

Abstract
A major challenge for RNA-seq analysis of gene expression is to achieve sufficient coverage of informative nonribosomal transcripts. In eukaryotic samples, this is typically achieved by selective oligo(dT)-priming of messenger RNAs to exclude ribosomal RNA (rRNA) during cDNA synthesis. However, this strategy is not compatible with prokaryotes in which functional transcripts are generally not polyadenylated. To overcome this, we adopted DASH (depletion of abundant sequences by hybridization), initially developed for eukaryotic cells, to improve both the sensitivity and depth of bacterial RNA-seq. DASH uses the Cas9 nuclease to remove unwanted cDNA sequences prior to library amplification. We report the design, evaluation, and optimization of DASH experiments for standard bacterial short-read sequencing approaches, including software for automated guide RNA (gRNA) design for Cas9-mediated cleavage in bacterial rDNA sequences. Using these gRNA pools, we effectively removed rRNA reads (56%–86%) in RNA-seq libraries from two different model bacteria, the Gram-negative pathogen Salmonella enterica and the anaerobic gut commensal Bacteroides thetaiotaomicron. DASH works robustly, even with subnanogram amounts of input RNA. Its efficiency, high sensitivity, ease of implementation, and low cost (∼$5 per sample) render DASH an attractive alternative to rRNA removal protocols, in particular for material-constrained studies where conventional ribodepletion techniques fail.
Keywords: bacterial RNA-seq, DASH, ribosomal RNA, Cas9, CRISPR, Salmonella, Bacteroides
INTRODUCTION
The advent of high-throughput RNA sequencing (RNA-seq) has revolutionized the field of bacterial RNA biology (Croucher and Thomson 2010; Hör et al. 2018). RNA-seq has shown that bacterial transcriptomes, once believed to be simple in terms of structure and regulation, can be almost as complex as their eukaryotic counterparts (Sorek and Cossart 2010), and helped to realize that bacteria amply use post-transcriptional control to regulate gene expression (Hör et al. 2018). In RNA-seq, the experimental steps prior to sequencing are universal and consist mainly of RNA extraction, enzymatic digestion of genomic DNA, depletion of ribosomal RNA (rRNA), and conversion of the remaining RNA pool into complementary DNA (cDNA) libraries. The removal of rRNA (typically ∼90% of the total cellular RNA) is important as it much increases coverage of messenger RNA (mRNA) and regulatory noncoding RNA. A straightforward method to avoid rRNA reads in eukaryotic RNA-seq studies harnesses oligo(dT) oligonucleotides that anneal to the poly(A) tail of mRNAs to selectively prime reverse transcription (RT). However, prokaryotic transcripts are not normally polyadenylated (Dreyfus and Regnier 2002), which necessitates the development of alternative rRNA removal strategies.
The most popular methods for rRNA depletion from prokaryotic samples follow a “pull-out” strategy whereby rRNA molecules are depleted from a sample with complementary oligonucleotides coupled to magnetic beads. This strategy underlies several commercial ready-to-use kits, especially the popular MICROBExpress and RiboMinus kits (Thermo Fisher Scientific) as well as the Ribo-Zero technology (Illumina). While it generally achieves excellent results for model bacteria such as Escherichia coli, these kits are associated with both, high cost ($60–80 per sample) and a limited efficiency for nonmodel species such as the vast majority of those >1000 different bacteria in the human microbiota. In addition, Ribo-Zero—long considered the gold standard for bacterial rRNA depletion (Giannoukos et al. 2012; Petrova et al. 2017)—was recently discontinued. This has renewed efforts in the community to develop in-house solutions for rRNA depletion (Kim et al. 2019; Culviner et al. 2020).
Available alternative bacterial rRNA removal strategies use RNase H-mediated digestion of RNA:DNA hybrids (Huang et al. 2020), size-selection of mRNAs through liquid chromatography (Castro et al. 2013), or selective exclusion of rRNAs from cDNA conversion with “not-so-random” hexamers (Hirakawa et al. 2011; Chugani et al. 2012); the latter is also available as part of the Universal Prokaryotic RNA-Seq kit (NuGen). Overall, these methods achieve good to excellent (∼70%–99%) rRNA depletion rates. However, each of these protocols depletes rRNA at the RNA level, that is, prior to multiplexing. This limits the applicability of these techniques for studies with low-input material or high-throughput analysis including single-cell RNA-seq. Some recent library preparation alternatives such as RNAtag-seq (Shishkin et al. 2015) barcode RNA samples and pool them for joint rRNA depletion, thereby reducing cost. However, since barcoding at the RNA level requires high input amounts, it too, is little suited for low-input bacterial RNA-seq.
Instead of removing rRNA from the input sample, rRNA fragments might as well be removed at the cDNA level, that is, following library preamplification and multiplexing. An early study digested rRNA-derived cDNAs with a double strand-specific DNase after melting and reannealing (Yi et al. 2011). However, this method requires substantial optimization of the reannealing conditions. Moreover, the protocol is yet to be combined with multiplexing of cDNA. In other words, it still requires nanogram amounts of input RNA, while its efficiency of rRNA read depletion is inferior to the above-described methods (Yi et al. 2011; Giannoukos et al. 2012). A second study introduced cDNA-level depletion of rRNAs through probe-directed degradation (PDD; [Archer et al. 2014]), in which DNA probes are annealed to rRNA-derived cDNAs and degraded with double strand-specific DNase. While promising, this method requires circularization of the cDNA fragments, hampering its introduction in common RNA-seq protocols.
