

Abstract
ASD is a largely heritable, multistage, prenatal disorder that impacts a child’s ability to perceive and react to social information. Most ASD risk genes express prenatally and fall into two categories: broadly-expressed regulatory genes that express in the brain and other organs and brain-specific genes. In trimesters 1 to 3 (Epoch-1), one set of broadly-expressed (the majority) and brain-specific risk genes disrupt cell proliferation, neurogenesis, migration and cell fate, while in trimester 3 and early postnatal (Epoch-2) another set (the majority being brain-specific) disrupt neurite outgrowth, synaptogenesis and “wiring” of cortex. A proposed model is that upstream highly interconnected regulatory ASD gene mutations disrupt transcriptional programs or signaling pathways resulting in dysregulation of downstream processes such as proliferation, neurogenesis, synaptogenesis and neural activity. Dysregulation of signaling pathways is correlated with ASD social symptom severity. Since the majority of ASD risk genes are broadly-expressed, many ASD individuals may benefit by being treated as a broader medical disorder. An important future direction is the non-invasive study of ASD cell biology.
Keywords: Autism, prenatal, regulatory, broadly-expressed, brain-specific, gene, proliferation, synapse
A QUIET REVOLUTION: UPENDING CONVENTIONAL BELIEFS ABOUT ASD
ASD impacts an individual’s ability to both perceive and react to social information[1–6]. It has captured the attention of clinicians, scientists and the lay public because of its debilitating social, emotional, language and cognitive symptoms and unexplained clinical heterogeneity. First described >75 years ago[7], its causes and neural developmental origins remained elusive for many decades. However, research this past decade has revolutionized our understanding of ASD, and many previous unfounded speculations and misconceptions have been disproven. For example, it is now clear that ASD is not caused by parents, nor by vaccines. It is also not rare, nor does it start in early childhood. Instead, it is clinically detectable and highly diagnostically stable by 14 months of age[8]. In most individuals, ASD is not due to a single stage of deviate development. It also is not due to abnormality in a single brain region, and remarkably, in some subjects might not even be exclusively a brain disorder.
Instead, recent computational, cell line model, animal model, clinical, genetic and postmortem findings show that ASD is a highly heritable, multistage, multi-process progressive, brain-wide disorder of prenatal and early postnatal development, with prenatal subtypes leading to clinical outcome heterogeneity[9]. Rather than a single stage of deviant development, multiple stages are involved (Fig. 1A) including cell proliferation[10], neurogenesis, migration[11], laminar organization[11], and neurite outgrowth during the 1st and 2nd trimesters, and spine development, synaptogenesis, and synapse functioning in the 3rd trimester and early postnatal life[9, 12–20].
Figure 1. ASD is a progressive multi-stage disorder of prenatal and early postnatal development.
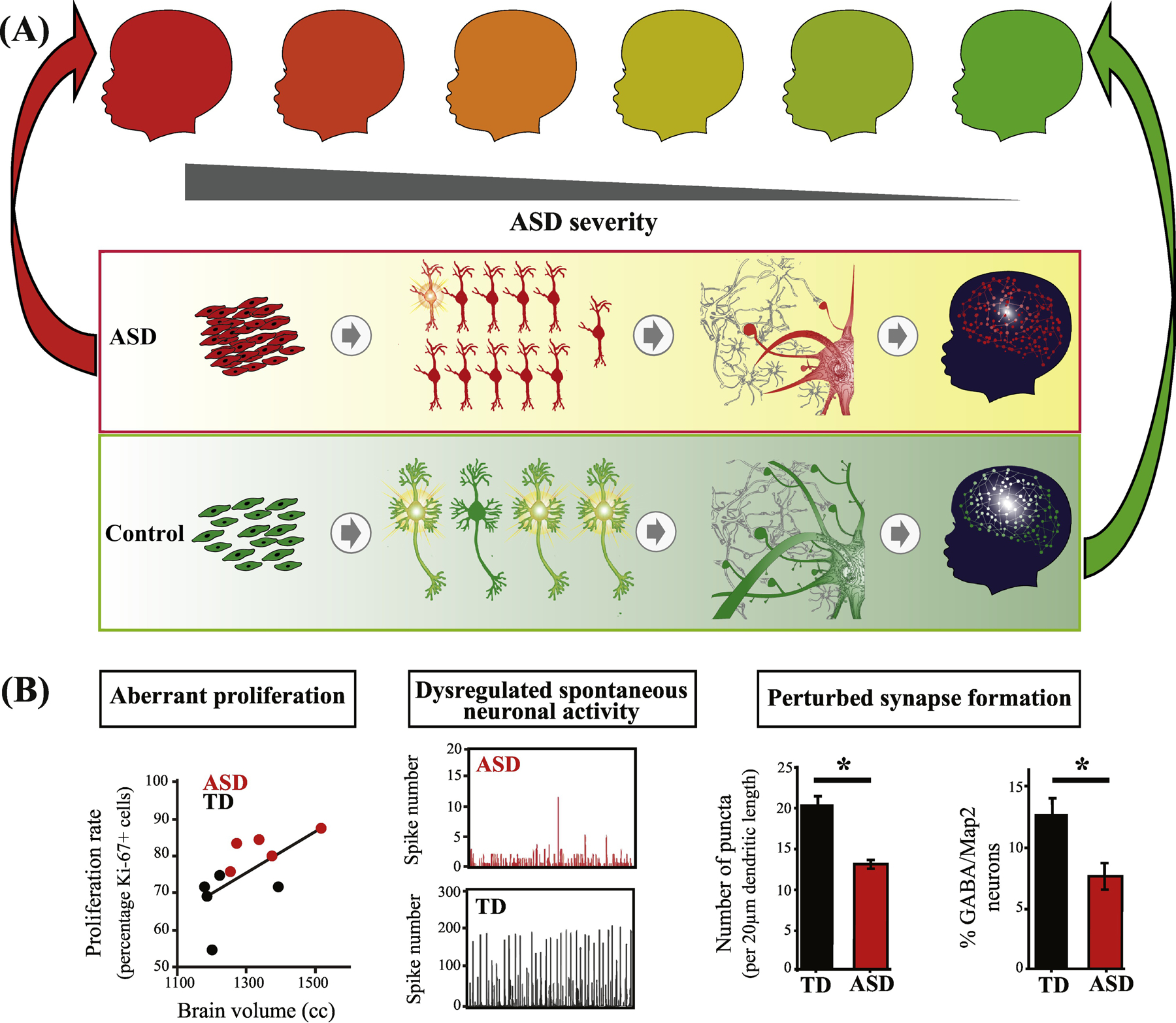
(A) ASD involves multiple stages of prenatal cortical maldevelopment as compared with typical development (from[9]). ASD begins with aberrant cortex formation that involves disruption of cell cycle processes and excess cell proliferation (left panel); neuronal maturation, neurite outgrowth and spontaneous neural activity (next panel); and continues with disruption of later prenatal and postnatal synaptogenesis and neural network expansion (third panel); and “wiring” of cortex and coordinated neural circuits activity (right panel)(from[9]). Upper series of child icons from red to green, illustrates the hypothesis that ASD outcome severity (darker red) may be related to severity of dysregulation in some or all of these prenatal and postnatal stages. (B) In one study, ASD subject-derived iPS cell (iPSC) models show that abnormal increases in cell proliferation are correlated with each child’s early brain overgrowth (left panel). iPSC-derived ASD neurons also show reduced spontaneous neuronal activity (middle panel) and deviant synapse formation (right panel) (from[20]). See Box 1 for more details.
Here, we first review new biological and genetic as well as direct postmortem findings, and, from these, we identify two often undiscussed attributes of the new findings that open new concepts and directions for ASD genetic, genomic and cellular research.
ASD BEGINS IN PRENATAL LIFE: It Involves Multiple Prenatal Stages, Processes and Brain Regions
iPSC studies of idiopathic ASD individuals implicate multiple prenatal stages and processes.
Recent ASD subject-derived inducible pluripotent stem cells (iPSC) studies have provided insights into prenatal origins in idiopathic ASD[20–23]. In the largest study to date (n=8 ASD and n=5 healthy controls), every ASD child studied showed disruptions in multiple prenatal-stages including proliferation, maturation, synaptogenesis and neural activity (Fig. 1B). ASD displayed high rates of cell proliferation, G1/S shortening, reduced differentiation and neuronal maturation, abnormal inhibitory and excitatory synaptic maturation, and reduced neural activity (Fig. 1B). Excess proliferation and abnormal differentiation, neurite and synapse development were also found[21] during iPSC to neuron differentiation in 4 ASD adults with brain enlargement. An iPSC study of 6 idiopathic ASD males and 3 controls found dysregulation of genes involved in neuronal differentiation, axon guidance, cell migration, and regional patterning[22], and another small sample iPSC study of individuals with idiopathic ASD and those with risk gene or CNV deletions also reported delayed neuronal differentiation[23].
Confirming ASD postmortem findings (Text Box 1) and extending them to living ASD subjects, iPSC-derived molecular and cellular evidence depicts idiopathic ASD as a multistage disorder of prenatal development. The disrupted stages begin with shortening cell cycle G1-S and excess cell proliferation. It then continues with deviant neuronal differentiation, maturation, migration, synaptogenesis and neural network activity[9]. Abnormal cell proliferation explains reports of overabundance of cortical neurons[24], excess brain weight[24–26], and early brain overgrowth in ASD[27–30]. Excess prenatal neurogenesis causes an overabundance of upper layer cortical neurons, imbalance of excitation and inhibition, altered neural functioning and ASD-like behavior in ASD animal models[31].
TEXT BOX 1: Postmortem evidence implicates multiple prenatal stages.
