

Abstract
Exposure to arsenic in contaminated drinking water is a worldwide public health problem that affects more than 200 million people. Protein quality control constitutes an evolutionarily conserved mechanism for promoting proper folding of proteins, refolding of misfolded proteins, and removal of aggregated proteins, thereby maintaining homeostasis of the proteome (i.e., proteostasis). Accumulating lines of evidence from epidemiological and laboratory studies revealed that chronic exposure to inorganic arsenic species can elicit proteinopathies that contribute to neurodegenerative disorders, cancer, and type II diabetes. Here, we review the effects of arsenic exposure on perturbing various elements of the proteostasis network, including mitochondrial homeostasis, molecular chaperones, inflammatory response, ubiquitinproteasome system, autophagy, as well as asymmetric segregation and axonal transport of misfolded proteins. We also discuss arsenic-induced disruptions of post-translational modifications of proteins, for example, ubiquitination, and their implications in proteostasis. Together, studies in the past few decades support that disruption of protein quality control may constitute an important mechanism underlying the arsenic-induced toxicity.
Graphical Abstract
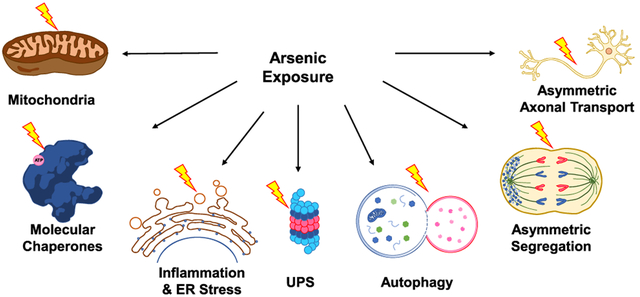
1. INTRODUCTION
Natural occurrence and anthropogenic activities render arsenic species ubiquitously present in the environment.1 Arsenic contamination in drinking water is a major public health concern in the modern world, where exposure to inorganic arsenic (iAs) in contaminated drinking water or agricultural products impacts approximately 200 million people in over 70 nations.1 Along this line, exposure to arsenic is thought to contribute to the etiology of many human diseases, including cancer,2,3 neurodegenerative diseases,4,5 and type II diabetes.6,7 Multiple mechanisms are thought to contribute to arsenic-elicited human diseases including binding to cysteine sulfhydryl groups in proteins, induction of reactive oxygen species, disruption of DNA repair, and perturbation of epigenetic pathways of gene regulation, etc.3,8,9
In cells, proteins need to be properly folded into their native three-dimensional structures so as to execute their biological functions. This is a challenging task, especially in the context that numerous nascently synthesized polypeptides must fold properly in crowded intracellular environment and they must maintain appropriate folding under a wide range of physiological and environmental stress conditions.10–12 To maintain homeostasis of the proteome (i.e., proteostasis), cells are equipped with sophisticated, yet highly conserved protein quality control machineries, collectively known as the proteostasis network.10,11
Proteostasis network comprises cellular machineries regulating the production, folding, trafficking, degradation, and clearance of proteins.10,11 In this vein, approximately 30% of proteins in higher eukaryotes possess extensive intrinsically unstructured regions (>30 amino acids in length), which render these proteins metastable and toxic upon aggregation.13 Therefore, a robust proteostasis network is particularly critical for maintaining correct folding and minimizing aggregation of proteins.
2. METABOLIC TRANSFORMATIONS OF ARSENIC SPECIES
Toxicity of inorganic arsenic (iAs), in both trivalent (iAs3+) and pentavalent (iAs5+) states, in mammals depends largely on their metabolic transformations. The majority of ingested iAs (As3+ or As5+) is absorbed by the gastrointestinal tract.14 In liver, As5+ can be reduced by glutathione (GSH) to yield iAs3+, which can undergo iterative oxidative methylation, catalyzed by arsenite methyltransferase (As3MT), and GSH-mediated reduction to yield organic arsenic species, including monomethylarsonic acid (MMAV), monomethylarsonous acid (MMAIII), dimethylarsinic acid (DMAV), and dimethylarsinous acid (DMAIII).15 The different chemical forms of arsenic exhibit variations in cellular uptake, efflux, and retention.16–18 For instance, higher cytotoxicity of MMAIII and DMAIII over iAs3+ is associated with greater cellular uptake and retention of the methylated arsenic species.16 As a result, it is important to consider both inorganic arsenic species and their methylated metabolites when considering arsenic toxicity.
3. ARSENIC-INDUCED DISRUPTION OF PROTEOSTASIS NETWORK
Chronic exposure to arsenic species was shown to induce aberrant folding and aggregation of proteins,19,20 which may overwhelm the capacity of proteostasis network and engender a self-propagating, vicious cycle of proteotoxic stress. In the following sections, we review the various protein quality control machineries that can be disrupted in cells upon arsenic exposure (Figure 1).
Figure 1.
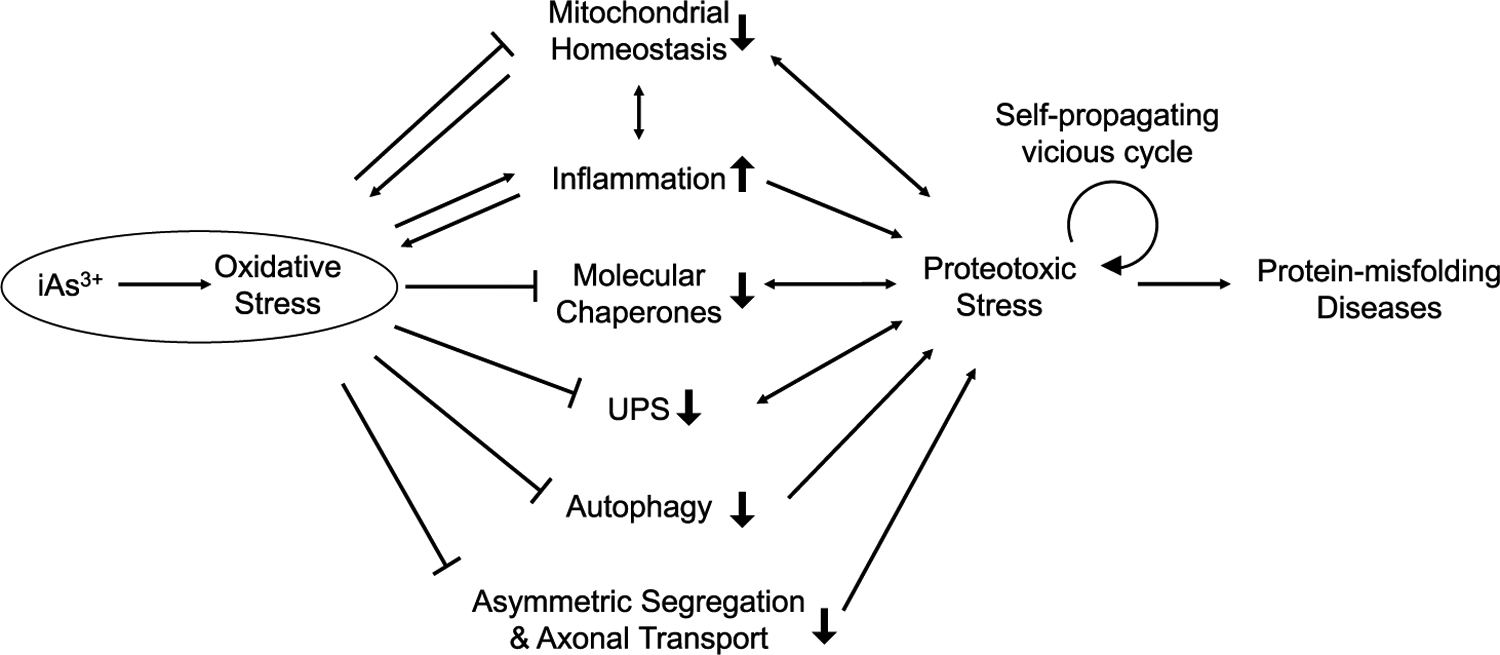
Schematic diagram illustrating the molecular mechanisms through which iAs3+-elicited oxidative stress induces proteotoxicity via targeting various elements of the proteostasis network, resulting in protein-misfolding diseases. Double-headed arrows denote the mutual interaction between the designated components of the proteostasis network and proteotoxic stress.
3.1. Mitochondrial Homeostasis.
Mitochondria are critical organelles in cellular proteostasis owing to their multiple roles in cellular physiology and in shaping cellular decisions for life or death.21 Mitochondria are important sources of intracellular reactive oxygen species (ROS).22,23 Given the detrimental effects of oxidative stress on proteostasis, mitochondrial homeostasis is intimately linked with the proteostasis network,21 as manifested by the observations of mitochondrial dysfunctions in human diseases associated with proteotoxic stress including cancer and neurodegenerative diseases.24
Growing lines of evidence from epidemiological, animal, and cellular studies revealed that exposure to iAs can result in mitochondrial dysfunctions,25–28 which can occur via (i) mitochondrial DNA damage,25,26,29 (ii) uncoupling of mitochondrial respiration through metabolic reprogramming,27,30,31 (iii) excessive production of ROS,25,28,32 and(iv) augmented proton leak from ROS-induced depolarization and damage of mitochondrial membrane (Table 1).27
Table 1.
