

Abstract
Background and purpose:
KRAS and BRAF are mutated in 35% and 10% of colorectal cancers, respectively. However, data specifically for locally advanced rectal cancers are scarce, and the frequency of KRAS mutations in codons 61 and 146 remains to be established.
Materials and methods:
DNA was isolated from pre-therapeutic biopsies of 94 patients who were treated within two phase-III clinical trials receiving preoperative chemoradiotherapy. Mutation status of KRAS exons 1–3 and BRAF exon 15 was established using the ABI PRISM Big Dye Sequencing Kit and subsequently correlated with clinical parameters.
Results:
Overall, KRAS was mutated in 45 patients (48%). Twenty-nine mutations (64%) were located in codon 12, 10 mutations (22%) in codon 13, and 3 mutations (7%) in codons 61 and 146. No V600E BRAF mutation was detected. The presence of KRAS mutations was correlated neither with tumor response or lymph node status after preoperative chemoradiotherapy nor with overall survival or disease-free survival. When KRAS exon 1 mutations were separated based on the amino-acid exchange, we again failed to detect significant correlations (p = 0.052). However, G12V mutations appeared to be associated with higher rates of tumor regression than G13D mutations (p = 0.012).
Conclusion:
We are the first to report the mutation status of KRAS and BRAF in pre-therapeutic biopsies from locally advanced rectal cancers. The high number of KRAS mutations in codons 61 and 146 emphasizes the importance to expand current mutation analyses, whereas BRAF mutations are not relevant for rectal carcinogenesis. Although the KRAS mutation status was not correlated with response, the subtle difference between G12V and G13D mutations warrants analysis of a larger patient population.
Keywords: Rectal cancer, Preoperative chemoradiotherapy, KRAS, BRAF, Response
The MAP kinase (MAPK) pathway is a fundamental signal transduction pathway with impact on cellular functions such as proliferation, differentiation and apoptosis, and is hyper-activated in about 30% of human cancers [ 1 ]. KRAS and BRAF are two important members of this pathway and are mutated in 30–40% and 5–10% in colorectal cancer (CRC), respectively [2].
KRAS is a membrane-bound G protein and is activated by receptor tyrosine kinases. Mutations within the gene increase the kinase activity and correlate with a more aggressive biological behavior [3]. In patients with stage IV CRC receiving anti-epidermal growth factor receptor (EGFR) agents mutations in this gene are associated with poor treatment response [4,5]. Several studies analyzed the role of KRAS mutations for outcome prediction in patients treated solely with 5-FU with conflicting results. However, in a multicenter study conducted on 3439 patients sub-analysis of the mutations revealed a decreased failure-free survival and overall survival for the glycine to valine mutation in codon 12 [6].
A thymidine to adenosine transversion at nucleotide 1799 accounts for about 90% of all the BRAF mutations and is located within the kinase domain of the gene leading to an elevated activity compared to the wild type [7,8]. To some extent BRAF mutations and KRAS mutations can be considered as equivalent in their tumorigenic effect [9,10], and at least the T1799A transversion seems to be inversely correlated with the frequency of KRAS mutations [7,9,11]. These findings are in line with the recently published data indicating that the usefulness of anti-EGFR therapy in metastatic CRC depends on the presence of BRAF wild type [12].
Since rectal cancers represent a clinical entity that is distinct from that of colon cancers, we focused our analysis on this tumor type. Beside the well-known mutations in codons 12 and 13, which are located in exon 1, we aimed to detect the less frequent mutations including those in codons 61 (exon 2) and 146 (exon 3) using bidirectional sequence analysis. To estimate the BRAF mutation frequency we focused on exon 15 which was analyzed in its entire length. We then explored a possible correlation of KRAS and BRAF mutations with pertinent clinical parameters, including response to preoperative chemoradiotherapy and survival data.
Materials and methods
Selection of patients, study design and treatment
All 94 patients were enrolled in the CAO/ARO/AIO-94 [13] or CAO/ARO/AIO-04 (EudraCT-Number: 2006-002385-20) [14] trial of the German Rectal Cancer Study Group; 64 were males and 30 were females with a median age of 62.3 years (range: 35–81 years). They were exclusively treated at the Department of General and Visceral Surgery, University Medicine Göttingen, Germany. Preoperative CT/RT, surgical resection and pathological work-up were standardized according to the guidelines of these randomized phase-III trials. Pre-therapeutic staging included rigid rectoscopy and endorectal ultrasound, colonoscopy, abdominal and pelvic computed tomography and chest X-ray. Only locally advanced adenocarcinomas (cUICC II/III) located within 12 cm from the anocutaneous verge were included (Table 1). All patients subsequently received a total radiation dose of 50.4 Gy (single dose of 1.8 Gy) accompanied by either (n = 57) a 120-h continuous intravenous application of 5-FU (1000 mg/m2/day on days 1–5 and days 29–33) or (n = 37) a combination of an intravenous infusion of oxaliplatin (50 mg/m2 on days 1, 8, 22 and 29 over 2 h) and a continuous infusion of 5-FU (250 mg/m2/day on days 1–14 and days 2235). Four to six weeks after the completion of preoperative CT/RT, standardized surgery was performed including total mesorectal excision [15]. The patients were followed up within these two clinical trials and data of recurrence (local relapse or distant metastases) or death (tumor-associated death or death from other reasons) were recorded.
