

Abstract
The Cancer Genome Atlas consortium brought us terabytes of information about genetic alterations in different types of human tumors. While many cancer-driver genes have been identified through these efforts, interrogating cancer genomes has also shed new light on tumor complexity. Mutations were found to vary tremendously in their allelic frequencies within the same tumor. Based on those variant allelic frequencies grouping, an estimate of genetically distinct “clones” of cancer cells can be determined for each tumor. It was estimated that 4–8 clones are present in every human tumor. Presence of distinct clones, cells that differ in their genotype and/or phenotype, is one of the roots for the major challenge of effectively curing cancer patients. Any given treatment applied to a heterogeneous mixture of cancer cells will yield distinct responses in different cells and may be ineffective in killing particular clones. Moreover, in highly heterogeneous tumors, stochastically, there is a higher chance of presence of traits, such as point mutations in key receptor tyrosine kinases, that drive drug resistance. Thus, intratumor heterogeneity is like an arsenal, providing a variety of weapons for self-defense against cancer-targeted therapy. However, in this arsenal the supplies are constantly changing, as cancer cells are accumulating new mutations. What is also changing is the battlefield-the tumor microenvironment including all noncancerous cells within the tumor and surrounding tissue, which also contribute to the diversification of cancer’s forces. In order to design more effective therapies that would target this ever-changing landscape, we need to learn more about the two elusive variables that shape the tumor ecosystem: the space-how could we exploit the organization of tumor microenvironment? and the time-how could we predict the changes in heterogeneous tumors?
Sources of heterogeneity in cancer
The abnormal morphology of cancer cells is the basis for cancer diagnosis in current medical practice. The morphological diversity of cancer cells within a single cancer specimen has been known since the first pathology studies by Rudolf Vichrov in 1920s. Early days of cancer research were focused on genetic aberrations that can initiate malignant transformation and contribute to disease progression. The classic view of tumorigenesis assumes that cancer arises from a single cell that acquires an oncogenic mutation and defies the rules governing normal tissue homeostasis and divides uncontrollably [1]. As this initial cell divides, it accumulates additional mutations and gains new properties, such as apoptosis resistance and invasiveness, resulting in more aggressive tumor growth and metastasis [2]. However, mistakes in DNA replication that result in generation of mutations are stochastic and can arise in any daughter cell derived from the initial cancer cell, resulting in cellular diversification. Clonal populations become more and more genetically distant, as individual cells accumulate mutations at random during tumor growth. Thus, stochasticity of mutational processes is one of the causes of intratumor heterogeneity. This randomness of new genetic aberrations would result in an infinite number of cells with different aberrations. The original work by Peter Nowell described cancer progression as a process of Darwinian evolution, in which randomly generated clones have different levels of fitness and undergo environment-driven selection [3]. In this view, a tumor is a mixture of genetically distinct populations of cells with variable ability to proliferate or metastasize. Of note, the definition of a clone with regards to tumor heterogeneity depends on measures taken and relates to common ancestry with shared features, rather than a precise genetic replica.
Beyond genotype, phenotypic plasticity of some cancer cell populations is another important driver of tumor heterogeneity. In several tumor types, including breast, lung, and brain cancer, cells within a tumor display a diversity that resembles hierarchical organization of a normal tissue, with stem-like cells that can give rise to more differentiated progeny [4]. Despite having the same genetic aberrations as their more differentiated counterparts, cancer stem cells are usually more quiescent and resistant to treatments. These cells express transcriptional programs inherent to normal stem cells and maintain higher level of phenotypic plasticity due to a more promiscuous chromatin organization [5]. Thus, differences in epigenetic states of distinct populations of cancer cells, defined by DNA methylation, chromatin accessibility or histone modifications, contribute to intratumor heterogeneity [6].
