

Abstract
Since their discovery, neutrophil extracellular traps (NETs) have been implicated in a broad array of functions, both beneficial and detrimental to the host. Indeed, NETs have roles in infection, sepsis, wound healing, thrombotic disease, and cancer propagation, all of which are directly implicated in the care of surgical patients. Here we provide an updated review on the role of NETs in the perioperative period with specific emphasis on perioperative infections, wound healing, vascular complications, cancer propagation, as well as discussing ongoing, and future therapeutic targets. Surgeons will benefit from understanding the latest discoveries in neutrophil biology and how these novel functions affect the care of surgical patients. Furthermore, novel anti-NET therapies are being developed which may have profound effects on the care of surgical patients.
Keywords: NETs, neutrophil extracellular traps, surgery
NEUTROPHIL EXTRACELLULAR TRAPS: WHAT SURGEONS NEED TO KNOW
Neutrophils are the surgeon's best friend, and perhaps also his or her worst enemy. None of what we do is possible without their classically known functions as central mediators of innate immunity. Surgeons have, in fact, been central to many of our most important discoveries pertaining to neutrophil biology, largely because of the intimate role they play in all aspects of surgical stress and recovery.1 Indeed, neutrophilic function ranges from normal postoperative wound healing and inflammatory responses and extends to being central mediators of postoperative sepsis and neoplastic dissemination. This double-edged nature of neutrophilic function is the result of uncontrolled neutrophilic cellular activity, which at the cellular level involves a slew of mechanisms ranging from the production of reactive oxygen species to phagocytosis.2
For a little more than a decade, an entirely new mechanism of action has been described to occur in neutrophils. Various states of inflammation trigger nuclear DNA decondensation with active fusion of the nuclear and cytoplasmic membranes resulting in the release of web-like chromatin filaments termed neutrophil extracellular traps (NETs).3 Since their discovery in 2004, NETs have been implicated in a broad array of functions, both beneficial and detrimental to the host.3 Indeed, NETs have roles in infection, sepsis, wound healing, thrombotic disease, and cancer propagation, all of which are directly implicated in the care of surgical patients.3 However, to understand the implications of NETs for surgeons, a brief recapitulation of the process of NET formation, or NETosis, is necessary.
In response to certain stimuli, neutrophils release a meshwork of decondensed DNA into the extracellular space.3 Instigating factors come in the form of circulating molecules such as cytokines and antibodies, but can also include cells and their components, specifically endothelial cells and platelets.3 The complete extent of initiators of NETosis is unknown, and many new triggers are constantly discovered. NETs are primarily composed of DNA and histones arranged in a web form on which a vast amount of neutrophil-derived enzymes are bound, the likes of which include neutrophil elastase (NE), myeloperoxidase (MPO), and cathepsin G.3 In addition, NETs provide a scaffold on which other proteins and cells can bind, including chemotactic factors, activating factors, growth factors, and many others.3
The exact cellular mechanism of NETosis is unknown, however several key intracellular players have been identified. Membrane receptors such as Toll-Like Receptor 2 and 4, the antibody crystallizable fragment (Fc) receptors as well as the complement receptors represent some of the players in the early cascade leading to NETosis.3 Subsequently, a signal cascade results in the activation of the nuclear enzyme protein-arginine deaminase type 4 (PAD4), which results in hypercitrullination and thus decondensation of nuclear chromatin, an integral step in NET formation.3,4 Indeed, several studies have demonstrated that PAD4 knockdown or inhibition result in NET abrogation.5–7 This culminates in the excretion of webs of decondensed chromatin via vesicular export and release, a process that can either occur with or without neutrophil cell lysis.3 The specifics of these mechanisms are ill-defined and remain the subject of intensive research.
To summarize, neutrophils exposed to triggers ranging from soluble molecules to cell surface proteins activate signaling cascades via several receptors, in turn activating PAD4 activity, culminating in the release of chromatin webs layered with an extensive array of active proteins, which can activate extracellular cascades as well as serve as a substrate or scaffold for other proteins and cells alike.
NETs AND PERIOPERATIVE INFECTIONS
Neutrophils are a primary line of defense in the innate immune response to microbial pathogens in the postoperative period. Phagocytosis and subsequent killing through antimicrobial peptides and reactive oxygen species production have extensively been described in the literature. However, most recently, the release of NETs has been identified as a novel mechanism in the antimicrobial function of neutrophils.8 Indeed, it was in the context of infection that NETs were first discovered.8
NETs form during infection and contribute to bacterial clearance. Neutrophils cocultured in the presence of pathogenic bacteria such as S aureus and Shigella resulted in the formation of NETs which directly interacted and reduced the concentration of bacteria-derived virulence factors in vitro.8 In fact, in-vitro models in which neutrophils are either deficient in NET production, or in which NETs, composed primarily of double-stranded DNA, are dismantled with DNase treatment, demonstrate that abrogation of NETs is linked to reduced ability of neutrophils to kill bacteria.8 These findings were further corroborated in vivo where depletion of NETs by DNase administration in a mouse model of polymicrobial sepsis resulted in higher rates of systemic microbial dissemination.9 However, when scrutinized, the results shown in these studies demonstrated that the difference in susceptibility to systemic infection was small in early time points, with no significant difference in mortality at subsequent time points.9 Subsequent studies showed that PAD4-deficient mice, an enzyme central to NETosis, did not have increased mortality over their wild-type (WT) counterparts.6 Furthermore, when treated with antibiotics, PAD4 deficiency was not associated with increased bacteremia.6 As such, although evidence is present to suggest a role for NETs in immunity both in vitro and in vivo, it may only represent one of many redundant mechanisms in which neutrophils mediate antibacterial immunity.