Table 1.
Pre- and postoperative tumor stage, T-level and lymph node (LN) status.
| Stage | T-level | LN status | |||
|---|---|---|---|---|---|
| Preoperative | |||||
| II | 31 | uT2 | 2 | N+ | 2 |
| uT3 | 90 | N− | 31 | ||
| III | 63 | N+ | 59 | ||
| uT4 | 2 | N+ | 2 | ||
| Postoperative | |||||
| 0 | 11 | pT0 | 13 | N− | 11 |
| N+ | 2 | ||||
| I | 32 | pT1 | 13 | N− | 2 |
| N+ | 11 | ||||
| II | 20 | pT2 | 24 | N− | 3 |
| N+ | 21 | ||||
| III | 28 | pT3 | 37 | N− | 17 |
| N+ | 20 | ||||
| IV | 3 | pT4 | 7 | N− | 3 |
| N+ | 4 | ||||
| T-level downsizing | Downstaging | ||||
| Yes | 51 | 57 | |||
| No | 43 | 37 | |||
| Tumor regression gradinga | |||||
| TRG 0 | 1 | ||||
| TRG 1 | 12 | ||||
| TRG 2 | 30 | ||||
| TRG 3 | 38 | ||||
| TRG 4 | 12 | ||||
For one case tumor regression grading was not assessable.
Ascertainment of tumor biopsies and DNA isolation
From each patient, we prospectively collected pre-therapeutic biopsies from representative adjacent areas of the tumors, adhering to the guidelines set by the Local Ethical Review Board. The first one was used for histopathological confirmation of tumor diagnosis; the second one was immediately stored in RNAlater (Ambion, Austin, TX) for subsequent extraction of nucleic acids. DNA isolation was performed using TRIZOL (Invitrogen, Carlsbad, CA) following standard procedures as previously described [16] (details can be found at http://www.riedlab.nci.nih.gov/protocols.asp).
Mutation analysis
Sequence analysis of KRAS exons 1, 2, and 3, and BRAF exon 15 was performed with genomic tumor DNA. Considered regions were amplified by multiplex PCR and the obtained fragments were subjected to direct sequencing in both directions on an ABI-3100 Sequencer. Primer sequences for KRAS were as follows: 1F: 5’-TCCCAAGGAAAGTAAAGTTCCCATATTAATG-3’, 1R: 5’-CGCAGAACAGCAGTCTGGCTATTTAG-3’, 2F: 5’-CACTGCTCTAATCCCCCAAGAACTTC-3’, 2R: 5’-GGAGCAGGAACAATGTCTTTTCAAGTC-3’, 3F: 5’-CAAAGCCA AAAGCAGTACCATGGA-3’, and 3R: 5’-AGCCAAATTTTATGACAAAAGTTGTGGACAG-3’. The sequences for BRAF were F15: 5’-GTGGATCACACCTGCCTTAAATTGC-3’ and R15: 5’-GAGAATATCTGGGCCTACATTGCTAAAATC-3’. Briefly, multiplex PCR using Qiagen Multiplex Kit (Qiagen, Hilden, Germany) was carried out according to the protocol using 40 cycles at an annealing temperature of 64 °C. Product contamination was monitored using negative controls in each PCR run. PCR products were analyzed on a 2% agarose gel (NuSieve® 3:1 Agarose, Lonza, Rockland, USA). Non-incorporated primers and nucleotides were digested with 3 and 5 U of shrimp alkaline phosphatase and Escherichia coli exonuclease I (USB, Staufen, Germany), respectively, per 10 μl of multiplex PCR product. Direct sequencing was then carried out using ABI PRISM Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA). Sequencing primers were identical to PCR-Primers except for BRAF 15F where 5’-CAGCATCTCAGGGCCAAAAATTTAATC-3’ was used. DNAs from colorectal cancer cell lines SW480 and HT29, known for KRAS and BRAF mutations, were used as positive controls. Mutation analysis was performed using the STADEN package (http://staden.sourceforge.net/).