The interplay between the genetic and epigenetic heterogeneity is a topic of high interest in current studies. Advances in single-cell transcriptomic [7] and epigenomic technologies [8] enable studies that integrate the contribution of mutations and epigenetic and transcriptomic plasticity in tumor heterogeneity. Single-cell DNA methylation assay coupled with whole transcriptome sequencing and targeted sequencing of known somatic mutations revealed that in chronic lymphoblastic leukemia (CLL) changes in DNA methylation, termed epimutations, drive transcriptional entropy, and phenotypic diversification [9]. This integrated approach has also shown that wile driver mutation in SF3B1 is a late subconal event in CLL, it can still drive evolutionary branching of the mutant clones by significant transcriptional changes. Another recent study focusing on integration of genetic and epigenetic diversity in glioblastoma has shown that copy number amplifications in distinct subpopulations are associated with distinct cellular states, which recapitulate the diversity of neural cell types [10]. The mechanisms and timing of genetic and epigenetic diversification are not yet well understood and may differ between different tumor types. Nevertheless, the abovementioned examples show that integrative approach can help delineate the principles of tumor evolution.
Tumor heterogeneity is not only governed by the intrinsic properties of cancer cells. The “fitness: of a cancer cell can be viewed as the ability to survive and/or proliferate more rapidly than other cells in a particular environment. Selection processes that eliminate less fit cancer cells are thus inherently connected with tumor microenvironment. Just like in any ecosystem, environmental changes, such as hypoxia in a growing tumor mass, will lead to death of cells unable to adapt or respond to change, for example by secreting pro-angiogenic factor to promote new vessel formation and increase oxygen and nutrient supply. Immune cells within the tumor microenvironment can also prune certain clones, and select for cancer cells with fewer neoantigens or cells that actively dampen the immune response [11]. It still debatable as to whether the presence of genetically distinct clones in a late-stage lesion is a result of positive selection or natural drift [12]. It may be that certain tumor types, such as colorectal cancer, are much less dependent on environmental constrains and early genomic instability could be the main driver of heterogeneity in those tumors. However, in many other cancer types both the cancer cells and the normal cells surrounding and infiltrating the tumor affect tumor evolution by shaping clonal dynamics.
Spatial heterogeneity: The space conquerors
The term “tumor microenvironment” encompasses all of the noncancer cells that may be found intermixed within the tumor or in a direct contact with cancer cells at the edge of a malignant lesion. These “normal” cells, such as leukocytes, fibroblasts, and endothelial cells, can interact with cancer cells promoting antitumorigenic response, but can also be reprogrammed to support growth of the tumor and suppress immune response against cancer cells. Tumor microenvironment can be also defined as a set of variables, such as blood vessel density, and extracellular matrix stiffness, which contribute to the overall conditions that the cancer cells need to thrive in and migrate through. All of these components can vary substantially within different areas of the tumor. Thus, from the ecological perspective, tumor microenvironment creates specific niches, in which distinct cellular phenotypes might be necessary for survival. As such, heterogeneity in tumor microenvironment would play a vital role in selecting distinct subpopulations of cancer cells from randomly mutated cells in a heterogeneous pool.
