

Abstract
Despite numerous demonstrations that the immune system is activated in heart failure, negatively affecting patients' outcomes, no definitive treatment strategy exists directed to modulate the immune system. In this review, we present the evidence that B cells contribute to the development of hypertrophy, inflammation, and maladaptive tissue remodelling. B cells produce antibodies that interfere with cardiomyocyte function, which culminates as the result of recruitment and activation of a variety of innate and structural cell populations, including neutrophils, macrophages, fibroblasts, and T cells. As B cells appear as active players in heart failure, we propose here novel immunomodulatory therapeutic strategies that target B cells and their products.
Keywords: Heart failure, B cells, Inflammation, Therapeutics
Introduction
Heart failure (HF) is accompanied by a systemic pro‐inflammatory state, 1 in which both the innate and adaptive immune system participate. 2 , 3 Despite mounting evidence linking inflammation and HF, specific immunomodulatory therapies for HF have not been successfully developed. This is explained in part because not all the components of the immune system have been thoroughly investigated in the context of HF, as is the specific role that each immune cell plays in the heart. Germane to this discussion is the fact that the accumulated evidence shows that B cells, both directly (by secreting antibodies) and indirectly (by antigen presentation and cytokines/chemokines secretion), play an essential role in the progression of HF. Therefore, we suggest that addressing B cells more than B‐cell products in HF patients may serve as therapeutic alternatives for patients with treatment‐refractory HF. In this review, we present current evidence of the role of B cells in adverse cardiac remodelling, highlighting that this role is independent of aetiology, and introduce our ongoing investigations into novel immunomodulatory therapeutic strategies that target B cells and their products.
Antibody‐mediated mechanisms that contribute to cardiac dysfunction
Antibody‐mediated contribution to cardiac injury includes, on the one hand, direct consequences of anti‐cardiac antibodies binding to target cells and, on the other hand, the activation of the complement system following the formation of antigen–antibody complexes.
Direct effects of anti‐cardiac antibodies
In a study of end‐stage failing human myocardium tissue, we reported the presence of immunoglobulin G (IgG) deposits in up to 70% of heart tissue evaluated. Approximately 50% of biopsies stained positive for the IgG of the type 3 subclass, and a smaller proportion was also positive for C3c deposition. 4 As the IgG3 subclass exhibit the most effective complement‐fixating activity, 5 its presence in heart tissue can provide powerful signals of cell injury and to recruit inflammatory cells. Remarkably, the presence of IgG3 and C3c in the myocardium correlated significantly with the length and severity of illness. 4 , 6 The evidence provided by this large cohort and others demonstrates a strong association of failing myocardium with B‐cell activation and potentially B cell‐mediated injury in HF in humans. 4 , 6 , 7 An experimental model of ischaemic cardiac injury further supports the role of cardiac autoantibodies. 8 In this model, ischaemic injury in control mice produced myocardial infarction (MI) and depressed ejection fraction, while infarct size was reduced, with cardiac function improved in Ig‐deficient mice. 8
Autoantibodies might cause injury either by directly interacting with target receptors on heart cells or by triggering signals via their Fc‐binding domain when it is interacting with Fc γ‐receptors (FcγR) present on the surface of a variety of cells. 9 Activation of FcγR via the antibody Fc‐binding domain implies that antibody specificity is irrelevant for this pathway. Therefore, a large proportion of, or perhaps all, anti‐cardiac antibodies have these potential effects. FcγRs are present on cardiac fibroblasts 10 and cardiomyocytes. 11 FcγR signal transduction promotes fibrosis in cardiomyocytes and reduced calcium transients, cell shortening, and induction of cardiac cell death through activation of apoptotic pathways in myocytes 8 , 10 , 11 (Figure 1 A ).
Figure 1.
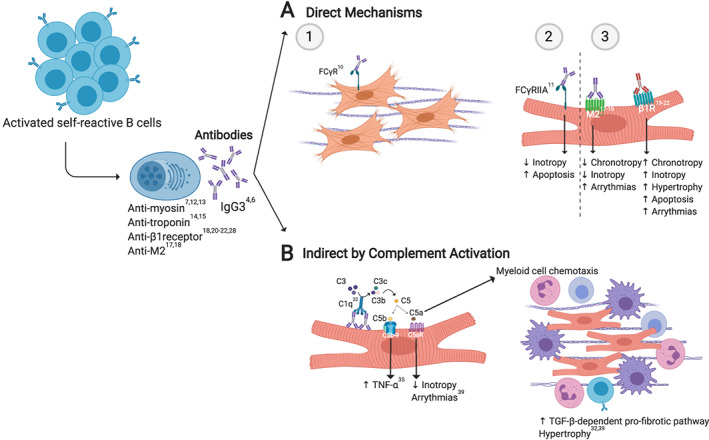
Antibody‐dependent mechanisms. (A) IgG3 antibodies can exert their function by being recognized through their Fc by Fc (fragment crystallizable region) γ‐receptor (FCγR) (1, 2) or by binding to specific surface receptors influencing their activity, as occurs with M2‐adrenergic and β1‐adrenergic receptors (3). (B) Antibody‐mediated disease may also induce the activation of the complement system via the classical pathway ending in membrane attack complex (C5b‐9) formation and chemotaxis of myeloid cells, allowing inflammation, fibrosis, and tissue dysfunction promoting hypertrophy and arrhythmias. C5aR, C5a receptor; TNF‐α, tumour necrosis factor‐α; TGF‐β, transforming growth factor‐β. Original image created with BioRender®.
Autoantibodies target a variety of proteins. In the context of HF, most identified antigens are present on the cell surface. However, a smaller number of intracellular proteins are also known as anti‐cardiac antibodies targets, such as sarcomere proteins (e.g. actin, myosin, and troponin). 7 , 12 , 13 , 14 , 15 The consequences of anti‐cardiac antibodies presence on heart physiology depend on their target and also on how they modulate activity (Table 1 ). For instance, the most extensively studied antibodies are those that target G protein‐coupled membrane receptors. 28 Anti‐M2 receptor antibodies have been associated with negative chronotropism at the sinoatrial level, 23 , 24 negative inotropism, 29 and supraventricular arrythmias. 24 In contrast, antibodies specific to the β1‐adrenergic receptor induce positive chronotropism and inotropism, 20 , 29 cardiac hypertrophy, desensitization to catecholamines, 21 and cardiomyocyte apoptosis. 22 Clinically, anti‐β‐receptor antibodies are the best characterized and are associated with compromised left ventricular function, 30 increased incidence of ventricular arrythmias, 21 , 23 and mortality in patients with HF. 21 , 31 (Figure 1 A ).
Table 1.
The specificity of cardiac autoantibodies identified in patients with HF
| Antibody specificity | Aetiology | Findings | Reference |
|---|---|---|---|
| Heart mitochondria: M7 (sarcosine dehydrogenase) | DCM, HCM, and acute myocarditis | React with heart mitochondria | Klein et al. 16 |
| Laminin | DCM and myocarditis | Unknown | Wolff et al. 17 |
| Hsp60 | DCM and AMI | Unknown | Latif et al. 7 |
| Actin, tropomyosin, and myosin light chain | DCM | Unknown | Latif et al. 7 |
| Adenine nucleotide translocator | DCM | Cytotoxic damage and enhanced calcium current in cardiac myocytes | Liao et al. 18 |
| β1‐ARs | DCM | Related to ventricular arrhythmias and sudden death, altering calcium management, modifying action potential, and apoptotic cell death | Liao et al. 19 ; Christ et al. 20 ; Iwata et al. 21 ; Jane‐wit et al. 22 ; and Chiale et al. 23 |
| M2‐muscarinic acetylcholine receptors | DCM | Play a role in mediating the development of atrial fibrillation probably by a sinus node dysfunction | Baba et al. 24 ; Chiale et al. 23 |
| Sarcolemmal Na‐K‐ATPase | DCM | Associated to ventricular tachycardia | Baba et al. 25 |
| Cardiac myosin | DCM and children myocarditis | Impair myocyte contractility and suggest being associated with protein kinase A activation and non‐recovery | Warraich et al. 12 ; Simpson et al. 13 |
| cTnI | DCM and AMI | Less ventricular function after acute myocardial infarction than patients with negative titres and associated with improved survival in patients with chronic DCM, but not ICM | Leuschner et al. 14 ; Doesch et al. 15 |
| KChIP2 | DCM and AMI | Associates with cell death on in vitro assays | Landsberger et al. 26 |
| ATP synthase β‐subunit | End‐stage HF | Unknown | Youker et al. 4 |
| CS | End‐stage HF | IgG CS autoantibodies in transplanted hearts of patients vs. natural IgM autoantibodies in healthy controls | Petrohai et al. 27 |
DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; AMI, acute myocardial infarction; β1‐ARs, β1‐adrenergic receptors; HF, heart failure; cTnI, cardiac troponin I; KChIP2, Kv channel‐interacting protein 2; CS, citrate synthase.