NETs exert antimicrobial activity both by creating a physical barrier and delivering high concentrations of neutrophil-derived antimicrobial peptides in the microenvironment. The bactericidal activity of NET components such as histones, NE, MPO, and cathepsin G is well established in the current literature.10–12 In their groundbreaking article, Brinkmann et al8 attributed the antibacterial function of NETs primarily to histones, which had already been well established as having antimicrobial activity. Monoclonal antibodies targeting the H2A-H2B-DNA complex resulted in a complete loss of killing of S aureus.8 Furthermore, co-culturing of several bacteria with H2A protein demonstrated that even low concentrations of H2A resulted in effective bactericidal activity. Brinkmann et al8 also demonstrated that NET-bound NE was responsible for extracellular targeting of bacterial virulence factors. These findings are supported by previous in vitro studies in which NE successfully inactivated virulence factors of Shigella, Salmonella, and Yersinia bacteria, an effect that was lost when NE was inactivated either pharmacologically or genetically.13 These findings are echoed in several other studies in which NE demonstrates potent antimicrobial activity.14
Most recently, however, the DNA found within NETs was identified as a novel antimicrobial agent. Halverson et al15 demonstrated that the DNA backbone is an independent contributor to the antibacterial activity of NETs in P Aeruginosa infections, a mechanism mediated in part by its ability to sequester surface bound cations and in turn disrupt bacterial cell membranes to induce lysis. Brinkmann et al8 had suggested that the physical structure of NETs was key in mediating bacterial cell death (as seen by the loss of function with DNase treatment) but had not identified a mechanism via which this was executed, only suggesting that neutrophil-derived DNA created a mesh that sequestered bacteria in a microenvironment rich in extracellular antibacterial enzymes. Building on this, Halverson's findings identify DNA as both a physical restraint but even more interesting, a direct contributor to the antimicrobial activity of NETs.15 These findings are complemented by studies in which certain microbes such as Group A Streptococcus enhance their virulence through expression of DNase enzymes.16 Inhibition of these bacterial-derived enzymes results in rapid clearance of infection by neutrophils.16
Although NETs appear to have an antibacterial role in early response to bacterial infection, emerging studies have identified a double-edged facet of NET function in the context of severe bacterial sepsis. Excessive NETosis has been linked to “bystander” end-organ damage in the liver, lungs, and kidneys.17 As such, although NETs appear to play a role in limiting the dissemination of bacterial infections, unregulated NETosis in the context of more severe postoperative sepsis may have clinically deleterious consequences.
NETs AND WOUND HEALING
Wound healing, and its associated complications, is a source of significant mortality and morbidity in the postoperative period. The role of neutrophils in wound healing and infection has extensively been documented.18 Indeed, neutrophils are the first leukocytes recruited and represent the predominant cell-type in the skin during wound healing.18 Their primary function is to trap and kill microbes, a process in part mediated by the production of NETs.8 However, the effect of neutrophils and NETs on wound healing remain incompletely understood. Interestingly, studies done in neutrophil-depleted mice have shown that under sterile conditions, absence of neutrophils resulted in faster epidermal wound healing.19 Further to this, diabetic neutropenic mice showed impressive rates of wound closure as compared to their WT counterparts, an exciting discovery given that diabetes is one of the most common comorbidities in the surgical patient population.19 These results establish neutrophils as central players in wound healing, with a double-edged function with regards to infection prevention and wound closure.
The role of NETs in wound healing was first established in a groundbreaking report by Wong et al20 which showed that neutrophils in diabetic mice were primed for NETosis, and that wounds in these mice had significantly elevated levels of NETs as compared to their WT counterparts. The increase in NETosis was mediated by PAD4 and resulted in elevated extracellular NET histone levels at the wound site.20 Interestingly, disruption of NETs with DNAse 1 improved healing in both diabetic and normoglycemic mice, suggesting that NETs were responsible for delayed wound healing.20 To further establish this, Wong et al used a PAD4-deficient mouse model. PAD4 has previously been shown to be central to NETosis, without compromising neutrophil function or mouse viability, thus making it a good model to study function of NETs.6 The authors demonstrated that PAD4-deficient mice not only heal faster but demonstrated faster reepithelialization than control mice.20 Furthermore, diabetic PAD4-deficient mice showed no impairment in wound healing.20 These findings were corroborated in human samples by Fadini et al21 in a report on diabetic ulcers, which showed that nonhealing diabetic wounds showed overexpression of NET components, specifically extracellular DNA, histones, and NE levels. Similarly, they demonstrated in a diabetic mouse model that PAD4 inhibition with Cl-amidine resulted in a decrease in NETosis at the wound site with a concomitant restoration of diabetic wound healing.21 These reports together clearly establish that NETs have a deleterious effect on wound closure—a feature of biology underappreciated and yet so important to the surgical community.