Recently, programmed DNA cleavage by the CRISPR-associated nuclease Cas9 has been introduced as a novel technology to deplete with high sequence specificity “unwanted” fragments from eukaryotic cDNA libraries. In this so-called DASH (depletion of abundant sequences by hybridization) approach (Gu et al. 2016), a pool of single-guide RNAs (sgRNAs) is used to direct Cas9 cleavage of rRNA-derived cDNA molecules during library preparation. Since the cleaved fragments are not amplified in the subsequent PCR step, intact non-rRNA fragments become enriched (Fig. 1). To illustrate the power of DASH, a pool of tiled (roughly every 50 bases) sgRNAs against human mitochondrial rRNAs reduced the corresponding cDNA reads by more than 1000-fold, while it concomitantly increased coverage of nonribosomal transcripts by ∼2.4-fold. DASH has also been used to increase coverage of nonabundant transcripts in human small RNA-seq libraries by targeting adapter dimers and tissue-specifically highly expressed microRNAs (Hardigan et al. 2019) and to deplete hemoglobin transcripts from polar bear peripheral blood RNA samples before long- and short-read sequencing (Byrne et al. 2019). Similar to DASH of eukaryotic cDNA, a thermostable Cas9 variant was recently used to cleave E. coli 16S rRNA sequences in cDNA during PCR-mediated library amplification (Schmidt et al. 2019).
FIGURE 1.

Overview of the bacterial DASH workflow. Schematic of the principle behind DASH-mediated removal of rRNA-derived cDNA fragments from sequencing libraries. The individual steps in the bacterial DASH pipeline are indicated in the zoom-in at the bottom.
The promise of DASH to provide a generic approach notwithstanding, the method is yet to be fully established for bacterial transcriptomics. In this study, we evaluate DASH for bacterial short-read RNA-seq for two intensely studied bacteria: Salmonella enterica serovar Typhimurium (henceforth, Salmonella), which is a major model species of both, bacterial RNA biology and pathogenesis; and Bacteroides thetaiotaomicron as an example of an abundant human microbiota species. The presented sgRNA design software and optimized wet-lab protocols bear great potential for efficient, sensitive, and economic removal of unwanted rRNA sequences for RNA-seq analysis of any bacterium of interest.
RESULTS
Proof-of-concept of bacterial DASH and optimization of Cas9 reaction conditions
The well-studied Gram-negative bacterium Salmonella has been the subject of many RNA-seq studies (Kroger et al. 2013; Westermann et al. 2016). Unless depleted, Salmonella rRNA typically represents ∼95% of all cDNA reads in a library (Fig. 2A; Betin et al. 2019). Salmonella rRNA is transcribed from seven ribosomal operons (rrn) across the chromosome, giving rise to each seven homologs of the 16S (genes rrsA-G) and 23S (rrlA-G), and eight copies of 5S rRNA (the rrnD locus carries two 5S rRNA-encoding rrf genes). Generally speaking, the sequences of the rRNA genes are highly conserved across all loci, but they do show indels and point mutations.
FIGURE 2.

Initial DASH run on Salmonella total RNA. (A) Composition of total RNA extracted from exponentially growing Salmonella as deduced from RNA-seq reads. (B) Conservation of Salmonella rRNAs genes and location of the identified sgRNA target sites. The per-base conservation of each rRNA gene across all Salmonella rRNA homologs is plotted. Above each consensus sequence (represented as a gray rectangle), the designed sgRNAs target sites are indicated by a black triangle. (C) Pilot run of DASH-mediated rRNA depletion in Salmonella. (Left) The fraction of ribosomal-derived reads over the total number of mapped reads for a control and a DASH-treated library. (Right) Same data are expressed as efficiency of DASH-mediated rRNA depletion.
To deplete ribosomal fragments in cDNA from Salmonella total RNA, we initially designed a pool of sgRNAs targeting the consensus sequence of each rRNA gene. To this end, we wrote a Python script that identifies SpCas9 target sites within rRNA genes based on the following selection criteria: (i) perfect complementarity to at least five out of the seven (16S and 23S) or six out of the eight (5S) rRNA copies, (ii) a GC content of 35%–70%, (iii) a predicted low tendency to form secondary structures, and (iv) followed by the “NGG” protospacer adjacent motif (PAM) for recognition by the SpCas9 nuclease (Jinek et al. 2012). The script aimed at identifying one target site that satisfied the set criteria on average every ∼50 bp along the target space, as this density proved sufficient in eukaryotic DASH (Fig. 2B; see Materials and Methods for details on sgRNA design; Gu et al. 2016). As a result, our script proposed 113 sgRNAs in total. We then ordered DNA oligonucleotides, each comprising a T7 promoter, a single target site, and the first part of the SpCas9 sgRNA scaffold. To these sense oligonucleotides, we added a universal, partially overlapping reverse DNA oligonucleotide containing the remaining portion of the sgRNA scaffold. The resulting annealing products were filled up with the corresponding nucleobases, giving rise to the double-stranded DNA templates that were subjected to in vitro transcription with T7 RNA polymerase to yield the sgRNA pool.
Salmonella cDNA was preamplified in two PCR cycles and subsequently incubated with the in vitro-transcribed sgRNA pool and SpCas9 nuclease for 2 h at 37°C. We used a molar sgRNA:Cas9:cDNA ratio of 1000:100:1—as inferred as optimal in the original DASH protocol (Gu et al. 2016). Following digestion, we removed Cas9 with a silica-based column purification kit and proceeded with 16 cycles of PCR to amplify the uncleaved cDNA fragments. Sequencing of the resulting library revealed that, even after Cas9 cleavage, ∼83% of the obtained Salmonella reads derived from rRNA, thus a mere ∼13% reduction over the control library (Fig. 2C).