Summary of Key Findings Associated with Arsenic-Induced Compromised Protein Quality Control
| components of the proteostasis network | experimental system | Reagent used | concentration; exposure time | refs |
|---|---|---|---|---|
| Mitochondrial Homeostasis | ||||
| mitochondrial DNA damage | AL human-hamster hybrid cells, CHOK1 cells stably expressing a single copy of human chromosome 11 | NaAsO2 | 0.5–1 μg/mL; 60 d | 26 |
| uncoupling of mitochondrial respiration through metabolic reprogramming | C. elegans | NaAsO2 | 50, 250, or 500 μM; 12–48 h | 27 |
| Cultured human cells (BEAS-2B, RWPE-1, A549, HUC and HDF) | NaAsO2 | 0, 1, or 5 μM; 1–2 wk | 30 | |
| BEAS-2B, cancer stem-like cells | NaAsO2 | 0.25 μM; 3–7 d | 31 | |
| excessive production of ROS | AL hybrid cells | NaAsO2 | 2 μg/mL; 5 min | 25 |
| 3xTgAD mouse model | NaAsO2 | 3 ppm (0.2 mg/kg/day); 6 mo | 28 | |
| elevated proton leak by ROS-induced depolarization and lipid peroxidation | C. elegans | NaAsO2 | 0, 50, or 500 μM; 48 h | 27 |
| Molecular Chaperones | ||||
| interference with substrate binding to chaperone proteins | S. cerevisiae | NaAsO2 | 1 mM; 1 h | 42 |
| disruption of activities of ATP-dependent chaperones | HeLa cells | NaAsO2 | 50, 100, 200, 400, 500 μM; 1–4 h | 43 |
| induction of protein aggregation of molecular chaperones | S. cerevisiae | NaAsO2 | 50, 100, 200, 400, 600 μM; 120 min | 41 |
| 1-day-old male chickens/cocks | As2O3 | 7.5, 15, and 30 mg/kg | 45 | |
| In vitro (e.g., murine NIH-3T3 cells) and in vivo (e.g., rodent) | NaAsO2 | 1–300 uM; 1–16 h (in vitro); 0.8–6 mg/kg; 1–24 h (in vivo) | 44 | |
| GM00637 human fibroblast cells | NaAsO2 | 5 μM; 24 h | 46 | |
| C. elegans | NaAsO2 | 100 mM; 4 d | 47 | |
| Pro-inflammatory Pathways and ER Stress | ||||
| activation of inflammatory response | porcine cells from fresh aortas | NaAsO2 | 0.5, 2, 5 μM; 0.5–2 h | 53 |
| circulating lymphocytes from arsenic-exposed human subjects | Blood arsenic | 0–4.32, 4.64–9.00, 9.60–46.50 μg/L | 54 | |
| human infants born to arsenic-exposed mothers | Arsenic | maternal toenail ≥0.5 μg/g | 55 | |
| female C57BL/6 mice (6–7 weeks old) | NaAsO2 | 0, 2.5, 5, 10 mg/kg; 24 h | 56 | |
| male Sprague-Dawley rats | NaAsO2 | 0, 5, 10, 20, 50, 100 ppb; 7, 14, or 28 d | 57 | |
| reduction of phagocytic receptors on immune cells | exposed rural women from districts of 24 Parganas (south) | arsenic | 11–50 μg/L; lifetime (22–45 yr) | 58 |
| induction of ER stress | RIN-m5F rat insulinoma pancreatic β-cell line | As2O3 | 0, 2, 5 μM; 8, 16, 24 h | 61 |
| SKH-1 hairless mice | NaAsO2 | 50, 100, 200 ppm; 1 mo | 62 | |
| RAW 264.7 mouse macrophage cells | As2O3 | 0, 0.2, 2 μM; 1 or 3 h | 63 | |
| Neuro-2a murine neuroblastoma cells | As2O3 | 5 μM; 6, 12, or 24 h | 64 | |
| SVEC4–10 mouse vascular endothelium cells | As2O3 | 5 or 7.4 μM; 16 h | 65 | |
| enhanced APP processing and Aβ oligomerization | cholinergic SN56.B5.G4 cells, primary neuronal cells derived from transgenic Tg2576 mice overexpressing human APPswe | NaAsO2 or DMA | 5 or 10 μM; 12 or 24 h | 66 |
| male Wistar rats | NaAsO2 | 3.80 ppm; 68 d | 67 | |
| Ubiquitin-Proteasome System | ||||
| impairment of enzymatic activities of ZnF-containing E3 ubiquitin ligases | GM00637, HeLa cells | NaAsO2 | 1, 2, 4, 5 μM; 24 h | 73 |
| HEK293T cells | NaAsO2 | 1, 3, 5 μM; 24 h | 74 | |
| GM00637, IMR90, and HEK293T cells | NaAsO2 | 5 or 20 μM; 8 or 24 h | 75 | |
| NB4 APL cells | As2O3 | 1–2 μM; 1–24 h | 77 | |
| compromising functions of p97 and proteasome | NIH3T3, DTC25 cells | arsenate | 50 or 250 μM; 0–25 min or 16 h | 72 |
| Autophagy | ||||
| impairment of TORC1-mediated protein sequestration to autophagosome | BEAS-2B cells | NaAsO2 | 0.25 μM; 24 h, 1 wk or 16 wk | 85 |
| S. cerevisiae | NaAsO2 | 50, 100, 200, 500 μM; 5, 10, or 30 min | 88 | |
| inhibition of autophagic flux via sustained Nrf2 activation | HEK293T cells | NaAsO2 | 1, 3, 5 μM; 24 h | 74 |
| BEAS-2B, HBE cells | NaAsO2 | 125, 250, 500 nM, 1, 4, or 10 uM; 4 or 16 h | 84 | |
| HaCaT human keratinocytes | NaAsO2 | 8 uM; 6 h | 89 | |
| suppression of autophagosome-lysosome Fusion | NIH3T3 cells | NaAsO2 | 0.25, 0.5, 1, or 2 μM; 2 or 4 h | 86 |
| HeLa, HEK293T, NIH3T3, iBMKs cells | NaAsO2 | 0, 0.5, 1, 2 μM; 4 h | 87 | |
| sustained overproduction of IL-6 | BEAS-2B cells | NaAsO2 | 0.25 μM; 24 h, 1 wk or 16 wk | 85 |
| Asymmetric Segregation and Axonal Transport of Damaged Proteins | ||||
| disturbed mitotic progression and aberrant positioning of mitotic spindles | G2-enriched HFW cells | NaAsO2 | 5 μM; 18 h | 99 |
| HeLa-S3, CGL2 cells | NaAsO2 | 1 or 3 μM; 20 h | 100 | |
| attenuated microtubule dynamics and abnormal mitotic morphologies | HeLa-S3 cells | NaAsO2 | 5, 10, 20, 50 μM; 1, 2, or 24 h | 102 |
| Asymmetric Segregation and Axonal Transport of Damaged Proteins | ||||
| inhibition of TRiC for folding and oligomerization of cytoskeleton components | S. cerevisiae | NaAsO2 | 1 mM; 3 h | 42 |
| decreased light subunit of neurofilament | male Wistar rats | NaAsO2 | 15 or 20 mg/kg; 3, 6, or 9 h | 106 |
| augmented phosphorylation of tau | Chinese hamster ovary (CHO) T40 cells | NaAsO2 | 500 μM; 2 h | 107 |
Multiple studies documented that exposure to iAs3+ could elicit mitochondrial DNA damage, which could be alleviated by treating cells with an oxygen radical scavenger.25,26,29 Arsenite was shown to augment glycolysis, diminish the function of electron transport chain, and disrupt pyruvate metabolism in nematodes and human cells.27,30 This so-called Warburg-like effect may originate from inhibition of mitochondrial metabolic enzymes, for example, pyruvate dehydrogenase, through arsenite-induced ROS.33 The arsenate-mediated impairment of oxidative phosphorylation may also stem, in part, from the formation of ADP-arsenate.34
Trivalent arsenicals were shown to trigger the production of mitochondrial ROS in human cells and Caenorhabditis elegans by uncoupling mitochondrial respiration and eliciting mitochondrial dysfunction including altered metabolism, diminished intracellular ATP, and increased proton leak.27,30 In this vein, As3+ exposure was shown to lead to reduced steady-state ATP levels in cells, partly through the aforementioned disruption of pyruvate metabolism.27,33
Several rodent studies indicated that subchronic and chronic exposure to arsenite (i.e., 1–3 months) reduced the activities of mitochondrial antioxidant enzymes, decreased intracellular glutathione level, and augmented lipid peroxidation and protein oxidation.35,36 This suggests that arsenite exposure triggers oxidative stress, which inactivates mitochondrial enzymes35 and damages mitochondrial membrane via protein oxidation and lipid peroxidation,36,37 thereby inducing proton leak.27
Apart from enzyme inhibition and membrane damage in mitochondria, chronic (i.e., 90 days) exposure to low concentration of arsenite elicits a dose-dependent elevation in expression of mitochondrial transcription factor A (mtTFA) in RWPE-1 cells.38 This mtTFA-mediated alteration in mitochondrial signaling, which is believed to arise from arsenic-elicited oxidative stress, leads to mitochondrial DNA damage and elevated levels of mitochondrial ROS while enhancing the survival of cells under proteotoxic stress.38
Lastly, mitochondria are interconnected with other components of the protein quality control (PQC) systems.21
3.2. Molecular Chaperones.
Molecular chaperones are key modulators of the proteostasis network by sensing and proof-reading misfolded polypeptides to prevent misfolded proteins from aggregation and to actively refold non-native structural ensembles into their correct folding state.39 This chaperone network is also involved in triage decisions of misfolded proteins for de novo folding, refolding, or sorting to other PQC machineries.40
Exposure to trivalent arsenic has been shown to disrupt the functions of molecular chaperones.41 This may proceed through: (i) interfering with the binding of chaperone proteins with their substrates42 and (ii) disrupting the activities of ATP-dependent chaperones through inhibition of ATP production in mitochondria (Table 1).43
As part of the cellular stress response, iAs3+ was found to induce the expression of chaperone proteins, especially heat shock proteins HSP70 and sHSPs.44–46 Additionally, chronic arsenite exposure was shown to up-regulate heat shock proteins via activation of DAF-16/FOXO transcription factor through oxidative stress.47
Exposure to iAs3+ was observed to suppress protein folding and stimulate the formation of protein aggregates in yeast cells.41 iAs3+ was also shown to bind to cysteine residues in reduced forms of those proteins without correct folding, thereby inhibiting spontaneous oxidative protein folding (i.e., through disulfide bond formation) and suppressing chaperone-mediated disaggregation/refolding of proteins in vitro.41,48,49 Additionally, this inhibition of oxidative refolding can be aggravated by iAs3+-induced depletion of intracellular reduced glutathione, which binds to iAs3+ and suppresses its interaction with cysteine sulfhydryl groups in proteins.48
Arsenic exposure may also perturb the functions of molecular chaperones by inducing their aggregation.41 For instance, Hsp104p in yeast cells was found to undergo aggregation and redistribution upon exposure to iAs3+, which may exacerbate protein aggregation by disrupting cells’ ability in dissolving misfolded protein aggregates in the presence of free iAs3+.41
As noted above, arsenic exposure can disrupt mitochondrial homeostasis, which stimulates ROS production and attenuates intracellular ATP levels. Therefore, iAs3+ can inhibit the binding of substrates to chaperones and diminish ATP supply for ATP-dependent chaperones, as observed for the TRiC chaperonin complex in Saccharomyces cerevisiae.42 Along this line, we reason that DnaJ, which harbors two Cys4-type zinc fingers,50 may also be inhibited by iAs3+ binding and ROS, thereby impairing its recognition of denatured protein substrates and its regulation of HSP70.51