These reports represent recent evidence linking NETs to wound healing. However, the role of NET components, such as NE and MPO, has long been reported in the literature. Indeed, NE is found to be overexpressed in fluid isolated from chronic wounds, and its inhibition is associated with more rapid reepithelialization of wounds.22 NE can lead to poor wound healing in several ways. First, NE induces degradation of the wound matrix, both directly and through its activation of metalloproteinases, which are often found to be overactive in nonhealing wounds.23–25 Second, NE is responsible in part for the proteolytic cleavage of such factors as platelet-derived growth factor or vascular endothelial growth factor, both which form part of the normal healing process.24 In addition, NE has been shown to mediate the conversion of proepithelin, another growth factor, to epithelin, whose function is to recruit and activate neutrophils, likely contributing to an excessive inflammatory milieu in nonhealing wounds.26 These results therefore directly implicate NE in the pathophysiology surrounding poor wound healing. However, to date, it is unclear whether the source of NE in wound healing is within the context of NETs, or from granules released by neutrophils via alternative mechanisms, or both.
In parallel to this, neutrophil-associated MPO is also associated with delayed wound healing. Specifically, extracellular and surface-bound MPO derived from infiltrating neutrophils in the vicinity of intestinal epithelial cells resulted in delayed wound healing both in vitro and in vivo in a colonoscopic biopsy mouse model; concomitantly, inhibition of MPO resulted in a significant acceleration of wound healing.27 In this report, the authors report that activation of infiltrating neutrophils at the injury site was associated with decreased total MPO level intracellularly, suggesting that in part, MPO was released into the extracellular space. Although no specific assay was conducted, it remains a possibility that MPO was released as part of extracellular NETs in this context.
Although these studies link key NET components to delayed wound healing, no study to date exists describing these functions in the specific context of NETs. Indeed, reports have suggested that these molecules could be alternatively released through neutrophil degranulation, a completely separate process from NETosis.21 Further study is therefore warranted before specific conclusions can be drawn. Nonetheless, it is clear that these molecules and the process of NETosis are key therapeutic targets to address improved wound healing in a number of clinical situations.
IMPACT ON THE VASCULAR SYSTEM
Atherosclerosis is a leading cause of morbidity and mortality worldwide, one that is intimately linked to surgical practice. Atherosclerosis is the result of chronic inflammation inducing endothelial dysfunction which culminates in the formation of macrophage-derived foam cells and proliferation of smooth muscle within the vascular wall. Recent studies have implicated NETs in the pathophysiology and sequelae of atherosclerosis and atherothrombosis alike. First, Warnatsch et al28 demonstrated that cholesterol crystals induced NET formation in vitro, and that NETs were identified in the atherosclerotic lesions in vivo. Further to this, they demonstrated that loss of NET function, both in a NET-deficient mouse model and a control model treated with DNase, resulted in a 3-fold reduction in plaque size.28 In line with these findings, Knight et al29 demonstrated that PAD4 inhibition in a mouse model of atherosclerosis resulted in a loss of NET formation with concomitant reduction in size of plaques and rate of thrombosis. The function of NETs has also been implicated in the promotion of endothelial dysfunction as well as recruitment of macrophages to atherosclerotic plaques.28 Together, these findings implicate NETs in the pathogenesis of atherosclerosis.
Atherothrombosis is a critical complication of atherosclerosis and represents a central pillar of vascular and cardiac surgical practice. Several studies have found neutrophils and NETs to be key components of thrombi found in acute ischemic events such as myocardial infarct or ischemic stroke.30–32 In a study of ST-segment acute coronary syndromes, Mangold et al33 found that higher NET levels were associated with increased infarct size and prolonged time to ST-segment resolution, whereas higher endogenous DNase levels were linked with lower infarct size and a shortened time to ST-segment resolution. These findings suggest that NETs play a role in acute thrombosis; however, the mechanism via which this occurs remains poorly understood. Furthermore, there is evidence to suggest that NETs are involved in injury following reperfusion. In a mouse model of induced limb ischemia-reperfusion, Oklu et al34 demonstrated that reduction in the level of NETs via TLR4 inhibition was associated with a reduction in reperfusion injury to skeletal muscle. Other studies have similarly implicated NETs in the ischemic injury of several organs including kidney, brain, myocardium, and intestine.35,36 To assess whether limiting NET levels in ischemic tissues could minimize deleterious effects on end-organ function, Wang et al36 designed a study in which mice undergoing clamping of the superior mesenteric artery were treated with DNase-1 intravenously. The results demonstrated that DNase treatment resulted in a decrease in the proinflammatory milieu of intestinal reperfusion injury and histologically resulted in reduced intestinal mucosal injury with maintenance of the epithelial barrier junction proteins.36 Boettcher et al37 showed similar improved outcomes in rats that had undergone DNase treatment in a model of midgut volvulus. Interestingly, their study found that although both DNase and tissue-plasminogen activator could be used to improve reperfusion injury, DNase was the only treatment that was not associated with prolonged bleeding time.37 Additional reports have demonstrated similar benefits of NET targeting in models of reperfusion injury in kidneys, and myocardial reperfusion.38,39 These findings together implicate NETs in the full spectrum of arterial disease, from plaque formation, to acute thrombus formation, and finally to the subsequent reperfusion injury that may occur following intervention. Together, these findings put forth NETs as a potential target for therapy within the perioperative period.