Classification of response
Response was defined as T-category downsizing or as UICC downstaging. Both methods compared the pre-therapeutic assessment determined by endorectal ultrasound with the histopathological diagnosis after surgery (Table 1). As previously described, tumors exhibiting a T-level downsizing or a UICC downstaging of at least one category were considered responsive [17]. Furthermore, response was assessed using a histopathological tumor regression grading (TRG) as assessed by an experienced pathologist. Based on the residual tumor mass, chemoradiotherapy-in-duced fibrotic changes and irradiation vasculopathy, the resection specimen was evaluated semi-quantitatively according to a five-point grading system, a modification of the tumor regression grading (TRG) as described by Gavioli et al. [18]. Briefly, tumor samples without any fibrosis/regression were considered as TRG 0, whereas complete regression (TRG 4) was defined as the absence of viable tumor cells in the primary tumor. The tumor samples with more than 50% viable tumor cells (less than 50% fibrosis) were considered as TRG 1. A regression within 50–70% was classified as TGR 2, while samples were scored as TRG 3 if tumor regression exceeded 70%.
Statistical analysis
Response levels and clinical parameters were compared between patients without and those with distinct KRAS mutations by the Kruskal-Wallis test. Additionally, TRG levels were compared by subsequent pairwise Wilcoxon rank-sum tests. The global significance level was set to α = 5%. Due to non-significant differences in the all-group-comparisons, the significance level was adjusted for the pairwise comparisons by the Bonferroni method. Correlation of disease-free survival and KRAS mutation was done using Kaplan-Meier Curves. All analyses were performed with the free software R (version 2.8, www.r-project.org).
Results
BRAF mutation status
Activating BRAF mutations are known to be predominantly located within codon 600, which resides in exon 15. To detect rare mutations the entire exon 15 was sequenced. Surprisingly, none of the 94 patients showed a typical V600E mutation. However, a single patient showed a G1780A mutation resulting in the amino acid exchange G594N. The known mutation in HT29 cells, which served as positive control, was consistently detected.
KRAS mutation status
For KRAS mutation analysis the entire exons 1, 2, and 3 were sequenced to detect the mutation hotspots at codons 12 and 13 as well as to screen for rare mutations such as those in codons 61 and 146 or additional rare ones previously described in the literature. In total, 45 (48%) mutations were found in 94 patients, whereas 29 (64%) were located in codon 12, 10 (22%) in codon 13, and 3 (7%) in codons 61 and 146, respectively (Table 2). Only one patient exhibited more than one mutation (34G>T and 36T>G).
Table 2.
Types of KRAS mutations within 94 locally advanced rectal cancer biopsies.
| Codon | Number | Nucleotide exchange | Amino acid |
|---|---|---|---|
| 12 (n = 29) | 1 | 34G>A | Gly12Ser |
| 1 | 34G>T | Gly12Cys | |
| 1 | 34G>T + 36T>G | Gly12Trp | |
| 15 | 35G>A | Gly12Asp | |
| 3 | 35G>C | Gly12Ala | |
| 8 | 35G>T | Gly12Val | |
| 13 (n = 10) | 10 | 38G>A | Gly13Asp |
| 61 (n = 3) | 2 | 182A>T | Gln61Leu |
| 1 | 182A>G | Gln61Arg | |
| 146 (n = 3) | 2 | 436G>A | Ala146Thr |
| 1 | 436G>C | Ala146Pro |
KRAS mutations and clinical parameters
Therapy response levels (TRG, T-level downsizing and UICC downstaging) as well as postoperative T-category and lymph node status (ypT and ypN) were compared between patients without KRAS mutation and those with a mutation in either codons 12, 13, 61 or 146. None of these comparisons showed a significant difference between the groups (Table 3).
Table 3.
Comparison of response levels and histopathological parameters between patients without KRAS mutation and those with a mutation on either codons 12, 13, 61 or 146.
| Parameter | Level |
KRAS mutation |
p | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No (n = 49a) |
Codon 12 (n = 29) |
Codon 13 (n = 10) |
Codon 61 (n = 3) |
Codon 146 (n = 3) |
||||||||
| n | % | n | % | n | % | n | % | n | % | |||
| TRG | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 33 | 0 | 0 | 0.22 |
| 1 | 4 | 8 | 6 | 21 | 0 | 0 | 1 | 33 | 1 | 33 | ||
| 2 | 18 | 38 | 10 | 34 | 2 | 20 | 0 | 0 | 0 | 0 | ||
| 3 | 20 | 42 | 11 | 38 | 6 | 60 | 0 | 0 | 1 | 33 | ||
| 4 | 6 | 12 | 2 | 7 | 2 | 20 | 1 | 33 | 1 | 33 | ||
| T-level downsizing (uT-yT) | 0 | 20 | 41 | 18 | 62 | 2 | 20 | 1 | 33 | 2 | 67 | 0.14 |
| 1 | 29 | 59 | 11 | 38 | 8 | 80 | 2 | 67 | 1 | 33 | ||
| UICC downstaging (uUICC-yUICC) | 0 | 24 | 49 | 10 | 34 | 1 | 10 | 0 | 0 | 2 | 67 | 0.07 |
| 1 | 25 | 51 | 19 | 66 | 9 | 90 | 3 | 100 | 1 | 33 | ||
| ypT | 0 | 7 | 14 | 2 | 7 | 2 | 20 | 1 | 33 | 1 | 33 | 0.19 |
| 1 | 8 | 16 | 2 | 7 | 2 | 20 | 1 | 33 | 0 | 0 | ||
| 2 | 13 | 27 | 7 | 24 | 4 | 40 | 0 | 0 | 0 | 0 | ||
| 3 | 18 | 37 | 16 | 55 | 2 | 20 | 0 | 0 | 1 | 33 | ||
| 4 | 3 | 6 | 2 | 7 | 0 | 0 | 1 | 33 | 1 | 33 | ||
| ypN | 0 | 28 | 57 | 22 | 76 | 9 | 90 | 3 | 100 | 1 | 33 | 0.81 |
| 1 | 14 | 29 | 6 | 21 | 1 | 10 | 0 | 0 | 2 | 67 | ||
| 2 | 7 | 14 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | ||
For one patient TRG was not assessable.