3.3. Induction of Pro-inflammatory Pathways and ER Stress.
Inflammation is an immune response triggered by pathogen infection or tissue injury; chronic inflammatory response not only activates innate and adaptive immune cells but also releases ROS.52 A wide range of cellular, animal, and epidemiological studies showed that exposure to iAs3+ can activate pro-inflammatory signaling pathways, which aggravate oxidative stress and compromise proteome integrity.53–57 Acute (i.e., 24 h) and chronic (i.e., 90 days) exposure of laboratory animals to iAs3+ induces pro-inflammatory modulators while reducing the expression of phagocytic receptors on immune cells (e.g., monocytes and granulocytes) and CD4+ T cell subpopulation in serum, lung, brain, spleen, and thymus.56–58 Likewise, cellular and epidemiological studies revealed that exposure to low doses of iAs3+ and MMAIII can lead to persistent inflammation.53,54,59,60 Moreover, Fry et al.55 observed that prenatal arsenic exposure results in NF-κB activation in infants (Table 1).
Molecular mechanisms have been proposed to account for the arsenic-elicited inflammatory response. This inflammatory effect is believed to originate from iAs3+- or MMAIII-induced oxidative stress, which up-regulates ERK1/2, JNK, p38, and their downstream transcription factors including NF-κB, AP-1, and Nrf2.56,60 Prolonged inflammation also initiates ER stress and unfolded protein response (UPR), where iAs3+-induced up-regulations of GRP78, IRE1, ATF4, and ATF6α exacerbate protein misfolding and aggregation.61–65
Last but not least, several cellular studies reveal that exposure to iAs3+ or DMAV significantly augments the expression of APP gene, which is accompanied by diminished expression of peroxisome proliferator-activated receptor-γ (PPARγ), a negative regulator of BACE-1 (β-secretase).66–68 Furthermore, iAs3+-elicited oxidative stress induces a prooxidant and inflammatory environment, which facilitates APP processing and increases Aβ oligomerization, thereby exacerbating amyloidogenesis and pathogenesis of Alzheimer’s disease (Table 1).68
3.4. Impairment of Protein Clearance and Degradation Machineries.
Exposure to iAs3+ in drinking water also impairs the protein clearance and degradative machineries in the proteostasis network through disrupting the ubiquitinproteasome system (UPS), autophagy, as well as asymmetric segregation and axonal transport of damaged proteins (Table 1).
3.4.1. UPS.
UPS is the major cytoplasmic machinery responsible for the degradation of the majority of short-lived, damaged, or misfolded proteins in eukaryotic cells.69 UPS consists of complex ubiquitination cascades mediated by a concerted network consisting of E1, E2, and E3 enzymes, ubiquitin-modifying enzymes (e.g., deubiquitinating enzymes, SUMO-dependent ubiquitin ligases), and the 26S proteasome.69 In this pathway, misfolded proteins destined for turnover are conjugated with ubiquitin, especially with the K48-linked polyubiquitin chains, recognized by p97 (i.e., an AAA+ ATPase) in complex with ubiquitin adaptors, and degraded by the 20S proteasome.70,71
Arsenic species were shown to perturb the efficient turnover of misfolded and aggregated proteins through disrupting the functions of the UPS.72 This may proceed through (i) impairing the enzymatic activities of zinc finger (ZnF)-containing E3 ubiquitin ligases73–75 and (ii) compromising the functions of p97 and proteasome (Table 1).72
iAs3+ can bind directly and selectively to Cys4- and Cys3His1-type ZnF motifs, and this binding displaces their bound Zn2+ ions.76 In addition, iAs3+ interacts directly with a number of RING finger (i.e., a specific type of ZnF) proteins, some of which are involved in protein ubiquitination.73–75,77 In this vein, an acute 24-h exposure to arsenite diminishes, in a dose-dependent manner, the enzymatic activities of RNF20-RNF40, Rbx1, and FANCL RING finger E3 ubiquitin ligases, as manifested by the decreased ubiquitinations of their substrate proteins.73–75 This raises the possibility that exposure to iAs3+ may inhibit other ZnF-containing regulatory proteins in the UPS, especially RING finger E3 ubiquitin ligases.78
Arsenate exposure also perturbs the functions of p97 and proteasome.72 The ATP-dependent segregase activity of p97 is critical for extracting ubiquitinated, misfolded proteins for ATP-driven protein unfolding prior to proteasomal degradation.71 The optimal functions of p97 and proteasome, which depend on a well-timed ATPase cycle, are crucial for the maintenance of cellular proteostasis,71 as manifested by the observations of impairment of p97 in neurodegenerative diseases.79,80 Interestingly, arsenate enhances the ATPase activities of p97, which augments ATP hydrolysis rate and diminishes the activity of p97.72 Moreover, iAs3+ exposure leads to depletion of cellular ATP pool (vide supra),43 which may disrupt cells’ ability to empower the ATP-dependent clearance of misfolded proteins via the UPS.
3.4.2. Autophagy.
Autophagy is an evolutionarily conserved process in eukaryotes, where long-lived proteins, macro-molecular assemblies, and dysfunctional organelles (e.g., mitochondria) are eliminated through segregation into autophagosomes and subsequent degradation in lysosomes.81 Autophagy, as another critical degradative machinery of the proteostasis network,82 is often activated to enhance protein turnover under stress conditions, and loss of autophagy can contribute to neurological diseases.83
Growing lines of evidence indicate that arsenic exposure can disrupt autophagy,84–87 and this occurs through (i) impairment of TORC1-mediated protein sequestration into autophagosome,85,88 (ii) inhibition of autophagic flux via sustained Nrf2 activation,84,89 and (iii) suppression of autophagosome-lysosome fusion (Table 1).86,87
TORC1 is a protein kinase known for its regulatory role in eliminating insoluble protein aggregates through sequestration into autophagic vesicles (a.k.a. autophagosomes) in cells.88,90 Interestingly, acute exposure of S. cerevisiae to high concentrations of iAs3+ (50–200 μM, 10 min) and chronic exposure of BEAS-2B cells to a low dose of iAs3+ (250 nM, 16 weeks) both lead to TORC1 inhibition.85,88
iAs3+ has also been proposed to inhibit autophagic flux, which perturbs proteostasis through inefficient elimination of protein aggregates.84 Acute exposure to low doses of iAs3+ (125–500 nM, 4 h) was shown to dysregulate autophagic influx in various human cell lines through prolonged activation of Nrf2, and this process depends on p62, but independent of Cys151 in Keap1.84 Interestingly, Nrf2 protein can also be activated upon a 24-h exposure to 5 μM iAs3+, which involves the direct binding of As3+ to the RING finger domain of Rbx1 and the ensuing inhibition of the activity of the Keap1-Cul3-Rbx1 E3 ubiquitin ligase complex.74 This gives rise to diminished ubiquitination and turnover of its substrate protein(i.e., Nrf2).74
Low concentration of iAs3+ inhibits the fusion between autophagosome and lysosome,87 which involves three SNARE proteins (STX17, SNAP29, and VAMP8).91 O-GlcNAcylation of SNAP29 suppresses autophagosome-lysosome fusion and results in aberrant autophagic flux by suppressing the formation of the active SNARE complex (STX17-SNAP29-VAMP8).91 In addition, Dodson et al.87 demonstrated that acute iAs3+ exposure (0.5–2 μM, 4 h) augments the OGlcNAcylation of SNAP29, which subsequently perturbs the formation of the SNARE complex and suppresses autophagic flux. Lastly, iAs3+-induced inhibition of autophagy also proceeds through sustained overproduction of IL-6, which enhances the interaction between Mcl-1 and Beclin-1 through STAT3.85
3.4.3. Asymmetric Segregation and Axonal Transport of Damaged Proteins.
Under normal circumstances, the above mentioned cellular PQC systems have adequate capacity to dispose and eliminate misfolded or damaged proteins. However, once the amount of misfolded proteins exceeds the degradative capacity of the proteostasis network in higher eukaryotes, these misfolded proteins can aggregate and be partitioned into aggresomes at the microtubule-organizing center (MTOC) in the forms of juxta nuclear quality control compartment (JUNQ) and insoluble protein deposit (IPOD).92,93 Interestingly, during cytokinesis, damaged (e.g., carbonyl modified) and aggregated proteins in aggresomes are asymmetrically segregated to mother cells, whereas daughter cells are devoid of these aberrant proteins through a polarisome- and tropomyosin-dependent polarized flow of misfolded proteins.94,95 Therefore, this spatial PQC confers cellular fitness and prevents clonal senescence at the expense of mother cells under stress conditions.96,97
Proper asymmetric segregation of protein aggregates during cell division largely depends on the correct orientation and position of the mitotic spindle.98 iAs3+ is well documented to disturb mitotic progression.99–101 Specifically, exposure to iAs3+ attenuates microtubule dynamics and induces abnormal mitotic morphologies including augmented polar distance and abnormal patterns of assembly/disassembly of mitotic spindles in exposed cells.102 Additionally, acute exposure to low levels of iAs3+ disrupts the positioning of mitotic spindles through the PIP4KIIγ/Rho pathways (Table 1).99
Asymmetric axonal transport of misfolded and damaged proteins is also critical to proteostasis in neurons, as reflected by aberrant accumulation of damaged organelles and proteins in human neurodegenerative diseases manifesting axonal pathologies.103 Axonal integrity is maintained by neurofilaments, microtubules, and actin filaments, which were shown to be impaired upon iAs3+ exposure.104,105 As noted above, the eukaryotic chaperonin TRiC can be inhibited by iAs3+, and this inhibition perturbs the folding and subsequent oligomerization of cytoskeleton components (e.g., α/β-tubulin and actin) into microtubules and actin filaments.42 A single exposure of rodents to iAs3+ via intravenous injection leads to a time- and dose-dependent diminution in light subunit of neurofilaments, which compromises axonal transport.104,105 Therefore, iAs3+-elicited disruption of cytoskeletal protein components in axons impairs protein clearance by perturbing asymmetric axonal transport.