Deep venous thrombosis (DVT) remains a leading cause of preventable mortality and morbidity in the perioperative window. Broadly speaking, DVT formation is initiated by endothelial dysfunction and culminates with the activation of coagulation cascades and the promotion of platelet recruitment and activation, resulting in thrombus propagation; as such, targeting these processes with generalized anticoagulants was and remains the mainstay of prophylactic therapy today. This presents a dilemma in the surgical environment as these same broad mechanisms of thrombosis concurrently apply to postoperative hemostasis. In light of this, identifying specific molecular mechanisms underlying DVT formation remains crucial in the development of targeted therapies. Studies over the years have identified innate immune cells as key players, with emphasis on the interaction of platelets with monocytes and neutrophils.40 With regards to neutrophils, a significant body of literature has amassed suggesting that they are central to the establishing of DVTs.
To establish a role for NETs in DVT formation, it is necessary to clarify the currently accepted mechanism of DVT initiation. Endothelial cells maintain vascular homeostasis acting as a barrier, preventing circulating coagulation factors from interacting with tissue-sequestered tissue factor (TF), an initiator of the coagulation cascade.41 Additionally, endothelial cells contain Weibel-Palade bodies (WPBs), intracellular organelles rich in factors such as vWF, P-Selectin, and interleukin (IL)-8 whose primary function is to induce local inflammation and activate coagulation when released.42 Thrombosis is initiated by endothelial cell apoptosis resulting in exposure of TF as well as release of WPBs.42 Subsequently there is recruitment of monocytes, neutrophils, and platelets which enhance both the inflammatory response and hold key roles in coagulation.42 This process culminates in the deposition of fibrin, trapping circulating cells and factors and resulting in vessel thrombosis.41 NETs are intimately linked with many of these steps during the process, from initiation to propagation.
NETs promote thrombus organization by enhancing endothelial dysfunction, inducing release of WPBs and subsequently exposing TF to the intravascular milieu.42 Indeed, in-vitro coculturing of activated endothelial cells with neutrophils resulted in NET formation, a process in part mediated by IL-8 released from endothelial cells.43 Furthermore, prolonged coculturing resulted in an increase in endothelial cell death, a process that was disrupted when NETosis was inhibited.43–45 Further characterization identified that NET-mediated cytotoxicity was predominantly on the activity of histones and MPO found in NETs.45 Indeed, histones, but not DNA, were subsequently shown to mediate release of vWF from WBPs both in vitro and in a mouse model that received injections of histones or DNA.46 Thus, not only can endothelial dysfunction promote NET formation, but NETs, and their internal components, in turn exert a positive feedback mechanism to increase local inflammation and endothelial cell death, both of which represent key processes in the initial organization of DVTs.
Beyond exposure of TF, NETs are intimately linked to thrombosis through direct modulation of coagulation cascades. Specifically, NETs within thrombi have been shown to bind and activate Factor XII, thus directly initiating the intrinsic pathway.40,47 Second, the enzymatic activity of NE and MPO found on NETs results in inactivation of tissue-factor pathway inhibitor and thrombomodulin, effectively enhancing the activity of the extrinsic coagulation pathway.48,49 Lastly, NETs, DNA, and histones significantly slowed plasminogen-driven lysis of plasma clots in vitro, an effect that was reversed by treatment with DNase.50 Together, these results suggest that NETs modulate coagulation, both as promoters of the extrinsic and intrinsic coagulation pathways, while concurrently inhibiting factors involved in regulation or breakdown of thrombi.
Furthermore, NETs were shown to enhance platelets interactions leading to propagation of thrombi. Initially, NETs directly bind vWF released from WPBs, serving as a substrate for subsequent recruitment of platelets.51 NE and other proteases found in NETs result in activation of platelet receptors, further enhancing platelet aggregation.52 Histones within NETs induce platelet activation, a process mediated via TLR2 and TLR4 signaling, and result in downstream increases in thrombin generation through a platelet-dependent mechanism.53 Furthermore, activated platelets cocultured with neutrophils are capable of inducing NET release, suggesting that a positive-feedback mechanism exists in which NETosis not only enhances, but is enhanced by platelet activation.54 These results together demonstrate clearly that NETs are key mediators in recruiting and activating platelets during DVT formation.