Using the same sgRNA pool, we then tested whether different reaction conditions would increase depletion efficiency. However, higher Cas9 concentrations only made rRNA depletion less efficient, suggesting that—among the concentrations tested—the above sgRNA:Cas9 ratio of 1000:100 was optimal for bacterial DASH, too (Fig. 3A). However, preincubation of Cas9 with the sgRNA pool, meant to allow for more time for the ribonucleoprotein complex to form prior to addition of the cDNA substrate, did increase rRNA depletion efficiency to ∼20% (Fig. 3B). Based on this finding, a Cas9:sgRNA preincubation step was included in all further reactions.
FIGURE 3.

Optimization of DASH reaction conditions. (A) rRNA depletion efficiency upon DASH cleavage with varying amounts of Cas9 and the sgRNA pool. Cas9 and sgRNA amounts are indicated as molar excess over a single fragment of the cDNA library. (B) Impact of preincubation of sgRNA and Cas9 prior to DASH on rRNA depletion efficiency. (C) Length distribution of the mapped portions of reads aligning to rRNAs after DASH compared with an untreated control. Frequency values are expressed as fraction (%) of the total number of reads mapping to rRNAs. (D) Same as in C but for reads aligning to non-rRNA genes.
Maximizing sgRNA density
The above-described initial ∼20% depletion of rRNA reads from Salmonella total RNA libraries was a far cry from the >99% depletion previously reported for human RNA samples (Gu et al. 2016). Inspection of the read length distribution obtained from our “DASHed” Salmonella libraries (1000:100 sgRNA:Cas9 ratio) confirmed efficient depletion of rRNA-derived reads >50 nt (Fig. 3C). However, shorter rRNA reads were even enriched over the untreated control sample. For comparison, the DASH treatment hardly affected the read length distribution of nonribosomal reads (Fig. 3D). This suggested that short ribosomal cDNA fragments evaded Cas9 cleavage, presumably because they were less likely to contain a full-length sgRNA target sequence.
To test whether covering more sites within the rRNA sequences would more efficiently remove those refractory short rRNA reads, we modified our design tool to obtain a sgRNA pool with the maximal number of target sites (Fig. 4A). This new sgRNA pool targeted all copies of the rRNA genes individually rather than just their consensus sequences. Discarding sgRNAs with potential off-target effects, this final pool consisted of 797 sgRNAs. With the same molar ratio of 1000:100:1 (sgRNA:Cas9:cDNA) as above, this pool brought rRNA depletion efficiency to 38% (Fig. 4B).
FIGURE 4.

Improved DASH efficiency by maximizing sgRNA target site density. (A) Workflow of the software developed for the design of high-density sgRNA pools. The number of sgRNAs passing each step is indicated to the right. (B) rRNA depletion efficiency using the 797 sgRNA pool in different ratios of Cas9 and sgRNA. Cas9 and sgRNA amounts are indicated as molar excess over a single fragment of the cDNA library. (C) Correlation of transcript abundances in the control versus DASHed (3500:35,000 excess of Cas9:sgRNA) libraries. Red dots (n = 22) represent rRNA transcripts, blue ones (n = 1511) the genetic features with at least 15 reads in the control and 150 reads in the DASH library, and gray dots (n = 3469) the genetic features below this threshold. The regression line and correlation coefficient were computed for the blue dots only. (D) Sequence read coverage of a representative rRNA locus and the dnaK, pinT, invR, and chiX genes in the control and DASH libraries (3500:35,000 excess of Cas9:sgRNA).
Given the much larger number of sgRNAs in this new pool, we speculated that higher molar excess of Cas9:sgRNA over cDNA could further improve depletion efficiency. Indeed, changing the molar ratio to 35,000:3500:1 (sgRNA:Cas9:cDNA) further increased the depletion efficiency, up to 56% (Fig. 4B). Consequently, the number of detected (RPKM > 1) Salmonella transcripts at the set sequencing depths of ∼5–12 M reads increased from 2724 in the untreated sample to 3875 in the DASH-treated library (Supplemental Fig. S1A).
To assess whether any sgRNA in this larger pool showed signs of off-targeting, we compared gene-wise read counts between the DASH-treated and cognate untreated libraries. Excluding rRNA, read counts for all genetic features showed very high correlation between the two libraries (Fig. 4C). Likewise, read coverage across rRNA genes decreased upon DASH, liberating more informative reads that mapped to mRNAs (e.g., dnaK) or regulatory small RNA (sRNA) sequences (PinT, InvR, ChiX; Fig. 4D). Together, this demonstrates that our design pipeline indeed selects sgRNAs with very little off-targeting and so enables specific removal of rRNA reads.
Increasing the sensitivity of DASH
One key advantage of rRNA depletion at the cDNA level is that it does not lower the amount of starting material for library preparation. Therefore, we tested whether DASH could improve the sensitivity of our RNA-seq protocol by systematically decreasing the quantity of input RNA from ∼800 ng to ∼0.4 ng (Fig. 5A). Using a fixed 1000:100:1 molar ratio of sgRNA:Cas9:cDNA, we observed efficient rRNA depletion with each of the four amounts tested, albeit efficiency varied from ∼30% to ∼50% (Fig. 5A). Importantly, the number of detected genetic features (which is a more robust readout) with RPKM > 1 was stable in the two highest RNA input amounts and decreased only in the lowest one (Supplemental Fig. S1B, top), likely due to the stochastic loss of low-abundance transcripts. For high-abundance transcripts (RPKM > 25), DASH increased the number of detected genes irrespective of input amount (Supplemental Fig. S1B, bottom).
FIGURE 5.

DASH of low-input RNA samples. (A) DASH efficiency for steadily decreasing input RNA amounts and for different Cas9 removal methods. DASH was performed on 1/5th of the cDNA resulting from reverse transcription. Cas9 and the sgRNA pool were used in a 1000 and 10,000 excess over a single fragment of the cDNA library, respectively. The indicated RNA amounts correspond to the reverse transcription input, while the cDNA amounts refer to what was used for DASH. (B) Depletion efficiency of DASH (3500:35,000 excess of Cas9:sgRNA) when combined with different library preparation kits. The indicated RNA amounts correspond to the reverse transcription input. (C) RNA class distribution of sequencing reads in the control and DASH samples shown in panel B.