Postmortem cellular, molecular and genetic evidence shows that ASD is a multistage, multi-process disorder of prenatal development,[9] and that it involves one or a combination of common gene variants, de novo variants, and somatic mosaicism in a subset of subjects[11, 66]. While most postmortem findings reflect neuropathology pertinent to the age at death of the case and not to early ASD pathobiological origins, a few key postmortem studies of idiopathic ASD children as young as 2 years find definitive signs of prenatal 1st, 2nd and 3rd trimester maldevelopment[9, 11, 15, 16, 18, 24]. In prefrontal cortex at young ages, there are excess numbers of neurons in each child studied. On average, ASD children have 67% more prefrontal neurons than controls[24], 30% more overall in cortex at young ages (see Table S2 in[9]), and 57% more von Economo neurons in cortex involved in socioemotional processes[67]. Because proliferation of cortical neurons is exponential between 10 and 20 weeks of gestation[68, 69] and does not occur postnatal, pathological neuron excess shows that neuropathology of ASD has a prenatal origin, likely by the 1st or 2nd trimester. In utero brain overgrowth occurs in ASD[70]. In animal models, cell cycle disruption leading to excess prenatal cortical cell proliferation causes brain overgrowth and ASD-like symptoms[31]. In postmortem brains from young ASD cases, cell cycle, differentiation, and DNA damage detection genes and pathways are dysregulated in prefrontal cortex[71]. Also, living ASD toddlers with enlarged brain size have dysregulation of cell cycle gene expression indicative of G1/S shortening[54].
Disruption of 2nd and 3rd trimester development in ASD is signalled by focal disorganization of cortical layers and nearby clusters of mis-migrated neurons in prefrontal and temporal cortices[11, 72–75]. Multiregional dysregulation of neurogenesis, neuronal mis-migration and multifocal cerebral dysplasias is reported in several ASD cases[72]. Dysregulation of maturation and neuronal growth are signalled by reduced neuron size and dendritic arbors in many postmortem ASD studies[73] [24, 76, 77]. In living ASD toddlers, evidence is also consistent with reduced axon size and growth but increased axon numbers in the cerebrum (likely from excess small neurons)[40, 78].
GWAS evidence further implicates early prenatal corticogenesis in idiopathic ASD.
A recent GWAS study implicated multiple genetic loci in strong association with idiopathic ASD[32]. Analyses of genes at these loci identified 34 genes. These genes are involved in prenatal corticogenesis during late 1st and during 2nd and 3rd trimesters but are not strongly expressed postnatally (Fig. 2A, B). Peak expression is during proliferation, neurogenesis, migration, cell fate and cortical layer formation. These results are predicted by the iPSC studies and, as discussed below, by the finding of a strong expression of high confidence ASD (hcASD) risk genes (see Glossary) during early prenatal corticogenesis.
Figure 2. ASD genetics uniformly implicate prenatal beginnings in multiple brain regions and multiple functional processes.
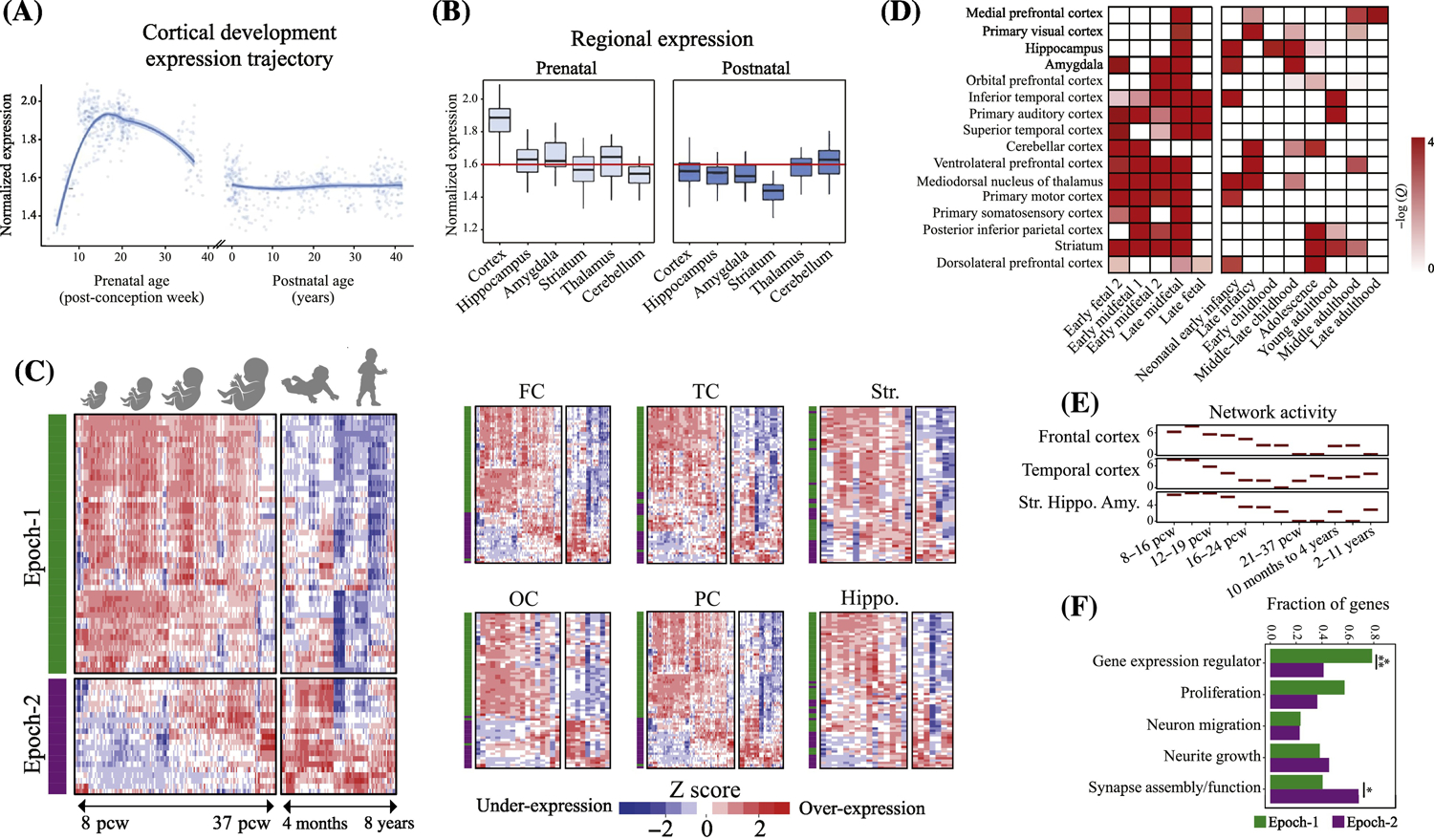
(A-B) Genes implicated in ASD by a recent GWAS study show strong expression during prenatal corticogenesis but not during postnatal cortex development (from[32]). (C) Neocortex expression pattern of 69 hcASD genes during brain development. 94% of 69 hcASD risk genes express at prenatal ages across multiple brain regions (from[9]). The heatmaps also show there are two major groups of hcASD genes: Epoch-1 (green bar in heatmaps) consisting of 68% of hcASD genes and Epoch-2 (purple bar in heatmaps) consisting of 32% (from[9]). FC: Frontal cortex; OC: Occipital cortex; PC: Parietal cortex; TC: Temporal cortex; Hippo: Hippocampus; Str: Striatum. (D) Similar to hcASD genes, an extended set of ASD risk genes show peak expression at prenatal ages across multiple brain regions (from[17]). (E) Co-expression activity of genes perturbed in leukocytes of individuals with ASD implicate prenatal neurodevelopmental stage across frontal and temporal cortices and the striatum, hippocampus and amygdala (Str.Hippo.Amy)(from[51]). (F) hcASD genes are highly pleotropic and are involved in multiple stages of prenatal neural development. Epoch-1 (green bars) is heavily dominated by regulatory hcASD genes (79% of Epoch-1 genes) that can disrupt proliferation, neurogenesis and other early stages of cortical formation, while Epoch-2 (purple bars) is dominated by brain-specific hcASD genes (>60% of Epoch-2 genes) that may disrupt late prenatal and postnatal synaptogenesis and “wiring” of cortex. Percentages across functional clusters add up to more than 100% because the majority of hcASD gene mutations affect more than one developmental process.
ASD risk gene mutations strongly implicate prenatal beginnings in multiple brain regions.
Most ASD risk genes are expressed in neocortex during prenatal life (Fig. 2A, B, C and D)[9, 13, 14, 17, 19, 33–36]. This effect has remained robust and unchanged despite the expanding list of ASD risk gene mutations over the years. For example, our recent analysis showed 94% of hcASD genes are expressed in prenatal development (Fig.2C). In another recent ASD gene discovery study of 102 putative ASD risk genes, 98 had their highest expression in prenatal cortex as compared with postnatal[36]. Moreover, prenatal expression of ASD risk genes is consistently reported for most ASD-implicated brain regions including cortex, cerebellum, amygdala, hippocampus and striatum (Fig. 2C and D)[9, 17]. The expression of hcASD genes fall into two prenatal developmental periods, Epoch-1 (68% of ASD genes) and Epoch-2 (32% of ASD genes), as shown in Fig.2C[9]. Thus, risk gene evidence shows ASD is a prenatal disorder of every major brain region implicated to date by anatomical and functional imaging[10, 37–42] and postmortem studies (see Box 1).
ASD risk genes point to multiple prenatal stages, diverse biological processes, pleiotropy and neuron types.
To understand the prenatal pathobiology and mechanisms leading to ASD, many hcASD genes are functionally characterized through cell line and animal models. A recent literature survey of several hundreds of published reports found that ASD involves multiple prenatal stages [9]. Specifically, of the 58 hcASD genes with characterized neurofunctional roles, 33 (57%) are involved in proliferation, 15 (26%) in migration and cell fate specification, 30 (52%) in neurite outgrowth and 34 (59%) in synaptogenesis and synapse functioning (Fig. 2F)[9]. About two-thirds of hcASD genes are pleiotropic, influencing two or more processes. This evidence adds a genetic explanation to the developmental pathobiology revealed by postmortem, neuroimaging and iPSC evidence. It shows that ASD risk genes are involved in excess cell proliferation, disrupted neurogenesis and maturation, mis-migration, disrupted synaptic development and function, and deviant neurofunctional activity and connectivity. While excess proliferation and dysregulated differentiation appear to begin ASD prenatal maldevelopment, the pleiotropic nature of most hcASD genes and their involvement in successive prenatal stages, serves to emphasize that ASD is a multifaceted, progressive disorder beginning with dysregulation of cell numbers, neurogenesis, migration, cell fate and early neural maturation, but then transitions to a layered prenatal to postnatal path where effects of ASD-synaptic risk genes interact with activity- and experience-dependent postnatal factors. Supporting a possible successive propagation of the perturbations, it is found that aberrant excess excitatory neurogenesis is sufficient to cause ASD neurophysiology and behavior in animal models due to a cascade of maldevelopment triggered by the early excess (see Box 2).