Anti‐cardiac tissue antibodies as activators of complement
Activation of the complement system is a well described outcome of the presence of antibodies. The complement system is an integral part of the innate immune response activated in HF through three pathways. The classical pathway is mediated by IgG and IgM antibodies, while the mannose‐binding lectin pathway and the alternative pathway depend on properdin (positive activator of complement activation) and plasma factor D. 4 , 32 , 33 , 34 All three pathways lead to the activation of plasma proteins in a coordinated manner by forming an enzymatic complex requiring the sequential formation of protein fragments. Activated complement may exhibit three downstream consequences: (i) induction of leukocyte chemotaxis by anaphylatoxins (C3a and C5a) through their respective receptors (C3aR and C5aR); (ii) opsonization (C3b, iC3b, and C3d) to facilitate transport and removal of immune complexes; and (iii) formation of the terminal membrane attack complex (C5b‐9) to directly lyse targeted cells 32 (Figure 1 B ).
As mentioned earlier, we have evidenced the presence of C3c in the myocardium, which correlated with the duration and severity of illness. 4 In a different study, HF patients exhibited increased circulating levels of the cleavage end product of complement activation C5b‐9 compared with healthy controls, and this similarly associated with severity. C5b‐9 induced tumour necrosis factor‐α (TNF‐α) expression in cardiomyocytes, 35 a cytokine known to contribute to cardiomyocyte hypertrophy, cardiac fibrosis, and apoptosis, all of which are critical components of injury in HF. 36 , 37 Interestingly, C5b‐9 deposits were associated with IgG deposition and TNF‐α expression in failing myocardium of patients with dilated cardiomyopathy (DCM). 35
The anaphylatoxin C5a also plays a direct role in inotropic dysfunction via C5aR‐mediated signalling in cardiomyocytes, as evidenced in murine sepsis models. 38 C5a appears to have an essential role in adverse cardiac remodelling, as C5aR antagonism decreased cardiac hypertrophy and perivascular fibrosis in a murine model of hypertension. 39 Additionally, C5a is a potent chemokine that attracts myeloid cells to sites of damage 32 and is capable of activating TGF‐β‐dependent pro‐fibrotic pathways in the heart 39 (Figure 1 B ).
Triggers of B‐cell activation and anti‐cardiac antibodies in heart failure
About 10% of B cells are present in healthy hearts, as demonstrated in various mouse models. 40 , 41 , 42 There, B cells are involved in modulating the myocardial immune cell traffic as well as left ventricular structure and function. 42 Similarly, in patients with failing heart tissue, B cells are present in the intravasculature and in close contact with the endothelium. 42 Following cardiac damage, damage‐associated molecular patterns (DAMPs) are released from damaged cardiac cells, interacting with antigen‐presenting cells such as B cells. 2 , 43 Therefore, B cells have an important role in cardiac tissue and can undergo DAMP‐mediated activation, which in turn activates T cells, overall contributing to the pro‐inflammatory milieu. In mouse cardiac tissue, B cells are present in the same proportion as neutrophils. 41 Neutrophils are the leading infiltrating cells during MI 2 , 44 and are the most abundant cells in peripheral blood counts of patients along with the progression of ischaemic HF. 45 It has been reported that B cells and neutrophils act cooperatively, 46 , 47 allowing an antibody response, 46 but B cell‐helper neutrophil interactions in the heart remain to be studied. However, there are at least three mechanisms for the formation of anti‐cardiac cell autoantibodies. First, autoreactive naïve B cells evade negative selection mechanisms in the bone marrow, which then capture, process, and present cardiac antigens (cAgs) through major histocompatibility complex‐II molecules to activate autoreactive T helper cells. 48 , 49 Second, large antigens with repetitive sequences can generate a T cell‐independent humoral response, a mechanism previously proposed for the formation of myosin autoantibodies. 50 Third, memory B cells could be activated by contact with low doses of cAgs re‐encounters, causing T‐cell activation and their differentiation to long‐lived antibody‐producing plasma cells. 51 This final mechanism is particularly relevant because it would allow maintaining a constant autoantibodies production. All of these findings are consistent with the clinical observation that a higher number of B cells, naïve and memory, during an ST‐segment elevation MI (STEMI), are associated with increased mortality. 52
Although the presence of self‐reactive lymphocytes is a prerequisite for the development of autoimmunity, the presence of inflammatory milieu is necessary to avoid peripheral tolerance while promoting autoreactive cell full activation. 53 HF can be thought as chronic inflammatory state, 1 , 54 in which increased levels of pro‐inflammatory cytokines trigger immune activation upon cAgs encounter. This inflammatory state becomes more severe in terminal stages of the disease, as TNF‐α levels increase substantially with advancing New York Heart Association (NYHA) stages. The rise in TNF‐α levels is explained as TNF‐α is released from the failing myocardium into circulation. 37 , 55 Chronic inflammation is further promoted by peripheral nuclear factor‐κB activation secondary to widespread tissue hypoxia and free radical generation in advanced HF. 54 This concept has been documented previously, in the context of diabetes mellitus type 1, where increased local concentrations of TNF‐α in pancreatic islets resulted in enhanced T‐cell autoreactivity to β cells. 56 Furthermore, considering that TNF‐α is produced in Th1 inflammatory reactions, the increase in Th1:Th2 ratio is associated with adverse cardiac remodelling and impaired function following MI 57 and in decompensated HF. 58 This is consistent with results showing that peripheral Th cells are associated with left ventricular dysfunction. 59 Interestingly, interferon‐γ (IFN‐γ) was significantly elevated in patients with NYHA III–IV compared with NYHA I–II patients 30 days after MI. 57 Th1 response facilitates the activation of B cells. 60 , 61 IFN‐γ induces IgG3 expression, 62 which is consistent with IgG3 deposits in failing myocardium. 4 , 6 Finally, IgG3 deposits in patients failing heart tissue are accompanied by mixed inflammatory infiltrates of B cells, T cells, and macrophages. 6
Cardiac antigens may be a consequence of the expression of neo‐antigens and/or the failure of the mechanisms of self‐tolerance post‐cardiac injury. However, there is evidence of cross‐reactivity from antibodies towards non‐cardiac proteins, which also interact with the myocardium and alter its function. A well‐documented example is the case of anti‐Sjögren's syndrome, where related antigen A (anti‐SSA or Ro) antibodies are typically present in autoimmune disorders like Sjögren's syndrome and systemic lupus erythematosus (SLE). These antibodies can cross‐react with both T‐type Ca2+ channels (CaV3.1 and CaV3.2) and L‐type Ca2+ channels (CaV1.2 and CaV1.3), causing conduction disorders such as sinus bradycardia and atrioventricular block. 63 Similar observations have been documented for autoantibodies recognizing the NaV1.5 sodium channel and those targeting the KV11.1, KV1.4, or KV7.1 potassium channels. 63 This alternative mechanism is not necessarily caused by autoreactivity, as it can occur in response to infection, which generates antibody‐mediated cardiac injury by molecular mimicry. For instance, in patients with rheumatic heart disease, antibodies against streptococcal N‐acetylglucosamine and α‐helical coiled‐coil M proteins cross‐react with cardiac myosin and can produce myocardial damage. 64
In patients with HF, mechanisms involving both autoreactivity and molecular mimicry may be taking place following exposure to antigens that are normally ‘hidden' from the immune system (as occurs in myocardial injury from any cause), a context in which autoantibodies, as well as other cellular and soluble inflammatory mediators, may arise and cause damage. 28
Antibody‐independent mechanisms
B cells can alter the cardiac function and induce remodelling by secreting factors that influence cardiomyocytes, cardiac fibroblasts, and leukocytes. B‐cell depletion in mouse models of HF resulted in a decrease in infarct size, adverse ventricular remodelling, and protected ventricular function, 65 , 66 as well as an attenuated hypertensive response. 67 The protective consequences on B‐cell depletion were not recapitulated when T cells were eliminated in this model. 66 B‐cell depletion was accompanied by a significantly decrease in TNF‐α, interleukin (IL)‐1β, IL‐18, and BNP serum levels, myocardium apoptosis, and IgG depositions 65 , 66 , 67 in which the pro‐inflammatory, pathological phenotype was restored when B cells were reintroduced. 66 , 67
A recent report demonstrated that cardiac fibrosis was dependent on the direct modulation of a specific cardiac B‐cell subset (CD19+CD11b−). 40 In this mouse model of HF, it was previously demonstrated that the splenic plasma cells (CD19+CD138+) and activated B cells (CD19+CD86+) are increased, 67 which could migrate to the heart. 42 Activated B cell secretes cytokines and chemokines that may be directly involved in pathways that lead to adverse cardiac remodelling such as increased recruitment of monocytes, higher differentiation of pro‐fibrotic macrophages, and increased expression of TGF‐β, collagen‐I, and IL‐1β by fibroblast and macrophages. 65 , 67 Activated B cells recruited inflammatory monocytes (Ly6C+) to the myocardium in a CCL7‐dependent fashion contributing to adverse ventricular remodelling 65 that is impaired by B cell‐activating factor (BAFF) neutralization, which promotes B‐cell depletion. Patients' CCL‐7 serum levels positively correlated with increased risk of death and recurrent infarctions after acute MI. 65 Therefore, systemic depletion of B cells is likely to reduce macrophage‐induced myocardial damage. As antigen‐presenting cells, activated B cells can activate CD4+ T cells and promote their differentiation into the Th1 phenotype. In turn, Th1 cells may stimulate cardiac fibrosis through direct cell‐to‐cell interaction with cardiac fibroblasts, favouring their transition to TGF‐β and collagen‐producing myofibroblasts. 68 Consequently, B cells promote a full expression of HF and downstream cardiac injury by mediating immune cells chemotaxis and activation, and these are impaired in the absence of B cells.
