

Dilated cardiomyopathy (DCM) is a genetic heart disease that frequently leads to end‐stage heart failure and is an important cause of mortality and morbidity in individuals <45 years of age. 1 Patients with DCM are prone to arrhythmias with 30% suffering sudden cardiac death (SCD) events. Current guidelines recommend the use of implantable cardioverter defibrillators (ICDs) for the primary prevention of SCD in DCM patients with New York Heart Association (NYHA) Class II to III heart failure and a left ventricular ejection fraction (LVEF) ≤35%. 2 However, most cases of SCD occur in DCM patients with preserved or only slightly reduced LVEF. 3 Notably, a sizeable proportion of those with an ICD will never receive appropriate device therapy while being exposed to a significant risk of device‐related complications, which occur in ~9% of patients. 4 , 5 Thus, there is a need for a bespoke, individualized, precision medicine approach to improve patient selection for ICD with tangible improvement of outcomes.
During the past four decades, a multitude of studies has tried to identify clinical parameters that would more reliably predict sustained ventricular arrhythmias (VA) in this arrhythmia‐susceptible population. 6 Many of these variables showed inconsistent results in different populations, with limited reliability, validity, and reproducibility, while others were challenging to translate to clinical practice. 6 An ideal biomarker should be (i) sensitive; (ii) specific; (iii) cost‐effective; (iv) widely available; (v) non‐invasive; (vi) quantifiable; (vii) correlate temporally and with severity of disease; (vii) able to offer early detection; (viii) validated and reliable; and (ix) reproducible. 7 Many of these biomarkers have fallen short on most of these aspects, leaving LVEF as the most reliable prognostic biomarker. However, most SCD events occur in those with no prior history of VA or an LVEF > 40%, and SCD can be the sentinel tragic and lethal event. Given the challenging task of decision making for ICD implantation in DCM patients, it is important to understand the collective evidence for previously proposed predictors and their potential role in DCM risk stratification. The increased implementation of next‐generation genetic sequencing technologies in clinical practice results in larger genotyped DCM populations with distinct genetic backgrounds and ensuing heterogeneous profiles, which may pave the way for individualizing risk. 8
To this end, in this issue of ESC Heart Failure, Sammani et al. 9 performed a systematic review and meta‐analysis of studies that investigated the predictors of sustained VA in DCM. The authors report an annual risk of 4.5% of sustained VA, which demonstrates the importance of arrhythmic SCD prevention in this population. Younger age, presence of hypertension, male sex, history of non‐sustained VA, decreased LVEF, left ventricular dilation, late gadolinium enhancement (LGE) on cardiac magnetic resonance imaging, and presence of mutations in phospholamban (PLN), lamin A/C (LMNA), and filamin C (FLNC) genes were significant predictors of sustained VA. The authors suggest that future clinical decision support tools for ICD implantation in DCM should incorporate many clinical parameters to improve risk prediction and perform competing event analyses.
The findings of this meta‐analysis are interesting in many aspects. First, in the context of the published literature and clinical knowledge, they once again attest that predictors of sustained VA in DCM are hard to identify, and their additive value is currently low to moderate when compared with LVEF. This challenge can be attributed to several factors: (i) DCM is a heterogeneous disease and an end manifestation of many conditions (many of which remain under‐recognized) with different arrhythmia risk patterns 1 ; (ii) arrhythmias in DCM result from different mechanisms (e.g. re‐entry propagated by myocardial fibrosis, and primary arrhythmias due to genetic defects or secondary remodelling in ion channels, desmosomes, or connexins) 10 ; (iii) the role of neural circulatory control and its impact on initiation and maintenance of arrhythmias are under‐investigated; (iv) the natural course of the disease can influence the effect of arrhythmic risk factors over time, with some predictors being valid only at a certain disease stage 11 ; and (v) lack of sufficient data for identifying potential predictors of sustained VAs.