The aforementioned evidence puts forth the targeting of NETs as a novel therapeutic approach in DVT prophylaxis. Indeed, inhibition of NET formation with DNase 1 treatment resulted in abrogation of thrombus formation both in in vitro and in vivo.51,55 Interestingly, DNase appears to be the endogenous way that the body regulates NET levels, as demonstrated by Jimenez-Alcazar et al56 whose report identified 2 host DNases that were directly implicated in controlling vascular occlusion by NETs. The report went beyond this in identifying that NET clots were sufficient by themselves to obstruct vessels and that these did not respond to standard antithrombotic treatment; in fact, DNase reconstitution was the only effective way to inhibit NET clot formation, suggesting that our current DVT prophylaxis may be insufficient.56 Parallel to this work, other studies have sought to identify and target proteins central to NETosis as potential therapeutic targets. Li et al4 demonstrated that PAD4-deficient neutrophils were unable to form NETs with appropriate stimulation, a finding that was later demonstrated in in-vivo models. Kusunoki et al took this one step further by demonstrating that pharmacological targeting of PAD4 in vivo with a pan-PAD inhibitor, Cl-amidine, resulted in reduced capacity to form NETs, a finding corroborated in several other in-vivo models.5,7,57,58 In several studies on arterial thrombosis, treatment with Cl-amidine reduced thrombus formation and end-organ damage.29,57,59,60 Although the mechanism of atherothrombosis and DVT is not identical, several parallels can be drawn to suggest that therapeutic targeting of PAD4 could be viable in DVT prevention. Together these findings establish NETs as a feasible therapeutic target, one that warrants further investigation in the context of DVT prophylaxis.
NETs AND CANCER
Neutrophils are often found in high numbers in proximity to human tumors and in mouse models of cancer. In the last several years, there has been accumulating evidence that NETs also have a role in cancer. Indeed, a prospective study on 957 patients by Grilz et al61 demonstrated that elevated levels of citrullinated histone H3, a marker of NET formation, was associated with elevated mortality in cancer patients, a finding which has been previously replicated in smaller studies. Both experimental and clinical evidence also lend support to the idea that surgery, which is intended to be a curative option to remove and reduce tumor mass, can paradoxically augment the development and growth of metastases.62,63 The neutrophil influx that follows surgical trauma thus has the potential to promote tumor growth and may in part explain the tumor promoting effects of surgery and its attendant complications.
Handling of the tumor can result in a 10-fold rise in circulating tumor cells.64 In addition to the dissemination of circulating cells, several postoperative changes, including NET formation, can help the cancer cells to establish metastatic foci.65 In addition to their mechanical function, the DNA strands released with NET formation are studded with a variety of proinflammatory molecules that are crucial to the capture of tumor cells.66 The inhibition of NETs after surgery powerfully inhibits the previously observed accelerated development of new metastatic disease.65,66 In humans undergoing resection of hepatic colorectal metastases, the greater the serum evidence of NET formation the higher the risk of recurrence.67 In addition, in a model of peritoneal metastases from colon cancer, mice treated with intraperitoneal DNase I showed an 88% decrease in the number of peritoneal metastases, suggesting that NETs are central to peritoneal spread, and secondly, that targeting NETs may be an effective strategy to minimize recurrence after cytoreductive surgeries.68
Furthermore, metastatic cancer cells may leave the primary tumor early during its development and form clinically undetectable micrometastases at distant sites.67 These islands of clinically undetectable micrometastases can remain in a dormant equilibrium between cellular proliferation and apoptosis.67 The local and systemic inflammatory events associated with surgical trauma, such as NET formation, can unpredictably unleash their potential for growth.67 NETs resulting from surgery can persist for weeks and induce growth of residual disease by activating Stat3 and NF-kB.67 In addition, recent evidence by Albrengues et al has shown a direct role of NETs in awakening dormant cancer cells by releasing NET-associated proteases, neutrophil elastase, and matrix metalloproteinase which sequentially cleaved laminin. The proteolytically remodeled laminin induced proliferation of dormant cancer cells by activating integrin α3β1 signaling.69
In addition to the previously mentioned changes directly related to surgical treatment, there are countless perioperative variables that can alter the oncological outcomes. Postsurgical infections in patients with cancer have been associated with adverse oncological outcomes independent of the morbidity associated with the infectious insult.65 This phenomenon has been observed across a broad range of malignancies, including lung, esophageal, breast, ovarian, and colorectal cancer; severe postoperative infectious complications are significantly associated with an increased rate of death from metastatic disease.62 In mouse models, sepsis is a strong stimulus for formation of NETs that promote early adhesion of tumor cells to distant organ sites and facilitate metastatic disease progression.65,66 However, what may prove to be most interesting are recent reports that demonstrate that cancer cells are capable of inducing NETs that support metastases in the absence of a septic stimuli. Indeed, Park et al70 were the first to demonstrate that breast cancer cells, in the absence of infection, were able to promote metastatic lung foci in a process mediated via PAD4 and NADPH oxidase. This effect was abrogated by DNAse I treatment.70 Subsequently, Albrengues et al69 similarly demonstrated in a mouse model that tobacco smoke, a noninfectious stimuli, was sufficient to convert dormant cancer cells to aggressive lung metastases. Similarly, treatment with DNase inhibited the conversion of dormant cells to metastatic foci.69 Together, these findings suggest that NETs are a pivotal component in the progression of metastatic disease in cancer treatment, and that this role can occur both in the context of infectious and noninfectious stimuli alike.