B cells undergo activation following acute decompensation of HF, as indicated by the increased expression of CD69 in patients. 69 Higher concentrations of BAFF correlated with increased risk of death or reinfarction. 65 Furthermore, TNF‐α‐secreting B cells in patients with DCM are associated with enhanced cardiac fibrosis, as demonstrated by late enhancement on cardiac magnetic resonance imaging and higher levels of serum pro‐collagen type III. 36 Thus, B cells may also directly participate in cardiac remodelling through up‐regulation of TGF‐β and IL‐6 and further maintenance of a detrimental inflammatory environment via TNF‐α, IL‐1β, and IL‐6 production (Figure 2 ).
Figure 2.
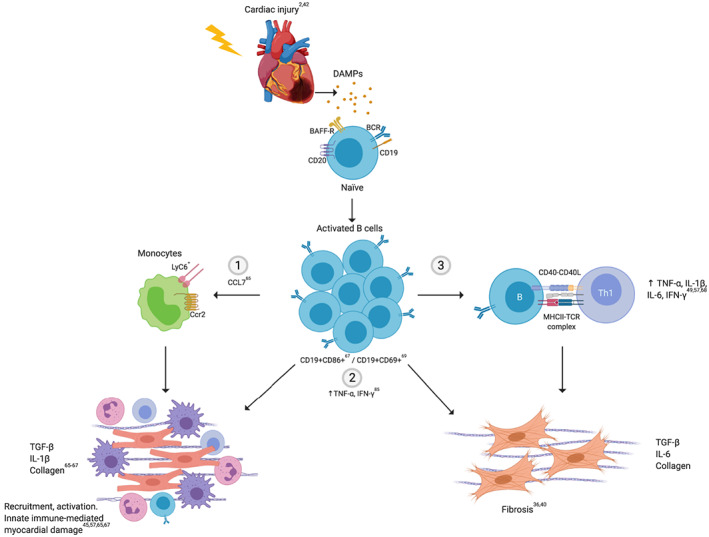
Antibody‐independent mechanisms. After cardiac ischaemic and non‐ischaemic injury, B cells become activated and proliferate in response to damage‐associated molecular patterns (DAMPs) that are released (from damaged cells and tissues) in response to cardiac injury. Their activation has been associated with chemotaxis of LyC6 + CCR2+ monocytes, which are involved in pathogenic remodelling and inflammation (1), though CCL7. The secretion of pro‐inflammatory cytokines is associated with fibrosis and detrimental function (2) as well as with proper cell activation and cell differentiation. The promotion of T‐cell activation and differentiation to the Th1 phenotype might be mediated by antigen cell presentation by B cells. This response contributes to the inflammatory milieu and may subsequently stimulate cardiac fibrosis through cardiac fibroblasts (3). BAFF‐R, B cell‐activating factor receptor; BCR, B cell receptor; CCL7, C–C motif chemokine ligand 7; CCR2, C–C chemokine receptor type 2; CD, cluster of differentiation (CD19 and CD20); IFN‐γ, interferon‐γ; IL‐1β, interleukin‐1β; Th1, type 1 helper T cell; TGF‐β, transforming growth factor‐β; TNF‐α, tumour necrosis factor‐α. Original image created with BioRender®.
Altogether, B cells initiate a self‐perpetuating cycle of cardiac injury through antibody‐independent and antibody‐dependent mechanisms (Figure 3 ), in which damage causes leakage of intracellular proteins and activation of self‐reactive B cells against other myocardial components. This damage is further exacerbated by the phenomenon of epitope spreading. 70 Cardiac autoantigens are ubiquitously present in the myocardium and, as such, cannot be fully cleared by the ensuing inflammatory reaction. Therefore, it seems logical that the inflammatory response continues indefinitely and is amplified with every new insult to the myocardium (e.g. a new infarct or acute decompensation of HF). Accordingly, antibody and complement deposits tend to be more frequent in the late stages of HF. 4 Importantly, this may partially explain why HF tends to follow an adverse natural history independently of current treatment strategies, which only target neuroendocrine components of the disease.
Figure 3.
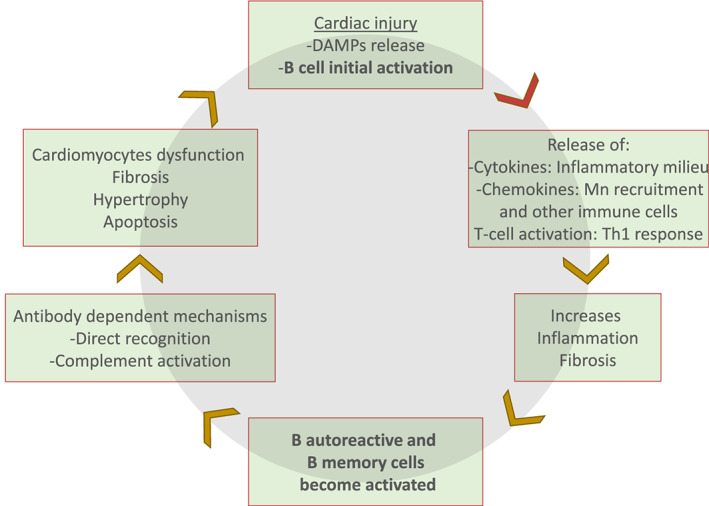
B cells play a central role in heart failure. After the cardiac injury, damage‐associated molecular patterns (DAMPs) are recognized, processed, and presented by resident B cells that become activated in a T‐dependent mechanism. In response, B cells secrete cytokines and chemokines that contribute with the inflammatory milieu along with the activated T cells mainly by a Th1 response (tumour necrosis factor‐α and interferon‐γ). Cell recruitment as monocytes (Mn) become activated, promoting fibrosis, hypertrophy, and tissue remodelling. The inflammatory milieu allows autoreactive B cells to become fully activated and differentiated into a memory B cell or plasma cell that produces mainly IgG3 antibodies against cardiac proteins. This induces further myocardial damage by antibody‐dependent mechanisms.
Potential endogenous modulators of B‐cell response in heart failure
There are at least two possible mechanisms that regulate B‐cell function in the HF setting. The interaction of the neuroendocrine system and immune cells and the role of regulatory B and T cells (Bregs and Tregs, respectively).
First, both T and B cells express β2‐adrenergic receptors, 71 and activation of T and B cells in the presence of norepinephrine enhances cytokine and IgG secretion, respectively. 72 This implies that high levels of catecholamines may exacerbate immune activation in HF. In accordance, B cells from patients with congestive HF display intracellular Ca2+ leak due to ryanodine receptor (RyR1) phosphorylation, possibly as consequence of catecholamine overproduction, although their functional role in this context has not been evaluated, intracellular Ca2+ is fundamental to B‐cell activation. 73 Interestingly, it has also been shown that catecholamines enhance monocyte mobilization from the bone marrow following MI, 74 which may be synergistic with B cell‐derived CCL7 in generating myocardial damage 65 and promoting vascular disease.
Second, Bregs have a role in suppressing self‐reactive B cells in various autoimmune diseases, and dysregulation of Bregs has been proposed as a mechanism of autoimmunity. Although specific surface marker or transcription factor profiles for Bregs subsets have not been clearly defined, these cells are primarily characterized by the expression of the anti‐inflammatory cytokine IL‐10 and the induced suppression of T‐cell responses, including the impairment of cytokine secretion by Th1 and Th17 lymphocytes. 75 , 76 This implies that Bregs may protect against contractile dysfunction by interfering with the pro‐inflammatory environment. Consistently, decreased levels of IL‐10 have worsened cardiac remodelling in murine models of angiotensin II‐induced cardiac injury. 77 However, evidence of the role of Bregs in human HF has only been addressed by two contradictory reports. The first study showed increased numbers of circulating Bregs (CD19+CD5+CD1d+IL10+) in patients with DCM. 78 The second study reported opposite results, albeit in a different Breg cell subpopulation (CD19+CD24hiCD27+IL10+), which also showed a decreased potential to suppress the TNF‐α production by T cells. 79 Therefore, further studies to evaluate the role of Bregs in HF are warranted.
