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Vassilev PM et al. Polycystin-2 is a novel cation channel implicated in defective intracellular Ca(2+) homeostasis in polycystic kidney disease. Biochem Biophys Res Commun 282, 341–350, doi: 10.1006/bbrc.2001.4554 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Qian Q et al. Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum Mol Genet 12, 1875–1880 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Brill AL & Ehrlich BE Polycystin 2: A calcium channel, channel partner, and regulator of calcium homeostasis in ADPKD. Cell Signal 66, 109490, doi: 10.1016/j.cellsig.2019.109490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pablo JL, DeCaen PG & Clapham DE Progress in ciliary ion channel physiology. J Gen Physiol 149, 37–47, doi: 10.1085/jgp.201611696 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winyard P & Jenkins D Putative roles of cilia in polycystic kidney disease. Biochim Biophys Acta 1812, 1256–1262, doi: 10.1016/j.bbadis.2011.04.012 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Guay-Woodford LM et al. Filling the holes in cystic kidney disease research. Clin J Am Soc Nephrol 9, 1799–1801, doi: 10.2215/CJN.03410414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleene SJ & Kleene NK The native TRPP2-dependent channel of murine renal primary cilia. Am J Physiol Renal Physiol 312, F96–F108, doi: 10.1152/ajprenal.00272.2016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao M, Cao E, Julius D & Cheng Y Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112, doi: 10.1038/nature12822 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hildebrandt F, Benzing T & Katsanis N Ciliopathies. N Engl J Med 364, 1533–1543, doi: 10.1056/NEJMra1010172 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waters AM & Beales PL Ciliopathies: an expanding disease spectrum. Pediatr Nephrol 26, 1039–1056, doi: 10.1007/s00467-010-1731-7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerdes JM, Davis EE & Katsanis N The vertebrate primary cilium in development, homeostasis, and disease. Cell 137, 32–45, doi: 10.1016/j.cell.2009.03.023 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torres VE, Harris PC & Pirson Y Autosomal dominant polycystic kidney disease. Lancet 369, 1287–1301, doi: 10.1016/S0140-6736(07)60601-1 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Bergmann C et al. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat 23, 453–463, doi: 10.1002/humu.20029 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Kaimori JY et al. Polyductin undergoes notch-like processing and regulated release from primary cilia. Hum Mol Genet 16, 942–956, doi: 10.1093/hmg/ddm039 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Y et al. Kinesin-2 mediates physical and functional interactions between polycystin-2 and fibrocystin. Hum Mol Genet 15, 3280–3292, doi: 10.1093/hmg/ddl404 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Yoshiba S et al. Cilia at the node of mouse embryos sense fluid flow for left-right determination via Pkd2. Science 338, 226–231, doi: 10.1126/science.1222538 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Praetorius HA & Spring KR The renal cell primary cilium functions as a flow sensor. Curr Opin Nephrol Hypertens 12, 517–520, doi: 10.1097/01.mnh.0000088730.87142.d1 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Mangolini A, de Stephanis L & Aguiari G Role of calcium in polycystic kidney disease: From signaling to pathology. World J Nephrol 5, 76–83, doi: 10.5527/wjn.v5.i1.76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boulter C et al. Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc Natl Acad Sci U S A 98, 12174–12179, doi: 10.1073/pnas.211191098 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim K, Drummond I, Ibraghimov-Beskrovnaya O, Klinger K & Arnaout MA Polycystin 1 is required for the structural integrity of blood vessels. Proc Natl Acad Sci U S A 97, 1731–1736 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu W et al. Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat Genet 17, 179–181, doi: 10.1038/ng1097-179 (1997). [DOI] [PubMed] [Google Scholar]
- 42.Wu G et al. Trans-heterozygous Pkd1 and Pkd2 mutations modify expression of polycystic kidney disease. Hum Mol Genet 11, 1845–1854 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Wu G & Somlo S Molecular genetics and mechanism of autosomal dominant polycystic kidney disease. Mol Genet Metab 69, 1–15, doi: 10.1006/mgme.1999.2943 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL & Germino GG A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med 13, 1490–1495, doi: 10.1038/nm1675 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davenport JR et al. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol 17, 1586–1594, doi: 10.1016/j.cub.2007.08.034 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shibazaki S et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet 17, 1505–1516, doi: 10.1093/hmg/ddn039 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li A et al. Human polycystin-2 transgene dose-dependently rescues ADPKD phenotypes in Pkd2 mutant mice. Am J Pathol 185, 2843–2860, doi: 10.1016/j.ajpath.2015.06.014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nonaka S et al. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 95, 829–837, doi: 10.1016/s0092-8674(00)81705-5 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Brennan J, Norris DP & Robertson EJ Nodal activity in the node governs left-right asymmetry. Genes & development 16, 2339–2344, doi: 10.1101/gad.1016202 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bangs F & Anderson KV Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb Perspect Biol 9, doi: 10.1101/cshperspect.a028175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singla V & Reiter JF The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science 313, 629–633, doi: 10.1126/science.1124534 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Reiter JF & Leroux MR Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol 18, 533–547, doi: 10.1038/nrm.2017.60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Helou J et al. Mutation analysis of NPHP6/CEP290 in patients with Joubert syndrome and Senior-Loken syndrome. Journal of medical genetics 44, 657–663, doi: 10.1136/jmg.2007.052027 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leigh MW et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genetics in medicine : official journal of the American College of Medical Genetics 11, 473–487, doi: 10.1097/GIM.0b013e3181a53562 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McIntyre JC et al. Gene therapy rescues cilia defects and restores olfactory function in a mammalian ciliopathy model. Nature medicine 18, 1423–1428, doi: 10.1038/nm.2860 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venkatachalam K & Montell C TRP channels. Annual review of biochemistry 76, 387–417, doi: 10.1146/annurev.biochem.75.103004.142819 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu X et al. Polycystin-2 is an essential ion channel subunit in the primary cilium of the renal collecting duct epithelium. eLife 7, doi: 10.7554/eLife.33183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Orts-Del’Immagine A et al. A single polycystic kidney disease 2-like 1 channel opening acts as a spike generator in cerebrospinal fluid-contacting neurons of adult mouse brainstem. Neuropharmacology 101, 549–565, doi: 10.1016/j.neuropharm.2015.07.030 (2016). [DOI] [PubMed] [Google Scholar]
- 59.DeCaen PG, Liu X, Abiria S & Clapham DE Atypical calcium regulation of the PKD2-L1 polycystin ion channel. eLife 5, doi: 10.7554/eLife.13413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langenhan T et al. Latrophilin signaling links anterior-posterior tissue polarity and oriented cell divisions in the C. elegans embryo. Developmental cell 17, 494–504, doi: 10.1016/j.devcel.2009.08.008 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langenhan T, Aust G & Hamann J Sticky signaling--adhesion class G protein-coupled receptors take the stage. Science signaling 6, re3, doi: 10.1126/scisignal.2003825 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Kim E et al. Interaction between RGS7 and polycystin. Proc Natl Acad Sci U S A 96, 6371–6376, doi: 10.1073/pnas.96.11.6371 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Su X et al. Regulation of polycystin-1 ciliary trafficking by motifs at its C-terminus and polycystin-2 but not by cleavage at the GPS site. J Cell Sci 128, 4063–4073, doi: 10.1242/jcs.160556 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhoonderowa L et al. Polycystins and intercellular mechanotransduction: A precise dosage of polycystin 2 is necessary for alpha-actinin reinforcement of junctions upon mechanical stimulation. Exp Cell Res 348, 23–35, doi: 10.1016/j.yexcr.2016.08.021 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Kurbegovic A et al. Novel functional complexity of polycystin-1 by GPS cleavage in vivo: role in polycystic kidney disease. Molecular and cellular biology 34, 3341–3353, doi: 10.1128/MCB.00687-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gilder AL et al. Newly synthesized polycystin-1 takes different trafficking pathways to the apical and ciliary membranes. Traffic 19, 933–945, doi: 10.1111/tra.12612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Su Q et al. Structure of the human PKD1-PKD2 complex. Science 361, doi: 10.1126/science.aat9819 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Gainullin VG, Hopp K, Ward CJ, Hommerding CJ & Harris PC Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J Clin Invest 125, 607–620, doi: 10.1172/JCI76972 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cai Y et al. Altered trafficking and stability of polycystins underlie polycystic kidney disease. The Journal of clinical investigation 124, 5129–5144, doi: 10.1172/JCI67273 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Geng L et al. Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J Cell Sci 119, 1383–1395, doi: 10.1242/jcs.02818 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Field S et al. Pkd1l1 establishes left-right asymmetry and physically interacts with Pkd2. Development 138, 1131–1142, doi: 10.1242/dev.058149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wilkes M et al. Molecular insights into lipid-assisted Ca(2+) regulation of the TRP channel Polycystin-2. Nat Struct Mol Biol 24, 123–130, doi: 10.1038/nsmb.3357 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Grieben M et al. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2). Nat Struct Mol Biol 24, 114–122, doi: 10.1038/nsmb.3343 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Hulse RE, Li Z, Huang RK, Zhang J & Clapham DE Cryo-EM structure of the polycystin 2-l1 ion channel. eLife 7, doi: 10.7554/eLife.36931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Montell C The TRP superfamily of cation channels. Sci STKE 2005, re3, doi: 10.1126/stke.2722005re3 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Kottgen M et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol 182, 437–447, doi: 10.1083/jcb.200805124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bai CX et al. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep 9, 472–479, doi: 10.1038/embor.2008.29 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kleene SJ et al. The TRPP2-dependent channel of renal primary cilia also requires TRPM3. PLoS One 14, e0214053, doi: 10.1371/journal.pone.0214053 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vargas E et al. An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J Gen Physiol 140, 587–594, doi: 10.1085/jgp.201210873 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ng LCT, Vien TN, Yarov-Yarovoy V & DeCaen PG Opening TRPP2 (PKD2L1) requires the transfer of gating charges. Proceedings of the National Academy of Sciences of the United States of America 116, 15540–15549, doi: 10.1073/pnas.1902917116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Payandeh J, Scheuer T, Zheng N & Catterall WA The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358, doi: 10.1038/nature10238 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Su Q et al. Cryo-EM structure of the polycystic kidney disease-like channel PKD2L1. Nature communications 9, 1192, doi: 10.1038/s41467-018-03606-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zheng W et al. Hydrophobic pore gates regulate ion permeation in polycystic kidney disease 2 and 2L1 channels. Nature communications 9, 2302, doi: 10.1038/s41467-018-04586-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sula A et al. The complete structure of an activated open sodium channel. Nature communications 8, 14205, doi: 10.1038/ncomms14205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cao E, Liao M, Cheng Y & Julius D TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118, doi: 10.1038/nature12823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deng Z et al. Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nature structural & molecular biology 25, 252–260, doi: 10.1038/s41594-018-0037-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Binshtok AM, Bean BP & Woolf CJ Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 449, 607–610, doi: 10.1038/nature06191 (2007). [DOI] [PubMed] [Google Scholar]
- 88.Chung MK, Guler AD & Caterina MJ TRPV1 shows dynamic ionic selectivity during agonist stimulation. Nature neuroscience 11, 555–564, doi: 10.1038/nn.2102 (2008). [DOI] [PubMed] [Google Scholar]
- 89.Jara-Oseguera A, Huffer KE & Swartz KJ The ion selectivity filter is not an activation gate in TRPV1–3 channels. eLife 8, doi: 10.7554/eLife.51212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zubcevic L, Borschel WF, Hsu AL, Borgnia MJ & Lee SY Regulatory switch at the cytoplasmic interface controls TRPV channel gating. eLife 8, doi: 10.7554/eLife.47746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang X et al. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486, 130–134, doi: 10.1038/nature11054 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cordero-Morales JF, Jogini V, Chakrapani S & Perozo E A multipoint hydrogen-bond network underlying KcsA C-type inactivation. Biophysical journal 100, 2387–2393, doi: 10.1016/j.bpj.2011.01.073 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li J, Ostmeyer J, Cuello LG, Perozo E & Roux B Rapid constriction of the selectivity filter underlies C-type inactivation in the KcsA potassium channel. The Journal of general physiology 150, 1408–1420, doi: 10.1085/jgp.201812082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schmiege P, Fine M, Blobel G & Li X Human TRPML1 channel structures in open and closed conformations. Nature 550, 366–370, doi: 10.1038/nature24036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gout AM, Martin NC, Brown AF & Ravine D PKDB: Polycystic Kidney Disease Mutation Database--a gene variant database for autosomal dominant polycystic kidney disease. Hum Mutat 28, 654–659, doi: 10.1002/humu.20474 (2007). [DOI] [PubMed] [Google Scholar]
- 96.Sun M et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Human molecular genetics 9, 2471–2478, doi: 10.1093/hmg/9.17.2471 (2000). [DOI] [PubMed] [Google Scholar]
- 97.Catterall WA, Wisedchaisri G & Zheng N The chemical basis for electrical signaling. Nat Chem Biol 13, 455–463, doi: 10.1038/nchembio.2353 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yarov-Yarovoy V et al. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proceedings of the National Academy of Sciences of the United States of America 109, E93–102, doi: 10.1073/pnas.1118434109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tao X, Lee A, Limapichat W, Dougherty DA & MacKinnon R A gating charge transfer center in voltage sensors. Science 328, 67–73, doi: 10.1126/science.1185954 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu Y et al. Structural and molecular basis of the assembly of the TRPP2/PKD1 complex. Proc Natl Acad Sci U S A 106, 11558–11563, doi: 10.1073/pnas.0903684106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Allen MD, Qamar S, Vadivelu MK, Sandford RN & Bycroft M A high-resolution structure of the EF-hand domain of human polycystin-2. Protein science : a publication of the Protein Society 23, 1301–1308, doi: 10.1002/pro.2513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Petri ET et al. Structure of the EF-hand domain of polycystin-2 suggests a mechanism for Ca2+-dependent regulation of polycystin-2 channel activity. Proceedings of the National Academy of Sciences of the United States of America 107, 9176–9181, doi: 10.1073/pnas.0912295107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhu J et al. Structural model of the TRPP2/PKD1 C-terminal coiled-coil complex produced by a combined computational and experimental approach. Proc Natl Acad Sci U S A 108, 10133–10138, doi: 10.1073/pnas.1017669108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Z et al. The ion channel function of polycystin-1 in the polycystin-1/polycystin-2 complex. EMBO reports 20, e48336, doi: 10.15252/embr.201948336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hateboer N et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet 353, 103–107, doi: 10.1016/s0140-6736(98)03495-3 (1999). [DOI] [PubMed] [Google Scholar]
- 106.Delling M et al. Primary cilia are not calcium-responsive mechanosensors. Nature 531, 656–660, doi: 10.1038/nature17426 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Delling M, DeCaen PG, Doerner JF, Febvay S & Clapham DE Primary cilia are specialized calcium signalling organelles. Nature 504, 311–314, doi: 10.1038/nature12833 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]