Clinical consideration of spatial heterogeneity
Even without considering the role of the microenvironment, spatial heterogeneity is apparent in many tumor types. Phenotypic diversity of cancer cells can be readily observed in formalin-fixed paraffin embedded samples of human tumors without the need for specific markers. For example, glioblastoma multiforme, the most aggressive form of brain cancer, takes its name from the diverse shapes and sizes of cells readily visible with the basic hematoxylin and eosin counterstain. This cellular phenotypic variability is related to transcriptional variation present in these tumors [13]. Phenotypic diversity in different regions of a sample is also often causing challenges in pathologic assessment of markers used in clinical decision-making. In breast cancer, tumor classification used for treatment selection is based on immunohistochemical staining of the biopsies for presence of ER, PR, and HER2 proteins. Often, in different areas of the same biopsy distinct levels of marker expression can be found. This is especially true for HER2 and is underlined by cell-to-cell variation in copy number of the HER2 gene [14, 15] and could explain why even with HER2-targeted therapies only 40% of patients are in full remission. Other studies have shown cellular heterogeneity for KRAS and TP53, common driver mutations, as well as for mutations associated with emergence of resistance after targeted treatment [16–20]. Despite these studies, regional heterogeneity within a single biopsy is not yet included in standard pathology reports. Clinically approved cytogenetic techniques can reveal a divergence of gene copy number between different cells within a tumor. However, due to a laborious manual counting single-cell based variation is rarely added to clinical reports. An extreme case of cell-to-cell variation of clinically actionable targets was shown in glioblastoma [21]. Amplifications of receptor tyrosine kinases EGFR, MET, and PDGFR were found in different cells within the same tumor and were mutually exclusive. All those populations were viable and contributed to tumor growth and harbored mutations pointing to a common ancestor cell. Thus, evaluation of intratumor heterogeneity for therapeutic targets could be useful in stratifying patients to different arms of clinical trials, especially for inhibitors of receptor tyrosine kinases. Relationships between treatment-induced changes in single-cell based heterogeneity and long term patient outcomes were also reported. In a small cohort study of HER2+ breast cancer, patient survival correlated with changes in intratumor heterogeneity between different areas of the tumor sample [22].Techniques that allow for spatial characterization of intratumor heterogeneity, such as fluorescence in situ hybridization and in situ PCR and their combination, STAR-FISH, could help assess these changes in local tumor niches. Yet, limitations in combining multiple markers and requirement of selecting only a handful of genes of interest need to be addressed. In the following sections, different approaches to mapping cancer cell heterogeneity in intact tumor tissues are discussed.
Genetic heterogeneity, topology, and microenvironment
Next-generation sequencing revolutionized the field of cancer genetics, revealing multitude of new cancer-related genes and the breadth of mutations in individual tumors [23]. Deep sequencing of tumor DNA has also allowed for reconstruction of clonality based on mutant allele frequency quantification [24–27]. The vastness of intratumor heterogeneity was further reveled by next-generation sequencing of distinct fragments of a tumor (Fig. 1a). Multiregion sequencing of a kidney tumor was one of the first studies that showed significant genetic diversity present in different areas of the same tumor [28]. Each sequenced spatially separated area was characterized by private mutations, not found in any other fragment of the tumor. However, all samples also shared a set of mutations, again pointing to a common ancestor and later diversification. This study pioneered the attempts to reconstruct the phylogenetic trees of tumors with regards to their topological organization. It also highlighted the problem of vast underestimation of intratumor heterogeneity measured from a single biopsy. Similar conclusions came from multiregional sequencing of melanoma, breast, and lung tumors [29–31]. Multiregion sequencing has also been used for deconvolution of T cell immunoreactivity in distinct areas of the tumor [32–34]. Variable expression of tumor antigens, differential immune infiltration and T cell specificity between regions of the same tumor all point to divergent evolution of immune evasion. Regional variations and local immunological heterogeneity may thus have significant implications for any systemic treatment, including immunotherapy.
Fig. 1.