Regulatory T cells can regulate the activity of autoreactive B cells by two mechanisms: first, avoiding autoreactive B cells to become activated. Tregs have direct actions on B cells by inhibiting immunoglobulin class switching, inducing apoptosis, 80 and inhibiting their activation and proliferation via interaction between programmed death‐1 and programmed death‐1 ligand present on the surface of Tregs. 81 This could partially explain why lower circulating Tregs are associated with left ventricular dysfunction and poor prognosis in patients with HF, 82 , 83 as supported by the evidence in murine models of lupus, in which depletion of Tregs increases the production of autoantibodies while their administration has the opposite effect. 84 Second, Tregs can also acquire an inflammatory phenotype and contribute to adverse myocardial remodelling, as was shown in a murine model of ischaemic cardiomyopathy, by increasing the expression of IFN‐γ and TNF‐α. 85 This could create a permissive environment for autoreactive B cells to become activated to produce pro‐inflammatory cytokines and autoantibodies that exacerbate the initial insult. This evidence indicates that Tregs' interaction with B cells needs to be evaluated as Tregs could also represent a potential target for immunomodulatory therapies in HF.
B cells as a therapeutic target in heart failure
Immunomodulatory strategies for HF to date have mostly targeted autoantibodies and pro‐inflammatory cytokines and have produced inconclusive results with subtle condition improvements only. These strategies include selective inhibition of TNF‐α, immunoadsorption, intravenous immunoglobulin, therapeutic plasma exchange, and non‐specific immunomodulation with autologous apoptotic cells, which are all reviewed in detail elsewhere. 43 , 55 , 86
The reasons why these therapeutic approaches have not resulted in definite clinical improvement remain elusive, but some hypotheses in which B cells are involved can be drawn. TNF‐α blockers have been associated with worsening HF and mortality. Although interventions decreased levels of TNF‐α, myocardial function did not improve. 87 , 88 It was previously observed that treatment with anti‐TNF‐α blockers alters the distribution of peripheral blood B cells by increasing the frequency of pre‐switch IgD+CD27+ memory B cells in non‐responder patients with rheumatoid arthritis (RA). 89 This could favour cAgs recognition by these cells and results in their differentiation to antibody‐producing plasma cells. In addition, it is worth noting the signalling amplification of the immune activation that occurs in HF, in which many other pro‐inflammatory cytokines and mediators, not just TNF‐α (such as IL‐1β and IL‐6), are responsible for continued myocardial damage. 1 These other mediators, either directly, by inducing cardiac dysfunction, or indirectly, by activating effector functions of various classes of immune cells, including B cells, can also promote the progression of HF.
On the other hand, immunoadsorption, plasma exchange, and intravenous immunoglobulin, which non‐specifically deplete antibodies from the patient, have been associated mostly with significant improvements in cardiac function, in the short term. 69 , 90 , 91 However, the long‐term benefit is unclear, as cardiac autoantibodies reappear in a proportion of treated patients who eventually may require a heart transplant or a ventricular assist device. 91 This is consistent with the fact that these therapies target the damage‐mediating product but not its source. Indeed, eventual disease relapse may be related to the reappearance of anti‐cardiac antibodies produced by terminally differentiated B cells. This prompted the non‐specific immunomodulation strategy employed in the ACCLAIM trial, in which apoptotic autologous cells were injected to patients with the rationale that apoptotic cells would induce a systemic anti‐inflammatory response by polarizing phagocytic cells to an alternatively activated phenotype, promoting decreased inflammatory myocardial damage. 92 However, only a small subset of patients with non‐ischaemic HF demonstrated significant benefit. 92 It has been observed that some patients with chronic inflammation had impaired clearance of apoptotic cells that eventually leads to secondary necrosis and damage. 93
Novel strategies targeting other immune components are currently undergoing evaluation in clinical trials. 94 Here, considering the evidence described earlier, we discuss and evaluate the possibility of new treatment strategies that directly target B cells in HF.
Rituximab (RTX) is a chimeric monoclonal antibody that targets CD20, a membrane‐spanning protein present exclusively in the majority of B lineage cells (except pro‐B cells and plasma cells). 95 RTX exerts its B cell‐depleting action by binding to CD20 molecules on the B cell's membrane, activating the classical pathway of the complement system, facilitating cell‐mediated cytotoxicity and apoptosis. 96 Clinical experience with RTX is extensive, and it is currently approved for the treatment of haematological malignancies and prototypical autoimmune diseases, such as RA. 51 Although RTX clears virtually all CD20+ B cells from peripheral blood, B cells residing in secondary and tertiary lymphoid organs may survive depletion. 95 , 97 Notwithstanding, repopulation with beneficial B‐cell subsets may follow treatment with RTX. For instance, patients with thrombotic thrombocytopenic purpura treated with RTX demonstrated slow regeneration of memory B‐cell subsets, and BAFF‐R expression was reduced in all B‐cell subsets after RTX. 98 As BAFF is a crucial survival signal for B cells, the decreased in BAFF‐R expression could potentially hinder autoreactive B‐cell maturation in the periphery and hence could be a long‐lasting indirect benefit of RTX in HF patients. After RTX, B‐cell repopulation may also be characterized by higher proportions of IL‐10‐producing Bregs, 51 which may also benefit patients with HF. However, experience with the drug has revealed potential adverse effects and variable responses between patients, as well, differential susceptibility to RTX‐mediated depletion within B‐cell subpopulations. 95
Thus, the use of RTX as a therapeutic agent in HF could lead to a decrease in circulating cardiac autoantibodies, B cell‐derived cytokines, and activation of self‐reactive T cells, which jointly could potentially prevent further progression of HF.
Tschöpe et al. 99 have recently published a case series of six patients with refractory inflammatory DCM treated with RTX. Five patients had a favourable response to RTX, as indicated by decreased NYHA functional class at 8 weeks of follow‐up, improved left ventricular ejection fraction, and decreased B‐cell infiltrates in endomyocardial biopsies. However, one patient did not improve: NYHA class did not change, left ventricular ejection fraction remained significantly compromised, and, paradoxically, infiltrated B cells in the myocardium nearly doubled. Although the authors did not propose an explanation to this non‐responder phenotype, we hypothesize that it may be related to an unfavourable B‐cell subset that is resistant to depletion by RTX, as was described in patients with RA.
Our group designed a phase II, single‐centred prospective clinical trial. We propose that RTX may be safely used as adjunctive therapy in the management of treatment‐refractory HF. Our ongoing study includes patients with ejection fraction ≤40%, NYHA functional class III/IV, who are unresponsive to standard HF treatment and have not previously been treated with an immunosuppressive drug. RTX dose will mimic that already applied to post‐transplant patients and patients with RA, based on prior evidence of safety. 100
After RTX, B‐cell repopulation may also be characterized by higher proportions of IL‐10‐producing Bregs, 51 which may also benefit patients with HF. However, experience with RTX has revealed potential adverse effects and variable responses between patients, as well as differential susceptibility to RTX‐mediated depletion within B‐cell subpopulations. 95
The use of BAFF antagonists may be another therapeutic possibility in HF. The BAFF‐directed antibody belimumab is currently approved for treatment‐resistant SLE in adults, reducing flares and overall disease activity when used in combination with standard therapy. 101 However, it is known that belimumab only partially inhibits the production of IgG autoantibodies, as it depletes both naïve and activated B cells, but not memory B cells. 51 Considering that pathogenic autoantibodies in HF are mostly of the IgG3 class and that B‐cell subsets in this context are largely unknown, further research is necessary to justify its use in HF.
There are other B cell‐target therapies, but they are less extensively studied, only on patients with lymphoma or one autoimmune disease (SLE and RA), with recent available data (2018). 102 A comparative study between RTX and abatacept, a B–T cell co‐stimulatory inhibitor, showed that RTX had better outcomes in terms of failure defined as all cause death. 103
Determination of B‐cell subsets in heart failure patients
The strategies mentioned earlier exhibit varied depletion efficacy depending on B‐cell subsets. 95 , 101 If B cell‐depleting strategies are to be used for the treatment of HF, then the characterization of B‐cell profiles, as has been determined in the setting of prototypical autoimmune diseases, 101 could be useful from a clinical standpoint.