THERAPEUTIC IMPLICATIONS
Although NETs are clearly involved in almost every facet of surgical practice, from infection control to wound healing to vascular dysfunction and cancer propagation (Fig. 1), little attention has been paid to implementing anti-NET therapies to surgical practice. The preclinical evidence points to anti-NET therapies being safe and with limited effects on immunity from invading pathogens. Furthermore, several agents are currently approved for human use that have anti-NET activity (Table 1). One of those drugs is recombinant DNase (or Dornase α) that brakes the DNA backbone of NETs, leading to their degradation.8,71 DNase has been successful in treating cystic fibrosis (CF) by thinning lung secretions and hence decreasing the risk of infection.72,73 DNase has also shown great promise in any several other diseases of the lung and in multiple autoimmune diseases (Table 1). Now that NETs have been implicating in many more diseases, multiple clinical trials have been evaluating the effects of degrading NETs on diseases such as trauma (NCT03368092) and head and neck cancers (NCT-00536952). Another category of NET-targeted therapy is the use of NE inhibitors (NEi), which are serine protease inhibitors that have activity against NE, a critical enzyme involved in the formation of NETs.74 NEi have been trialed mainly in lung injuries, lung infections, chronic obstructive pulmonary disease (COPD), and CF (Table 1). Finally, a third promising category of NET-targeted therapies is CXCR2 inhibitors that inhibit CXCR2, a recently discovered NET regulator in COPD.75 CXCR2 inhibitors have been trialed mainly in COPD, asthma, and more recently in several metastatic solid tumors (Table 1).
FIGURE 1.
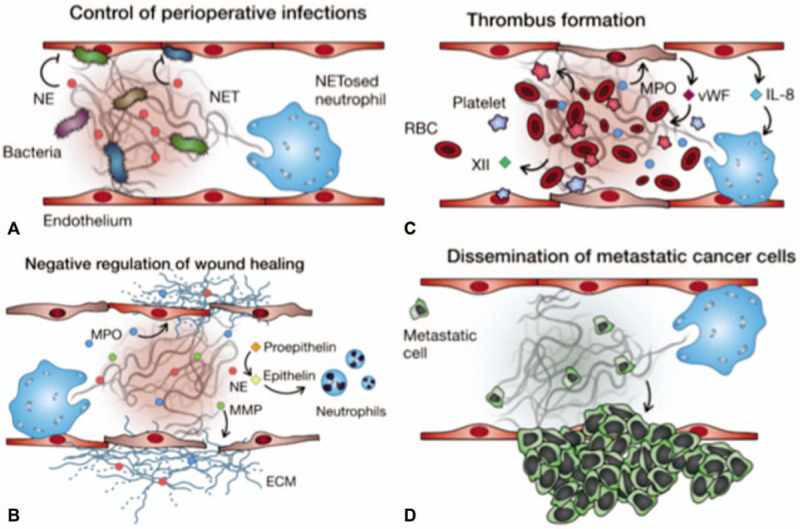
The multifacet role of neutrophil extracellular traps in disease. Shown is a diagrammatic representation of NETs in (A) control of perioperative infections, (B) negative regulation of wound healing, (C) thrombus formation, and (D) dissemination of metastatic cancer cells.
TABLE 1.
Clinically Available NET-targeted Therapies in Humans

CONCLUSIONS
Surgeons will benefit from understanding the latest discoveries in neutrophil biology and how these novel functions affect the care of surgical patients. Indeed, the surgical community is ideally positioned to trial these agents in a wide variety of contexts. Untangling the webs cast by neutrophils during surgical stress may improve a number of surgical outcomes and lead to novel therapeutic strategies. Indeed, neutrophils are the first-responders of our innate immune system and these soldiers continue to surprise us with their diverse armamentarium.
Footnotes
J.H.E. and S.T. contributed equally to the production of this manuscript.
The authors report no conflicts of interest.