Due to small sample sizes, further analyses of response levels were performed excluding patients that carried mutations in codons 61 and 146. Based on the change of amino acids codon 12 mutations were analyzed separately. In eight patients glycine was replaced by valine (G12V) and in fifteen patients by aspartate (G12D). In these analyses T-level downsizing and UICC downstaging again showed no significant association with KRAS mutation status (Table 4). However, TRG showed a high association with the individual mutations (p = 0.052, Fig. 1). Although statistically not significant this association was mainly due to the differences between G12V and G13D (p = 0.012) and the differences between the wild type and G12V (p = 0.04), respectively, when pairwise comparison was performed. This association did not maintain significance after the Bonferroni adjustment was applied (Bonferroni-adjusted significance level: 0.05/6 = 0.008).
Table 4.
Comparison of response levels between patients without KRAS mutation and those with the mutations G12D, G12V, and G13D.
| Parameter | Level |
KRAS mutation |
p | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No (n= 49)a |
G12D (n = 15) |
G12V (n = 8) |
G13D (n = 10) |
|||||||
| n | % | n | % | n | % | n | % | |||
| TRG | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.052 |
| 1 | 4 | 8 | 1 | 7 | 3 | 38 | 0 | 0 | ||
| 2 | 18 | 38 | 5 | 33 | 3 | 38 | 2 | 20 | ||
| 3 | 20 | 42 | 7 | 47 | 2 | 24 | 6 | 60 | ||
| 4 | 6 | 12 | 2 | 13 | 0 | 0 | 2 | 20 | ||
| T-level downsizing (uT-yT) | 1 | 20 | 41 | 8 | 54 | 6 | 75 | 2 | 20 | 0.11 |
| 1 | 29 | 59 | 7 | 47 | 2 | 25 | 8 | 80 | ||
| UICC downstaging (uUICC-yUICC) | 0 | 24 | 49 | 4 | 27 | 4 | 50 | 1 | 10 | 0.08 |
| 1 | 25 | 51 | 11 | 63 | 4 | 50 | 9 | 90 | ||
For one patient, TRG was not assessable.
Fig. 1.
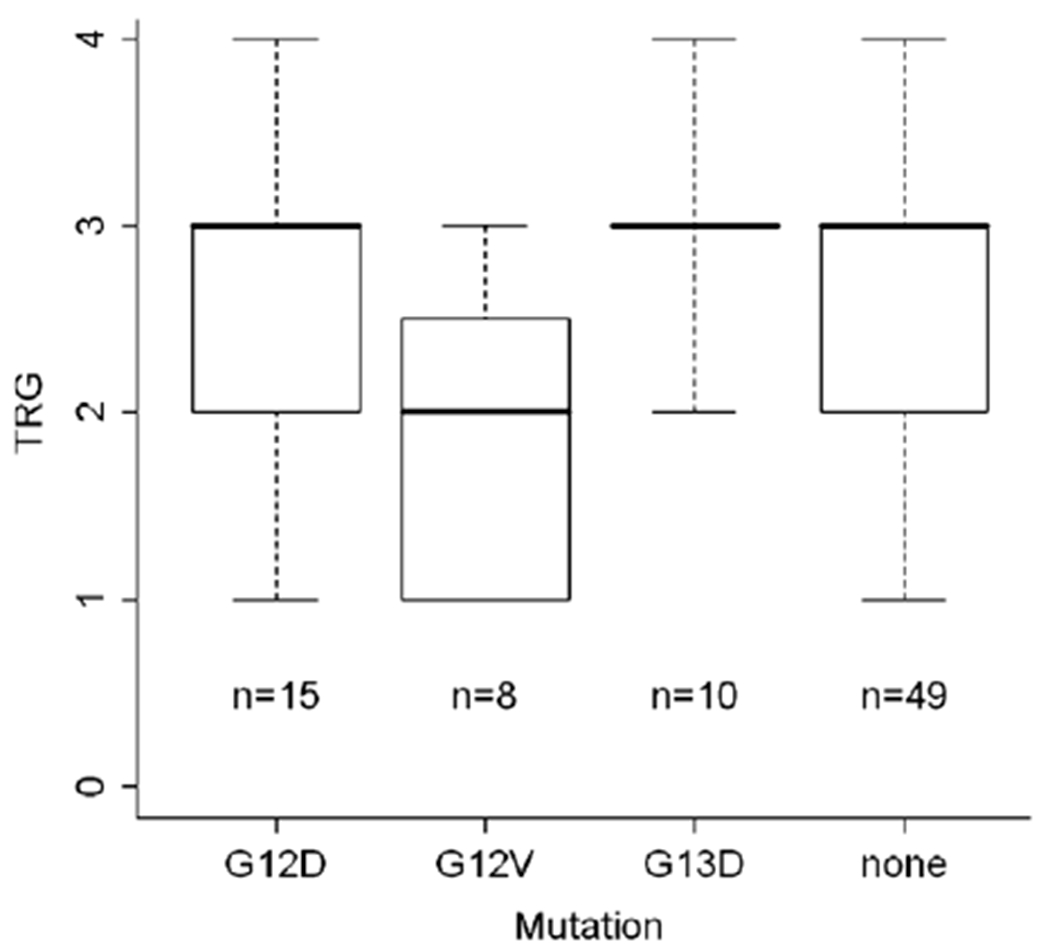
Distribution of the tumor regression grade in the distinct mutation groups.