First, it would help predict which subsets of patients are more likely to respond to treatment or have a sustained clinical response, a concept supported by studies in RA in which patients who had higher proportions of double‐negative naïve B cells (CD19+IgD−CD27−) and lower percentages of plasmablasts (CD19+IgD+CD27++) and memory B cells (CD19+IgD+CD27+CD95−; CD19+CD27+) were more likely to have a favourable clinical response with RTX. 104 , 105 Furthermore, the clinical response in RA patients treated with RTX correlated positively with the depletion of the pre‐switch memory B cells (CD19+IgD+CD27+CD95−), which is the increased cell population in response to anti‐TNF‐α therapy. 89 These findings suggest that patients with higher numbers of memory B cells or plasmablasts that might correspond to those with increased disease activity are more likely to be resistant to therapy. As discussed earlier, memory B cells are responsible for the swift humoral immune response that occurs upon re‐encounter with antigen. 51 However, plasmablasts are short lived in peripheral blood before they home to bone marrow, mucosal tissues, or sites of ongoing inflammation. 50 Therefore, their presence may imply current autoantigen presentation and B‐cell activation. In patients with HF, the relative abundance of these subsets could precipitate a detrimental feedback loop of myocardial injury, as described earlier. Supporting this concept, van den Hoogen et al. have recently reported that patients with HF had increased proportions of circulating plasmablasts and decreased transitional/regulatory B cells compared with healthy controls. Interestingly, stratified analysis revealed that these differences were more pronounced in patients with ischaemic HF, which likely reflects the nature of the insult to the myocardium. 6
Ischaemic damage results in massive cardiomyocyte necrosis and leak of cAgs to the circulation, while non‐ischaemic insults result in increased wall stress with a less significant release of cAgs. Although the observation of increased plasmablasts did not reach statistical significance, this was likely due to small sample size (n = 10), as we have also identified increased circulating plasmablasts in 21 patients with HF (unpublished data). Furthermore, patients admitted for STEMI had significantly higher total B‐cell counts than no‐STEMI and control groups, in which naïve and memory B cells demonstrated a strong positive correlation with troponin I and creatine kinase levels (plasmablasts were not measured). 52 Interestingly, the increased level of circulating memory B cells correlated with the 6 month probability of death from admission, as calculated with the GRACE score. 52 The fact that STEMI, by definition, is indicative of cardiomyocyte death and the release of intracellular products and that increased B‐cell counts were observed in this group, together with the observation that patients with ischaemic HF tend to have higher levels of plasmablasts, 6 further supports the involvement of B cells in myocardial damage, as previously reported in murine models. 65
Furthermore, knowledge of the relative proportions of B‐cell subsets in HF may explain why most, but not all, end‐stage HF patients have antibody‐dependent‐mediated myocardial damage. Indeed, a considerable fraction of end‐stage HF patients lacks antibody or complement deposits in the myocardium. 4 , 69 These deposits being observed more often in more severe HF patients or longer time since diagnosis 4 suggest that cumulative insults to the myocardium are bypassing peripheral tolerance mechanisms due to continued exposure of cardiac self‐antigens, resulting in higher proportions of pathogenic B cells. Hence, knowledge of which B‐cell subsets are altered throughout the spectrum of HF could be helpful to clarify this hypothesis.
Finally, evidence that specific B‐cell subsets cause disease progression would strongly support the claim for developing novel selective immunotherapies or treatments.
Conclusions
Heart failure is characterized by a maladaptive process of cardiac interstitial fibrosis and contractile dysfunction, which may be initiated and maintained following insults to the myocardium. Among many processes that lead to HF, B cells are highlighted as playing a prominent role in its progression, regardless of aetiology, through mechanisms that are dependent and independent of antibody production. These mechanisms include secretion of pro‐inflammatory cytokines, 36 monocyte recruitment following myocardial injury, 65 and interaction with CD4+ T helper cells, with a subsequent amplification of the inflammatory cascade. 49 On the other hand, the production of antibodies directed against cAgs, 4 predominantly of the IgG3 subtype, may fix and activate complement to promote myocardial inflammation, injury, and remodelling. 4 , 35 , 38 , 39 Then, apoptosis 66 and cardiac functional impairment alter cardiac contraction and heart rate 28 , 29 (Figure 3 ). Additionally, regulatory T‐cell 82 and B‐cell dysfunction 79 may further enhance self‐reactive B‐cell responses in HF. Further research must confirm which B‐cell subpopulations may mediate continued myocardial damage in HF, as this would support trials of more selective therapeutics, possibly decreasing the scope of adverse events linked to the currently available therapies.
Conflict of interest
None declared.
Funding
This work was partially supported by the GIEE Medicina Cardiovascular y Metabolomica (Tecnologico de Monterrey, 0020209M01) as well as the Consejo Nacional de Ciencia y Tecnología (CONACYT) grants 151136, 133591, and 269399, and Fronteras de la Ciencia grant (0682). A.M.G.‐G. and A.T.‐Q. were supported by the Graduate Student Fellowship of CONACYT.
García‐Rivas, G. , Castillo, E. C. , Gonzalez‐Gil, A. M. , Maravillas‐Montero, J. L. , Brunck, M. , Torres‐Quintanilla, A. , Elizondo‐Montemayor, L. , and Torre‐Amione, G. (2020) The role of B cells in heart failure and implications for future immunomodulatory treatment strategies. ESC Heart Failure, 7: 1387–1399. 10.1002/ehf2.12744.
References
- 1. Torre‐Amione G. Immune activation in chronic heart failure. Am J Cardiol 2005; 95: 3C–8C discussion 38C‐40C. [DOI] [PubMed] [Google Scholar]
- 2. Brenes‐Castro D, Castillo EC, Vázquez‐Garza E, Torre‐Amione G, García‐Rivas G. Temporal frame of immune cell infiltration during heart failure establishment: lessons from animal models. Int J Mol Sci 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 2015; 116: 1254–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Youker KA, Assad‐Kottner C, Cordero‐Reyes AM, Trevino AR, Flores‐Arredondo JH, Barrios R, Fernandez‐Sada E, Estep JD, Bhimaraj A, Torre‐Amione G. High proportion of patients with end‐stage heart failure regardless of aetiology demonstrates anti‐cardiac antibody deposition in failing myocardium: humoral activation, a potential contributor of disease progression. Eur Heart J 2014; 35: 1061–1068. [DOI] [PubMed] [Google Scholar]
- 5. Derer S, Rösner T, Lohse S, Peipp M, Valerius T. Switching from IgG1 to IgG3 isotype to boost complement‐activating capacities of therapeutic antibodies—impact of CD55 expression on the mode of action. Blood 2014; 124: 3506–3506. [Google Scholar]
- 6. van den Hoogen P, de Jager SCA, Huibers MMH, Schoneveld AH, Puspitasari YM, Valstar GB, Oerlemans MIFJ, de Weger RA, Doevendans PA, den Ruijter HM, Laman JD, Vink A, Sluijter JPG. Increased circulating IgG levels, myocardial immune cells and IgG deposits support a role for an immune response in pre‐ and end‐stage heart failure. J Cell Mol Med 2019; 23: 7505–7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Latif N, Baker CS, Dunn MJ, Rose ML, Brady P, Yacoub MH. Frequency and specificity of antiheart antibodies in patients with dilated cardiomyopathy detected using SDS‐PAGE and Western blotting. J Am Coll Cardiol 1993; 22: 1378–1384. [DOI] [PubMed] [Google Scholar]
- 8. Keppner L, Heinrichs M, Rieckmann M, Demengeot J, Frantz S, Hofmann U, Ramos G. Antibodies aggravate the development of ischemic heart failure. Am J Physiol Heart Circ Physiol 2018; 315: H1358–H1367. [DOI] [PubMed] [Google Scholar]