Hyperphosphorylated tau may also influence axonal transport.106 iAs3+ exposure significantly increases the phosphorylation levels of several amino acid residues in tau (i.e., Thr-181, Ser-202, Thr-205, Thr-231, Ser-262, Ser-356, Ser-396, and Ser-404).107 The hyperphosphorylated tau can trigger microtubule depolymerization, owing to its diminished affinity toward microtubules and augmented formation of amorphous tangles;108 this may increase the likelihood of developing Alzheimer’s disease upon arsenic exposure.109
Taken together, iAs3+ exposure compromises cytoskeleton functions in cells, which affect asymmetric axonal transport and partitioning of protein aggregates during cell division, thereby impairing the clearance of damaged proteins (Table 1).
4. CONCLUSIONS AND PERSPECTIVES
In this review, we surveyed recent epidemiological and laboratory-based studies on the effects of arsenic exposure on compromised protein quality control and discussed various pathways through which iAs exposure perturbs the proteostasis network and leads to pathogenesis of human diseases associated with protein misfolding. The physiological regulation of the proteostasis network involves a complex interplay of multiple pathways.10,11,40 Exposure to environmentally relevant levels of iAs3+ targets multiple elements of the proteostasis network, encompassing mitochondria, chaperone network, immune systems, ubiquitin-proteasome system, autophagosome, and cytoskeleton. Hence, chronic arsenic exposure can result in a progressive decline in the capacity of refolding and clearance of misfolded/aggregated proteins, and the robustness of stress response pathways (Figures 2 and 3), thereby resulting in protein-misfolding diseases.
Figure 2.
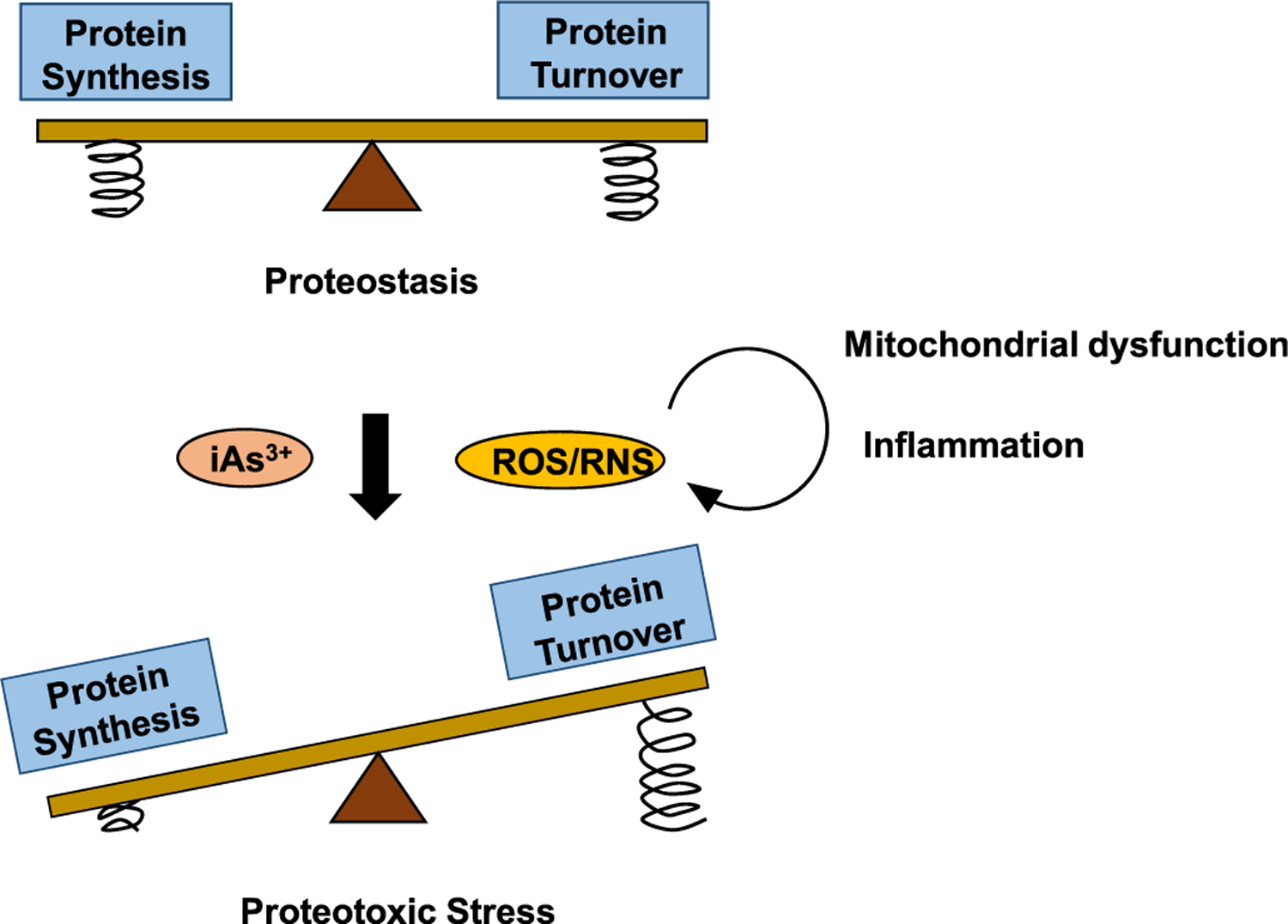
Proteostasis is achieved by sustaining a balance between protein synthesis and protein turnover, with arsenic exposure and arsenic-elicited overproduction of ROS/RNS tipping the equilibrium away through impairing the degradative capacity of the proteostasis network. Arsenic exposure diminishes the folding capacity of molecular chaperones, resulting in the increased formation of misfolded/aggregated proteins. In addition, arsenic exposure and ROS/RNS impair the UPS, autophagy, and asymmetric segregation and axonal transport of damaged proteins, reducing the degradative capacity of the proteostasis network. Mitochondrial dysfunction and inflammation induced by arsenic exposure form a vicious self-feeding cycle of excessive ROS/RNS production, further aggravating proteotoxic stress through the imbalance between protein synthesis and turnover. Compression spring in the image represents the feedback mechanisms regulating the PQC machineries of protein synthesis or those of protein turnover. Arsenic and ROS/RNS can perturb the feedback signaling pathways involved in the PQC.
Figure 3.
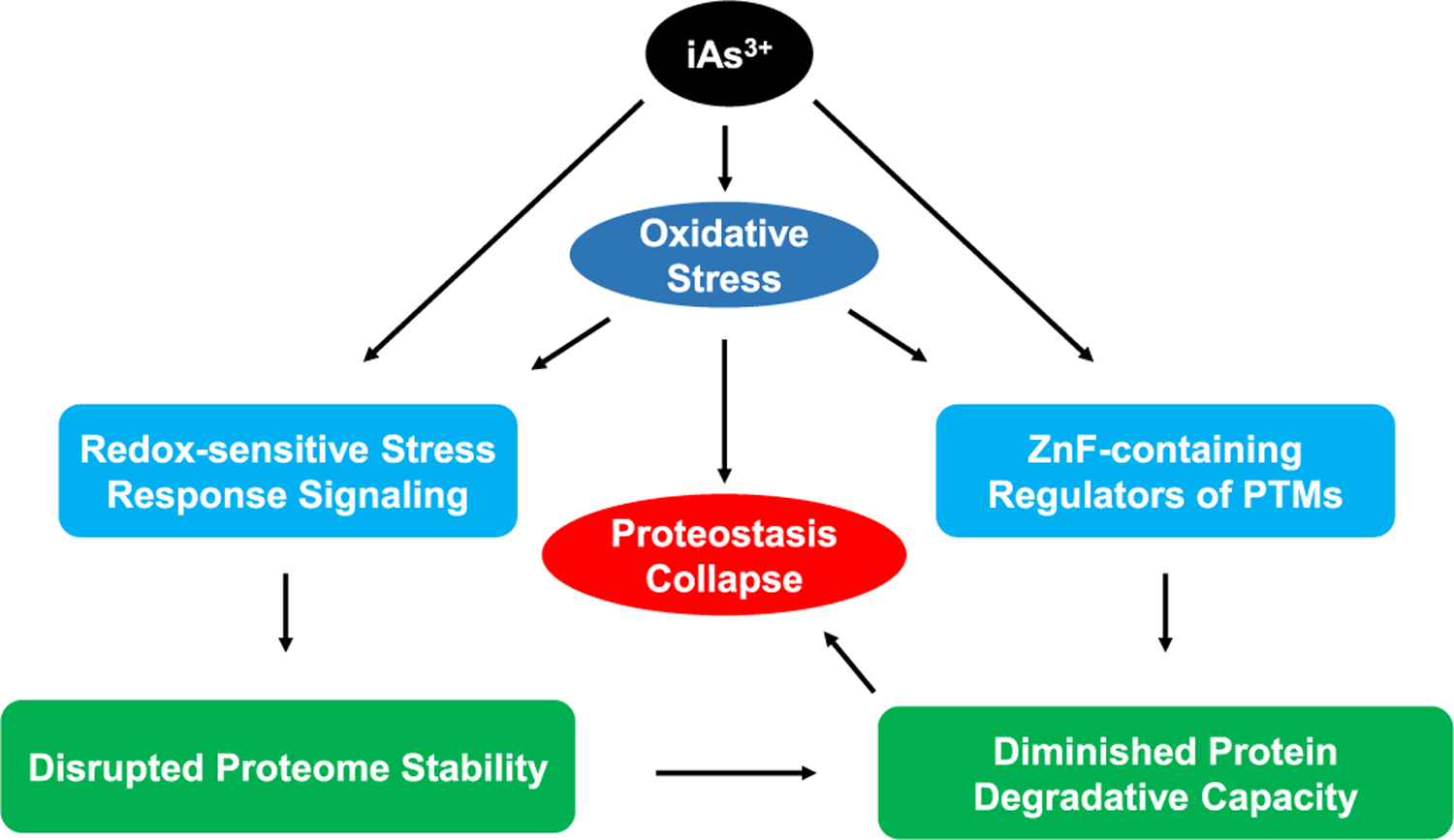
iAs3+ and iAs-induced oxidative stress elicit proteostasis collapse through disrupting the functions of ZnF proteins and redox-sensitive stress response signaling pathways. iAs-elicited oxidative stress and iAs3+ itself can disrupt post-translational modifications (e.g., ubiquitination, SUMOylation, and PARylation), which are mediated by ZnF-containing enzymes and critical for robust degradative capacity of the proteostasis network. Meanwhile, iAs3+ and ROS/RNS can impair redox-active stress response signaling (e.g., oxidative stress response, heavy metal response), which prolong cellular functions at the expense of disrupted proteome stability. Combined together, the diminished protein degradative capacity of the proteostasis network leads to proteotoxicity, ultimately resulting in protein-misfolding diseases.