In the original eukaryotic DASH protocol (Gu et al. 2016), the Cas9 enzyme is removed after the cleavage reaction by purification over a column, which runs the risk of losing cDNA as well. Here, we implemented digest of the Cas9 protein by proteinase K (Hardigan et al. 2019) as an alternative to column purification in bacterial DASH. Treatment with proteinase K did not affect the removal of rRNA reads (Fig. 5A), but resulted in increased cDNA yields after the post-DASH PCR amplification (Supplemental Table S1).
Lastly, we tested whether DASH works with a library preparation kit that is optimized for low input samples. Using the Takara SMARTer Stranded Total RNA-Seq kit with 1 ng total Salmonella RNA as input, we successfully removed more than half of the rRNA reads (Fig. 5B). Concomitantly, and similar to the above libraries generated with the NEBNext kit, the proportion of mRNA and sRNA reads increased by approximately ninefold (Fig. 5C). However, DASH treatment led to a slight increase in the fraction of reads <12 nt in length, which should not be a major concern since these reads are typically filtered out during read processing (Supplemental Fig. S2A). Taken together, this demonstrates that combining DASH with a library construction protocol optimized for minute RNA amounts enables robust RNA-seq analysis of low-input samples extracted from as few as ∼1000 bacteria, and potentially even fewer.
DASHing Bacteroides thetaiotaomicron RNA
To address generalizability of bacterial DASH, we selected a phylogenetically distant species. The Bacteroidia representative and human intestinal microbiota member B. thetaiotaomicron harbors five rRNA operons in its genome, each carrying one 23S, 16S, and 5S gene copy. Using our custom script, we designed 651 sgRNAs targeting all B. thetaiotaomicron rRNA operons individually (same selection criteria as for the optimized Salmonella rRNA depletion; see Fig. 4A). These sgRNAs were tested in parallel with the two different library construction kits and sgRNA:Cas9:cDNA molar ratios used for Salmonella samples (Figs. 4A, 5), on nanogram amounts of B. thetaiotaomicron RNA (Fig. 6A). We observed depletion of rRNA reads up to 86% for the standard library preparation kit (NEB) and up to 76% for the low-input protocol (Takara), with a corresponding increase in coverage of non-rRNA transcripts (Fig. 6B,D; Supplemental Fig. S2B). As before, there was a high correlation within abundance of genetic features between the untreated and the DASH samples (Fig 6C), arguing for negligible if any off-targeting by the sgRNAs.
FIGURE 6.
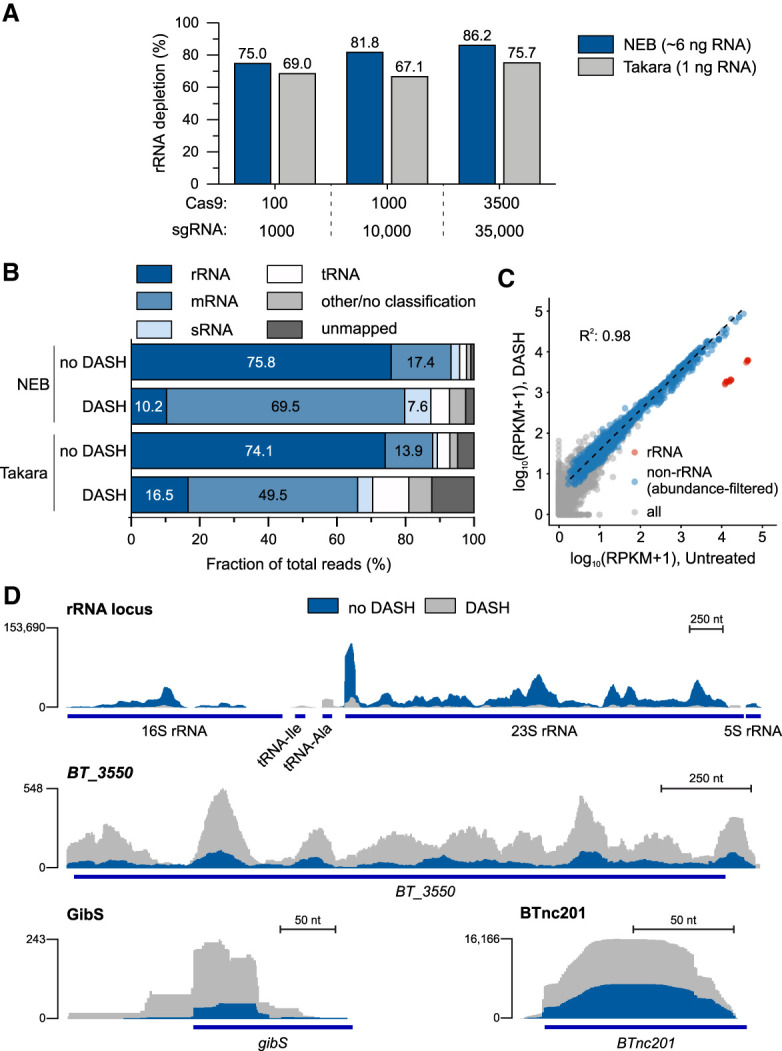
DASH-mediated removal of Bacteroides thetaiotaomicron rRNA. (A) rRNA depletion efficiency from a B. thetaiotaomicron cDNA library. DASH was combined with the different library preparation kits and using the indicated ratios of Cas9 and sgRNA over cDNA fragments. The indicated RNA amounts correspond to the reverse transcription input. (B) RNA class distribution of reads in control and DASH (3500:35,000 excess of Cas9:sgRNA) samples. (C) Correlation of transcript abundances in the control and DASH libraries shown in panel B. Red dots (n = 15) represent rRNA transcripts, blue ones (n = 2474) the genetic features with at least 15 reads in the control library and 150 reads in the DASH sample, and gray dots (n = 2658) the features below this cutoff. The regression line and correlation coefficient were computed for the blue dots only. (D) Sequence read coverage of a representative rRNA locus and BT_3550 (encoding a putative long-chain fatty acid-CoA ligase) and the gibS and BTnc201 regulatory RNA genes (bottom) in the control and DASH library of panels B and C.