- 9. Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM, Komorowski L, Luo J, Cabral‐Marques O, Hammers CM, Lindstrom JM, Lamprecht P, Fischer A, Riemekasten G, Tersteeg C, Sondermann P, Rapoport B, Wandinger K‐P, Probst C, El Beidaq A, Schmidt E, Verkman A, Manz RA, Nimmerjahn F. Mechanisms of autoantibody‐induced pathology. Front Immunol 2017; 8: 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haudek SB, Trial J, Xia Y, Gupta D, Pilling D, Entman ML. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc Natl Acad Sci U S A 2008; 105: 10179–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Staudt A, Eichler P, Trimpert C, Felix SB, Greinacher A. FcγReceptors IIa on cardiomyocytes and their potential functional relevance in dilated cardiomyopathy. J Am Coll Cardiol 2007; 49: 1684–1692. [DOI] [PubMed] [Google Scholar]
- 12. Warraich RS, Griffiths E, Falconar A, Pabbathi V, Bell C, Angelini G, Suleiman M‐S, Yacoub MH. Human cardiac myosin autoantibodies impair myocyte contractility: a cause‐and‐effect relationship. FASEB J Off Publ Fed Am Soc Exp Biol 2006; 20: 651–660. [DOI] [PubMed] [Google Scholar]
- 13. Simpson KE, Cunningham MW, Lee CK, Ward K, Tong A, Danon S, Simon C, Delaney JW, Canter CE. Autoimmunity against the heart and cardiac myosin in children with myocarditis. J Card Fail 2016; 22: 520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leuschner F, Li J, Göser S, Reinhardt L, Ottl R, Bride P, Zehelein J, Pfitzer G, Remppis A, Giannitsis E, Katus HA, Kaya Z. Absence of auto‐antibodies against cardiac troponin I predicts improvement of left ventricular function after acute myocardial infarction. Eur Heart J 2008; 29: 1949–1955. [DOI] [PubMed] [Google Scholar]
- 15. Doesch AO, Mueller S, Nelles M, Konstandin M, Celik S, Frankenstein L, Goeser S, Kaya Z, Koch A, Zugck C, Katus HA. Impact of troponin I‐autoantibodies in chronic dilated and ischemic cardiomyopathy. Basic Res Cardiol 2011; 106: 25–35. [DOI] [PubMed] [Google Scholar]
- 16. Klein R, Maisch B, Kochsiek K, Berg PA. Demonstration of organ specific antibodies against heart mitochondria (anti‐M7) in sera from patients with some forms of heart diseases. Clin Exp Immunol 1984; 58: 283–292. [PMC free article] [PubMed] [Google Scholar]
- 17. Wolff PG, Kühl U, Schultheiss H‐P. Laminin distribution and autoantibodies to laminin in dilated cardiomyopathy and myocarditis. Am Heart J 1989; 117: 1303–1309. [DOI] [PubMed] [Google Scholar]
- 18. Liao Y, Tu Y, Chen L, Dai S, Peng Y, Li S, Zhang J. Cardiac cytotoxic mechanism mediated by antibodies against myocardial mitochondrial ADP/ATP carrier in patients with dilated cardiomyopathy. Chin Med J (Engl) 1996; 109: 193–196. [PubMed] [Google Scholar]
- 19. Liao Y, Cheng L, Tu Y, Zhang J, Dong J, Li S, Tian Y, Peng Y. Mechanism of anti‐β‐adrenoceptor antibody mediated myocardial damage in dilated cardiomyopathy. J Tongji Med Univ Tong Ji Yi Ke Xue Xue Bao 1997; 17: 5–8. [DOI] [PubMed] [Google Scholar]
- 20. Christ T, Wettwer E, Dobrev D, Adolph E, Knaut M, Wallukat G, Ravens U. Autoantibodies against the β1 adrenoceptor from patients with dilated cardiomyopathy prolong action potential duration and enhance contractility in isolated cardiomyocytes. J Mol Cell Cardiol 2001; 33: 1515–1525. [DOI] [PubMed] [Google Scholar]
- 21. Iwata M, Yoshikawa T, Baba A, Anzai T, Nakamura I, Wainai Y, Takahashi T, Ogawa S. Autoimmunity against the second extracellular loop of β1‐adrenergic receptors induces β‐adrenergic receptor desensitization and myocardial hypertrophy in vivo. Circ Res 2001; 88: 578–586. [DOI] [PubMed] [Google Scholar]
- 22. Jane‐wit D, Altuntas CZ, Johnson JM, Yong S, Wickley PJ, Clark P, Wang Q, Popović ZB, Penn MS, Damron DS, Perez DM, Tuohy VK. β1‐Adrenergic receptor autoantibodies mediate dilated cardiomyopathy by agonistically inducing cardiomyocyte apoptosis. Circulation 2007; 116: 399–410. [DOI] [PubMed] [Google Scholar]
- 23. Chiale PA, Ferrari I, Mahler E, Vallazza MA, Elizari MV, Rosenbaum MB, Levin MJ. Differential profile and biochemical effects of antiautonomic membrane receptor antibodies in ventricular arrhythmias and sinus node dysfunction. Circulation 2001; 103: 1765–1771. [DOI] [PubMed] [Google Scholar]
- 24. Baba A, Yoshikawa T, Fukuda Y, Sugiyama T, Shimada M, Akaishi M, Tsuchimoto K, Ogawa S, Fu M. Autoantibodies against M2‐muscarinic acetylcholine receptors: new upstream targets in atrial fibrillation in patients with dilated cardiomyopathy. Eur Heart J 2004; 25: 1108–1115. [DOI] [PubMed] [Google Scholar]
- 25. Baba A, Yoshikawa T, Ogawa S. Autoantibodies produced against sarcolemmal Na‐K‐ATPase: possible upstream targets of arrhythmias and sudden death in patients with dilated cardiomyopathy. J Am Coll Cardiol 2002; 40: 1153–1159. [DOI] [PubMed] [Google Scholar]
- 26. Landsberger M, Staudt A, Choudhury S, Trimpert C, Herda LR, Klingel K, Kandolf R, Schultheiss H‐P, Kroemer HK, Völker U, Felix SB. Potential role of antibodies against cardiac Kv channel‐interacting protein 2 in dilated cardiomyopathy. Am Heart J 2008; 156: 92–99 e2. [DOI] [PubMed] [Google Scholar]
- 27. Petrohai A, Nagy G, Bosze S, Hudecz F, Zsiros E, Paragh G, Nyárády Z, Németh P, Berki T. Detection of citrate synthase‐reacting autoantibodies after heart transplantation: an epitope mapping study. Transpl Int Off J Eur Soc Organ Transplant 2005; 17: 834–840. [DOI] [PubMed] [Google Scholar]
- 28. Boivin‐Jahns V, Jahns R. GPCR‐autoantibodies in chronic heart failure. Front Biosci Landmark Ed 2018; 23: 2065–2081. [DOI] [PubMed] [Google Scholar]
- 29. Stavrakis S, Kem DC, Patterson E, Lozano P, Huang S, Szabo B, Cunningham MW, Lazzara R, Yu X. Opposing cardiac effects of autoantibody activation of beta‐adrenergic and M2 muscarinic receptors in cardiac‐related diseases. Int J Cardiol 2011; 148: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jahns R, Boivin V, Siegmund C, Inselmann G, Lohse MJ, Boege F. Autoantibodies activating human β1‐adrenergic receptors are associated with reduced cardiac function in chronic heart failure. Circulation 1999; 99: 649–654. [DOI] [PubMed] [Google Scholar]
- 31. Störk S, Boivin V, Horf R, Hein L, Lohse MJ, Angermann CE, Jahns R. Stimulating autoantibodies directed against the cardiac β1‐adrenergic receptor predict increased mortality in idiopathic cardiomyopathy. Am Heart J 2006; 152: 697–704. [DOI] [PubMed] [Google Scholar]
- 32. Nesargikar PN, Spiller B, Chavez R. The complement system: history, pathways, cascade and inhibitors. Eur J Microbiol Immunol 2012; 2: 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prohászka Z, Munthe‐Fog L, Ueland T, Gombos T, Yndestad A, Förhécz Z, Skjoedt M‐O, Pozsonyi Z, Gustavsen A, Jánoskuti L, Karádi I, Gullestad L, Dahl CP, Askevold ET, Füst G, Aukrust P, Mollnes TE, Garred P. Association of ficolin‐3 with severity and outcome of chronic heart failure. PLoS ONE 2013; 8: e60976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shahini N, Michelsen AE, Nilsson PH, Ekholt K, Gullestad L, Broch K, Dahl CP, Aukrust P, Ueland T, Mollnes TE, Yndestad A, Louwe MC. The alternative complement pathway is dysregulated in patients with chronic heart failure. Sci Rep 2017; 7: 42532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zwaka TP, Manolov D, Ozdemir C, Marx N, Kaya Z, Kochs M, Höher M, Hombach V, Torzewski J. Complement and dilated cardiomyopathy: a role of sublytic terminal complement complex‐induced tumor necrosis factor‐α synthesis in cardiac myocytes. Am J Pathol 2002; 161: 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu M, Wen S, Wang M, Liang W, Li H‐H, Long Q, Guo H‐P, Liao Y‐H, Yuan J. TNF‐α‐secreting B cells contribute to myocardial fibrosis in dilated cardiomyopathy. J Clin Immunol 2013; 33: 1002–1008. [DOI] [PubMed] [Google Scholar]
- 37. Torre‐Amione G, Kapadia S, Lee J, Durand JB, Bies RD, Young JB, Mann DL. Tumor necrosis factor‐α and tumor necrosis factor receptors in the failing human heart. Circulation 1996; 93: 704–711. [DOI] [PubMed] [Google Scholar]
- 38. Niederbichler AD, Hoesel LM, Westfall MV, Gao H, Ipaktchi KR, Sun L, Zetoune FS, Su GL, Arbabi S, Sarma JV, Wang SC, Hemmila MR, Ward PA. An essential role for complement C5a in the pathogenesis of septic cardiac dysfunction. J Exp Med 2006; 203: 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang C, Li Y, Wang C, Wu Y, Du J. Antagonist of C5aR prevents cardiac remodeling in angiotensin II‐induced hypertension. Am J Hypertens 2014; 27: 857–864. [DOI] [PubMed] [Google Scholar]