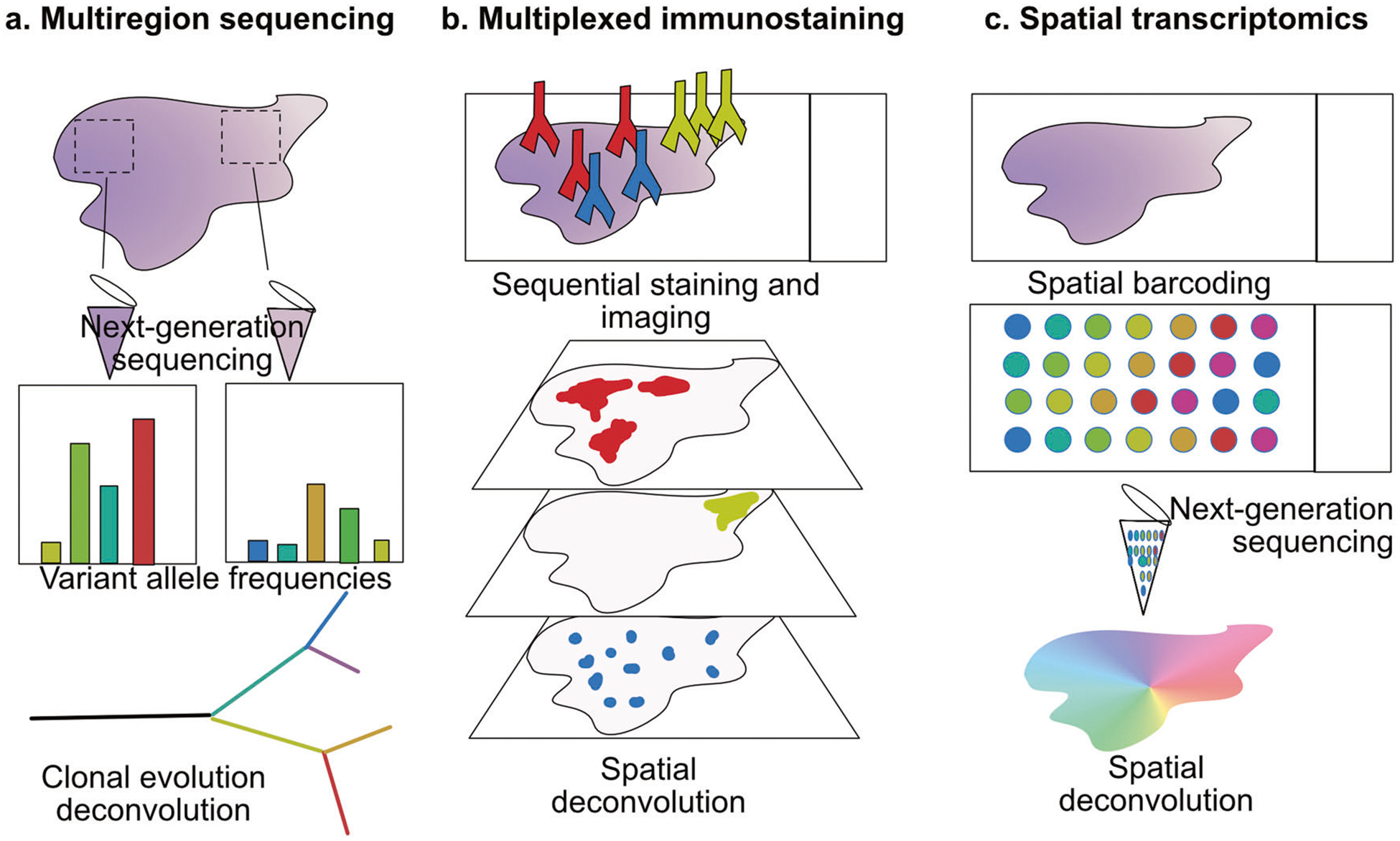
Dissecting spatial tumor heterogeneity. a Multiregion sequencing. Gross resection or microdissection of tissue fragments for distant areas of the tumor allow for the comparison of variant allelic frequencies. Mutational abundance is used to identify truncal and private events and reconstruct tumor evolutionary tree. b Multiplexed immunostaining. Labeled antibodies are used in multiple rounds of staining and acquired images are superimposed to generate multiplexed image of the tissue. c Spatial transcriptomics. Spatial barcoding during RNA reverse-transcription allows identification of tissue localization of sequenced transcripts
A closer look at the genetic diversity within a single biopsy and between histologically distinct areas was provided by tissue laser capture microdissection. Separating histologically distinct regions of tumor specimen and subsequent profiling of collected tissue fragments shed new light on the disease progression of breast and esophageal cancer [35, 36]. In these tumor types low-grade, noninvasive lesions are often found co-occurring with highly invasive malignant areas. Spatial separation at the tissue level thus provided evidence that invasive cancer-associated mutations may be present in the pre-malignant lesions. Further development of this approach into a single-cell microdissection and single-nucleus sequencing in breast cancer showed that individual invasive cells are present in the noninvasive carcinoma area and full-blown invasive ductal carcinoma could be founded by multiple clones migrating from distinct areas of ductal carcinoma in situ [37]. The fact that individual mutant malignant cells can be found within noninvasive lesions points to “the niche effect” in tumorigenesis. For an individual cancer cell acquisition of a new mutation, even in a cancer-driver gene, may not always lead to full-blown malignancy. This will depend on the surrounding cancer cells and on the tumor microenvironment. In fact, recent studies showing that point mutations in oncogenes are found at very low frequencies in almost all normal tissues suggest that the cancer-driver genes may not be enough to initiate tumorigenesis in vivo [38, 39].