TEXT BOX 2: Excess proliferation of excitatory cells in early prenatal life may be sufficient to cause ASD.
Fang et al[31] developed a unique ASD animal model showing prenatal generation of excess excitatory cortical neurons is sufficient to cause ASD-like brain overgrowth, imbalance in excitation and inhibition, aberrant cortical-cortical circuit formation and function, and abnormal social, communication and behavior. In a new study of 102 ASD risk genes, early maturing and mature excitatory cortical lineage neurons had the most robust enrichment among all cell types examined, with 71 of the 102 risk genes[36]. Furthermore, early excitatory cortical neurons expressed the most ASD risk genes; ASD genes were also expressed in striatal inhibitory lineage neurons, oligodendrocyte progenitors and astrocytes. A 67% overabundance of frontal cortical neurons is found postmortem at young ages in ASD[24]. An abnormal excess cell proliferation occurs in ASD toddler-derived iPSC models and correlates with early-age brain overgrowth in the ASD toddlers[20]. Thus, excess cell proliferation may be sufficient to cause ASD. This additionally evidence supports the long-standing theory that ASD cortical pathophysiology involves imbalance between cortical excitation and inhibition[79], and suggests the possibility that, in cortex, imbalance could be related to pathological excess excitatory cortical neurons.
Multiple prenatal biological processes also emerge from functional clustering of ASD risk genes[17]. Clusters included cell cycle G1-S and G2-M regulation; histone modification and chromatin remodeling; synapse-relevant processes and structures; and PI3K/AKT, RAS/ERK, and WNT/β-catenin, gamma interferon, TGFβ and TLR signaling. Importantly, many of the implicated signaling pathways are highly conserved and pleotropic, affecting multiple neurodevelopmental stages.
TWO PRENATAL EPOCHS OF RISK GENE EXPRESSION: Regulatory Genes Begin Prenatal Maldevelopment in ASD
By analysing the gene expression pattern of ASD risk genes in a systematic and unbiased way, one may better understand when ASD genetic dysregulation begins, what genes may be driving its initiation, and what biological mechanisms and processes may be involved. Different approaches have been used (see Box 3 and Fig. 3A and B).
TEXT BOX 3: Analyses of temporal patterns of expression of ASD risk genes during prenatal development.
One approach to identification of developmentally critical periods and pathobiological mechanisms in ASD has been to analyze temporal patterns of expression of ASD risk genes during prenatal development. Some studies first cluster genes into co-expression modules and next examine their enrichment for ASD risk genes (Fig. 3A)[14]. Other studies use a priori selection of specific gene types or gene modules/networks of interest and then ask how these selected genes change in expression during early development (Fig. 3B)[36]. Such analyses have commonly used BrainSpan normative data on prenatal and postnatal brain gene expression. Across studies, different lists of ASD risk genes have been examined, primarily due to the changing and expanding set of discovered and consensus ASD genes. Such gene lists have ranged from several dozen to about 100; recent studies have used substantially overlapping gene lists. See Supplementary Table S-1.
Two separate prenatal temporal patterns of gene expression trajectories have commonly been reported: one for regulatory ASD risk genes and a second for neural communication/synapse ASD risk genes, as shown in Figs. 3A and B. Findings can appear to support the interpretation of non-overlapping, non-interacting impact of these seemingly separate patterns of change in expression.
This is not fully supported by data from four studies shown in Fig. 3C. In these studies (which includes data from Fig. 3B), regulatory and neural communication/synapse-relevant hcASD genes are enriched in both prenatal Epoch-1 and Epoch-2 periods, contrary to the impression of the trajectories graphed in Fig. 3A and B. See Supplementary Table S-1.
Also at odds with the interpretation of two non-overlapping, non-interacting regulatory and synapse risk gene trajectories, is a recent detailed model of ASD risk gene-based synaptic developmental dysfunction [47] which thoroughly delineates the deeply intertwined and interactive importance of regulatory genes as well as calcium and sodium channel, GABAergic and glutaminergic receptor, cell adhesion and scaffolding protein, and other types of neural communication/synapse-relevant genes. This synaptic model suggests that theory-driven approaches as distinct from a priori selection approaches, may reveal different insights into prenatal trajectories and interactions of critical regulatory and synaptic processes.
Figure 3. Dysregulation of regulatory ASD genes dominate the earliest prenatal stages of ASD.
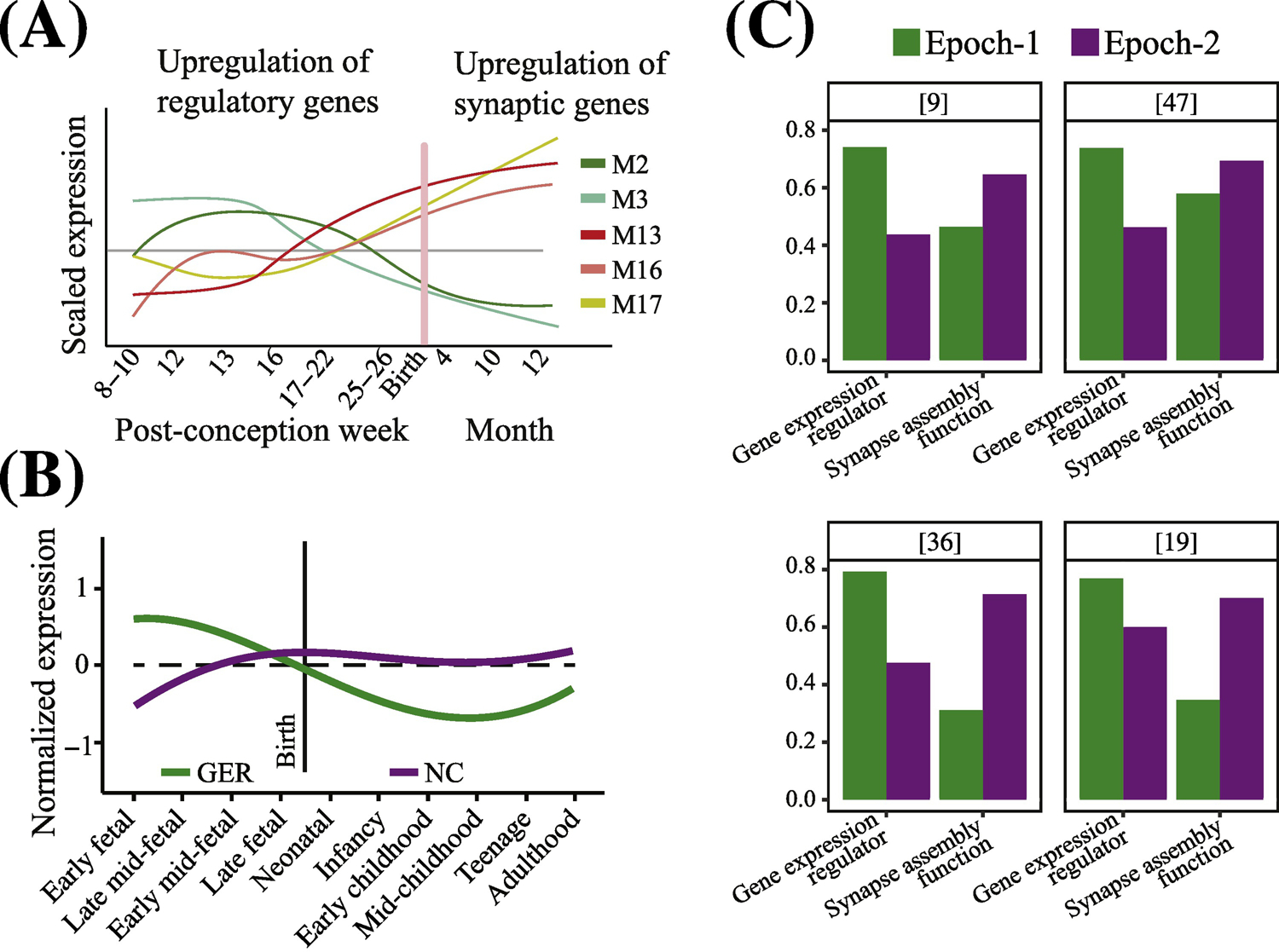
The identification of critical developmental periods and changes in pathobiological molecular mechanisms in ASD is of paramount importance. (A) In one approach, researchers first cluster genes according to co-expression patterns and next examine their enrichment for ASD risk genes (from[14]). M2 and M3 = modules enriched in gene expression regulator ASD risk genes; M13, M16 and M17 = modules enriched in synaptogenesis and synapse functioning ASD risk genes. (B) In a second approach, researchers make an a priori selection of genes, modules, functions or ontologies of interest from among all possible ones, and then analyze those selected for age-related changes in expression. A recent example is shown in this panel (from[36]). GER = gene expression regulator ASD risk genes; NC = neural communication ASD risk genes. Using such a priori selection of regulatory and synapse-relevant genes shows that ASD genes that are regulators normally upregulate expression as early as 1st and 2nd trimesters and then downregulate in perinatal and postnatal life, and the opposite pattern for synapse/neural communication genes, namely downregulation in early prenatal life followed by upregulation in later prenatal and postnatal life. Note that mutations in these ASD risk genes would therefore result in their dysregulation during these critical prenatal periods and disruption of these brain development processes. This approach has led some to view this as evidence of two largely non-overlapping and non-interacting types of genes, mechanisms and processes. (C) Bar graphs of hcASD genes from four recent studies[9, 19, 36, 47]. List of hcASD genes from each study were manually annotated by literature search (see Supplementary Table S-1). hcASD genes from each study were assigned to Epoch-1 or Epoch-2 based on their correlation with the neurodevelopmental time stages considering the time point as an ordinal variable. In all four studies, regulatory and synapse-relevant hcASD genes are present in both Epoch-1 and Epoch-2. In each study, Epoch-1 has a greater percentage of regulatory hcASD genes than does Epoch-2, while Epoch-2 has a greater percentage of synapse-relevant hcASD genes than does Epoch-1.