DISCUSSION
First systematic evaluation of DASH for bacterial transcriptomics
This study reports the adaptation of the DASH technology to prokaryotic RNA samples. Through Cas9-mediated cleavage of rRNA-derived cDNA fragments prior to library amplification, our protocol increases coverage of nonribosomal transcripts in Salmonella and Bacteroides total RNA samples by ∼12- or ∼3.8-fold, respectively. Only a few changes to the normal library preparation protocol were necessary to implement our DASH protocol into a standard Illumina short-read sequencing pipeline.
Since it removes rRNA fragments after RT, DASH offers the major advantage that an initial amplification of the cDNA library can be performed before Cas9 cleavage. This increases the overall amount of cDNA and minimizes stochastic fragment loss. For this reason, our approach reaches good depletion (∼50%–80%) even with minute RNA amounts well below the lower recommended limit of the common rRNA depletion kits and techniques (Supplemental Table S2). Importantly, the recommended minimal input amount of the “gold standard” Ribo-Zero kit was 500 ng RNA, whereas our DASH approach works robustly for ∼400 pg of input RNA (Fig. 5A). Given that we have succeeded in combining DASH with state-of-the-art library preparation kits used in eukaryotic single-cell transcriptomics, we expect to be able to further reduce the necessary amount of starting material in the future. Obviously, this would open bacterial RNA-seq to many exciting areas of microbiology; to give just one example, it would allow one to perform gene expression profiling on bacteria recovered from insect guts.
While the upfront investment for purchasing all DASH reagents is high (Supplemental Table S4), we estimate a cost of $3–7 per sample for RNA-seq libraries, which is >10-fold lower than for commercial rRNA depletion kits (Supplemental Table S2). In this regard, bacterial DASH will remain competitive even with a very recently released new Ribo-Zero kit (“Ribo-Zero Plus,” Illumina catalog number: 20037135), which despite now using enzymatic rRNA depletion instead of rRNA pull-out, still runs at ∼$80 per sample. What is more, DASH bears potential for further cost reduction, for example, through in-house production of the Cas9 protein or the T7 RNA polymerase for in vitro transcription.
Although optimized on Salmonella RNA samples, the conditions established here enabled us to successfully run DASH on a phylogenetically distant bacterium, B. thetaiotaomicron. This argues that our protocol is applicable to total RNA from diverse bacterial species and, potentially, even to organisms beyond the bacterial kingdom. In principle, the modularity of our DASH approach should allow for the design of combined sgRNA pools targeting different species for ribodepletion of samples derived from mixed populations, such as metatranscriptomic samples or RNA mixtures isolated from infected host cells and tissues.
Comparison to previous DASH protocols
The original description of DASH on eukaryotic samples (Gu et al. 2016) reported a reduction of the targeted fragments by 99%, substantially higher than what we achieved here in bacteria. How can this difference be explained? Both the original DASH (Gu et al. 2016) and a recently updated protocol (Dynerman et al. 2020) were combined with long-read sequencing where average insert size was ∼300 nt. Similarly, the previous DASH-like experiment with thermostable Cas9 was applied to an E. coli cDNA library with an average insert size of 300–400 nt (Schmidt et al. 2019). In contrast, we describe the application of DASH to a standard Illumina RNA-seq pipeline with a maximal read length of 75 nt. Obviously, the longer the inserts, the higher the number of targetable fragments, which biases DASH toward longer reads (Fig. 3C). However, short-read sequencing is the standard in the field of bacterial RNA-seq and we therefore predict our DASH version to be particularly useful for any transcriptomics approach that involves bacteria. Additionally, our improved DASH protocol omits the multiple phenol/chloroform extraction or column purification steps of eukaryotic DASH (Gu et al. 2016; Dynerman et al. 2020). Instead, we remove Cas9 with a simple proteinase K treatment prior to further library amplification. Since this minimizes the risk of cDNA loss from organic extraction or silica column purification, our protocol will be particularly suitable for low-input samples.
By using a thermostable Cas9, Quake and colleagues recently demonstrated that DASH could be performed simultaneously with cDNA library amplification (Schmidt et al. 2019). However, the thermostable Cas9 variant used in the study requires a complex, 6 nt-long PAM, which dramatically reduces the number of possible sgRNA sites within the rRNA sequence space. Using our Python script with this hexameric PAM, at most 115 sgRNAs (as compared to 797 sgRNAs for SpCas9) could be designed for Salmonella rRNA. This is about the difference between our initial (Fig. 3) and the final (Fig. 4) sgRNA pools, which translates in a threefold difference in rRNA read removal. Therefore, although the classical Cas9 from Streptococcus pyogenes requires cleavage and PCR amplification to occur subsequently (not in parallel), it has an advantage over thermostable Cas9 with respect to rRNA read depletion in short-read libraries.
sgRNA design tool
As part of this work, we developed a Python script for designing sgRNAs targeting the rRNAs of a selected species with known reference genome (including annotations for ribosomal genes) that outputs the sequences of the DNA oligonucleotides needed as templates to in vitro-transcribe the customized sgRNA pool. Importantly, however, our software can also be fed with manually entered coordinates of rRNA genes, which will be important for organisms that lack a complete transcriptome annotation, such as many relevant microbiota members and important environmental bacteria.