In situ profiling of cancer phenotypes and tumor microenvironment
Since tumor heterogeneity is not only reflected in the genotypic diversity, but may also be a result of epigenetic variation, studies targeting the transcriptomic diversity can provide more complete answer to functional roles of distinct populations of cancer cells within a tumor. They also allow to identify phenotypes associated with particular microenvironments. Differential protein expression in tumor tissues provided the first clues to the extent of intratumor heterogeneity, as discussed above. Profiling of tumor and its microenvironment based on protein marker expression can be performed by immunofluorescence, which can be easily applied to archival human specimen. However, comprehensive studies using this technique were limited by technical challenges. Spectral overlap between fluorophores used to distinguish different markers in a stained sample made simultaneous detection of more than six proteins impossible. Multiple innovative approaches have recently been introduced to overcome this obstacle. One of them uses multiple rounds of red, blue, and green fluorophore-conjugated antibody staining followed by fluorophore inactivation and another staining round [40, 41]. This technique, named cyclic immunofluorescence, requires overlaying multiple images of the same area, yet allows to obtain single-cell resolution of co-occurrence of distinct markers of cancer cells, immune cells and other tumor components (Fig. 1b). Another way to increase the throughput of proteins analyzed on a heterogeneous tissue slide in immunostaining is to replace the fluorophores with heavy metals [42, 43]. Imaging mass cytometry and MIBI methods rely on laser ablation of the tissue stained with those conjugates and identification of metals derived from a particular tissue location by mass cytometry. Over thirty distinct antigens can be simultaneously identified with this approach. It allows for single-cell reconstruction and is more quantitative than fluorescence-based techniques. Multiplexed immunofluorescence and mass cytometry-based analyses proved to be particularly useful in profiling leukocyte complexity and functional status in archival tissues, as the identity and activity of individual tumor-infiltrating immune cells can be assessed [44–46]. Immune infiltration and activity are tightly linked to success of cancer immunotherapy, which is one of the most promising treatment strategies of the recent years [47]. However, the interactions between diverse populations of cancer cells and distinct types of leukocytes remain poorly understood. These interactions may have important implications for successful immunotherapy for cancer patients. Thus, more studies with advanced spatial analysis and automated feature recognition will drive the focus on spatial distribution and proximity of different cells types.