In a recent approach, we utilized an unbiased analysis of high confidence ASD genes identified in large-scale genome-wide genetic samples reported by two sources[43, 44] (see Supplementary Table S–1). In this approach, we clustered all of these hcASD genes based on their temporal expression trajectories in neocortex at prenatal and early postnatal ages[9]. This unbiased approach revealed hcASD genes that have similar and dissimilar prenatal ages of expression, regardless of what type of gene it might be.
This approach revealed two separate prenatal neurodevelopmental Epochs: Epoch-1 in the 1st to 3nd trimesters, has 68% of the hcASD genes; Epoch-2 in the 3rd trimester and early postnatal life, has 32% (Figs. 2C, 4C). Importantly, it also found two categories of hcASD genes -- regulatory and brain-specific – that were both present in both Epoch-1 and Epoch-2 (Fig. 2F, 3C and 4C).
Figure 4, Key Figure. ASD as a multi-organ disorder: Most regulatory high confidence ASD genes express broadly in other organs and tissues as well as brain.
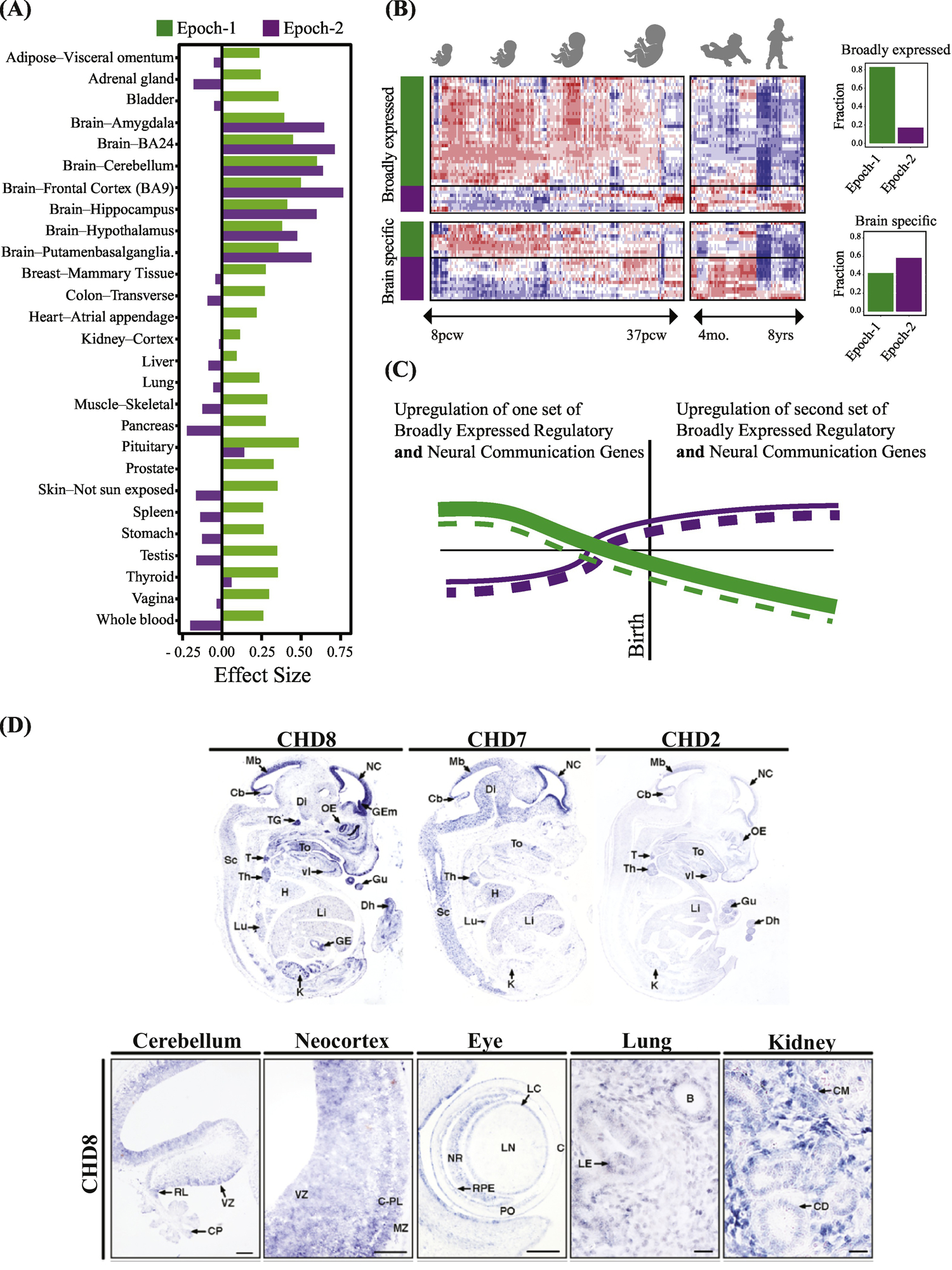
(A) The great majority of ASD regulatory genes are broadly-expressed in multiple organs and tissues in addition to the prenatal brain. The majority of all Epoch-1 hcASD genes are broadly-expressed regulators of gene expression in multiple body organs and tissues as well as brain. In contrast, the majority of Epoch-2 hcASD genes have largely brain-specific expression. The y-axis represents the effect size of median expression strength in Epoch-1 and Epoch-2 genes compared to all protein coding genes present in each tissue as measured by Cohen’s D. TPM-level median gene expressions each tissue were retrieved from the GTEx portal[64]. Positive values show stronger expression than expected and negative values show weaker expression than expected by chance. (B) Gene expression of three top ASD genes, Chd8, Chd7 and Chd2, in embryonic mouse models (from[45]). There is widespread expression of these genes during organogenesis, such as in the gut, lungs, kidney, thymus, thyroid, heart, kidney, lung, and eye as well as in cerebellum and neocortex (from[45]). Many other top ASD genes are also known to be involved in different organs, tissues and disorders involving the gastrointestinal system, heart, kidneys, eye, muscles, lungs, arterial maldevelopment, medulloblastoma, cone-rod dystrophy, facial dysmorphology, eye and vision defects, whole body overgrowth, finger clinodactyly, colorectal cancer, and leukemia. (C) Heat map of 69 hcASD genes shows broadly-expressed regulator and brain-specific hcASD genes in Epoch-1 and Epoch-2. Bar graphs show percentage of each type in each prenatal developmental Epoch. (D) Based on our interpretation of the heat map and bar graphs in this Panel C and in Fig. 3C, we propose a new model of how expression changes in broadly-expressed and brain-specific hcASD genes. Beginning in the 1st trimester, a large number of broadly expressed regulatory hcASD genes (represented by thick green lines) and a small number of brain-specific hcASD genes (thin dashed green lines) normally upregulate and later downregulate. Mutations in these genes thus would dysregulate Epoch-1 prenatal corticogenesis. Conversely, during Epoch-2, many brain-specific hcASD genes (thick dashed purple lines) and a smaller number of broadly-expressed hcASD genes (thin purple lines) upregulate in the 3rd trimester and early postnatal life. Thus, mutations in these later expressing genes would dysregulate later prenatal and early postnatal cortical wiring. This new model shows a pattern of prenatal and postnatal ASD risk gene dysregulation that differs from previous work: Namely, rather than regulator genes and brain-specific genes each having a separate, opposite and single expression trajectory, both categories of genes – broadly-expressed regulatory and brain-specific -- were both present in both Epoch-1 and Epoch-2; moreover, each Epoch had its own set of broadly-expressed regulator and brain-specific genes (compare to this Fig. 4D to Fig. 3A and B).
The set of regulatory and brain-specific hcASD genes in Epoch-1 impact cell proliferation, neurogenesis, migration, and cell fate, while the different set of regulatory and brain-specific hcASD genes in Epoch-2 impact neurite outgrowth, synaptogenesis and wiring of cortical functional networks. The great majority ---79%-- of Epoch-1 hcASD genes are regulatory (Fig. 2F and Fig. 3C), while the majority of Epoch-2 hcASD genes are brain-specific (Fig. 2F and Fig. 3C). Overall 67% of all Epoch-1 and Epoch-2 hcASD genes are gene expression regulators[9].
This high percentage of regulatory ASD risk genes is consistent with other recent work. For example, 34 high confidence common gene variants in idiopathic ASD[32] were largely regulatory and involved in prenatal corticogenesis (Fig. 2A)[32]. In another, 9 of the top 12 de novo recurrent ASD genes were regulatory, and across the top 38 in that study, 27 (71%) were regulatory[44]. In a recent study, 59% of 102 genome-wide significant ASD risk genes were regulatory and prenatal expressing in both ASD with and without intellectual disability[36]. Thus, across studies, high confidence ASD risk genes are most strongly enriched for regulatory genes, which play roles in chromatin modelling, signaling pathway modulation, and transcription programs (Figs. Fig. 3C and 4C) (Supplementary Table S–1).
The abrupt end of Epoch-1 just before birth (Figs. 2C and 4C), suggests that the functions of Epoch-1 hcASD genes are largely restricted to prenatal development. Meanwhile, Epoch-2 expression upregulation begins gradually late in the 2nd trimester but strongly upregulates just before birth when nearly all Epoch-1 genes normally downregulate. This switch coincides with a dramatic upsurge of cortical synaptogenesis and axonal and neuronal growth in the developing brain. Consistent with this timing, Epoch-2 is significantly and strongly enriched for brain-specific genes involved in synaptogenesis and synapse functioning (Fig. 2F). Building on this rapid increase in synaptogenesis and connectivity and in the context of the beginning of behavioral experiences, experience-dependent and experience-expectant neural plasticity rapidly accelerate during this early postnatal time.