Second, the independent predictive power of LGE and genetic findings, perhaps the two most promising of all potential VA/SCD prognostic biomarkers, may be increased manifold when more genotype–phenotype association data emerge for other DCM‐associated genes. 8 Stated otherwise, individual clinical parameters may pose risk for only certain genetic subtypes of DCM but be of no significant clinical value to others (e.g. marked LGE is a predictor for VA in FLNC‐mediated disease, 12 whereas VA/SCD in RBM20‐related DCM typically result from disturbed calcium handling in the absence of discernable structural remodelling 13 ). Recent advances in long‐term heart rate monitoring, high‐resolution cardiac magnetic resonance imaging, and next‐generation sequencing promise incremental progress in identifying disease subtypes over the coming years, facilitating clustering of DCM groups based on their genetic, morphological, and electrophysiological substrates (Figure 1 ). 14 , 15 , 16
Figure 1.
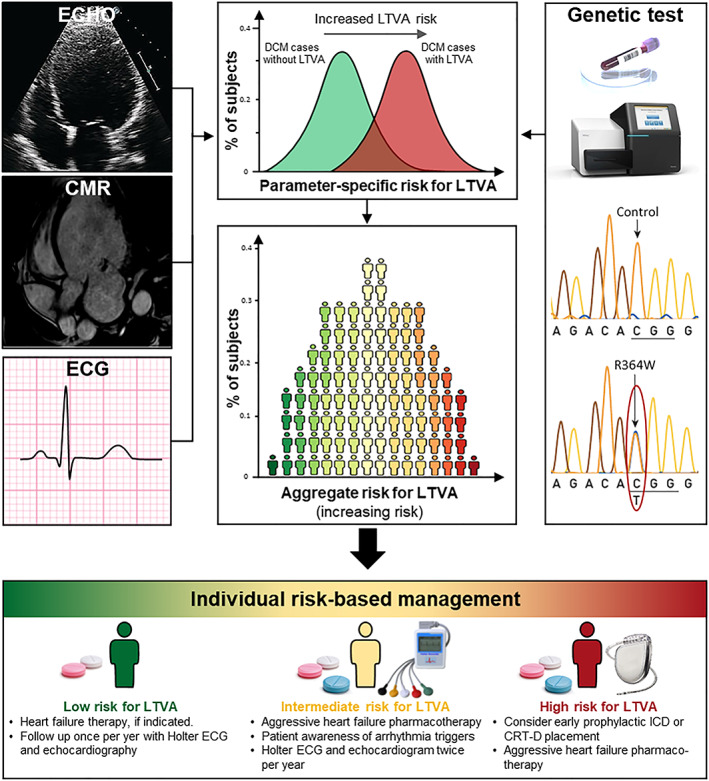
The principle for a future model for sustained ventricular arrhythmias/sudden cardiac death risk stratification in dilated cardiomyopathy. Risk factors should be determined among demographic, clinical, and genetic parameters by analysing separately for and compared in dilated cardiomyopathy (DCM) patients with and those without life‐threatening ventricular arrhythmias (LTVAs). Final determination of the aggregate arrhythmic risk should be based on a multi‐parametric risk model, which will allow to tailor the therapy to individual patient's needs and prevent sudden cardiac death in DCM patients. CMR, cardiac magnetic resonance imaging; CRT‐D, cardiac resynchronization therapy defibrillator; ECG, electrocardiogram; ICD, implantable cardioverter defibrillator.