In situ profiling of cancer cells using antibody-based approaches is challenging, as selection of antigens requires a priori knowledge of expected cell types. While a lot is known about specific markers of the tumor microenvironment, cancer cell subtypes within any given tumor type are much more elusive. Therefore, unbiased approaches to profile phenotypic diversity have been focused on transcriptomic profiling. Recent revolution in single-cell sequencing enabled RNA sequencing from thousands of individual cells. Both normal tissues and cancer samples after dissociation into single cells can be now characterized at an unprecedented resolution [7]. However, because of the dissociation, information about spatial localization of cells within the tissue is lost. This loss of tissue context may prevent our understanding of the interactions between cancer cells and the tumor microenvironment that may drive selection and tumor evolution. Several new approaches are being used to overcome this limitation. RNA in situ hybridization allows for detection of expression of several targeted transcripts, while maintaining the tissue topology. Due to its low throughput it is rarely used to profile heterogeneity in tumors. Yet, this approach was modified to allow detection of individual cells with mutant KRAS in colorectal cancer and the frequencies and distribution of the mutant cells pointed to early evolution of the subclone or high fitness advantage conferred by the mutation [48]. First attempts to increase throughput of transcript detection in situ, named FISSEQ, relied on in situ sequencing on the slide containing a tissue [49]. An alternative strategy lead to the development of a barcoding technique, where transcripts within a tissue are hybridized to a slide that contains spatially distinct barcodes and after an amplification step labeled transcripts can be pooled and processed in a standard next-generation sequencing protocol [50, 51]. This results in location-specific indexing of all transcripts (Fig. 1c). Further miniaturization of the beads carrying the barcodes will increase the resolution of these techniques, which is now limited to ~10 microns. Thus far only few studies applied spatial transcriptomics to cancer samples [31, 52]. However, recent commercialization of spatial transcriptomics platform will have high impact on our understanding of tumor heterogeneity. The unbiased approaches will reveal novel transcriptional profiles associated with the interplay between tumor cell and the immune system. In situ phenotypic analysis based on RNA will also provide new clues to the metabolic dependencies of cancer cells found in distinct microenvironments.
Tumor heterogeneity and evolution: The time travel
Moving back in time has been explored by science-fiction literature as a way to prevent disasters and change futures of the novel’s characters. Cancer research is also focusing on moving across the time axis to learn more about the initiation, development, and progression of the tumors in hopes to be able to predict patients’ response and change the course of the disease. Since tumor evolution depends on random events and selection of individual new traits, how tumors change in time is tightly linked to their internal heterogeneity and behavior of single cells. This is why many of the aspects of studying intratumor heterogeneity described in this review also have implications for tumor evolution studies.
Tumor history from DNA
To monitor changes within a tumor over time, longitudinal sampling of every individual malignancy would be required. However, for most tumor types, repeated sampling is difficult due to risks it could bring for the patient, that must be outweighed by potential benefit of the procedure. In several clinical trials repeated biopsies are now being collected before and after treatment, as it is clear that changes in the tumor short after neoadjuvant therapy could be useful in predicting the outcomes. However, it is much more difficult to sample a disease at an early stage. Thus, tumor evolution is often modeled based on a sampling from an advanced lesion. Deep sequencing of cancer tissues is used not only to quantify tumor clonality, but can be also used to infer evolutionary trajectory [24–26]. Mutations that occurred early in tumor development are present in all subclonal populations within a tumor and mutations in smaller fractions of alleles would correspond to a late development. Using this approach, evolutionary trees can be reconstructed from a single tumor sample subject to bulk sequencing. However, as mentioned before, the multiregion sequencing of tumors has shown that distinct samples from a single tumor can be clonally diverse [28–31]. Thus, drawing conclusions about tumor evolution based on a single sample may not be sufficiently powerful to distinguish subclonal from clonal events.
An alternative to a repeated biopsy acquisition from a tumor over time is the use of “liquid biopsies”. Circulating tumor cells and the cell-free DNA allows sampling of the tumor genome from the patient’s plasma and can thus be more readily implemented in clinical trials and patient follow-up [53]. These samples are also useful in monitoring metastasis [54, 55]. While CTC and cfDNA proved to help monitor the occurrence of resistance to treatment [56–58], it is still not clear how representative they are in respect to the whole tumor mass. Cancer cells in circulation and cell-free DNA could be representing only the most invasive or apoptotic fractions of the tumor subpopulations, respectively. Studies of spatial localization of CTC are required to address these questions.