KRAS mutations and follow-up data
After a median follow-up of 30.7 months (range: 3–86 months), three patients developed recurrent disease and died 15, 21, and 57 months after operation. Two patients died three and four months after the operation due to sudden cardiac death, respectively. Accordingly, we calculated a median overall survival (OS) of 20 months (range: 3–57 months). One of these three patients showed a KRAS mutation (G12W), the others were wild type. Twelve patients suffered tumor recurrence, three showed local recurrence, nine distant metastases in lung, liver, cerebrum and/or systemic lymph nodes resulting in a median disease-free survival (DFS) of 22.8 months (range: 2–50 months). In one of the three patients with local recurrence a single KRAS mutation (G12D) was found as well as in six of the nine patients with distant metastases (66.7%). However, none of the amino acid exchanges (2×G12D, G12C, G12W, G13D, and G12V) was overrepresented significantly. To further investigate if a KRAS mutation has an impact on DFS we used Kaplan-Meier Curves but failed to show a significant difference (p = 0.58, Fig. 2).
Fig. 2.
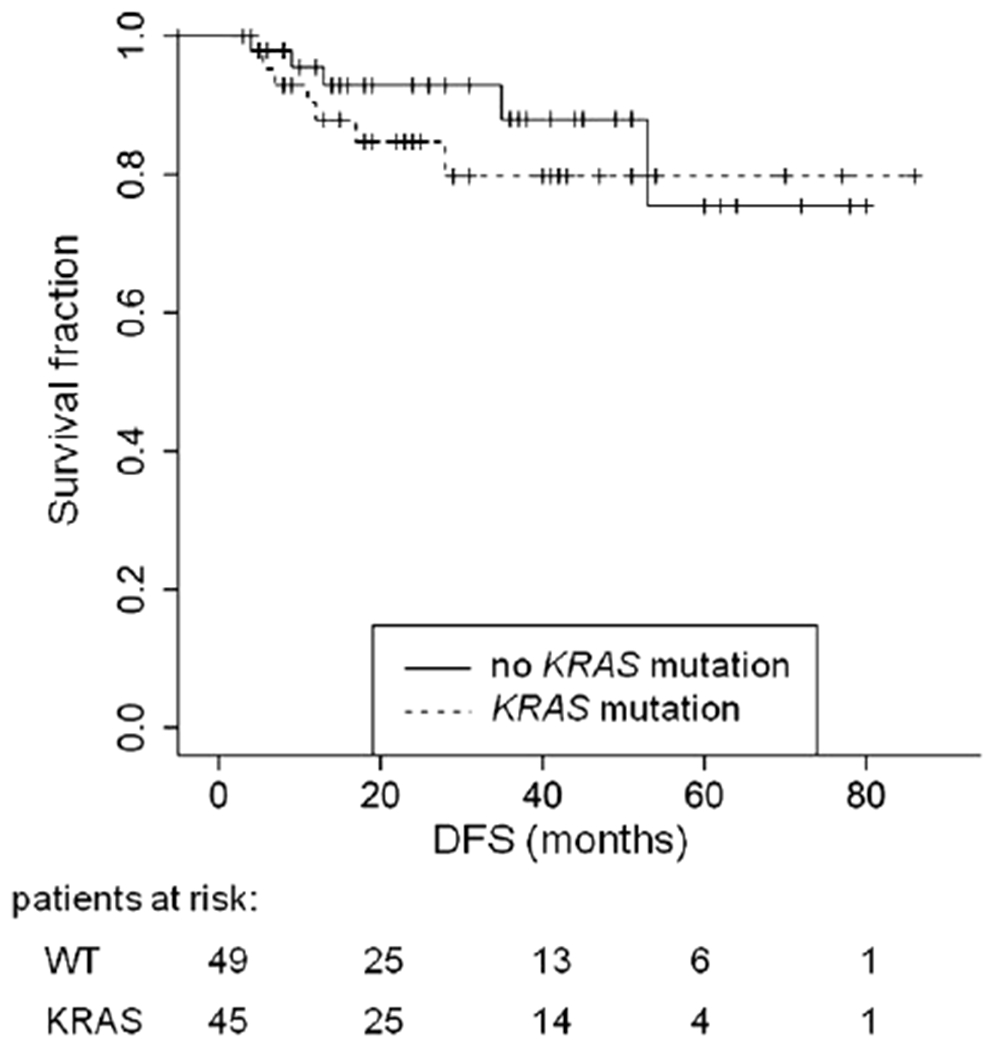
Kaplan-Meier Curves for disease-free survival (DFS) depending on KRAS mutation status (p = 0.58).