The authors chose a comprehensive approach by including known autonomic, ECG, and Holter‐based biomarkers of SCD, which have been plagued by lack of reproducibility and difficult implementation. 17 Most of these were unsurprisingly not predictive, reflective of the small number of studies and the high heterogeneity in the clinical parameters of included cohorts. Thus, although negative for most potential predictors, the quality of the meta‐analysis is only as good as the original data. Hence, these critical components of dysrhythmogenesis deserve detailed systematic re‐evaluation, especially with the advent of more reliable measures and wearable or implantable technologies (such as activity monitors, subcutaneous rhythm monitors, and CardioMEMS). 18
Given that the DCM phenotype is often the ultimate manifestation of etiologically heterogeneous groups of diseases, a broad definition of DCM (at times referred to as non‐ischemic DCM) reflecting inclusion of a rather mixed patient/disease population is an important limitation of this meta‐analysis. Furthermore, of all the monogenic cardiovascular diseases, the genetics of DCM is arguably the most complex, with DCM phenotypes seen in cases with genetic variants associated with other cardiomyopathies, cardiac ion channelopathies, muscular dystrophies, and mitochondrial disorders; therefore, another limitation of this paper is its focus on a limited number of genes, which have been reported to be associated with more arrhythmogenic DCM in an earlier pooled analysis published in 2017. 19 Additionally, similar to many previous studies, the right ventricular function was not included as a potential predictor of VA/SCD. Considering the recent trends in reshaping our approach to arrhythmogenic cardiomyopathies, as emphasized with the recent consensus document, 20 understanding the contribution of left‐dominant and bi‐ventricular arrhythmogenic cardiomyopathies to DCM caused by classical desmosomal gene variants is important. 21 This notion has been further supported by a recent study, showing higher arrhythmic burden in DCM patients with desmosomal gene variants. 14
This shift from traditional to a precision medicine approach has started with the recognition of cardiac laminopathies as a distinct clinical entity with specific risk parameters for SCD 22 and will likely be extended to other DCM subtypes in the near future. This knowledge will not only empower clinicians with tools for identifying candidates for ICD but will also allow us to provide assurance to those with benign DCM phenotypes who can afford not being implanted with an ICD. Thus, there is a need for well‐trained, competent subspecialists in clinical cardiovascular genetics able to interpret the swaths of information generated by ‘omics’ technologies. 23 Only then can the promise of precision be delivered and the peril avoided.
Asatryan, B. , and Chahal, C. A. A. (2020) Enhancing risk stratification for life‐threatening ventricular arrhythmias in dilated cardiomyopathy: the peril and promise of precision medicine. ESC Heart Failure, 7: 1383–1386. 10.1002/ehf2.12886.
References
- 1. Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, Fonarow GC, Francis GS, Lenihan D, Lewis EF, McNamara DM, Pahl E, Vasan RS, Ramasubbu K, Rasmusson K, Towbin JA, Yancy C, American Heart Association Committee on Heart Failure and Transplantation of the Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Epidemiology and Prevention; and Council on Quality of Care and Outcomes Research . Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 2016; 134: e579–e646. [DOI] [PubMed] [Google Scholar]
- 2. Priori SG, Blomstrom‐Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez‐Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ, Group ESCSD . ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2015; 36: 2793–2867. [DOI] [PubMed] [Google Scholar]