The different trajectories of ASD risk genes exemplified in Fig. 3A and B (see also Box 3), have led to various interpretations including that they reflect different ASD subtypes or that ASD may involve separate and non-interacting molecular mechanisms with severity being related to additive risk from one or both types of gene disruption.
However, the neurodevelopmental expression patterns in Fig. 3C and Fig. 4C (Key Figure) lead to a new view of prenatal ASD gene trajectories that is illustrated in Fig. 4D. The figure illustrates that ASD prenatal development is not strictly defined by a switch from regulatory to brain-specific hcASD gene processes, but instead from one combination of regulatory and brain-specific processes in Epoch-1 to a second set in Epoch-2 (Fig. 4C and D). In Epoch-1, we hypothesize, the first set may together disrupt early prenatal cortical formation, while the second set may disrupt later prenatal and postnatal cortical “wiring”. Thus, evidence supports a yet-to-be-tested hypothesis that ASD may be the consequence of dysregulation of different sets of regulatory and brain-specific genes acting in spatiotemporal combination across two successive, overlapping prenatal Epochs (Fig. 4D). Regardless, the evidence supports the conclusion that dysregulation of regulatory ASD risk genes dominate and drive the first Epoch of prenatal maldevelopment in ASD.
ASD AS A MULTI-ORGAN DISORDER: Most Regulatory High Confidence ASD Genes Express Broadly Across Organs and Tissues
Most early prenatal ASD regulatory genes are broadly-expressed in multiple organs and tissues in addition to the developing prenatal brain (Fig. 4A,). Thus, broadly-expressed regulatory genes might be both a major driver of prenatal neural maldevelopment in ASD and affect development and function of other organs and tissues. For example, prenatal mouse models of three hcASD genes, Chd8, Chd7 and Chd2, show wide-spread expression during organogenesis, such as in the gut, lungs, kidney, thymus, thyroid, heart, kidney, lung, and eye as well as in cerebellum and neocortex (Fig. 4B)[45]. Many other hcASD genes are involved in diverse organs, tissues and disorders involving the gastrointestinal system, heart, kidneys, eye, muscles, lungs, arterial maldevelopment, medulloblastoma, cone-rod dystrophy, facial dysmorphology, eye and vision defects, whole body overgrowth, finger clinodactyly, colorectal cancer, and leukemia (e.g., ADNP, ANK2, ARID1B, BCL11A, CHD8, MED13L, RIMS1, TBR1, TCF7L2)[46]. These observations predict clinical comorbidities from negligible to syndromic, across genes and ASD subjects in terms of tissues and organs affected. Thus, not only is ASD a prenatal multistage, regulatory disorder of multiple developmental brain molecular processes, but it is also a broadly-expressed, multi-organ, multi-tissue disorder among some ASD cases.
ASD IS A REGULATORY DISORDER OF CORE BRAIN PROCESSES: A Unifying Model of ASD Prenatal Development
The above evidence provides insights on when and what neural development is disrupted in autism. The other key question is how do these many Epoch-1 and Epoch-2 hcASD genes operate to cause prenatal-age abnormalities and ASD symptoms? As described above, hcASD genes do not all fall into an obvious pattern of mechanisms, but rather represent a multitude of neural developmental functions and mechanisms, including, in many instances, those impacting non-brain organs and tissues. For instance, while some broadly-expressed hcASD genes are involved in chromatin remodelling, some brain-specific hcASD genes encode proteins precisely tuned to calcium channels.
In the face of this ambiguity, many theories and models have emerged. Some theories and models focus on Epoch-2 synapse genes, ignoring the large percentage of early Epoch-1 regulatory genes, due to the ease of single synapse-theory of ASD in which synaptic dysfunction is attributed to the behavioral dysfunction in individuals with ASD. Other speculations arose that perhaps each ASD risk gene is a specific form of autism; this gene-centric view appears to simplify molecular modelling but massively multiplies the number of different “autism” disorders to tackle and explain. Moreover, ASD in most individuals is a result of the combination of genetic aberrations and, except for a hand full of genes, ASD risk genes are not fully penetrant to the disorder, further challenging such a gene-centric view of ASD. Consequently, some have hypothesized that diverse hcASD genes converge on a yet-to-be-identified common pathway or that the whole group of heterogeneous hcASD genes might form a coherent single network in specific neuronal cell types and at a specific spatiotemporal space to perform a specific biological function (eg. [47]). Such views are challenged by the above compelling evidence that ASD is a multi-stage, progressive disorder involving two major prenatal developmental Epochs, one generating the cortex and the second creating functional wiring of the cortex. Together, these Epochs span much of prenatal and early-age neural development and many brain-specific mechanisms. Such speculations are likewise challenged by evidence of microglia[48] as well as neuronal pathology and the involvement of multiple organs and tissues in many in the ASD population.
Yet other perspectives, such as the oligogenic or polygenic models, posit the complexity of the ASD genetic landscape is attributable to the existence of hundreds of ASD risk genes, each with a small effect on molecular processes controlling an ASD neural or clinical trait. The polygenic model, for instance, hypothesizes existence of a set of specific risk genes for a complex trait. However, identified risk loci by genome-wide association studies (GWAS) and other genetic studies (e.g., de novo damaging mutations or CNVs) explain only a small portion of heritability under the polygenic model, and a large fraction of heritability remains “missing”[49]. For ASD, the missing heritability could partly be explained by identification of novel risk variants through analysis of far larger cohorts and utilization of more comprehensive genome sequencing approaches than previous work. However, based on lessons learned from other complex diseases, there is a possibility that a large fraction of heritability will remain elusive even after vastly expanded efforts[50].
Recently, Gazestani et al[51] hypothesized that an individual ASD risk gene per se is not a function or mechanism standing alone, but rather each is a participant with many other risk genes in multiple early developmental functional molecular networks and pathways (Box 4). To test this hypothesis, they examined the connection of ASD risk genes with molecular perturbations present in leukocytes from ASD toddlers. Importantly, their analyses showed many ASD risk genes are upstream regulators of key signaling pathways (e.g., PI3K/AKT, RAS/ERK, and WNT/β-catenin[9, 52]), and converge on these pathways through chromatin remodelers, transcriptional factors, and ubiquitination and protease complexes[9, 51, 52] (Fig. 5A). They also found that processes downstream of these signaling pathways are perturbed at the transcriptome level in ASD. Thus, in leukocytes, they show many prenatal-stage regulatory ASD risk genes are not at the core of neural developmental processes, but rather target these key signaling pathways thereby disrupting multiple core prenatal neurodevelopmental processes. The study[51] further showed disrupted signaling networks enrich early Epoch-1 prenatal-stage networks, and the degree of dysregulation in these signaling pathways correlate with ASD social symptom severity in the ASD toddlers. This ASD toddler RNA expression evidence[51] is consistent with the hypothesis from our recent review of the large ASD animal, cell and genetic literature[9] that dysregulation of these signaling pathways disrupts a succession of prenatal stages implicated in Epochs-1 and Epoch-2, namely cell proliferation, neurogenesis, cell fate determination, migration, growth and synaptogenesis and synaptic wiring (Fig. 5B). These pathways are highly pleiotropic, regulating multiple core neural developmental processes, while also impacting other organs and tissues[9, 51, 53].
TEXT BOX 4: Leukocyte studies unite ASD genetics with molecular dysregulations and clinical outcomes.
New studies of leukocyte transcriptomics in toddlers[51, 56] provide evidence that gene expression dysregulation associated with ASD genetics is present in tissues other than brain. This is in line with the finding that hcASD risk genes with regulatory roles (e.g., CHD8, CHD2, PTEN, ADNP) are broadly-expressed and functional across tissues (Figs. 2 to 4).
Blood transcriptomic analyses allow for the recruitment of large ASD cohorts from the general pediatric population. The observation that dysregulation in these ASD cohorts is connected to ASD risk genes suggests that there exist common molecular and cellular mechanisms between subjects with and without hcASD mutations[51]. This is consistent with the molecular and cellular similarities seen in iPSC lines from idiopathic ASD individuals[21], such as seen with subjects harboring deleterious SHANK2 mutations[80]. Similarly, a postmortem transcriptome study found similar transcriptional dysregulations in postmortem brain samples from idiopathic subjects and those with 15q11.2–13.1 duplication syndrome[81].
In the blood transcriptome studies, hcASD genes were not differentially expressed, but their downstream regulatory targets were significantly enriched among the differentially expressed genes[51, 54]. Similar patterns have been observed in the transcriptomic analysis of ASD postmortem brain samples[81–84]. These findings suggest that ASD risk genes, rather than being directly involved in the core brain processes related to ASD, are the main regulators of such processes. In the omnigenic model, each ASD subject has a unique genetic architecture, while showing high similarities in the downstream brain processes. In the omnigenic model, the core genes have the strongest disease effect when damaged, but mutations in these may be extraordinarily rare due to their highly deleterious effects. Thus, more peripheral genes (i.e., genes with regulatory impact that is expressed in the right time in the right tissue) may drive the disease as they more subtly dysregulate core brain genes in a dose dependent manner.
Findings from blood and hcASD neural cell models transcriptome suggest that a few ASD risk genes including CHD8 and FMR1 could act as “master regulators” by directly modulating the expression levels of the perturbed network. Mutations in such genes can have dramatic impact on the core brain processes, resulting in a more pronounced phenotype.
Except for a handful of genes, the common belief is that ASD risk genes are not fully penetrant to ASD. Blood transcriptome studies suggest that the regulatory impact of a large number of ASD risk genes is channelled through signaling pathways[51]. Congruently, studies on cell line and animal models of hcASD genes demonstrated the convergence of hcASD genes on these signaling pathways[9]. Moreover, transcriptomic studies reproducibly find the dysregulation of these pathways and their downstream processes in ASD blood[51, 53, 63]. These signaling pathways are known to be highly preserved and active across human tissues. Such multi-functionalities could be the reason that their dysregulation was detected in leukocytes of toddlers with ASD[51].
Figure 5. A unifying genetic model of ASD and its connection with the ASD molecular perturbations.