- 40. Adamo L, Staloch LJ, Rocha‐Resende C, Matkovich SJ, Jiang W, Bajpai G, Weinheimer CJ, Kovacs A, Schilling JD, Barger PM, Bhattacharya D, Mann DL. Modulation of subsets of cardiac B lymphocytes improves cardiac function after acute injury. JCI Insight 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu Y‐RA, O'Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER, Que L, Gunn MD. A protocol for the comprehensive flow cytometric analysis of immune cells in normal and inflamed murine non‐lymphoid tissues. PloS One 2016; 11: e0150606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adamo L, Rocha‐Resende C, Lin C‐Y, Evans S, Williams J, Dun H, Li W, Mpoy C, Andhey PS, Rogers BE, Lavine K, Kreisel D, Artyomov M, Randolph GJ, Mann DL. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight American Society for Clinical Investigation 2020: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sánchez‐Trujillo L, Vázquez‐Garza E, Castillo EC, García‐Rivas G, Torre‐Amione G. Role of adaptive immunity in the development and progression of heart failure: new evidence. Arch Med Res 2017; 48: 1–11. [DOI] [PubMed] [Google Scholar]
- 44. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol 2013; 62: 24–35. [DOI] [PubMed] [Google Scholar]
- 45. Groot HE, van Blokland IV, Lipsic E, Karper JC, van der Harst P. Leukocyte profiles across the cardiovascular disease continuum: a population‐based cohort study. J Mol Cell Cardiol 2020; 138: 158–164. [DOI] [PubMed] [Google Scholar]
- 46. Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, Comerma L, Chorny A, Shan M, Xu W, Magri G. B cell‐helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol 2011; 13: 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parsa R, Lund H, Georgoudaki A‐M, Zhang X‐M, Ortlieb Guerreiro‐Cacais A, Grommisch D, Warnecke A, Croxford AL, Jagodic M, Becher B, Karlsson MCI, Harris RA. BAFF‐secreting neutrophils drive plasma cell responses during emergency granulopoiesis. J Exp Med 2016; 213: 1537–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pierce SK, Morris JF, Grusby MJ, Kaumaya P, van Buskirk A, Srinivasan M, Crump B, Smolenski LA. Antigen‐presenting function of B lymphocytes. Immunol Rev 1988; 106: 149–180. [DOI] [PubMed] [Google Scholar]
- 49. Smith SC, Allen PM. Expression of myosin‐class II major histocompatibility complexes in the normal myocardium occurs before induction of autoimmune myocarditis. Proc Natl Acad Sci U S A 1992; 89: 9131–9135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. O'Donohoe TJ, Schrale RG, Ketheesan N. The role of anti‐myosin antibodies in perpetuating cardiac damage following myocardial infarction. Int J Cardiol 2016; 209: 226–233. [DOI] [PubMed] [Google Scholar]
- 51. Hofmann K, Clauder A‐K, Manz RA. Targeting B cells and plasma cells in autoimmune diseases. Front Immunol 2018; 9: 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Feldtmann R, Kuemmel A, Abdiu E, Chamling B, Gross S, Doerr M, Felix SB, Busch R, Strohbach A. P1693. Elevated counts of naive and memory B cells might be associated with the severity of myocardial damage in the setting of acute myocardial infarction. Eur Heart J 2018; 39. [Google Scholar]
- 53. Theofilopoulos AN, Kono DH, Baccala R. The multiple pathways to autoimmunity. Nat Immunol 2017; 18: 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hasper D, Hummel M, Kleber FX, Reindl I, Volk HD. Systemic inflammation in patients with heart failure. Eur Heart J 1998; 19: 761–765. [DOI] [PubMed] [Google Scholar]
- 55. Flores‐Arredondo JH, García‐Rivas G, Torre‐Amione G. Immune modulation in heart failure: past challenges and future hopes. Curr Heart Fail Rep 2011; 8: 28–37. [DOI] [PubMed] [Google Scholar]
- 56. Skak K, Guerder S, Picarella DE, Brenden N, Flavell RA, Michelsen BK. TNF‐α impairs peripheral tolerance towards β‐cells, and local costimulation by B7.1 enhances the effector function of diabetogenic T cells. Eur J Immunol 2003; 33: 1341–1350. [DOI] [PubMed] [Google Scholar]
- 57. Cheng X, Liao Y‐H, Ge H, Li B, Zhang J, Yuan J, Wang M, Liu Y, Guo Z, Chen J, Zhang J, Zhang L. TH1/TH2 functional imbalance after acute myocardial infarction: coronary arterial inflammation or myocardial inflammation. J Clin Immunol 2005; 25: 246–253. [DOI] [PubMed] [Google Scholar]
- 58. Fukunaga T, Soejima H, Irie A, Sugamura K, Oe Y, Tanaka T, Kojima S, Sakamoto T, Yoshimura M, Nishimura Y, Ogawa H. Expression of interferon‐γ and interleukin‐4 production in CD4+ T cells in patients with chronic heart failure. Heart Vessels 2007; 22: 178–183. [DOI] [PubMed] [Google Scholar]
- 59. Brunetti ND, D'Antuono C, Rana M, D'Arienzo G, De Gennaro L, Di Biase M. Lymphocyte subset characterization in patients with early clinical presentation of coronary heart disease. J Thromb Thrombolysis 2012; 34: 475–482. [DOI] [PubMed] [Google Scholar]
- 60. Smith KM, Pottage L, Thomas ER, Leishman AJ, Doig TN, Xu D, Liew FY, Garside P. Th1 and Th2 CD4+ T cells provide help for B cell clonal expansion and antibody synthesis in a similar manner in vivo. J Immunol American Association of Immunologists 2000; 165: 3136–3144. [DOI] [PubMed] [Google Scholar]
- 61. Vazquez MI, Catalan‐Dibene J, Zlotnik A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine 2015; 74: 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Snapper CM, McIntyre TM, Mandler R, Pecanha LM, Finkelman FD, Lees A, Mond JJ. Induction of IgG3 secretion by interferon gamma: a model for T cell‐independent class switching in response to T cell‐independent type 2 antigens. J Exp Med 1992; 175: 1367–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lazzerini PE, Laghi‐Pasini F, Boutjdir M, Capecchi PL. Cardioimmunology of arrhythmias: the role of autoimmune and inflammatory cardiac channelopathies. Nat Rev Immunol 2019; 19: 63–64. [DOI] [PubMed] [Google Scholar]
- 64. Guilherme L, Kalil J, Cunningham M. Molecular mimicry in the autoimmune pathogenesis of rheumatic heart disease. Autoimmunity 2006; 39: 31–39. [DOI] [PubMed] [Google Scholar]
- 65. Zouggari Y, Ait‐Oufella H, Bonnin P, Simon T, Sage AP, Guérin C, Vilar J, Caligiuri G, Tsiantoulas D, Laurans L, Dumeau E, Kotti S, Bruneval P, Charo IF, Binder CJ, Danchin N, Tedgui A, Tedder TF, Silvestre J‐S, Mallat Z. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med 2013; 19: 1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cordero‐Reyes AM, Youker KA, Trevino AR, Celis R, Hamilton DJ, Flores‐Arredondo JH, Orrego CM, Bhimaraj A, Estep JD, Torre‐Amione G. Full expression of cardiomyopathy is partly dependent on B‐cells: a pathway that involves cytokine activation, immunoglobulin deposition, and activation of apoptosis. J Am Heart Assoc 2016; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, Kim HA, Krishnan SM, Lewis CV, Salimova E, Tipping P, Vinh A, Samuel CS, Peter K, Guzik TJ, Kyaw TS, Toh B‐H, Bobik A, Drummond GR. Obligatory role for B cells in the development of angiotensin II‐dependent hypertension. Hypertens Dallas Tex 1979 2015; 66: 1023–1033. [DOI] [PubMed] [Google Scholar]
- 68. Tania N, Salvador Ane M, Anna G‐P, Andrew K, Francisco V, Mark A, Kapur Navin K, Karas Richard H, Blanton Robert M, Pilar A. Left ventricular T‐cell recruitment contributes to the pathogenesis of heart failure. Circ Heart Fail 2015; 8: 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Torre‐Amione G, Orrego CM, Khalil N, Kottner‐Assad C, Leveque C, Celis R, Youker KA, Estep JD. Therapeutic plasma exchange a potential strategy for patients with advanced heart failure. J Clin Apher 2010; 25: 323–330. [DOI] [PubMed] [Google Scholar]
- 70. Cornaby C, Gibbons L, Mayhew V, Sloan CS, Welling A, Poole BD. B cell epitope spreading: mechanisms and contribution to autoimmune diseases. Immunol Lett 2015; 163: 56–68. [DOI] [PubMed] [Google Scholar]
- 71. Sanders VM. The beta2‐adrenergic receptor on T and B lymphocytes: do we understand it yet? Brain Behav Immun 2012; 26: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Padro CJ, Sanders VM. Neuroendocrine regulation of inflammation. Semin Immunol 2014; 26: 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kushnir A, Santulli G, Reiken SR, Coromilas E, Godfrey SJ, Brunjes DL, Colombo PC, Yuzefpolskaya M, Sokol SI, Kitsis RN, Marks AR. Ryanodine receptor calcium leak in circulating B‐lymphocytes as a biomarker in heart failure. Circulation 2018; 138: 1144–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T, Majmudar MD, Lasitschka F, Etzrodt M, Waterman P, Waring MT, Chicoine AT, van der Laan AM, Niessen HWM, Piek JJ, Rubin BB, Butany J, Stone JR, Katus HA, Murphy SA, Morrow DA, Sabatine MS, Vinegoni C, Moskowitz MA, Pittet MJ, Libby P, Lin CP, Swirski FK, Weissleder R, Nahrendorf M. Myocardial infarction accelerates atherosclerosis. Nature 2012; 487: 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Flores‐Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, Mauri C. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med 2013; 5: 173ra23. [DOI] [PubMed] [Google Scholar]