- 3. Stecker EC, Vickers C, Waltz J, Socoteanu C, John BT, Mariani R, McAnulty JH, Gunson K, Jui J, Chugh SS. Population‐based analysis of sudden cardiac death with and without left ventricular systolic dysfunction: two‐year findings from the Oregon Sudden Unexpected Death Study. J Am Coll Cardiol 2006; 47: 1161–1166. [DOI] [PubMed] [Google Scholar]
- 4. Kadish A, Dyer A, Daubert JP, Quigg R, Estes NA, Anderson KP, Calkins H, Hoch D, Goldberger J, Shalaby A, Sanders WE, Schaechter A, Levine JH. Defibrillators in Non‐Ischemic Cardiomyopathy Treatment Evaluation I. Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N Engl J Med 2004; 350: 2151–2158. [DOI] [PubMed] [Google Scholar]
- 5. Ezzat VA, Lee V, Ahsan S, Chow AW, Segal O, Rowland E, Lowe MD, Lambiase PD. A systematic review of ICD complications in randomised controlled trials versus registries: is our ‘real‐world’ data an underestimation? Open Heart 2015; 2: e000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Halliday BP, Cleland JGF, Goldberger JJ, Prasad SK. Personalizing risk stratification for sudden death in dilated cardiomyopathy: the past, present, and future. Circulation 2017; 136: 215–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Institute of Medicine (US) Forum on Drug Discovery, Development, and Translation. Emerging Safety Science: Workshop Summary. Washington (DC): National Academies Press (US) ; 2008. 7. Qualifying biomarkers. Available from: https://www.ncbi.nlm.nih.gov/books/NBK4041/ [accessed 2020 May 10]. [PubMed]
- 8. Asatryan B, Medeiros‐Domingo A. Translating emerging molecular genetic insights into clinical practice in inherited cardiomyopathies. J Mol Med (Berl) 2018; 96: 993–1024. [DOI] [PubMed] [Google Scholar]
- 9. Sammani A, Kayvanpour E, Bosman LP, Sedaghat‐Hamedani F, Proctor T, Gi WT, Broezel A, Jensen K, Katus HA, Te Riele A, Meder B, Asselbergs FW. Predicting sustained ventricular arrhythmias in dilated cardiomyopathy: a meta‐analysis and systematic review. ESC Heart Fail 2020. 10.1002/ehf2.12689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marian AJ, Asatryan B, Wehrens XHT. Genetic basis and molecular biology of cardiac arrhythmias in cardiomyopathies. Cardiovasc Res 2020. 10.1093/cvr/cvaa116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stolfo D, Ceschia N, Zecchin M, De Luca A, Gobbo M, Barbati G, Gigli M, Mase M, Pinamonti B, Pivetta A, Merlo M, Sinagra G. Arrhythmic risk stratification in patients with idiopathic dilated cardiomyopathy. Am J Cardiol 2018; 121: 1601–1609. [DOI] [PubMed] [Google Scholar]
- 12. Hall CL, Akhtar MM, Sabater‐Molina M, Futema M, Asimaki A, Protonotarios A, Dalageorgou C, Pittman AM, Suarez MP, Aguilera B, Molina P, Zorio E, Hernandez JP, Pastor F, Gimeno JR, Syrris P, McKenna WJ. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int J Cardiol 2020; 307: 101–108. [DOI] [PubMed] [Google Scholar]
- 13. van den Hoogenhof MMG, Beqqali A, Amin AS, van der Made I, Aufiero S, Khan MAF, Schumacher CA, Jansweijer JA, van Spaendonck‐Zwarts KY, Remme CA, Backs J, Verkerk AO, Baartscheer A, Pinto YM, Creemers EE. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 2018138: 1330–1342. [DOI] [PubMed] [Google Scholar]
- 14. Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, Stolfo D, Haywood ME, Dal Ferro M, Altinier A, Ramani F, Brun F, Cocciolo A, Puggia I, Morea G, McKenna WJ, La Rosa FG, Taylor MRG, Sinagra G, Mestroni L. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol 2019; 74: 1480–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Halliday BP, Gulati A, Ali A, Guha K, Newsome S, Arzanauskaite M, Vassiliou VS, Lota A, Izgi C, Tayal U, Khalique Z, Stirrat C, Auger D, Pareek N, Ismail TF, Rosen SD, Vazir A, Alpendurada F, Gregson J, Frenneaux MP, Cowie MR, Cleland JGF, Cook SA, Pennell DJ, Prasad SK. Association between midwall late gadolinium enhancement and sudden cardiac death in patients with dilated cardiomyopathy and mild and moderate left ventricular systolic dysfunction. Circulation 2017; 135: 2106–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Di Marco A, Anguera I, Schmitt M, Klem I, Neilan TG, White JA, Sramko M, Masci PG, Barison A, McKenna P, Mordi I, Haugaa KH, Leyva F, Rodriguez Capitan J, Satoh H, Nabeta T, Dallaglio PD, Campbell NG, Sabate X, Cequier A. Late gadolinium enhancement and the risk for ventricular arrhythmias or sudden death in dilated cardiomyopathy: systematic review and meta‐analysis. JACC Heart Fail 2017; 5: 28–38. [DOI] [PubMed] [Google Scholar]
- 17. Wellens HJ, Schwartz PJ, Lindemans FW, Buxton AE, Goldberger JJ, Hohnloser SH, Huikuri HV, Kaab S, La Rovere MT, Malik M, Myerburg RJ, Simoons ML, Swedberg K, Tijssen J, Voors AA, Wilde AA. Risk stratification for sudden cardiac death: current status and challenges for the future. Eur Heart J 2014; 35: 1642–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sana F, Isselbacher EM, Singh JP, Heist EK, Pathik B, Armoundas AA. Wearable devices for ambulatory cardiac monitoring: JACC state‐of‐the‐art review. J Am Coll Cardiol 2020; 75: 1582–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kayvanpour E, Sedaghat‐Hamedani F, Amr A, Lai A, Haas J, Holzer DB, Frese KS, Keller A, Jensen K, Katus HA, Meder B. Genotype‐phenotype associations in dilated cardiomyopathy: meta‐analysis on more than 8000 individuals. Clin Res Cardiol 2017; 106: 127–139. [DOI] [PubMed] [Google Scholar]
- 20. Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, Estes NAM 3rd, Hua W, Indik JH, Ingles J, James CA, John RM, Judge DP, Keegan R, Krahn AD, Link MS, Marcus FI, McLeod CJ, Mestroni L, Priori SG, Saffitz JE, Sanatani S, Shimizu W, van Tintelen JP, Wilde AAM, Zareba W. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019; 16: e301–e372. [DOI] [PubMed] [Google Scholar]
- 21. Asatryan B, Servatius H. Revisiting the approach to diagnosis of arrhythmogenic cardiomyopathy: stick to the arrhythmia criterion! Circ Genom Precis Med 2019; 12: 455–457. [DOI] [PubMed] [Google Scholar]
- 22. Wahbi K, Ben Yaou R, Gandjbakhch E, Anselme F, Gossios T, Lakdawala NK, Stalens C, Sacher F, Babuty D, Trochu JN, Moubarak G, Savvatis K, Porcher R, Laforet P, Fayssoil A, Marijon E, Stojkovic T, Behin A, Leonard‐Louis S, Sole G, Labombarda F, Richard P, Metay C, Quijano‐Roy S, Dabaj I, Klug D, Vantyghem MC, Chevalier P, Ambrosi P, Salort E, Sadoul N, Waintraub X, Chikhaoui K, Mabo P, Combes N, Maury P, Sellal JM, Tedrow UB, Kalman JM, Vohra J, Androulakis AFA, Zeppenfeld K, Thompson T, Barnerias C, Becane HM, Bieth E, Boccara F, Bonnet D, Bouhour F, Boule S, Brehin AC, Chapon F, Cintas P, Cuisset JM, Davy JM, De Sandre‐Giovannoli A, Demurger F, Desguerre I, Dieterich K, Durigneux J, Echaniz‐Laguna A, Eschalier R, Ferreiro A, Ferrer X, Francannet C, Fradin M, Gaborit B, Gay A, Hagege A, Isapof A, Jeru I, Juntas Morales R, Lagrue E, Lamblin N, Lascols O, Laugel V, Lazarus A, Leturcq F, Levy N, Magot A, Manel V, Martins R, Mayer M, Mercier S, Meune C, Michaud M, Minot‐Myhie MC, Muchir A, Nadaj‐Pakleza A, Pereon Y, Petiot P, Petit F, Praline J, Rollin A, Sabouraud P, Sarret C, Schaeffer S, Taithe F, Tard C, Tiffreau V, Toutain A, Vatier C, Walther‐Louvier U, Eymard B, Charron P, Vigouroux C, Bonne G, Kumar S, Elliott P, Duboc D. Development and validation of a new risk prediction score for life‐threatening ventricular tachyarrhythmias in laminopathies. Circulation 2019; 140: 293–302. [DOI] [PubMed] [Google Scholar]
- 23. Ahmad F, McNally EM, Ackerman MJ, Baty LC, Day SM, Kullo IJ, Madueme PC, Maron MS, Martinez MW, Salberg L, Taylor MR, Wilcox JE. Establishment of specialized clinical cardiovascular genetics programs: recognizing the need and meeting standards: a scientific statement from the American Heart Association. Circ Genom Precis Med 2019; 12: e000054. [DOI] [PubMed] [Google Scholar]