Based on the assumption that maximizing sgRNA density improves depletion efficiencies, our pipeline predicts all possible sgRNAs and filters out only those sequences with extreme GC content (<30% or >80%) or predicted off-target effects. However, our algorithm does not remove guides with low predicted on-target activities, as we postulate that—as long as free Cas9 molecules are not the rate-limiting factor—individual sgRNAs with low on-target activity would not negatively impact ribosomal depletion efficiency by the entire pool. In this respect, our software differs from the many CRISPR design tools that have been developed for genome editing (Liu et al. 2020) and search for the “best” sgRNA per each target gene/locus.
Perspective
Further optimization of the DASH approach could include testing alternative Cas nucleases (Gonatopoulos-Pournatzis et al. 2020; Wessels et al. 2020), for example, high-fidelity Cas versions and enzymes with altered PAM preference or elevated thermostability (Schmidt et al. 2019). Moreover, a better understanding of the minimal sgRNA density for saturated depletion efficiency could help to reduce both, the cost of the sgRNA template pool and the amounts of Cas9 and sgRNA per reaction. Among the tested intervals, we identified a Cas9:sgRNA ratio of 1:10 as optimal; however, evaluating more refined, intermediate ratios in the future could result in more efficient target cleavage. Finally, it is likely that multiple rounds of DASH on the same sample (Dynerman et al. 2020) lead to more efficient depletion.
MATERIALS AND METHODS
RNA isolation
Bacterial RNA was isolated from an in vitro culture of S. enterica serovar Typhimurium strain SL1344 (Stocker et al. 1983) grown in Lennox broth (LB) medium to an optical density at 600 nm (OD600) of 2.0 or from a culture of B. thetaiotaomicron VPI-5482 grown in TYG medium to an OD600 of 0.5. To this end, cells were harvested and total RNA extracted using the TRIzol reagent (Invitrogen) according to the manufacturer's recommendations. To remove contaminating genomic DNA, samples were further treated with 0.25 U of DNase I (Fermentas) per 1 µg of RNA for 45 min at 37°C, followed by phenol–chloroform extraction and ethanol precipitation of the RNA transcripts. RNA quality was checked on an Agilent 2100 Bioanalyzer (Agilent Technologies).
sgRNA design and synthesis
Target sequences within Salmonella rRNA genes were identified and selected with two versions of a custom Python script. In the first version (Figs. 2–4), all rRNA copies were aligned with MUSCLE (Edgar 2004) and the consensus sequence of each gene was generated so that all positions identical in at least six (16S, 23S) or seven (5S) rRNA copies were maintained. All 20-nt potential gRNA targets were identified by searching both strands for the presence of the “NGG” PAM and then filtered to remove those sites with an extreme GC content (i.e., GC < 35% or >70%) or strong predicted secondary structures (MFE < −5, as computed with RNAfold [Lorenz et al. 2011]). Within the remaining pool, gRNAs were then selected to be ∼50 nt distant from each other, starting from the one closest to the 5′ end of the rRNA sequence. The resulting 113 sequences were purchased from IDT as a unique oligo pool, each with the following structure (5′ to 3′): T7 promoter (TTCTAATACGACTCACTATA) + gRNA sequence + scaffold (GTTTTAGAGCTAGAAATAGC). Since activity of the T7 promoter is enhanced when two G's are present at the transcription start site, one or two G's were added immediately after the T7 promoter in oligos derived from gRNAs starting with a single or no G, respectively.
The second version of the Python script (Figs. 4, 5) designed all possible gRNA target sequences, independent of the conservation and structuredness of each region. The only filtering criteria were GC content between 30%–80% and a low predicted off-target probability, defined as the absence of sequences in the Salmonella chromosome or plasmids that aligned to the gRNA with up to three mismatches (identified with Bowtie [Langmead et al. 2009]), followed by a valid PAM. The resulting 979 sequences were purchased from IDT as an “oPools Oligo Pool” with a similar structure than above, except that the scaffold was GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT and was followed by a sequence for fill-in reactions (ACGATGTCGCAGAGTATGCC). The primer used for filling-in was 5′-GGCATACTCTGCGACATCGT-3′. Design of the B. thetaiotaomicron pool was done as above, resulting in 651 sequences. The script is freely available on https://github.com/gprezza/DASH_rRNA_depletion.
dsDNA templates for in vitro transcription were generated in a fill-in reaction performed with the KAPA HiFi HotStart ReadyMix (KAPA Biosystems). The first pool reaction was primed with 5′-AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3′ and consisted of denaturation at 95°C for 3 min, annealing, and extension from 95°C to 30°C at 0.1°C/sec with 10 sec pause every 10°C and a final extension at 72°C for 1 min. The second pool was filled-in with the 5′-GGCATACTCTGCGACATCGT-3′ primer and denaturation at 95°C for 3 min, annealing at 60°C for 20 sec, and extension at 72°C for 1 min.
sgRNA pools were in vitro-transcribed from 300 ng of column-purified dsDNA template with the MEGAshortscript T7 Transcription Kit (Thermo Fisher Scientific) and then purified with the Monarch RNA Cleanup Kit (NEB). SpCas9 protein was purchased from NEB (M0386M).