Apart from excessive ROS production, the findings cited in this review also suggest that the binding of iAs3+ with ZnF proteins, especially those RING finger E3 ubiquitin ligases involved in protein ubiquitination,73–75,77 constitutes one of the molecular mechanisms underlying arsenic-induced loss of proteostasis. In addition, exposure to iAs3+ induces ROS and reactive nitrogen species (RNS), including NO, may result in inadvertent S-nitrosylation of redox-sensitive cysteine thiolates in critical proteins, for example, poly(ADP-ribose) polymerase 1.110 Thus, arsenite-elicited ROS/RNS, in conjunction with direct iAs3+ binding to sulfhydryl groups of critical cysteines in proteins, may also represent an important mechanism for the exacerbated proteostasis capacity in arsenic-elicited protein-misfolding diseases (Figure 4).
Figure 4.
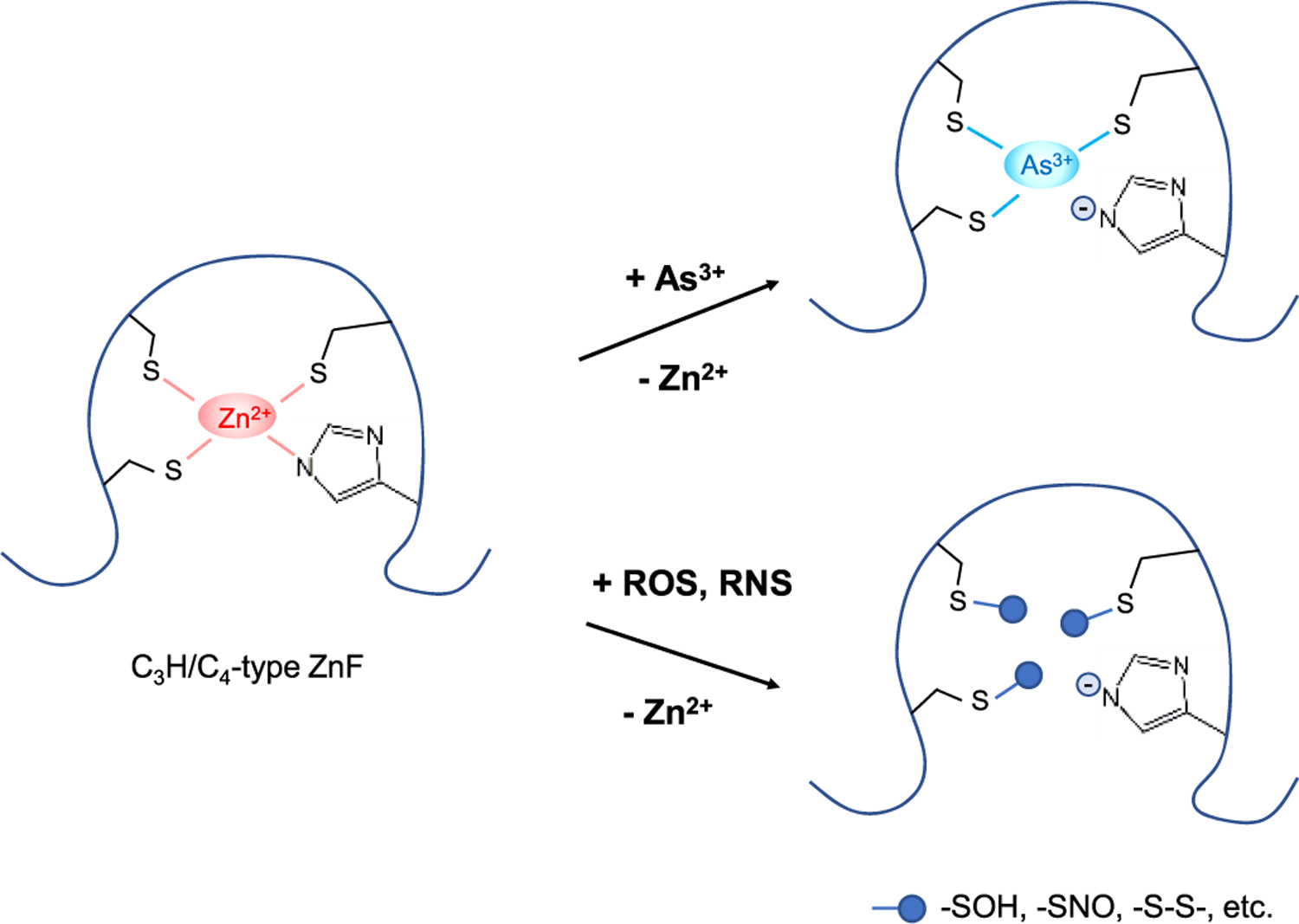
Modes of action of iAs3+ and iAs-elicited ROS/RNS in compromising the enzymatic activities of ZnF proteins. iAs3+ and ROS/RNS can target vicinal cysteines within the zinc coordination spheres of these proteins: (i) iAs3+ binds directly to these cysteines more tightly than Zn2+; (ii) ROS modify these cysteines to yield a series of oxidation products, such as −SOH and –S–S–; (iii) RNS, especially NO and peroxynitrite, can S-nitrosylate these cysteines. In all these cases, Zn2+ ions are released from the ZnF domains, which alter the conformations of ZnF proteins and perturbs their enzymatic activities.
Although a lot has been learned about the arsenic-elicited disruption of the proteostasis network, much remains to be done to further explore their implications in public health and to illustrate the underlying molecular mechanisms. First, a majority of the previously published studies were conducted by employing moderate to high concentrations of arsenic species (i.e., over 5 μM in 24 h) in cell-based systems. It will be important to examine whether the findings from these previous studies can be extended to exposure conditions using lower concentrations of arsenic species that are more commonly encountered during environmental exposure or cancer therapy.111 In this vein, it will be vital to differentiate the perturbations of the proteostasis network arising from the cytotoxic effects of high-dose exposure from those emanating from physiologically relevant doses of arsenic exposure. Moreover, inorganic arsenic species are known to undergo metabolic transformations to yield organic arsenic species in cells and tissues, as discussed above.15 Hence, some of observed proteotoxic effects revealed from cellular and animal studies with the use of inorganic arsenic species may arise, in part, from their methylated metabolites.
Second, ribosome-associated protein quality control (RQC) pathway was recently found to be initiated and modulated by regulatory ubiquitinations mediated through several E3 ubiquitin ligases, which enable quality control of translational infidelity at ribosomes.112–115 RQC is an evolutionarily conserved and energetically beneficial pathway for cells to predict, detect, and remove any potential errors at the earliest time points and locations from which potentially erroneous nascent polypeptides originate.116 On the basis of the well-documented interactions between As3+ and ZnF proteins, it will be important to examine if iAs can also perturb RQC by binding to and impairing important E3 ubiquitin ligases in this pathway (e.g., ZNF598 and listerin).116 Along this line, a recent study showed that iAs3+ binds to ZNF598 and perturbs its regulatory ubiquitinations of ribosomal proteins RPS10 and RPS20, thereby promoting ribosomal read-through of the stalling poly(adenosine) sequence in the coding region.117
Third, prolonged exposure to arsenite was shown to accelerate the natural processes of aging, as manifested by significant decrease of lifespan in C. elegans mediated by the DAF-16/FOXO transcription factor.47 The close interconnection between FOXO transcription factors and sirtuin proteins,118 along with the known disruption of sirtuin 1 and DAF-16/FOXO by chronic low-dose iAs3+ exposure,47,119 suggests a mechanistic basis for the iAs3+-induced disruption of proteostasis through the common pathways associated with longevity and stress response.
Lastly, many ZnF proteins are critical regulators of protein PTMs, especially ubiquitination, SUMOylation, and PARylation;120 thus, it will be important to systematically investigate how exposure to iAs3+ modulates these PTMs at the entire proteome level. It will also be important to examine how arsenic exposure perturbs proteostasis by compromising the cross-talk among ubiquitination, SUMOylation, and PARylation of proteins.121 For instance, SUMOylation can modulate proteostasis by not only altering a protein’s solubility but also cooperating with, complementing, and balancing the ubiquitinproteasome system.121 These studies will offer comprehensive insights into how As3+ exposure compromises the proteostasis network by modulating PTMs of cellular proteins.