REFERENCES
- 1.Cools-Lartigue J, Spicer J, Najmeh S, et al. Neutrophil extracellular traps in cancer progression. Cell Mol Life Sci 2014; 71:4179–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar V, Sharma A. Neutrophils: Cinderella of innate immune system. Int Immunopharmacol 2010; 10:1325–1334. [DOI] [PubMed] [Google Scholar]
- 3.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 2018; 18:134–147. [DOI] [PubMed] [Google Scholar]
- 4.Li P, Li M, Lindberg MR, et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 2010; 207:1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kusunoki Y, Nakazawa D, Shida H, et al. Peptidylarginine Deiminase Inhibitor Suppresses Neutrophil Extracellular Trap Formation and MPO-ANCA Production. Front Immunol 2016; 7:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinod K, Fuchs TA, Zitomersky NL, et al. PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood 2015; 125:1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rohrbach AS, Slade DJ, Thompson PR, et al. Activation of PAD4 in NET formation. Front Immunol 2012; 3:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science 2004; 303:1532–1535. [DOI] [PubMed] [Google Scholar]
- 9.Meng W, Paunel-Gorgulu A, Flohe S, et al. Depletion of neutrophil extracellular traps in vivo results in hypersusceptibility to polymicrobial sepsis in mice. Crit Care 2012; 16:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 2006; 6:173–182. [DOI] [PubMed] [Google Scholar]
- 11.Kawasaki H, Iwamuro S. Potential roles of histones in host defense as antimicrobial agents. Infect Disord Drug Targets 2008; 8:195–205. [DOI] [PubMed] [Google Scholar]
- 12.Parker H, Winterbourn CC. Reactive oxidants and myeloperoxidase and their involvement in neutrophil extracellular traps. Front Immunol 2012; 3:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinrauch Y, Drujan D, Shapiro SD, et al. Neutrophil elastase targets virulence factors of enterobacteria. Nature 2002; 417:91–94. [DOI] [PubMed] [Google Scholar]
- 14.Belaaouaj A. Neutrophil elastase-mediated killing of bacteria: lessons from targeted mutagenesis. Microbes Infect 2002; 4:1259–1264. [DOI] [PubMed] [Google Scholar]
- 15.Halverson TW, Wilton M, Poon KK, et al. DNA is an antimicrobial component of neutrophil extracellular traps. PLoS Pathog 2015; 11:e1004593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buchanan JT, Simpson AJ, Aziz RK, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 2006; 16:396–400. [DOI] [PubMed] [Google Scholar]
- 17.Czaikoski PG, Mota JM, Nascimento DC, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS One 2016; 11:e0148142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res 2018; 371:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dovi JV, He LK, DiPietro LA. Accelerated wound closure in neutrophil-depleted mice. J Leukoc Biol 2003; 73:448–455. [DOI] [PubMed] [Google Scholar]
- 20.Wong SL, Demers M, Martinod K, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med 2015; 21:815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fadini GP, Menegazzo L, Rigato M, et al. NETosis delays diabetic wound healing in mice and humans. Diabetes 2016; 65:1061–1071. [DOI] [PubMed] [Google Scholar]
- 22.Lee SK, Lee SS, Song IS, et al. Paradoxical effects of elastase inhibitor guamerin on the tissue repair of two different wound models: sealed cutaneous and exposed tongue wounds. Exp Mol Med 2004; 36:259–267. [DOI] [PubMed] [Google Scholar]
- 23.Owen CA, Campbell MA, Sannes PL, et al. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol 1995; 131:775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trengove NJ, Stacey MC, MacAuley S, et al. Analysis of the acute and chronic wound environments: the role of proteases and their inhibitors. Wound Repair Regen 1999; 7:442–452. [DOI] [PubMed] [Google Scholar]
- 25.Eming SA, Kaufmann J, Lohrer R, et al. [Chronic wounds. Novel approaches in research and therapy]. Hautarzt 2007; 58:939–944. [DOI] [PubMed] [Google Scholar]
- 26.Zhu J, Nathan C, Jin W, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell 2002; 111:867–878. [DOI] [PubMed] [Google Scholar]
- 27.Slater TW, Finkielsztein A, Mascarenhas LA, et al. Neutrophil microparticles deliver active myeloperoxidase to injured mucosa to inhibit epithelial wound healing. J Immunol 2017; 198:2886–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warnatsch A, Ioannou M, Wang Q, et al. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015; 349:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knight JS, Luo W, O’Dell AA, et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res 2014; 114:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Boer OJ, Li X, Teeling P, et al. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb Haemost 2013; 109:290–297. [DOI] [PubMed] [Google Scholar]
- 31.Pertiwi KR, van der Wal AC, Pabittei DR, et al. Neutrophil extracellular traps participate in all different types of thrombotic and haemorrhagic complications of coronary atherosclerosis. Thromb Haemost 2018; 118:1078–1087. [DOI] [PubMed] [Google Scholar]
- 32.Laridan E, Denorme F, Desender L, et al. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol 2017; 82:223–232. [DOI] [PubMed] [Google Scholar]
- 33.Mangold A, Alias S, Scherz T, et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res 2015; 116:1182–1192. [DOI] [PubMed] [Google Scholar]
- 34.Oklu R, Albadawi H, Jones JE, et al. Reduced hind limb ischemia-reperfusion injury in Toll-like receptor-4 mutant mice is associated with decreased neutrophil extracellular traps. J Vasc Surg 2013; 58:1627–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim SW, Lee H, Lee HK, et al. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol Commun 2019; 7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, Xie T, Sun S, et al. DNase-1 treatment exerts protective effects in a rat model of intestinal ischemia-reperfusion injury. Sci Rep 2018; 8:17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boettcher M, Eschenburg G, Mietzsch S, et al. Therapeutic targeting of extracellular DNA improves the outcome of intestinal ischemic reperfusion injury in neonatal rats. Sci Rep 2017; 7:15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ge L, Zhou X, Ji WJ, et al. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol 2015; 308:H500–H509. [DOI] [PubMed] [Google Scholar]