cDNA library generation, Cas9 cleavage, library amplification, and Illumina sequencing
Bacterial total RNA was fragmented at 94°C for 2.75 min using the NEBNext Magnesium RNA Fragmentation Module (NEB), dephosphorylated at the 3′ end, phosphorylated at the 5′ end and decapped using 10 U T4-PNK ± 40 nmol ATP and 5 U RppH, respectively (NEB). After each step, RNA was purified with the Zymo RNA Clean & Concentrator kit (Gu et al. 2016). cDNA libraries were generated with the NEBNext Multiplex Small RNA Library Prep Kit (NEB) and preamplified with two cycles of PCR. Following purification with the Oligo Clean & Concentrator kit (Zymo Research), DASH treatment was performed similar to Gu et al. (2016). Specifically, the purified cDNA library was incubated with the Cas9-sgRNA complex for 2 h at 37°C at the indicated molar ratios. Where mentioned, Cas9 and the sgRNA pool were preincubated at 37°C for 15 min before addition to the cDNA. After the digest, Cas9 was removed from the reaction by column purification with the Oligo Clean & Concentrator kit (Zymo Research) or treatment with 0.8 U (∼20 µg) proteinase K (NEB) for 15 min at 37°C, followed by heat-inactivation (15 min at 95°C). The resulting DASHed samples were PCR amplified for 12–24 cycles to select for nonribosomal, undigested cDNAs and purified with MagSi-NGSprep Plus beads (Steinbrenner Laborsysteme).
Alternatively, cDNA libraries were generated from bacterial total RNA using the Takara SMARTer Stranded Total RNA-Seq Kit v2 with 4 min RNA fragmentation at 94°C and five cycles of PCR for cDNA library preamplification. After column purification, DASH was performed as described above. The resulting DASHed samples were column purified, PCR amplified with Takara's SeqAmp DNA Polymerase for 18 cycles and further purified with AMPure XP beads (Beckman Coulter).
Sequencing of libraries, spiked with 5% PhiX control library, was performed in single-end mode on the Illumina NextSeq 500 platform with the Mid Output Kit v2.5 (75 cycles). A summary of all sequenced samples and the respective reaction conditions is reported in Supplemental Table S3.
Demultiplexed FASTQ files were generated with bcl2fastq2 v2.20.0.422 (Illumina). The sequencing data is currently being uploaded at NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under the accession number GSE147155.
Data analysis
Reads were trimmed for NEBNext or Illumina TruSeq (Takara kit) adapter sequences using Cutadapt version 2.5 with default parameters and the –nextseq-trim=20 switch to handle two color sequencing chemistry. Reads that were trimmed to length 0 were discarded.
Processed reads were mapped to the Salmonella (NC_016810.1, NC_017718.1, NC_017719.1, NC_017720.1) or Bacteroides (NC_004663.1, NC_004703.1) reference sequences. We modified the NC_016810.1 Salmonella chromosome annotation to include an updated sRNA annotation (Hör et al. 2020). The B. thetaiotaomicron sRNA annotation stems from D Ryan, L Jenniches, S Reichardt, et al. (in prep.). Mapping was performed with READemption version 0.4.3 (Förstner et al. 2014) with the argument –a 80 (NEB samples) or –a 80 –R (Takara samples) and with segemehl 0.2.0 (Hoffmann et al. 2009). Gene quantification was done with the READemption subcommand gene_quanti with arguments –a –o 10. Coverage plots were generated with the subcommand coverage and visualized with IGV (Robinson et al. 2011). Read length distribution was analyzed with SAMtools (Li et al. 2009). rRNA depletion efficiency was defined as
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Elena Katzowitsch from the Core Unit SysMed at the University of Würzburg for excellent technical support. This work was supported by the Interdisziplinäre Zentrum für Klinische Forschung (IZKF) at the University of Würzburg (project Z-6). G.P. was supported by a grant of the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.075945.120.
Freely available online through the RNA Open Access option.
REFERENCES
- Archer SK, Shirokikh NE, Preiss T. 2014. Selective and flexible depletion of problematic sequences from RNA-seq libraries at the cDNA stage. BMC Genomics 15: 401 10.1186/1471-2164-15-401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betin V, Penaranda C, Bandyopadhyay N, Yang R, Abitua A, Bhattacharyya RP, Fan A, Avraham R, Livny J, Shoresh N, et al. 2019. Hybridization-based capture of pathogen mRNA enables paired host-pathogen transcriptional analysis. Sci Rep 9: 19244 10.1038/s41598-019-55633-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne A, Supple MA, Volden R, Laidre KL, Shapiro B, Vollmers C. 2019. Depletion of hemoglobin transcripts and long-read sequencing improves the transcriptome annotation of the polar bear (Ursus maritimus). Front Genet 10: 643 10.3389/fgene.2019.00643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro TL, Seyffert N, Ramos RT, Barbosa S, Carvalho RD, Pinto AC, Carneiro AR, Silva WM, Pacheco LG, Downson C, et al. 2013. Ion torrent-based transcriptional assessment of a Corynebacterium pseudotuberculosis equi strain reveals denaturing high-performance liquid chromatography a promising rRNA depletion method. Microb Biotechnol 6: 168–177. 10.1111/1751-7915.12020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugani S, Kim BS, Phattarasukol S, Brittnacher MJ, Choi SH, Harwood CS, Greenberg EP. 2012. Strain-dependent diversity in the Pseudomonas aeruginosa quorum-sensing regulon. Proc Natl Acad Sci 109: E2823–E2831. 10.1073/pnas.1214128109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher NJ, Thomson NR. 2010. Studying bacterial transcriptomes using RNA-seq. Curr Opin Microbiol 13: 619–624. 10.1016/j.mib.2010.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culviner PH, Guegler CK, Laub MT. 2020. A simple, cost-effective, and robust method for rRNA depletion in RNA-sequencing studies. MBio 11: e00010–e00020. 10.1128/mBio.00010-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus M, Regnier P. 2002. The poly(A) tail of mRNAs: bodyguard in eukaryotes, scavenger in bacteria. Cell 111: 611–613. 10.1016/S0092-8674(02)01137-6 [DOI] [PubMed] [Google Scholar]