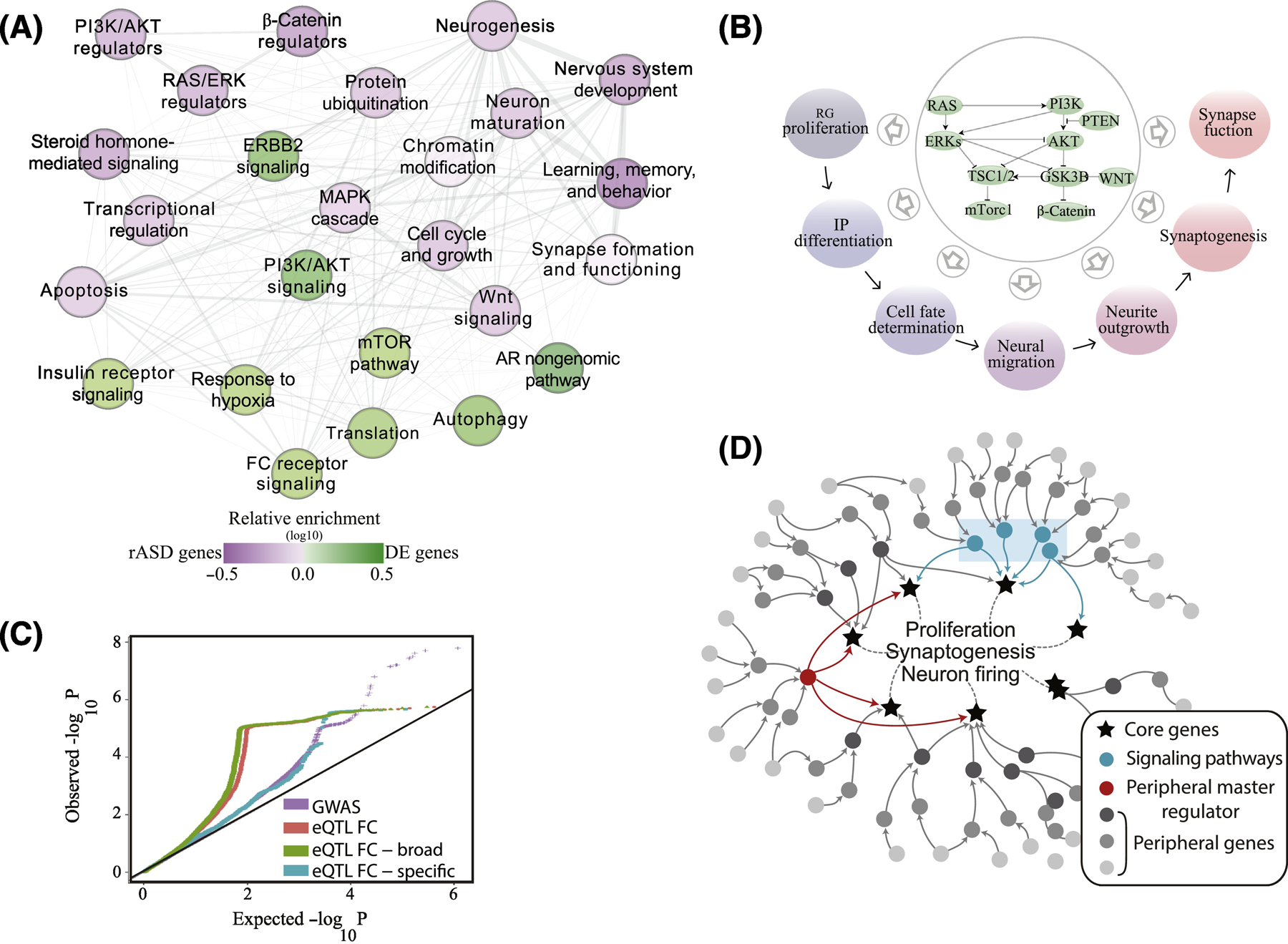
(A) In a recent study of large samples of ASD and typical toddlers, analyses of leukocyte gene expression identified a highly interconnected network of differentially expressed genes in ASD. In the panel (from[51]), each node represents a biological process that is significantly enriched in an expanded ASD network that included the differentially expressed genes and ASD risk genes. Nodes in green are signaling pathways (eg., PI3K/AKT, RAS/ERK, mTOR) and prenatal processes that are perturbed transcriptionally in leukocytes from toddlers with ASD. The purple nodes are enriched with ASD risk genes including risk ASD regulator genes that target the signaling pathways. Thus, many ASD risk genes and common variants are not directly involved in core prenatal processes underpinning ASD, but instead dysregulate such processes in ASD at critical prenatal and early postnatal time points that are relevant to ASD. Fig. 2E shows this same differentially expressed ASD gene network displays high gene coexpression across brain regions during 1st and 2nd trimester of development. (B) The PI3K/AKT, RAS/ERK, WNT/β-catenin signaling pathways that are dysregulated in leukocytes of ASD toddlers and correlate with their social symptom severity, are, in fact, highly conserved and their signaling can modulate multiple, Epoch-1 and Epoch-2 neurodevelopmental processes. (C) ASD liability resides in SNPs with broadly-expressed regulatory genes with roles across multiple tissues. GWAS data were retrieved from Grove et al[32]. Frontal cortex (FC) eQTLs were retrieved from the GTEx portal[64]. A frontal cortex eQTL was deemed as broadly functional if it was significantly correlated with gene expression levels in ≥50% of diverse tissues[64]. A similar pattern was observed from available GWAS data on schizophrenia[65]. (D) Illustration of proposed genetic architecture of ASD where regulatory ASD genes and common variants are not directly involved in core prenatal processes underpinning ASD, but instead are upstream in highly interconnected “peripheral” regulatory networks (dark and medium gray dots) that regulate downstream core brain genes (stars) and processes (eg., proliferation, synaptogenesis) at critical prenatal and early postnatal time points relevant to ASD[51]. Peripheral network dysregulations due to mutations in these regulatory ASD genes may propagate through gene regulatory networks to disrupt transcriptional programs or signaling pathways (blue rectangle) which impact downstream core brain-specific genes and processes related to ASD. Master regulators, such as CHD8, are proximal to and regulate directly key brain genes and processes and, so, their mutation can have dramatic impact on them.
Since Epoch-1 hcASD genes are predominantly broadly-expressed regulators, there are major implications on how ASD is perceived and studied. In contrast to neurons, which have a limited time window to proliferate and mature, other cell types constantly regenerate in postnatal life. Thus, molecular dysregulation associated with the genetics of ASD could in part, reoccur at postnatal ages in non-brain tissues. Indeed, in addition to [51], ASD blood transcriptome studies found dysregulations overlapping with dysregulated processes from iPSC studies[20, 21, 53–56]. Blood signatures are also predictive of ASD, neurofunctional hypoactivation to language stimuli in ASD, and early postnatal brain sizes[54–56]. Furthermore, genetic variations in neurodevelopmental genes correlate with their blood expression patterns in large cohorts of healthy individuals[57, 58].
To further investigate the influence of gene regulatory networks on ASD, we utilized available GWAS data on ASD[32] and overlaid it here with adult tissue expression quantitative loci (eQTL) data from the GTEx consortium. Consistent with the importance of gene regulatory networks in ASD genetic liability, we found eQTLs from the adult frontal cortex have a higher impact on the liability of ASD (Fig. 5C). These results are consistent with observations in schizophrenia wherein genetic risk loci alter gene expression in brain and other tissues (e.g., blood)[59]. Thus, genetic aberrations associated with the broadly-expressed genes substantially influence ASD liability.
A proposed new model of ASD prenatal development.
By demonstrating how the heterogeneous genetics of ASD may dysregulate signaling networks to influence prenatal brain development and, thereby, the severity of early-age ASD social symptoms, Gazestani et al[51] propose a new regulatory/core-process mechanistic model of ASD prenatal development: Upstream highly interconnected “peripheral” regulatory ASD risk gene mutations and variants --through either direct transcriptional regulation or modulation of interconnected signaling pathways (PI3K/AKT, RAS/ERK, WNT/β-catenin)-- dysregulate downstream core genes and developmental processes (Fig. 5D). A small number of “master” regulatory ASD gene mutations, such as CHD8 (Fig. 5D), that more directly dysregulate core prenatal neurodevelopmental genes and processes can also be sufficient to cause ASD symptoms.
These peripheral regulatory networks are governed by developmental- and tissue-specific spatiotemporal programs, making them ASD prenatal-brain critical but also potentially relevant to other organs and tissues in prenatal as well as adult life. This new hypothesis awaits empirical analyses and raises many questions about whether there may indeed be a unified mechanistic model of ASD and whether such a model explains ASD early-age clinical heterogeneity and outcome (see Outstanding Questions).
OUTSTANDING QUESTIONS BOX.
How do broadly-expressed regulatory and brain-specific ASD risk genes interact with each other during Epoch-1 and/or Epoch-2 to cause prenatal-age molecular and cellular abnormalities, early postnatal pathobiology, and initial clinical symptoms?
What are the critical developmental periods and processes for such pathogenic perturbations to lead to ASD?
What are the Epoch-1 and Epoch-2 genetic, genomic and cellular bases for ASD clinical heterogeneity, treatment responsiveness and outcome, and what are the differential roles of abnormal broad and brain-specific gene expression in this clinical heterogeneity?
Does the genetic structure of ASD result in molecular perturbations in non-neuronal cells in the brain? Are such perturbations due to the broadly expressed genes and do they overlap with the processes perturbed in ASD neurons?
How do dysregulations in broadly-expressed ASD risk genes perturb transcriptional programs in tissues and organs other than the brain?
Do early Epoch-1 dysregulations in broadly-expressed ASD risk genes have larger impact on brain development than brain-specific dysregulations?
Do findings of when, what and how of ASD prenatal Epoch- and Epoch-2 pathobiology apply at the individual ASD child level, and if so, what are the predictive individual-level signatures?
Does a unified model of prenatal genomic and cellular development underlie idiopathic ASD as well as ASD with risk mutations or do they have distinctly different developmental mechanisms?
At the molecular and cellular levels of prenatal cortical development, are there subtypes or a continuum of pathobiology, and how do effects relate to early-age clinical presentation, progression, treatment response and outcome?