ACKNOWLEDGMENTS
The authors would like to thank the National Institutes of Health for supporting this research (R35 ES031707).
Biographies
Lok Ming Tam earned his B.S. degree in Cell and Molecular Biology from Chinese University of Hong Kong (Hong Kong SAR) in 2014 and studied Environmental Toxicology at the University of California Davis for 1 year. He subsequently joined the graduate program in Environmental Toxicology at the University of California Riverside. He worked under the guidance of Prof. Yinsheng Wang and received his Ph.D. degree in late 2019. His research interests include protein quality control and etiology of human diseases, with a special focus on the arsenic impairment of proteostasis.
Yinsheng Wang is currently a Distinguished Professor in the Chemistry Department and the Environmental Toxicology graduate program at the University of California Riverside. His research interest includes DNA damage and mutagenesis, proteomics, and epigenetics. In recent years, he also developed a strong interest in understanding the molecular mechanisms through which arsenic exposure elicits toxicity and carcinogenicity. His co-workers employ an multidisciplinary approach, encompassing mass spectrometry-based bioanalytical chemistry, organic chemistry, molecular biology, genetics, and genomics, to tackle their research projects.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.chemrestox.0c00107
The authors declare no competing financial interest.
Contributor Information
Lok Ming Tam, Environmental Toxicology Graduate Program, University of California, Riverside, California 92521, United States.
Yinsheng Wang, Environmental Toxicology Graduate Program and Department of Chemistry, University of California, Riverside, California 92521, United States.
REFERENCES
- (1).Ravenscroft P, Brammer H, and Richards K (2011) Arsenic pollution: a global synthesis, Vol. 94, John Wiley & Sons. [Google Scholar]
- (2).Smith AH, Hopenhayn-Rich C, Bates MN, Goeden HM, Hertz-Picciotto I, Duggan HM, Wood R, Kosnett MJ, and Smith MT (1992) Cancer risks from arsenic in drinking water. Environ. Health Perspect 97, 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kitchin KT (2001) Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol. Appl. Pharmacol 172, 249–261. [DOI] [PubMed] [Google Scholar]
- (4).Vahidnia A, van der Voet GB, and de Wolff FA (2007) Arsenic neurotoxicity–a review. Hum. Exp. Toxicol 26, 823–832. [DOI] [PubMed] [Google Scholar]
- (5).Chen Y, Parvez F, Gamble M, Islam T, Ahmed A, Argos M, Graziano JH, and Ahsan H (2009) Arsenic exposure at low-to-moderate levels and skin lesions, arsenic metabolism, neurological functions, and biomarkers for respiratory and cardiovascular diseases: review of recent findings from the Health Effects of Arsenic Longitudinal Study (HEALS) in Bangladesh. Toxicol. Appl. Pharmacol 239, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tseng CH, Tai TY, Chong CK, Tseng CP, Lai MS, Lin BJ, Chiou HY, Hsueh YM, Hsu KH, and Chen CJ (2000) Long-term arsenic exposure and incidence of non-insulin dependent diabetes mellitus: a cohort study in arseniasis-hyper-endemic villages in Taiwan. Environ. Health Perspect 108, 847–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Navas-Acien A, Silbergeld EK, Pastor-Barriuso R, and Guallar E (2008) Arsenic exposure and prevalence of type 2 diabetes in US adults. JAMA 300, 814–822. [DOI] [PubMed] [Google Scholar]
- (8).Shen S, Li XF, Cullen WR, Weinfeld M, and Le XC (2013) Arsenic binding to proteins. Chem. Rev 113, 7769–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tam LM, Price NE, and Wang Y (2020) Molecular mechanisms of arsenic-induced disruption of DNA repair. Chem. Res. Toxicol 33, 709–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Labbadia J, and Morimoto RI (2015) The biology of proteostasis in aging and disease. Annu. Rev. Biochem 84, 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Balchin D, Hayer-Hartl M, and Hartl FU (2016) In vivo aspects of protein folding and quality control. Science 353, No. aac4354. [DOI] [PubMed] [Google Scholar]
- (12).Klaips CL, Jayaraj GG, and Hartl FU (2018) Pathways of cellular proteostasis in aging and disease. J. Cell Biol 217, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dunker AK, Silman I, Uversky VN, and Sussman JL (2008) Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol 18, 756–764. [DOI] [PubMed] [Google Scholar]
- (14).Vahter M, and Norin H (1980) Metabolism of 74As-labeled trivalent and pentavalent inorganic arsenic in mice. Environ. Res 21, 446–457. [DOI] [PubMed] [Google Scholar]
- (15).Thomas DJ, Styblo M, and Lin S (2001) The cellular metabolism and systemic toxicity of arsenic. Toxicol. Appl. Pharmacol 176, 127–144. [DOI] [PubMed] [Google Scholar]
- (16).Drobna Z, Waters SB, Devesa V, Harmon AW, Thomas DJ, and Styblo M (2005) Metabolism and toxicity of arsenic in human urothelial cells expressing rat arsenic (+3 oxidation state)-methyltransferase. Toxicol. Appl. Pharmacol 207, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Calatayud M, Gimeno J, Velez D, Devesa V, and Montoro R (2010) Characterization of the intestinal absorption of arsenate, monomethylarsonic acid, and dimethylarsinic acid using the Caco-2 cell line. Chem. Res. Toxicol 23, 547–556. [DOI] [PubMed] [Google Scholar]
- (18).Calatayud M, Barrios JA, Velez D, and Devesa V (2012) In vitro study of transporters involved in intestinal absorption of inorganic arsenic. Chem. Res. Toxicol 25, 446–453. [DOI] [PubMed] [Google Scholar]
- (19).Tamas MJ, Sharma SK, Ibstedt S, Jacobson T, and Christen P (2014) Heavy metals and metalloids as a cause for protein misfolding and aggregation. Biomolecules 4, 252–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cholanians AB, Phan AV, Ditzel EJ, Camenisch TD, Lau SS, and Monks TJ (2016) Arsenic induces accumulation of α-synuclein: implications for synucleinopathies and neurodegeneration. Toxicol. Sci 153, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Andreasson C, Ott M, and Buttner S (2019) Mitochondria orchestrate proteostatic and metabolic stress responses. EMBO Rep. 20, No. e47865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Balaban RS, Nemoto S, and Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120, 483–495. [DOI] [PubMed] [Google Scholar]
- (23).Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem. J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet 39, 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Liu S-X, Davidson MM, Tang X, Walker WF, Athar M, Ivanov V, and Hei TK (2005) Mitochondrial damage mediates genotoxicity of arsenic in mammalian cells. Cancer Res. 65, 3236–3242. [DOI] [PubMed] [Google Scholar]
- (26).Partridge MA, Huang SX, Hernandez-Rosa E, Davidson MM, and Hei TK (2007) Arsenic induced mitochondrial DNA damage and altered mitochondrial oxidative function: implications for genotoxic mechanisms in mammalian cells. Cancer Res. 67, 5239–5247. [DOI] [PubMed] [Google Scholar]
- (27).Luz AL, Godebo TR, Bhatt DP, Ilkayeva OR, Maurer LL, Hirschey MD, and Meyer JN (2016) Arsenite uncouples mitochondrial respiration and induces a Warburg-like effect in Caenorhabditis elegans. Toxicol. Sci 152, 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Niño SA, Morales-Martínez A, Chi-Ahumada E, Carrizales L, Salgado-Delgado R, Perez-Severiano F, Díaz-Cintra S, Jiménez-Capdeville ME, and Zarazúa S (2019) Arsenic exposure contributes to the bioenergetic damage in an Alzheimer’s disease model. ACS Chem. Neurosci 10, 323–336. [DOI] [PubMed] [Google Scholar]
- (29).Hei TK, Liu SX, and Waldren C (1998) Mutagenicity of arsenic in mammalian cells: role of reactive oxygen species. Proc. Natl. Acad. Sci. U. S. A 95, 8103–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhao F, Severson P, Pacheco S, Futscher BW, and Klimecki WT (2013) Arsenic exposure induces the Warburg effect in cultured human cells. Toxicol. Appl. Pharmacol 271, 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Li L, Bi Z, Wadgaonkar P, Lu Y, Zhang Q, Fu Y, Thakur C, Wang L, and Chen F (2019) Metabolic and epigenetic reprogramming in the arsenic-induced cancer stem cells. Semin. Cancer Biol 57, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Flora SJ (2011) Arsenic-induced oxidative stress and its reversibility. Free Radical Biol. Med 51, 257–281. [DOI] [PubMed] [Google Scholar]
- (33).Samikkannu T, Chen C-H, Yih L-H, Wang AS, Lin S-Y, Chen T-C, and Jan K-Y (2003) Reactive oxygen species are involved in arsenic trioxide inhibition of pyruvate dehydrogenase activity. Chem. Res. Toxicol 16, 409–414. [DOI] [PubMed] [Google Scholar]
- (34).Gresser MJ (1981) ADP-arsenate. Formation by submitochondrial particles under phosphorylating conditions. J. Biol. Chem 256, 5981–5983. [PubMed] [Google Scholar]
- (35).Dwivedi N, Mehta A, Yadav A, Binukumar B, Gill KD, and Flora SJ (2011) MiADMSA reverses impaired mitochondrial energy metabolism and neuronal apoptotic cell death after arsenic exposure in rats. Toxicol. Appl. Pharmacol 256, 241–248. [DOI] [PubMed] [Google Scholar]
- (36).Kadeyala PK, Sannadi S, and Gottipolu RR (2013) Alterations in apoptotic caspases and antioxidant enzymes in arsenic exposed rat brain regions: reversal effect of essential metals and a chelating agent. Environ. Toxicol. Pharmacol 36, 1150–1166. [DOI] [PubMed] [Google Scholar]