- 39.Peer V, Abu Hamad R, Berman S, et al. Renoprotective effects of DNAse-I treatment in a rat model of ischemia/reperfusion-induced acute kidney injury. Am J Nephrol 2016; 43:195–205. [DOI] [PubMed] [Google Scholar]
- 40.von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 2012; 209:819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med 2008; 359:938–949. [DOI] [PubMed] [Google Scholar]
- 42.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood 2014; 123:2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta AK, Joshi MB, Philippova M, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett 2010; 584:3193–3197. [DOI] [PubMed] [Google Scholar]
- 44.Kessenbrock K, Krumbholz M, Schonermarck U, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med 2009; 15:623–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saffarzadeh M, Juenemann C, Queisser MA, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One 2012; 7:e32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lam FW, Cruz MA, Parikh K, et al. Histones stimulate von Willebrand factor release in vitro and in vivo. Haematologica 2016; 101:e277–e279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reyes-Garcia AML, Aroca A, Arroyo AB, et al. Neutrophil extracellular trap components increase the expression of coagulation factors. Biomed Rep 2019; 10:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glaser CB, Morser J, Clarke JH, et al. Oxidation of a specific methionine in thrombomodulin by activated neutrophil products blocks cofactor activity. A potential rapid mechanism for modulation of coagulation. J Clin Invest 1992; 90:2565–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Massberg S, Grahl L, von Bruehl ML, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 2010; 16:887–896. [DOI] [PubMed] [Google Scholar]
- 50.Varju I, Longstaff C, Szabo L, et al. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb Haemost 2015; 113:1289–1298. [DOI] [PubMed] [Google Scholar]
- 51.Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 2010; 107:15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuchs TA, Brill A, Wagner DD. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol 2012; 32:1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011; 118:1952–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 2007; 13:463–469. [DOI] [PubMed] [Google Scholar]
- 55.Brill A, Fuchs TA, Savchenko AS, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost 2012; 10:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jimenez-Alcazar M, Rangaswamy C, Panda R, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017; 358:1202–1206. [DOI] [PubMed] [Google Scholar]
- 57.Novotny J, Chandraratne S, Weinberger T, et al. Histological comparison of arterial thrombi in mice and men and the influence of Cl-amidine on thrombus formation. PLoS One 2018; 13:e0190728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Biron BM, Chung CS, O’Brien XM, et al. Cl-Amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun 2017; 9:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knight JS, Subramanian V, O’Dell AA, et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis 2015; 74:2199–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ham A, Rabadi M, Kim M, et al. Peptidyl arginine deiminase-4 activation exacerbates kidney ischemia-reperfusion injury. Am J Physiol Renal Physiol 2014; 307:F1052–F1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grilz E, Mauracher LM, Posch F, et al. Citrullinated histone H3, a biomarker for neutrophil extracellular trap formation, predicts the risk of mortality in patients with cancer. Br J Haematol 2017; 186:1548–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tohme S, Simmons RL, Tsung A. Surgery for cancer: a trigger for metastases. Cancer Res 2017; 77:1548–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murthy SM, Goldschmidt RA, Rao LN, et al. The influence of surgical trauma on experimental metastasis. Cancer 1989; 64:2035–2044. [DOI] [PubMed] [Google Scholar]
- 64.Liotta LA, Kleinerman J, Saidel GM. Quantitative relationships of intravascular tumor cells, tumor vessels, and pulmonary metastases following tumor implantation. Cancer Res 1974; 34:997–1004. [PubMed] [Google Scholar]
- 65.Cools-Lartigue J, Spicer J, McDonald B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Najmeh S, Cools-Lartigue J, Rayes RF, et al. Neutrophil extracellular traps sequester circulating tumor cells via beta1-integrin mediated interactions. Int J Cancer 2017; 140:2321–2330. [DOI] [PubMed] [Google Scholar]
- 67.Tohme S, Yazdani HO, Al-Khafaji AB, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res 2016; 76:1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Al-Haidari AA, Algethami N, Lepsenyi M, et al. Neutrophil extracellular traps promote peritoneal metastasis of colon cancer cells. Oncotarget 2019; 10:1238–1249. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Albrengues J, Shields MA, Ng D, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018; 361:eaao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park J, Wysocki RW, Amoozgar Z, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 2016; 8:361ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Caudrillier A, Kessenbrock K, Gilliss BM, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest 2012; 122:2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Suri R. The use of human deoxyribonuclease (rhDNase) in the management of cystic fibrosis. BioDrugs 2005; 19:135–144. [DOI] [PubMed] [Google Scholar]
- 73.Pressler T. Review of recombinant human deoxyribonuclease (rhDNase) in the management of patients with cystic fibrosis. Biologics 2008; 2:611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Papayannopoulos V, Metzler KD, Hakkim A, et al. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 2010; 191:677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pedersen F, Waschki B, Marwitz S, et al. Neutrophil extracellular trap formation is regulated by CXCR2 in COPD neutrophils. Eur Respir J 2018; 51:1700970. [DOI] [PubMed] [Google Scholar]