This new ASD prenatal developmental model focusing on hcASD genes underlying regulation of core prenatal processes, may also speak to and serve as a disorder-specific example of more general emerging models, such as the omnigenic model. The omnigenic model of the genetics of complex traits suggest that regulatory gene mutations and epigenetic changes often propagate their effects through peripheral regulatory networks and converge on tissue and cell specific core pathways relevant to complex traits[60, 61] (Box 4). In the omnigenic model, regulatory networks are so interconnected that large numbers of genes contribute to a phenotype through expression in relevant cells[60, 61]. While displaying interesting attributes of the omnigenic model, here we theorize ASD results from dysregulation of developmentally delimited and transiently activated regulatory networks such as those described here and elsewhere[51].
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Recent research has revolutionized our understanding of ASD early-age origins. We now know ASD is largely an inherited, progressive disorder of prenatal and early postnatal development (Figs. 1–4) and is highly diagnostically stable from very early ages[8]. Broadly-expressed regulatory genes appear to be major drivers in Epoch-1 prenatal stages of ASD in both children with hcASD mutations (Figs. 2–4) and in idiopathic ASD (Fig. 2). Furthermore, two major hcASD gene expression Epochs exist. Epoch-1 is dominated by broadly-expressed regulatory risk genes that may disrupt early stages of corticogenesis, while Epoch-2 is dominated by brain-specific risk genes that may disrupt late prenatal and postnatal synaptogenesis and wiring of cortex. It remains to be studied if disruption of these two epochs occur independently, resulting in distinct subtypes of ASD, or if ASD emerges when disruptions of both converge on similar neurodevelopmental processes that generate ASD pathobiology. Alternatively, they could result in both similar and distinct perturbations, each contributing to the different dimensions of the autism spectrum, while the similarities contribute to some phenotypic overlap.
Evidence reviewed here leads to the conclusion that ASD begins as a prenatal-age regulatory disorder of multiple brain developmental processes. One model proposes a modular structure for ASD where mutations in regulatory ASD risk genes combine to disrupt transcriptional programs or signaling pathways (eg, PI3K/AKT, RAS/ERK, and WNT/β-catenin) and thereby dysregulate multiple downstream prenatal processes such as proliferation, neurogenesis, maturation, neurite outgrowth, synaptogenesis and functional neural network assembly (Fig. 5). Interestingly, some hcASD genes, such as CHD8 and FMR1, appear to directly regulate such core prenatal processes, suggesting a high impact of their genetic aberrations on ASD [62] (Fig. 5D). While the combined impact of child-specific genetic aberrations may not be easily detected or measured, dysregulation channelling onto downstream prenatal brain processes that are commonly perturbed across individuals, can be detected and correlated with symptom severities[51, 54, 56] (Box 4). While additional innovative experiments are needed to further test this model, it provides a novel perspective about how ASD genetics may lead to multiple stages of prenatal pathobiological processes and initial ASD clinical symptoms and neurofunctional outcome heterogeneity.
It is therefore critical to discover how these ASD prenatal processes are mechanistically linked with early-age ASD neural and clinical features. As a multistage, complex disorder spanning prenatal and early postnatal brain development, ASD could stem from disruption of multiple key processes with each contributing to postnatal heterogeneity ASD clinical outcomes. Since multiple prenatal processes are expected to be commonly dysregulated during Epochs-1 and −2 but to varying degrees, this model suggests that functional genomic experiments on large ASD cohorts are needed to systematically parse molecular and cellular heterogeneity, identify the bases of variation in prenatal processes, and examine how they account for very early-age ASD clinical heterogeneity. Therefore, large-sample ASD subject-derived cortical organoid technologies become critical for cell biology research into neurodevelopmental molecular and cellular bases for ASD and their connections with ASD genetics, early age clinical trajectories, and outcome[9].
In ASD children, insights into broadly-expressed regulatory genes and signaling pathways via non-invasive studies of accessible tissues, may shed light into ASD causes, mechanisms, development and interventions. Furthermore, this could identify molecular diagnostic, prognostic and treatment signatures from pathways and networks such as PI3K/AKT, RAS/ERK, and WNT/β-catenin in large cohorts of ASD infants and toddlers via various cell types and iPSC-based neural models[51, 53, 63]. Indeed, a correlation was found between signaling network level dysregulations and ASD social symptom severity[51, 56] (Box 4). It will be important to determine how broadly-expressed hcASD genes impact other tissues and organs during prenatal and early postnatal development, and thus connect comorbidities with ASD. Since broadly-expressed regulatory hcASD genes impact development in other cell and organ types[45], physiological comorbidities in ASD children may stem from the primary genetic etiology. Thus, ASD may also benefit by being treated as a broader medical disorder followed by a careful medical molecular, cellular, physiological and symptom assessments of other tissues. We suggest the cell biology of ASD, via non-invasive methods, should be a major future clinical translational research direction.
As a largely heritable disorder, ultimately precision medicine for most ASD children will require knowledge of the underlying genomic, molecular and cellular pathobiology and discovery of appropriately targeted therapeutics. Such precision medicine does not currently exist for ASD, but in principle it can be accomplished via the “ASD living biology” paradigm. This paradigm aims to derive clinical-relevant individual pathobiology information from living ASD children via non-invasive studies of ASD cell biology. This can be done through large-scale studies that leverage individual child derived-iPSC models and omics assays of accessible cell and tissue types. Such data will link neural-clinical-medical features in living ASD children with their own cell biology to drive personalized diagnosis, prognosis, and therapies[9].
Supplementary Material
HIGHLIGHTS.
ASD begins in prenatal life.
In Epoch-1 (1st to 3rd trimesters), cell proliferation, neurogenesis, cell fate and migration are disrupted. In Epoch-2 (3rd trimester and early postnatal life), cortical wiring is disrupted, including neurite outgrowth, synaptogenesis and neural network organization.
ASD is highly heritable. Most ASD risk genes express in prenatal life and fall into two major groups of broadly-expressed regulatory genes and brain-specific ones.
Broadly-expressed regulatory risk genes are the majority of ASD risk genes. In combination with brain-specific risk genes, they drive aberrant proliferation, neurogenesis, cell fate and migration in Epoch-1. A different set of broadly-expressed and brain-specific risk genes disrupt synaptogenesis and cortical wiring in Epoch-2.
Broadly-expressed risk genes may impact development and function of other organs as well as brain.
A new hypothesis proposes how Epoch-1 and Epoch-2 ASD risk genes operate to cause prenatal-age abnormalities: The effect of genetic aberrations in broadly expressed genes propagate through regulatory networks and key signaling pathways of PI3K/AKT, RAS/ERK, and WNT/β-catenin to converge upon and dysregulate multiple downstream core prenatal processes.
ACKNOWLEDGMENTS
Authors would like to thank Dr. Karen Pierce, Dr. Teresa Wen and Dr. Yaqiong Xiao for helpful comments on this manuscript. This work was supported by NIMH R01-MH110558 (E.C., N.E.L.), NIDCD R01-DC016385 (E.C.) and generous funding from the Novo Nordisk Foundation through Center for Biosustainability at the Technical University of Denmark (NNF10CC1016517 N.E.L.).
GLOSSARY
- Broadly-expressed genes
Broadly-expressed genes are strongly expressed in multiple organs and tissues
- Brain-specific genes
Brain-specific genes are expressed in the brain and are not strongly expressed in multiple other organs and tissue types
- Core brain processes
Core brain processes are critical for brain development during prenatal and early postnatal stages. Here, we speculate that ASD directly involves perturbation of several core brain processes
- Cortical organoid
Human cortical organoids are cellular models generated from human inducible pluripotent stem cells (iPSCs). They are organized into layers resembling those present in the developing human cerebral cortex. They recapitulate many cellular and molecular aspects of human prenatal neurodevelopmental stages. They can provide in vitro 3D cortical models for the study of neurological development and disease processes that are unique to the human nervous system
- High confidence ASD risk (hcASD) genes
High confidence ASD risk genes are recurrent, reported across multiple independent samples and not present in the typical, non-psychiatric population. Across different studies and criteria used, the number of hcASD genes vary from a few dozen to more than 100. There is not a fixed list or definition as such, and accrual of evidence may add new genes or remove previously considered genes. Examples of hcASD gene lists are shown in Supplementary Table S-1
- Omnigenic model
The omnigenic model is an expansion of the polygenic model and theorizes effects in every gene with spatiotemporal expression relevant to a complex trait, has a chance to propagate through highly interconnected peripheral regulatory gene networks to influence a set of core processes and genes connected with a trait of interest. Thus, while many genetic variants of peripheral regulatory genes do not meet statistical significance thresholds, they nevertheless make non-zero contributions to disease heritability
- Peripheral regulatory genes
Expression effects of peripheral genes propagate through gene regulatory networks to impact downstream core genes and processes. However, their genetic aberration is not necessary for a complex trait. These are expressed and functional in disease relevant cell types at relevant developmental time points. Master peripheral regulator genes can strongly dysregulate core processes and genes
- Signaling pathway
A group of molecules that interact in series to relay a message to drive a cellular response, such as neurogenesis or synaptogenesis. Alterations in the activation of a signaling pathway can change the cellular response and ultimately organism development and function
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Dawson G, et al. (2004) Early social attention impairments in autism: social orienting, joint attention, and attention to distress. Dev Psychol 40, 271–283 [DOI] [PubMed] [Google Scholar]
- 2.Fodstad JC, et al. (2009) Social and communication behaviours in infants and toddlers with autism and pervasive developmental disorder-not otherwise specified. Dev Neurorehabil 12, 152–157 [DOI] [PubMed] [Google Scholar]
- 3.Matson JL, et al. (2010) The effects of inattention/impulsivity and ASD symptom severity on social skills in toddlers. Dev Neurorehabil 13, 408–412 [DOI] [PubMed] [Google Scholar]
- 4.Elsabbagh M, et al. (2011) Social and attention factors during infancy and the later emergence of autism characteristics. Prog Brain Res 189, 195–207 [DOI] [PubMed] [Google Scholar]
- 5.Lombardo MV, et al. (2011) Specialization of right temporo-parietal junction for mentalizing and its relation to social impairments in autism. Neuroimage 56, 1832–1838 [DOI] [PubMed] [Google Scholar]
- 6.Pierce K, et al. (2016) Eye tracking reveals abnormal visual preference for geometric images as an early biomarker of an autism spectrum disorder subtype associated with increased symptom severity. Biol Psychiatry 79, 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanner L (1943) Autistic disturbances of affective contact. Nervous Child 2, 217–250 [PubMed] [Google Scholar]