- (37).Kumar MR, Flora S, and Reddy G (2013) Monoisoamyl 2, 3-dimercaptosuccinic acid attenuates arsenic induced toxicity: behavioral and neurochemical approach. Environ. Toxicol. Pharmacol 36, 231–242. [DOI] [PubMed] [Google Scholar]
- (38).Singh KP, Kumari R, Treas J, and DuMond JW (2011) Chronic exposure to arsenic causes increased cell survival, DNA damage, and increased expression of mitochondrial transcription factor A (mtTFA) in human prostate epithelial cells. Chem. Res. Toxicol 24, 340–349. [DOI] [PubMed] [Google Scholar]
- (39).Hartl FU, Bracher A, and Hayer-Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. [DOI] [PubMed] [Google Scholar]
- (40).Sontag EM, Samant RS, and Frydman J (2017) Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem 86, 97–122. [DOI] [PubMed] [Google Scholar]
- (41).Jacobson T, Navarrete C, Sharma SK, Sideri TC, Ibstedt S, Priya S, Grant CM, Christen P, Goloubinoff P, and Tamás MJ (2012) Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J. Cell Sci 125, 5073–5083. [DOI] [PubMed] [Google Scholar]
- (42).Pan X, Reissman S, Douglas NR, Huang Z, Yuan DS, Wang X, McCaffery JM, Frydman J, and Boeke JD (2010) Trivalent arsenic inhibits the functions of chaperonin complex. Genetics 186, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Yih LH, Huang HM, Jan KY, and Lee TC (1991) Sodium arsenite induces ATP depletion and mitochondrial damage in HeLa cells. Cell Biol. Int. Rep 15, 253–264. [DOI] [PubMed] [Google Scholar]
- (44).Del Razo LM, Quintanilla-Vega B, Brambila-Colombres E, Calderón-Aranda ES, Manno M, and Albores A (2001) Stress proteins induced by arsenic. Toxicol. Appl. Pharmacol 177, 132–148. [DOI] [PubMed] [Google Scholar]
- (45).Zhang K, Zhao P, Guo G, Guo Y, Li S, He Y, Sun X, Chai H, Zhang W, and Xing M (2016) Arsenic trioxide exposure induces heat shock protein responses in cock livers. Biol. Trace Elem. Res 170, 459–465. [DOI] [PubMed] [Google Scholar]
- (46).Zhang F, Xiao Y, and Wang Y (2017) SILAC-based quantitative proteomic analysis unveils arsenite-induced perturbation of multiple pathways in human skin fibroblast cells. Chem. Res. Toxicol 30, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Yu C-W, How CM, and Liao VH-C (2016) Arsenite exposure accelerates aging process regulated by the transcription factor DAF-16/FOXO in Caenorhabditis elegans. Chemosphere 150, 632–638. [DOI] [PubMed] [Google Scholar]
- (48).Ramadan D, Rancy PC, Nagarkar RP, Schneider JP, and Thorpe C (2009) Arsenic (III) species inhibit oxidative protein folding in vitro. Biochemistry 48, 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sharma SK, Goloubinoff P, and Christen P (2011) Non-native proteins as newly-identified targets of heavy metals and metalloids, in Cellular Effects of Heavy Metals, pp 263–274, Springer. [Google Scholar]
- (50).Szabo A, Korszun R, Hartl FU, and Flanagan J (1996) A zinc finger-like domain of the molecular chaperone DnaJ is involved in binding to denatured protein substrates. EMBO J. 15, 408–417. [PMC free article] [PubMed] [Google Scholar]
- (51).Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, and Bukau B (1999) Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. U. S. A 96, 5452–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Mittal M, Siddiqui MR, Tran K, Reddy SP, and Malik AB (2014) Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signaling 20, 1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Barchowsky A, Dudek EJ, Treadwell MD, and Wetterhahn KE (1996) Arsenic induces oxidant stress and NF-kB activation in cultured aortic endothelial cells. Free Radical Biol. Med 21, 783–790. [DOI] [PubMed] [Google Scholar]
- (54).Wu M-M, Chiou H-Y, Ho I-C, Chen C-J, and Lee T-C (2003) Gene expression of inflammatory molecules in circulating lymphocytes from arsenic-exposed human subjects. Environ. Health Perspect 111, 1429–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Fry RC, Navasumrit P, Valiathan C, Svensson JP, Hogan BJ, Luo M, Bhattacharya S, Kandjanapa K, Soontararuks S, and Nookabkaew S (2007) Activation of inflammation/NF-κB signaling in infants born to arsenic-exposed mothers. PLoS Genet. 3, e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Duan X, Gao S, Li J, Wu L, Zhang Y, Li W, Zhao L, Chen J, Yang S, Sun G, et al. (2017) Acute arsenic exposure induces inflammatory responses and CD4+ T cell subpopulations differentiation in spleen and thymus with the involvement of MAPK, NF-kB, and Nrf2. Mol. Immunol 81, 160–172. [DOI] [PubMed] [Google Scholar]
- (57).Zhao Y, Su X, Gao Y, Yin H, Wang L, Qiao R, and Wang S (2019) Exposure of low-concentration arsenic-initiated inflammation and autophagy in rat lungs. J. Biochem. Mol. Toxicol 33, No. e22334. [DOI] [PubMed] [Google Scholar]
- (58).Prasad P, and Sinha D (2017) Low-level arsenic causes chronic inflammation and suppresses expression of phagocytic receptors. Environ. Sci. Pollut. Res 24, 11708–11721. [DOI] [PubMed] [Google Scholar]
- (59).Vega L, Styblo M, Patterson R, Cullen W, Wang C, and Germolec D (2001) Differential effects of trivalent and pentavalent arsenicals on cell proliferation and cytokine secretion in normal human epidermal keratinocytes. Toxicol. Appl. Pharmacol 172, 225–232. [DOI] [PubMed] [Google Scholar]
- (60).Escudero-Lourdes C, Medeiros M, Cárdenas-González M, Wnek S, and Gandolfi J (2010) Low level exposure to monomethyl arsonous acid-induced the over-production of inflammation-related cytokines and the activation of cell signals associated with tumor progression in a urothelial cell model. Toxicol. Appl. Pharmacol 244, 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Lu TH, Su CC, Chen YW, Yang CY, Wu CC, Hung DZ, Chen CH, Cheng PW, Liu SH, and Huang CF (2011) Arsenic induces pancreatic beta-cell apoptosis via the oxidative stress-regulated mitochondria-dependent and endoplasmic reticulum stress-triggered signaling pathways. Toxicol. Lett 201, 15–26. [DOI] [PubMed] [Google Scholar]
- (62).Li C, Xu J, Li F, Chaudhary SC, Weng Z, Wen J, Elmets CA, Ahsan H, and Athar M (2011) Unfolded protein response signaling and MAP kinase pathways underlie pathogenesis of arsenic-induced cutaneous inflammation. Cancer Prev. Res 4, 2101–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Srivastava RK, Li C, Chaudhary SC, Ballestas ME, Elmets CA, Robbins DJ, Matalon S, Deshane JS, Afaq F, Bickers DR, et al. (2013) Unfolded protein response (UPR) signaling regulates arsenic trioxide-mediated macrophage innate immune function disruption. Toxicol. Appl. Pharmacol 272, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Lu TH, Tseng TJ, Su CC, Tang FC, Yen CC, Liu YY, Yang CY, Wu CC, Chen KL, Hung DZ, and Chen YW (2014) Arsenic induces reactive oxygen species-caused neuronal cell apoptosis through JNK/ERK-mediated mitochondria-dependent and GRP 78/CHOP-regulated pathways. Toxicol. Lett 224, 130–140. [DOI] [PubMed] [Google Scholar]
- (65).Weng C-Y, Chiou S-Y, Wang L, Kou M-C, Wang Y-J, and Wu M-J (2014) Arsenic trioxide induces unfolded protein response in vascular endothelial cells. Arch. Toxicol 88, 213–226. [DOI] [PubMed] [Google Scholar]
- (66).Zarazúa S, Bürger S, Delgado JM, Jiménez-Capdeville ME, and Schliebs R (2011) Arsenic affects expression and processing of amyloid precursor protein (APP) in primary neuronal cells overexpressing the Swedish mutation of human APP. Int. J. Dev. Neurosci 29, 389–396. [DOI] [PubMed] [Google Scholar]
- (67).Ashok A, Rai NK, Tripathi S, and Bandyopadhyay S (2015) Exposure to As-, Cd-, and Pb-mixture induces Abeta, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol. Sci 143, 64–80. [DOI] [PubMed] [Google Scholar]
- (68).Escudero-Lourdes C (2016) Toxicity mechanisms of arsenic that are shared with neurodegenerative diseases and cognitive impairment: role of oxidative stress and inflammatory responses. NeuroToxicology 53, 223–235. [DOI] [PubMed] [Google Scholar]
- (69).Ciechanover A (2005) Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol 6, 79–87. [DOI] [PubMed] [Google Scholar]
- (70).Saeki Y (2017) Ubiquitin recognition by the proteasome. J. Biochem 161, 113–124. [DOI] [PubMed] [Google Scholar]
- (71).van den Boom J, and Meyer H (2018) VCP/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol. Cell 69, 182–194. [DOI] [PubMed] [Google Scholar]
- (72).Tillotson J, Zerio CJ, Harder B, Ambrose AJ, Jung KS, Kang M, Zhang DD, and Chapman E (2017) Arsenic compromises both p97 and proteasome functions. Chem. Res. Toxicol 30, 1508–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Jiang J, Bellani M, Li L, Wang P, Seidman MM, and Wang Y (2017) Arsenite binds to the RING finger domain of FANCL E3 ubiquitin ligase and inhibits DNA interstrand crosslink repair. ACS Chem. Biol 12, 1858–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Jiang J, Tam LM, Wang P, and Wang Y (2018) Arsenite targets the RING finger domain of Rbx1 E3 ubiquitin ligase to inhibit proteasome-mediated degradation of Nrf2. Chem. Res. Toxicol 31, 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Zhang F, Paramasivam M, Cai Q, Dai X, Wang P, Lin K, Song J, Seidman MM, and Wang Y (2014) Arsenite binds to the RING finger domains of RNF20-RNF40 histone E3 ubiquitin ligase and inhibits DNA double-strand break repair. J. Am. Chem. Soc 136, 12884–12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Zhou X, Sun X, Cooper KL, Wang F, Liu KJ, and Hudson LG (2011) Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. J. Biol. Chem 286, 22855–22863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lallemand-Breitenbach V, Jeanne M, Zhang QY, Yang HY, Huang QH, Zhou GB, Tong JH, Zhang Y, Wu JH, Hu HY, de The H, Chen SJ, and Chen Z (2010) Arsenic trioxide controls the fate of the PMLRARs oncoprotein by directly binding PML. Science 328, 240–243. [DOI] [PubMed] [Google Scholar]