The evolution of metastasis
Timing in tumor development can have significant implications for metastasis, which if prevented would save many lives. The classic model of tumor evolution proposes that cells accumulate mutations that eventually lead to the acquisition of new traits, including the ability to invade and grow in a distant organ as a metastasis. Sequencing of metastatic lesions has shown that even the same tumor type can adopt different evolutionary models (reviewed in [59]). There is evidence for parallel evolution, where mutational burden points to long phylogenetic distances between primary and metastatic lesions [60, 61]. This can be associated with early seeding of the first metastatic clones or with very high mutation rates occurring in primary tumor and metastases [62]. On the other hand, there are also many examples of metastases that originate from a subclone that occurred late in the evolution of the primary lesion. Thus far, those distinct evolutionary pathways to metastasis were not linked to patient survival in any tumor type. It is possible that this variation is related to limited sampling, as in most studies a single primary tumor sample is compared with multiple metastatic lesions and multiregional sequencing could help decipher clonal relationships [37]. What is clearly emerging from the clonal reconstruction studies in metastatic cancers, is that multiple clones contribute to metastatic growth. This could be related to the clusters of CTC that are often found in the blood of cancer patients [55]. These clusters are oligoclonal and contribute to the generation of heterogeneous metastases. Therefore, a better understanding of the clonal dynamics in primary tumor and in clustering cells in circulation could help devise better strategies to prevent metastatic spread and outgrowth. In this perspective, the dynamics between heterogeneous populations of cancer cells may be more important that the timing of mutations associated with metastatic progression.
Models of tumor evolution
Since studying tumor evolution over time using patients’ samples poses a lot of technical and ethical challenges, in vitro cancer models are extensively used to track changes in clonal dynamics over time. Clones can be defined by simple overexpressed markers that can be detected via microscopy or fluorescence-activated cell sorting. While this approach is useful in targeting questions about clonal cooperation in defined systems [63–65], it is limited to only a handful of clones that could be discriminated. Genetic barcoding technologies enabled indexing of individual cells and enumeration of the barcodes via sequencing allows for detection of progeny of millions of cancer clones [66]. Barcoded cells can be used to track selection of distinct clones from highly heterogeneous population over time, in vitro and in vivo [67–69]. This approach enabled studies of emergence of therapy resistance. Combination of single-cell barcoding with CRISPR-based genetic perturbation screening will help identify evolutionary bottlenecks and collateral sensitivity to prevent posttreatment relapse.
Conclusions
The evolution of a full-blown malignancy from a single cell that gives rise to a heterogeneous population of cancer cells is a complex process. In the recent years technological advances increased the resolution at which heterogeneity can be observed (Table 1). As we get closer to the characterization of genome, epigenome, and transcriptome cell by cell, it becomes evident that the complexity of the interplay between the tumor and the surrounding normal cells and the immune system plays a central role. Thus, interrogation of intact cancer tissue, containing distinct cancer cell clones in their native microenvironment in which they evolved, will be key. The excitement brought by the novel spatial transcriptomics and single-cell profiling platforms will certainly bring multiple new answers to the question of how cancers are moving through space and time. As our detection methods become more sensitive, we also require novel approaches to coalesce the fine-grain single-cell data with observations at the tissue and organ level. Therefore, integration of cancer biology, systems biology and mathematic oncology will be needed to translate the finding from the microcosmos of single cells to successful therapy for a patient.
Table 1.
Summary of distinct features and techniques used to assess intratumor heterogeneity
| Heterogeneity source | Features | Tools and analyses | Resolution | Resolution | Ref. |
|---|---|---|---|---|---|
| Genetic | Mutations, CNV | Whole genome/exome seq | Whole genome | Tissue | [23–31] |
| CNV | Single-cell | [37] | |||
| Single-nucleus seq | Select CNV | Single-cell | [21] | ||
| FISH | Select mutations | Single-cell | [22] | ||
| STAR-FISH | |||||
| Epigenetic | DNA methylation Chromatin accessibility Histone modifications | Bisulfite seq | Whole genome | Single-cell | [9] |
| ATACseq | Single-cell | [8] | |||
| ChIPseq | Population | ||||
| Phenotypic | RNA expression | Transcript heterogeneity, single-cell RNAseq | Thousands genes/cell | Single-cell | [7] |
| [10] | |||||
| [49] | |||||
| FISSEQ | [50, 51] | ||||
| Spatial transcriptomics | |||||
| Protein expression | CyTOF, imaging CyTOF, MIBI, immunofluorescence CyCIF | Select markers | Single-cell | [40–46] |
Acknowledgements
I thank Dr Joseph Kissil (The Scripps Research Institute) and the Janiszewska lab members for insightful discussions. This work is supported by NIH R00 CA201606-01A1 and the startup funds provided by the Scripps Research Institute.