- 76. Maravillas‐Montero JL, Acevedo‐Ochoa E. Human B regulatory cells: the new players in autoimmune disease. Rev Investig Clin Organo Hosp Enfermedades Nutr 2017; 69: 243–246. [DOI] [PubMed] [Google Scholar]
- 77. Kwon W‐Y, Cha H‐N, Heo J‐Y, Choi J‐H, Jang BI, Lee I‐K, Park S‐Y. Interleukin‐10 deficiency aggravates angiotensin II‐induced cardiac remodeling in mice. Life Sci 2016; 146: 214–221. [DOI] [PubMed] [Google Scholar]
- 78. Guo Y, Cen Z, Wei B, Wu W, Zhou Q. Increased circulating interleukin 10‐secreting B cells in patients with dilated cardiomyopathy. Int J Clin Exp Pathol 2015; 8: 8107–8114. [PMC free article] [PubMed] [Google Scholar]
- 79. Jiao J, Lu Y‐Z, Xia N, Wang Y‐Q, Tang T‐T, Nie S‐F, Lv B‐J, Wang K‐J, Wen S, Li J‐Y, Zhou X‐D, Liao Y‐H, Cheng X. Defective circulating regulatory B cells in patients with dilated cardiomyopathy. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol 2018; 46: 23–35. [DOI] [PubMed] [Google Scholar]
- 80. Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J Immunol Baltim Md 1950 2005; 175: 4180–4183. [DOI] [PubMed] [Google Scholar]
- 81. Gotot J, Gottschalk C, Leopold S, Knolle PA, Yagita H, Kurts C, Ludwig‐Portugall I. Regulatory T cells use programmed death 1 ligands to directly suppress autoreactive B cells in vivo. Proc Natl Acad Sci U S A 2012; 109: 10468–10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tang T‐T, Yuan J, Zhu Z‐F, Zhang W‐C, Xiao H, Xia N, Yan X‐X, Nie S‐F, Liu J, Zhou S‐F, Li J‐J, Yao R, Liao M‐Y, Tu X, Liao Y‐H, Cheng X. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol 2012; 107: 232. [DOI] [PubMed] [Google Scholar]
- 83. Okamoto N, Noma T, Ishihara Y, Miyauchi Y, Takabatake W, Oomizu S, Yamaoka G, Ishizawa M, Namba T, Murakami K, Iwado Y, Ohmori K, Kohno M. Prognostic value of circulating regulatory T cells for worsening heart failure in heart failure patients with reduced ejection fraction. Int Heart J 2014; 55: 271–277. [DOI] [PubMed] [Google Scholar]
- 84. Seo S, Fields ML, Buckler JL, Reed AJ, Mandik‐Nayak L, Nish SA, Noelle RJ, Turka LA, Finkelman FD, Caton AJ, Erikson J. The impact of T helper and T regulatory cells on the regulation of anti‐double‐stranded DNA B cells. Immunity 2002; 16: 535–546. [DOI] [PubMed] [Google Scholar]
- 85. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD. Dysfunctional and proinflammatory regulatory T‐lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation 2019; 139: 206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand J‐L, Cohen‐Tervaert JW, Drexler H, Filippatos G, Felix SB, Gullestad L, Hilfiker‐Kleiner D, Janssens S, Latini R, Neubauer G, Paulus WJ, Pieske B, Ponikowski P, Schroen B, Schultheiss H‐P, Tschöpe C, Van Bilsen M, Zannad F, McCurray J, Shah AM. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2009; 11: 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, double‐blind, placebo‐controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor‐α, in patients with moderate‐to‐severe heart failure: results of the Anti‐TNF Therapy Against Congestive Heart failure (ATTACH) Trial. Circulation 2003; 107: 3133–3140. [DOI] [PubMed] [Google Scholar]
- 88. Mann DL, McMurray JJV, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, Krum H, Liu P, Nieminen M, Tavazzi L, van Veldhuisen DJ, Waldenstrom A, Warren M, Westheim A, Zannad F, Fleming T. Targeted anticytokine therapy in patients with chronic heart failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004; 109: 1594–1602. [DOI] [PubMed] [Google Scholar]
- 89. Souto‐Carneiro MM, Mahadevan V, Takada K, Fritsch‐Stork R, Nanki T, Brown M, Fleisher TA, Wilson M, Goldbach‐Mansky R, Lipsky PE. Alterations in peripheral blood memory B cells in patients with active rheumatoid arthritis are dependent on the action of tumour necrosis factor. Arthritis Res Ther 2009; 11: R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Aukrust P, Gullestad L, Lappegård KT, Ueland T, Aass H, Wikeby L, Simonsen S, Frøland SS, Mollnes TE. Complement activation in patients with congestive heart failure: effect of high‐dose intravenous immunoglobulin treatment. Circulation 2001; 104: 1494–1500. [DOI] [PubMed] [Google Scholar]
- 91. Dandel M, Englert A, Wallukat G, Riese A, Knosalla C, Stein J, Hetzer R. Immunoadsorption can improve cardiac function in transplant candidates with non‐ischemic dilated cardiomyopathy associated with diabetes mellitus. Atheroscler Suppl 2015; 18: 124–133. [DOI] [PubMed] [Google Scholar]
- 92. Torre‐Amione G, Anker SD, Bourge RC, Colucci WS, Greenberg BH, Hildebrandt P, Keren A, Motro M, Moyé LA, Otterstad JE, Pratt CM, Ponikowski P, Rouleau JL, Sestier F, Winkelmann BR, Young JB, Advanced Chronic Heart Failure CLinical Assessment of Immune Modulation Therapy Investigators . Results of a non‐specific immunomodulation therapy in chronic heart failure (ACCLAIM trial): a placebo‐controlled randomised trial. Lancet Lond Engl 2008; 371: 228–236. [DOI] [PubMed] [Google Scholar]
- 93. Szondy Z, Garabuczi É, Joós G, Tsay GJ, Sarang Z. Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front Immunol 2014; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Alvarez P, Briasoulis A. Immune modulation in heart failure: the promise of novel biologics. Curr Treat Options Cardiovasc Med 2018; 20: 26. [DOI] [PubMed] [Google Scholar]
- 95. Leandro MJ. B‐cell subpopulations in humans and their differential susceptibility to depletion with anti‐CD20 monoclonal antibodies. Arthritis Res Ther 2013; 15: S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Weiner GJ. Rituximab: mechanism of action. Semin Hematol 2010; 47: 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Thaunat O, Patey N, Gautreau C, Lechaton S, Fremeaux‐Bacchi V, Dieu‐Nosjean M‐C, Cassuto‐Viguier E, Legendre C, Delahousse M, Lang P, Michel J‐B, Nicoletti A. B cell survival in intragraft tertiary lymphoid organs after rituximab therapy. Transplantation 2008; 85: 1648–1653. [DOI] [PubMed] [Google Scholar]
- 98. Becerra E, Scully MA, Leandro MJ, Heelas EO, Westwood J‐P, De La Torre I, Cambridge G. Effect of rituximab on B cell phenotype and serum B cell‐activating factor levels in patients with thrombotic thrombocytopenic purpura. Clin Exp Immunol 2015; 179: 414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tschöpe C, Van Linthout S, Spillmann F, Posch MG, Reinke P, Volk H‐D, Elsanhoury A, Kühl U. Targeting CD20+ B‐lymphocytes in inflammatory dilated cardiomyopathy with rituximab improves clinical course: a case series. Eur Heart J Case Rep 2019; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sánchez‐Trujillo L, Jerjes‐Sanchez C, Rodriguez D, Panneflek J, Ortiz‐Ledesma C, Garcia‐Rivas G, Torre‐Amione G. Phase II clinical trial testing the safety of a humanised monoclonal antibody anti‐CD20 in patients with heart failure with reduced ejection fraction, ICFEr‐RITU2: study protocol. BMJ Open 2019; 9: e022826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Blair HA, Duggan ST. Belimumab: a review in systemic lupus erythematosus. Drugs 2018; 78: 355–366. [DOI] [PubMed] [Google Scholar]
- 102. Florentina P, Binder CJ. Impact of B‐cell‐targeted therapies on cardiovascular disease. Arterioscler Thromb Vasc Biol American Heart Association 2019; 39: 1705–1714. [DOI] [PubMed] [Google Scholar]
- 103. Gottenberg J‐E, Morel J, Perrodeau E, Bardin T, Combe B, Dougados M, Flipo R‐M, Saraux A, Schaeverbeke T, Sibilia J, Soubrier M, Vittecoq O, Baron G, Constantin A, Ravaud P, Mariette X. Comparative effectiveness of rituximab, abatacept, and tocilizumab in adults with rheumatoid arthritis and inadequate response to TNF inhibitors: prospective cohort study. The BMJ 2019; 364: l67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Brezinschek H‐P, Rainer F, Brickmann K, Graninger WB. B lymphocyte‐typing for prediction of clinical response to rituximab. Arthritis Res Ther 2012; 14: R161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nakou M, Katsikas G, Sidiropoulos P, Bertsias G, Papadimitraki E, Raptopoulou A, Koutala H, Papadaki HA, Kritikos H, Boumpas DT. Rituximab therapy reduces activated B cells in both the peripheral blood and bone marrow of patients with rheumatoid arthritis: depletion of memory B cells correlates with clinical response. Arthritis Res Ther 2009; 11: R131. [DOI] [PMC free article] [PubMed] [Google Scholar]