- (78).Lipkowitz S, and Weissman AM (2011) RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat. Rev. Cancer 11, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Hirabayashi M, Inoue K, Tanaka K, Nakadate K, Ohsawa Y, Kamei Y, Popiel AH, Sinohara A, Iwamatsu A, Kimura Y, Uchiyama Y, Hori S, and Kakizuka A (2001) VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 8, 977–984. [DOI] [PubMed] [Google Scholar]
- (80).Gitcho MA, Strider J, Carter D, Taylor-Reinwald L, Forman MS, Goate AM, and Cairns NJ (2009) VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J. Biol. Chem 284, 12384–12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Mizushima N (2007) Autophagy: process and function. Genes Dev. 21, 2861–2873. [DOI] [PubMed] [Google Scholar]
- (82).Ryter SW, Cloonan SM, and Choi AM (2013) Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol. Cells 36, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Menzies FM, Fleming A, and Rubinsztein DC (2015) Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci 16, 345–357. [DOI] [PubMed] [Google Scholar]
- (84).Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, and Zhang DD (2013) Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell. Biol 33, 2436–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Qi Y, Zhang M, Li H, Frank JA, Dai L, Liu H, Zhang Z, Wang C, and Chen G (2014) Autophagy inhibition by sustained overproduction of IL6 contributes to arsenic carcinogenesis. Cancer Res. 74, 3740–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Dodson M, de la Vega MR, Harder B, Castro-Portuguez R, Rodrigues SD, Wong PK, Chapman E, and Zhang DD (2018) Low-level arsenic causes proteotoxic stress and not oxidative stress. Toxicol. Appl. Pharmacol 341, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Dodson M, Liu P, Jiang T, Ambrose AJ, Luo G, de la Vega MR, Cholanians AB, Wong PK, Chapman E, and Zhang DD (2018) Increased O-GlcNAcylation of SNAP29 drives arsenic-induced autophagic dysfunction. Mol. Cell. Biol 38, No. e00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Hosiner D, Lempiäinen H, Reiter W, Urban J, Loewith R, Ammerer G, Schweyen R, Shore D, and Schüller C (2009) Arsenic toxicity to Saccharomyces cerevisiae is a consequence of inhibition of the TORC1 kinase combined with a chronic stress response. Mol. Biol. Cell 20, 1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Shah P, Trinh E, Qiang L, Xie L, Hu W-Y, Prins GS, Pi J, and He Y-Y (2017) Arsenic induces p62 expression to form a positive feedback loop with Nrf2 in human epidermal keratinocytes: implications for preventing arsenic-induced skin cancer. Molecules 22, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Peters TW, Rardin MJ, Czerwieniec G, Evani US, Reis-Rodrigues P, Lithgow GJ, Mooney SD, Gibson BW, and Hughes RE (2012) Tor1 regulates protein solubility in Saccharomyces cerevisiae. Mol. Biol. Cell 23, 4679–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovács AL, Zhang Z, et al. (2014) O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat. Cell Biol 16, 1215–1226. [DOI] [PubMed] [Google Scholar]
- (92).Ouellet J, and Barral Y (2012) Organelle segregation during mitosis: lessons from asymmetrically dividing cells. J. Cell Biol 196, 305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Ogrodnik M, Salmonowicz H, Brown R, Turkowska J, Średniawa W, Pattabiraman S, Amen T, Abraham A. c., Eichler N, Lyakhovetsky R, et al. (2014) Dynamic JUNQ inclusion bodies are asymmetrically inherited in mammalian cell lines through the asymmetric partitioning of vimentin. Proc. Natl. Acad. Sci. U. S. A 111, 8049–8054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Aguilaniu H, Gustafsson L, Rigoulet M, and Nyström T (2003) Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753. [DOI] [PubMed] [Google Scholar]
- (95).Liu B, Larsson L, Caballero A, Hao X,Öling D, Grantham J, and Nyström T (2010) The polarisome is required for segregation and retrograde transport of protein aggregates. Cell 140, 257–267. [DOI] [PubMed] [Google Scholar]
- (96).Lindner AB, Madden R, Demarez A, Stewart EJ, and Taddei F (2008) Asymmetric segregation of protein aggregates is associated with cellular aging and rejuvenation. Proc. Natl. Acad. Sci.U. S. A 105, 3076–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Erjavec N, Cvijovic M, Klipp E, and Nyström T (2008) Selective benefits of damage partitioning in unicellular systems and its effects on aging. Proc. Natl. Acad. Sci. U. S. A 105, 18764–18769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Kaltschmidt JA, Davidson CM, Brown NH, and Brand AH (2000) Rotation and asymmetry of the mitotic spindle direct asymmetric cell division in the developing central nervous system. Nat. Cell Biol 2, 7–12. [DOI] [PubMed] [Google Scholar]
- (99).Yih L-H, Ho I-C, and Lee T-C (1997) Sodium arsenite disturbs mitosis and induces chromosome loss in human fibroblasts. Cancer Res. 57, 5051–5059. [PubMed] [Google Scholar]
- (100).Yih L-H, Wu Y-C, Hsu N-C, and Kuo H-H (2012) Arsenic trioxide induces abnormal mitotic spindles through a PIP4KIIγ/Rho pathway. Toxicol. Sci 128, 115–125. [DOI] [PubMed] [Google Scholar]
- (101).States JC (2015) Disruption of mitotic progression by arsenic. Biol. Trace Elem. Res 166, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Huang S-C, and Lee T-C (1998) Arsenite inhibits mitotic division and perturbs spindle dynamics in HeLa S3 cells. Carcinogenesis 19, 889–896. [DOI] [PubMed] [Google Scholar]
- (103).De Vos KJ, Grierson AJ, Ackerley S, and Miller CC (2008) Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci 31, 151–173. [DOI] [PubMed] [Google Scholar]
- (104).Vahidnia A, van der Straaten R, Romijn F, Van Pelt J, van der Voet G, and De Wolff F (2007) Arsenic metabolites affect expression of the neurofilament and tau genes: An in-vitro study into the mechanism of arsenic neurotoxicity. Toxicol. In Vitro 21, 1104–1112. [DOI] [PubMed] [Google Scholar]
- (105).Vahidnia A, Romijn F, Tiller M, van der Voet GB, and de Wolff FA (2006) Arsenic-induced toxicity: effect on protein composition in sciatic nerve. Hum. Exp. Toxicol 25, 667–674. [DOI] [PubMed] [Google Scholar]
- (106).Rodriguez-Martin T, Cuchillo-Ibanez I, Noble W, Nyenya F, Anderton BH, and Hanger DP (2013) Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 34, 2146–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Giasson BI, Sampathu DM, Wilson CA, Vogelsberg-Ragaglia V, Mushynski WE, and Lee VM-Y (2002) The environmental toxin arsenite induces tau hyperphosphorylation. Biochemistry 41, 15376–15387. [DOI] [PubMed] [Google Scholar]
- (108).Busciglio J, Lorenzo A, Yeh J, and Yankner BA (1995) β-Amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14, 879–888. [DOI] [PubMed] [Google Scholar]
- (109).Savelieff MG, Lee S, Liu Y, and Lim MH (2013) Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem. Biol 8, 856–865. [DOI] [PubMed] [Google Scholar]
- (110).Zhou X, Cooper KL, Huestis J, Xu H, Burchiel SW, Hudson LG, and Liu KJ (2016) S-nitrosation on zinc finger motif of PARP-1 as a mechanism of DNA repair inhibition by arsenite. Oncotarget 7, 80482–80492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Wang ZY, and Chen Z (2008) Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 111, 2505–2515. [DOI] [PubMed] [Google Scholar]
- (112).Bengtson MH, and Joazeiro CA (2010) Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467, 470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Juszkiewicz S, Chandrasekaran V, Lin Z, Kraatz S, Ramakrishnan V, and Hegde RS (2018) ZNF598 is a quality control sensor of collided ribosomes. Mol. Cell 72, 469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Juszkiewicz S, and Hegde RS (2017) Initiation of quality control during poly(A) translation requires site-specific ribosome ubiquitination. Mol. Cell 65, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Sundaramoorthy E, Leonard M, Mak R, Liao J, Fulzele A, and Bennett EJ (2017) ZNF598 and RACK1 regulate mammalian ribosome-associated quality control function by mediating regulatory 40S ribosomal ubiquitylation. Mol. Cell 65, 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Brandman O, and Hegde RS (2016) Ribosome-associated protein quality control. Nat. Struct. Mol. Biol 23, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Tam LM, Jiang J, Wang P, and Wang Y (2020) Arsenite binds to ZNF598 to perturb ribosome-associated protein quality control. Chem. Res. Toxicol 33, DOI: 10.1021/acs.chemrestox.9b00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, and Auwerx J (2013) The NAD(+)/ sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Herbert KJ, Holloway A, Cook AL, Chin SP, and Snow ET (2014) Arsenic exposure disrupts epigenetic regulation of SIRT1 in human keratinocytes. Toxicol. Appl. Pharmacol 281, 136–145. [DOI] [PubMed] [Google Scholar]
- (120).Vilas CK, Emery LE, Denchi EL, and Miller KM (2018) Caught with one’s zinc fingers in the genome integrity cookie jar. Trends Genet. 34, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Liebelt F, and Vertegaal AC (2016) Ubiquitin-dependent and independent roles of SUMO in proteostasis. Am. J. Physiol.-Cell Physiol 311, C284–C296. [DOI] [PMC free article] [PubMed] [Google Scholar]