Discussion
The MAPK pathway plays a major role in cell proliferation and is involved in up to 30% of CRC [1]. Both KRAS and BRAFare the members of the signaling pathway and are known to be activated by oncogenic mutations. In contrast to the literature indicating a mutation frequency of about 10% in CRC, we only observed one mutation in 94 rectal cancer biopsies (1%). Furthermore, this single mutation was detected in codon 594, which, according to the published data, is very rarely affected [7]. The absence of V600E BRAF mutations in our group of locally advanced rectal cancer patients confirms the data from Kim et al. [19] who compared cancer of the right (n = 73) and left (n = 72) colon as well as rectal cancers (n = 79). Compromising UICC-stages I-IV they showed a significant reduction of mutational events between right colon and rectum as well as between right and left colon. In accordance with our data, no mutations were found in rectal cancers. In contrast, Di Nicolantonio et al. [12] found a single V600E allele in 43 rectal samples, whereas Fransen et al. [20] even found two mutations in 55 rectal cancers. However, the frequency of BRAF mutations decreases from the right to the left colon [19] and since our data represent only rectal cancer biopsies from the middle and the lower third of the rectum one could speculate that the mutation found was located within the upper third of the rectum. Taken together, these data clearly show that BRAF mutations only play a very minor role for rectal carcinogenesis compared to colon carcinogenesis.
EGFR targeting chemotherapeutics have recently been added to the preoperative treatment options of patients with rectal cancer [21–23]. Though BRAF mutations are considered as activating mutations of the MAPK pathway and recent findings indicate that response against anti-EGFR therapy requires the presence of the wild-type allele [12], testing for the mutation would have an impact on therapeutic outcome and on planning individualized therapy concepts. According to our data indicating the complete absence of V600E BRAF mutations in adenocarcinomas of the lower and middle rectum, this test would be superfluous.
Mutations in the KRAS gene are found in about 30–35% of CRC. This number varies depending on the extent of screening, but is mainly comprised of nucleotide transversions in codons 12 and 13. In our analysis 45 of 94 patients (48%) showed a mutant KRAS gene in the cancer biopsies. Focusing on the hotspot regions codons 12 and 13 still 39 (41.5%) cancers are non-wild type implicating that the frequency of mutations in locally advanced rectal cancer is comparable to colon cancers.
However, far not all publications about KRAS mutation include analyses of codon 61 (exon 2), which is expected to account for 1–5% of mutations. Furthermore, very little is known about the mutation frequency in codon 146 (exon 3), although a partial transforming activity could be shown [24]. To detect these and other rare KRAS mutations we sequenced exons 2 and 3 in their entire length and found three mutations (3.2%) in codons 61 and 146 each, counting for 13.3% of all mutations. For rectal cancer patients the relative high number of codon 146 mutations compared to codon 61 mutations has not been described previously. For CRC, Edkins et al. [25] investigated two different patient groups, one from the US and the other one from Hong Kong. In 94 patients from the US, they found two codon 146 mutations whereas in 126 patients from Hong Kong seven mutations were found. This emphasizes that mutation analysis for codon 146 should be included in future analyses.
KRAS mutations influence therapeutic outcome in patients treated with anti-EGFR agents [4,5,26,27] and potentially mediate resistance to ionizing radiation [28,29]. In patients with rectal cancer receiving preoperative chemoradiotherapy (CT/RT) in combination with anti-EGFR agents, an association between KRAS mutations and response has already been shown. In contrast, data for patients with locally advanced rectal cancer and preoperative treatment with non-EGFR agents are rare. In 2000 Luna-Perez et al. [30] found KRAS mutations in rectal cancer to be associated with longer DFS and OS. While these data were retrieved from the tissue taken after preoperative CT/RT, we analyzed 94 biopsies from patients with locally advanced rectal cancer that were taken prior to the therapy. We then aimed to correlate the KRAS mutation status with clinical parameters such as tumor regression grading, T-level downsizing and downstaging. These markers are of major interest because the degree of TRG is predictive for disease-free survival [31] and the prediction of response could therefore allow for an adjustment of therapy. The correlation of KRAS-mutated type versus wild type failed to show any significance for our response markers. The lack of significance remained even when adjusting the groups according to the codon which carried the mutation. Although preoperative treatment was slightly different our data support the recently published work from Zauber et al. [32] who showed in 53 patients that the mutation status of codons 12 and 13 does not predict response to preoperative CT/RT.