- Dynerman D, Lyden A, Quan J, Caldera S, McGeever A, Dimitrov B, King R, Cirolia G, Tan M, Sit R, et al. 2020. Designing and implementing programmable depletion in sequencing libraries with DASHit. bioRxiv 2020.01.12.891176.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förstner KU, Vogel J, Sharma CM. 2014. READemption—a tool for the computational analysis of deep-sequencing-based transcriptome data. Bioinformatics 30: 3421–3423. 10.1093/bioinformatics/btu533 [DOI] [PubMed] [Google Scholar]
- Giannoukos G, Ciulla DM, Huang K, Haas BJ, Izard J, Levin JZ, Livny J, Earl AM, Gevers D, Ward DV, et al. 2012. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol 13: R23 10.1186/gb-2012-13-3-r23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonatopoulos-Pournatzis T, Aregger M, Brown KR, Farhangmehr S, Braunschweig U, Ward HN, Ha KCH, Weiss A, Billmann M, Durbic T, et al. 2020. Genetic interaction mapping and exon-resolution functional genomics with a hybrid Cas9-Cas12a platform. Nat Biotechnol 10.1038/s41587-020-0437-z [DOI] [PubMed] [Google Scholar]
- Gu W, Crawford ED, O'Donovan BD, Wilson MR, Chow ED, Retallack H, DeRisi JL. 2016. Depletion of abundant sequences by hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol 17: 41 10.1186/s13059-016-0904-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardigan AA, Roberts BS, Moore DE, Ramaker RC, Jones AL, Myers RM. 2019. CRISPR/Cas9-targeted removal of unwanted sequences from small-RNA sequencing libraries. Nucleic Acids Res 47: e84 10.1093/nar/gkz425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa H, Oda Y, Phattarasukol S, Armour CD, Castle JC, Raymond CK, Lappala CR, Schaefer AL, Harwood CS, Greenberg EP. 2011. Activity of the Rhodopseudomonas palustris p-coumaroyl-homoserine lactone-responsive transcription factor RpaR. J Bacteriol 193: 2598–2607. 10.1128/JB.01479-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermuller J. 2009. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol 5: e1000502 10.1371/journal.pcbi.1000502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hör J, Gorski SA, Vogel J. 2018. Bacterial RNA biology on a genome scale. Mol Cell 70: 785–799. 10.1016/j.molcel.2017.12.023 [DOI] [PubMed] [Google Scholar]
- Hör J, Matera G, Vogel J, Gottesman S, Storz G. 2020. Trans-acting small RNAs and their effects on gene expression in Escherichia coli and Salmonella enterica. EcoSal Plus 9 10.1128/ecosalplus.ESP-0030-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Sheth RU, Kaufman A, Wang HH. 2020. Scalable and cost-effective ribonuclease-based rRNA depletion for transcriptomics. Nucleic Acids Res 48: e20 10.1093/nar/gkz1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IV, Ross EJ, Dietrich S, Doring K, Sanchez Alvarado A, Kuhn CD. 2019. Efficient depletion of ribosomal RNA for RNA sequencing in planarians. BMC Genomics 20: 909 10.1186/s12864-019-6292-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroger C, Colgan A, Srikumar S, Handler K, Sivasankaran SK, Hammarlof DL, Canals R, Grissom JE, Conway T, Hokamp K, et al. 2013. An infection-relevant transcriptomic compendium for Salmonella enterica Serovar Typhimurium. Cell Host Microbe 14: 683–695. 10.1016/j.chom.2013.11.010 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zhang Y, Zhang T. 2020. Computational approaches for effective CRISPR guide RNA design and evaluation. Comput Struct Biotechnol J 18: 35–44. 10.1016/j.csbj.2019.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26 10.1186/1748-7188-6-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova OE, Garcia-Alcalde F, Zampaloni C, Sauer K. 2017. Comparative evaluation of rRNA depletion procedures for the improved analysis of bacterial biofilm and mixed pathogen culture transcriptomes. Sci Rep 7: 41114 10.1038/srep41114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ST, Yu FB, Blainey PC, May AP, Quake SR. 2019. Nucleic acid cleavage with a hyperthermophilic Cas9 from an uncultured Ignavibacterium. Proc Natl Acad Sci 116: 23100–23105. 10.1073/pnas.1904273116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishkin AA, Giannoukos G, Kucukural A, Ciulla D, Busby M, Surka C, Chen J, Bhattacharyya RP, Rudy RF, Patel MM, et al. 2015. Simultaneous generation of many RNA-seq libraries in a single reaction. Nat Methods 12: 323–325. 10.1038/nmeth.3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Cossart P. 2010. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet 11: 9–16. 10.1038/nrg2695 [DOI] [PubMed] [Google Scholar]
- Stocker BA, Hoiseth SK, Smith BP. 1983. Aromatic-dependent “Salmonella sp.” as live vaccine in mice and calves. Dev Biol Stand 53: 47–54. [PubMed] [Google Scholar]
- Wessels H-H, Méndez-Mancilla A, Guo X, Legut M, Daniloski Z, Sanjana NE. 2020. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat Biotechnol 10.1038/s41587-020-0456-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann AJ, Förstner KU, Amman F, Barquist L, Chao Y, Schulte LN, Muller L, Reinhardt R, Stadler PF, Vogel J. 2016. Dual RNA-seq unveils noncoding RNA functions in host-pathogen interactions. Nature 529: 496–501. 10.1038/nature16547 [DOI] [PubMed] [Google Scholar]
- Yi H, Cho YJ, Won S, Lee JE, Jin Yu H, Kim S, Schroth GP, Luo S, Chun eJ. 2011. Duplex-specific nuclease efficiently removes rRNA for prokaryotic RNA-seq. Nucleic Acids Res 39: e140 10.1093/nar/gkr617 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.