- 8.Pierce K, et al. (2019) Evaluation of the Diagnostic Stability of the Early Autism Spectrum Disorder Phenotype in the General Population Starting at 12 Months. JAMA Pediatr 173, 578–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Courchesne E, et al. (2019) The ASD Living Biology: From cell proliferation to clinical phenotype. Molecular Psychiatry 24, 88–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Courchesne E, et al. (2011) Brain growth across the life span in autism: age-specific changes in anatomical pathology. Brain Res 1380, 138–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stoner R, et al. (2014) Patches of disorganization in the neocortex of children with autism. N Engl J Med 370, 1209–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Courchesne E and Pierce K (2005) Why the frontal cortex in autism might be talking only to itself: local over-connectivity but long-distance disconnection. Current Opinion in Neurobiology 15, 225–230 [DOI] [PubMed] [Google Scholar]
- 13.Willsey AJ, et al. (2013) Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parikshak NN, et al. (2013) Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Packer A (2016) Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci Biobehav Rev 64, 185–195 [DOI] [PubMed] [Google Scholar]
- 16.Kaushik G and Zarbalis KS (2016) Prenatal Neurogenesis in Autism Spectrum Disorders. Front Chem 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krishnan A, et al. (2016) Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat Neurosci 19, 1454–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donovan AP and Basson MA (2017) The neuroanatomy of autism - a developmental perspective. J Anat 230, 4–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuen RKC, et al. (2017) Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 20, 602–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marchetto MC, et al. (2017) Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry 22, 820–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mariani J, et al. (2015) FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 162, 375–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeRosa BA, et al. (2018) Convergent Pathways in Idiopathic Autism Revealed by Time Course Transcriptomic Analysis of Patient-Derived Neurons. Sci Rep 8, 8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adhya D, et al. (unpublished) Atypical neurogenesis and excitatory-inhibitory progenitor generation in induced pluripotent stem cell (iPSC) from autistic individuals. Biorxiv [Google Scholar]
- 24.Courchesne E, et al. (2011) Neuron number and size in prefrontal cortex of children with autism. JAMA 306, 2001–2010 [DOI] [PubMed] [Google Scholar]
- 25.Redcay E and Courchesne E (2005) When is the brain enlarged in autism? A meta-analysis of all brain size reports. Biol Psychiatry 58, 1–9 [DOI] [PubMed] [Google Scholar]
- 26.Bauman ML and Kemper TL (2005) Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci 23, 183–187 [DOI] [PubMed] [Google Scholar]
- 27.Courchesne E, et al. (2001) Unusual brain growth patterns in early life in patients with autistic disorder: An MRI study. Neurology 57, 245–254 [DOI] [PubMed] [Google Scholar]
- 28.Sacco R, et al. (2015) Head circumference and brain size in autism spectrum disorder: A systematic review and meta-analysis. Psychiatry Res 234, 239–251 [DOI] [PubMed] [Google Scholar]
- 29.Hazlett HC, et al. (2017) Early brain development in infants at high risk for autism spectrum disorder. Nature 542, 348–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen MD, et al. (2017) Increased Extra-axial Cerebrospinal Fluid in High-Risk Infants Who Later Develop Autism. Biol Psychiatry 82, 186–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang WQ, et al. (2014) Overproduction of upper-layer neurons in the neocortex leads to autism-like features in mice. Cell Rep 9, 1635–1643 [DOI] [PubMed] [Google Scholar]
- 32.Grove J, et al. (2019) Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51, 431–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu X, et al. (2014) Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J Neurosci 34, 1420–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang J, et al. (2015) Genotype to phenotype relationships in autism spectrum disorders. Nat Neurosci 18, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hormozdiari F, et al. (2015) The discovery of integrated gene networks for autism and related disorders. Genome Res 25, 142–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satterstrom FK, et al. (2020) Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584 e523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amaral DG, et al. (2008) Neuroanatomy of autism. Trends Neurosci 31, 137–145 [DOI] [PubMed] [Google Scholar]
- 38.Anagnostou E and Taylor MJ (2011) Review of neuroimaging in autism spectrum disorders: what have we learned and where we go from here. Mol Autism 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lombardo MV, et al. (2015) Different functional neural substrates for good and poor language outcome in autism. Neuron 86, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Solso S, et al. (2016) Diffusion Tensor Imaging Provides Evidence of Possible Axonal Overconnectivity in Frontal Lobes in Autism Spectrum Disorder Toddlers. Biol Psychiatry 79, 676–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piven J, et al. (2018) Toward a conceptual framework for early brain and behavior development in autism. Mol Psychiatry 23, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolff JJ, et al. (2018) The journey to autism: Insights from neuroimaging studies of infants and toddlers. Dev Psychopathol 30, 479–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.SFARI-Gene-Scoring (2019) https://gene-archive.sfari.org/database/human-gene/.
- 44.Kosmicki JA, et al. (2017) Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat Genet 49, 504–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kasah S, et al. (2018) Autism-linked CHD gene expression patterns during development predict multi-organ disease phenotypes. J Anat 233, 755–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gamsiz ED, et al. (2015) Discovery of Rare Mutations in Autism: Elucidating Neurodevelopmental Mechanisms. Neurotherapeutics 12, 553–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mullins C, et al. (2016) Unifying Views of Autism Spectrum Disorders: A Consideration of Autoregulatory Feedback Loops. Neuron 89, 1131–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morgan JT, et al. (2010) Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry 68, 368–376 [DOI] [PubMed] [Google Scholar]
- 49.Manolio TA, et al. (2009) Finding the missing heritability of complex diseases. Nature 461, 747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turner TN, et al. (2017) Genomic Patterns of De Novo Mutation in Simplex Autism. Cell 171, 710–722 e712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gazestani VH, et al. (2019) A perturbed gene network containing PI3K-AKT, RAS-ERK and WNT-beta-catenin pathways in leukocytes is linked to ASD genetics and symptom severity. Nat Neurosci 22, 1624–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y, et al. (2015) Pten Mutations Alter Brain Growth Trajectory and Allocation of Cell Types through Elevated beta-Catenin Signaling. J Neurosci 35, 10252–10267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faridar A, et al. (2014) Mapk/Erk activation in an animal model of social deficits shows a possible link to autism. Mol Autism 5, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pramparo T, et al. (2015) Cell cycle networks link gene expression dysregulation, mutation, and brain maldevelopment in autistic toddlers. Mol Syst Biol 11, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pramparo T, et al. (2015) Prediction of autism by translation and immune/inflammation coexpressed genes in toddlers from pediatric community practices. JAMA Psychiatry 72, 386–394 [DOI] [PubMed] [Google Scholar]
- 56.Lombardo MV, et al. (2018) Large-scale associations between the leukocyte transcriptome and BOLD responses to speech differ in autism early language outcome subtypes. Nat Neurosci 21, 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wright FA, et al. (2014) Heritability and genomics of gene expression in peripheral blood. Nat Genet 46, 430–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qi T, et al. (2018) Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun 9, 2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pardinas AF, et al. (2018) Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet 50, 381–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boyle EA, et al. (2017) An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu X, et al. (2019) Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell 177, 1022–1034 e1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beighley JS, et al. (2019) Clinical Phenotypes of Carriers of Mutations in CHD8 or Its Conserved Target Genes. Biol Psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kong SW, et al. (2012) Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PLoS One 7, e49475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Consortium GT (2015) Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schizophrenia Working Group of the Psychiatric Genomics, C. (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McConnell MJ, et al. (2017) Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Santos M, et al. (2011) Von Economo neurons in autism: a stereologic study of the frontoinsular cortex in children. Brain Res 1380, 206–217 [DOI] [PubMed] [Google Scholar]
- 68.Rabinowicz T, et al. (1996) Human cortex development: estimates of neuronal numbers indicate major loss late during gestation. J Neuropathol Exp Neurol 55, 320–328 [PubMed] [Google Scholar]
- 69.Gohlke JM, et al. (2007) Computational models of neocortical neuronogenesis and programmed cell death in the developing mouse, monkey, and human. Cereb Cortex 17, 2433–2442 [DOI] [PubMed] [Google Scholar]
- 70.Bonnet-Brilhault F, et al. (2018) Autism is a prenatal disorder: Evidence from late gestation brain overgrowth. Autism Res 11, 1635–1642 [DOI] [PubMed] [Google Scholar]
- 71.Chow ML, et al. (2012) Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet 8, e1002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wegiel J, et al. (2010) The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol 119, 755–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Varghese M, et al. (2017) Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hutsler JJ and Casanova MF (2016) Review: Cortical construction in autism spectrum disorder: columns, connectivity and the subplate. Neuropathol Appl Neurobiol 42, 115–134 [DOI] [PubMed] [Google Scholar]
- 75.Bailey A, et al. (1998) A clinicopathological study of autism. Brain 121 (Pt 5), 889–905 [DOI] [PubMed] [Google Scholar]
- 76.van Kooten IA, et al. (2008) Neurons in the fusiform gyrus are fewer and smaller in autism. Brain 131, 987–999 [DOI] [PubMed] [Google Scholar]
- 77.Jacot-Descombes S, et al. (2012) Decreased pyramidal neuron size in Brodmann areas 44 and 45 in patients with autism. Acta Neuropathol 124, 67–79 [DOI] [PubMed] [Google Scholar]
- 78.Wolff JJ, et al. (2012) Differences in white matter fiber tract development present from 6 to 24 months in infants with autism. Am J Psychiatry 169, 589–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rubenstein JL and Merzenich MM (2003) Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2, 255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zaslavsky K, et al. (2019) SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat Neurosci 22, 556–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parikshak NN, et al. (2016) Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Voineagu I, et al. (2011) Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gupta S, et al. (2014) Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun 5, 5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chow ML, et al. (2012) Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet 8, e1002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.