However, when we considered the nature of the nucleotide exchange in codons 12 (G12D and G12V) and 13 (G13D), we discovered an association (p = 0.052) between these subtypes and the TRG. This relation was mainly attributable to the differences between G12V and G13D (p = 0.012) as well as between wild type and G12V (p = 0.04) indicating that tumors with G12V mutations show less regression than tumors that carry G13D. However, the differences do not remain significant after applying the Bonferroni adjustment. In combination with the small sample size these associations have to be interpreted very carefully. Nevertheless, the observation that the G12V mutation may result in a more resistant tumor would be consistent with a large multicenter study on 3439 patients indicating that the subtype G12V is associated with shorter disease-free and overall survival in Dukes’ B and C CRC [6]. The increased kinase activity, and associated with this the increased activation of the ras pathway confirm the data reported by Guerrero and colleagues [33–36].
Interestingly, we did not observe significant associations between any KRAS mutation and survival data. However, these results need to be interpreted very carefully. First of all, the number of relapse events is very small and may represent a sample bias. Second, the follow-up period is far too short to draw definitive conclusions. Although patients from the CAO/ARO/AIO-94 trial have been followed up for more than 5 years, many patients from the ongoing CAO/ARO/AIO-04 trial were diagnosed less than 1 or 2 years ago.
In summary, we screened biopsies from 94 patients with locally advanced rectal cancers for KRAS and BRAF mutations. Interestingly, no V600E BRAF mutation was found suggesting limited relevance of BRAF mutations for rectal carcinogenesis. KRAS mutations in codons 12 and 13 showed comparable frequency to colon cancers but revealed a high number of mutations in codons 61 and 146. Consequently, these two codons should be included in future studies. While none of these mutations was significantly associated with response to preoperative CT/RT or as a predictor of relapse and/or survival, the G12V and G13D types of KRAS mutations revealed a tendency to positively correlate with the response to preoperative CT/RT. To further elucidate the impact of distinct KRAS nucleotide changes, analyses of a larger patient population will be performed.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (KFO 179). We are grateful to Dr. Hilka Rothe for histopathological work-up, and to Mr. Chan Rong Lai and Mrs. Monika Winkler for technical assistance.
References
- [1].Hoshino R, Chatani Y, Yamori T, et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 1999;18:813–22. [DOI] [PubMed] [Google Scholar]
- [2].Wang L, Cunningham JM, Winters JL, et al. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res 2003;63:5209–12. [PubMed] [Google Scholar]
- [3].Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008;9:517–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757–65. [DOI] [PubMed] [Google Scholar]
- [5].Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009;360:1408–17. [DOI] [PubMed] [Google Scholar]
- [6].Andreyev HJ, Norman AR, Cunningham D, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer 2001;85:692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54. [DOI] [PubMed] [Google Scholar]
- [8].Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004;116:855–67. [DOI] [PubMed] [Google Scholar]
- [9].Yuen ST, Davies H, Chan TL, et al. Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res 2002;62:6451–5. [PubMed] [Google Scholar]
- [10].Storm SM, Rapp UR. Oncogene activation: c-raf-1 gene mutations in experimental and naturally occurring tumors. Toxicol Lett 1993;67:201–10. [DOI] [PubMed] [Google Scholar]
- [11].Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002;418:934. [DOI] [PubMed] [Google Scholar]
- [12].Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008;26:5705–12. [DOI] [PubMed] [Google Scholar]
- [13].Sauer R, Becker H, Hohenberger W, et al. Preoperative versus postoperative chemoradiotherapy for rectal cancer. N Engl J Med 2004;351:1731–40. [DOI] [PubMed] [Google Scholar]
- [14].Rodel C, Sauer R. Integration of novel agents into combined-modality treatment for rectal cancer patients. Strahlenther Onkol 2007;183:227–35. [DOI] [PubMed] [Google Scholar]
- [15].Heald RJ, Ryall RD. Recurrence and survival after total mesorectal excision for rectal cancer. Lancet 1986;1:1479–82. [DOI] [PubMed] [Google Scholar]
- [16].Grade M, Ghadimi BM, Varma S, et al. Aneuploidy-dependent massive deregulation of the cellular transcriptome and apparent divergence of the Wnt/beta-catenin signaling pathway in human rectal carcinomas. Cancer Res 2006;66:267–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ghadimi BM, Grade M, Difilippantonio MJ, et al. Effectiveness of gene expression profiling for response prediction of rectal adenocarcinomas to preoperative chemoradiotherapy. J Clin Oncol 2005;23:1826–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gavioli M, Bagni A, Piccagli I, Fundaro S, Natalini G. Usefulness of endorectal ultrasound after preoperative radiotherapy in rectal cancer: comparison between sonographic and histopathologic changes. Dis Colon Rectum 2000;43:1075–83. [DOI] [PubMed] [Google Scholar]
- [19].Kim JC, Cho YK, Roh SA, et al. Individual tumorigenesis pathways of sporadic colorectal adenocarcinomas are associated with the biological behavior of tumors. Cancer Sci 2008;99:1348–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fransen K, Klintenas M, Osterstrom A, Dimberg J, Monstein HJ, Soderkvist P. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis 2004;25:527–33. [DOI] [PubMed] [Google Scholar]
- [21].Debucquoy A, Haustermans K, Daemen A, et al. Molecular response to cetuximab and efficacy of preoperative cetuximab-based chemoradiation in rectal cancer. J Clin Oncol 2009;27:2751–7. [DOI] [PubMed] [Google Scholar]
- [22].Horisberger K, Treschl A, Mai S, et al. Cetuximab in combination with capecitabine, irinotecan, and radiotherapy for patients with locally advanced rectal cancer: results of a phase II MARGIT trial. Int J Radiat Oncol Biol Phys 2009;74:1487–93. [DOI] [PubMed] [Google Scholar]
- [23].Rodel C, Grabenbauer GG, Papadopoulos T, Hohenberger W, Schmoll HJ, Sauer R. Phase I/II trial of capecitabine, oxaliplatin, and radiation for rectal cancer. J Clin Oncol 2003;21:3098–104. [DOI] [PubMed] [Google Scholar]
- [24].Feig LA, Cooper GM. Relationship among guanine nucleotide exchange, GTP hydrolysis, and transforming potential of mutated ras proteins. Mol Cell Biol 1988;8:2472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Edkins S, O’Meara S, Parker A, et al. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol Ther 2006;5:928–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tol J, Koopman M, Cats A, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med 2009;360:563–72. [DOI] [PubMed] [Google Scholar]
- [27].Lurje G, Nagashima F, Zhang W, et al. Polymorphisms in cyclooxygenase-2 and epidermal growth factor receptor are associated with progression-free survival independent of K-ras in metastatic colorectal cancer patients treated with single-agent cetuximab. Clin Cancer Res 2008;14:7884–95. [DOI] [PubMed] [Google Scholar]
- [28].Grana TM, Rusyn EV, Zhou H, Sartor CI, Cox AD. Ras mediates radioresistance through both phosphatidylinositol 3-kinase-dependent and Raf-dependent but mitogen-activated protein kinase/extracellular signal-regulated kinase kinase-independent signaling pathways. Cancer Res 2002;62:4142–50. [PubMed] [Google Scholar]
- [29].Gupta AK, Bakanauskas VJ, Cerniglia GJ, et al. The Ras radiation resistance pathway. Cancer Res 2001;61:4278–82. [PubMed] [Google Scholar]
- [30].Luna-Perez P, Segura J, Alvarado I, Labastida S, Santiago-Payan H, Quintero A. Specific c-K-ras gene mutations as a tumor-response marker in locally advanced rectal cancer treated with preoperative chemoradiotherapy. Ann Surg Oncol 2000;7:727–31. [DOI] [PubMed] [Google Scholar]
- [31].Rodel C, Martus P, Papadoupolos T, et al. Prognostic significance of tumor regression after preoperative chemoradiotherapy for rectal cancer. J Clin Oncol 2005;23:8688–96. [DOI] [PubMed] [Google Scholar]
- [32].Zauber NP, Marotta SP, Berman E, et al. Molecular genetic changes associated with colorectal carcinogenesis are not prognostic for tumor regression following preoperative chemoradiation of rectal carcinoma. Int J Radiat Oncol Biol Phys 2009;74:472–6. [DOI] [PubMed] [Google Scholar]
- [33].Bazan V, Migliavacca M, Zanna I, et al. Specific codon 13 K-ras mutations are predictive of clinical outcome in colorectal cancer patients, whereas codon 12 K-ras mutations are associated with mucinous histotype. Ann Oncol 2002;13:1438–46. [DOI] [PubMed] [Google Scholar]
- [34].Guerrero S, Figueras A, Casanova I, et al. Codon 12 and codon 13 mutations at the K-ras gene induce different soft tissue sarcoma types in nude mice. FASEB J 2002;16:1642–4. [DOI] [PubMed] [Google Scholar]
- [35].Guerrero S, Casanova I, Farre L, Mazo A, Capella G, Mangues R. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res 2000;60:6750–6. [PubMed] [Google Scholar]
- [36].Monticone M, Biollo E, Maffei M, et al. Gene expression deregulation by KRAS G12D and G12V in a BRAF V600E context. Mol Cancer 2008;7:92. [DOI] [PMC free article] [PubMed] [Google Scholar]