Footnotes
Conflict of interest
The author declares no conflict of interest.
References
- 1.Vogelstein B, Kinzler KW. The path to cancer-three strikes and you’re out. N Engl J Med. 2015;373:1895.-. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 3.Merlo LMF, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–35. [DOI] [PubMed] [Google Scholar]
- 4.Prager BC, Xie Q, Bao S, Rich JN. Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell. 2019;24:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinohara K, Polyak K. Intratumoral heterogeneity: more than just mutations. Trends Cell Biol. 2019;29:569–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanay A, Regev A. Scaling single-cell genomics from phenomenology to mechanism. Nature. 2017;541:331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shema E, Bernstein BE, Buenrostro JD. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat Genet. 2018;51:19–25. [DOI] [PubMed] [Google Scholar]
- 9.Gaiti F, Chaligne R, Gu H, Brand RM, Kothen-Hill S, Schulman RC, et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature. 2019;569:576–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019. 10.1016/j.cell.2019.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee C-H, Yelensky R, Jooss K, Chan TA. Update on tumor neoantigens and their utility: why it is good to be different. Trends Immunol. 2018;39:536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun R, Hu Z, Curtis C. Big bang tumor growth and clonal evolution. Cold Spring Harb Perspect Med. 2018;8:a028381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seol H, Lee HJ, Choi Y, Lee HE, Kim YJ, Kim JH, et al. Intratumoral heterogeneity of HER2 gene amplification in breast cancer: its clinicopathological significance. Mod Pathol. 2012;25:938–48. [DOI] [PubMed] [Google Scholar]
- 15.Rye IH, Trinh A, Saetersdal AB, Nebdal D, Lingjaerde OC, Almendro V, et al. Intratumor heterogeneity defines treatment-resistant HER2+ breast tumors. Mol Oncol. 2018;12:1838–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giaretti W, Monaco R, Pujic N, Rapallo A, Nigro S, Geido E. Intratumor heterogeneity of K-ras2 mutations in colorectal adenocarcinomas: association with degree of DNA aneuploidy. Am J Pathol. 1996;149:237–45. [PMC free article] [PubMed] [Google Scholar]
- 17.Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–73. [DOI] [PubMed] [Google Scholar]
- 18.Konishi N, Hiasa Y, Matsuda H, Tao M, Tsuzuki T, Hayashi I, et al. Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma. Am J Pathol. 1995;147:1112–22. [PMC free article] [PubMed] [Google Scholar]
- 19.Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. 2015;12:573–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soucheray M, Capelletti M, Pulido I, Kuang Y, Paweletz CP, Becker JH, et al. Intratumoral heterogeneity in EGFR-mutant NSCLC results in divergent resistance mechanisms in response to EGFR Tyrosine Kinase inhibition. Cancer Res. 2015;75:4372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011; 20:810–7. [DOI] [PubMed] [Google Scholar]
- 22.Janiszewska M, Liu L, Almendro V, Kuang Y, Paweletz C, Sakr RA, et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat Genet. 2015;47:1212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL, et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol. 2014;10:e1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11:396–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C, et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2016;22:105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endes-felder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015; 21:751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346:256–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thrane K, Eriksson H, Maaskola J, Hansson J, Lundeberg J. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 2018;78:5970–9. [DOI] [PubMed] [Google Scholar]
- 32.McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567:479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joshi K, de Massy MR, Ismail M, Reading JL, Uddin I, Woolston A, et al. Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat Med. 2019;25:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hernandez L, Wilkerson PM, Lambros MB, Campion-Flora A, Rodrigues DN, Gauthier A, et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J Pathol. 2012;227:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stachler MD, Taylor-Weiner A, Peng S, McKenna A, Agoston AT, Odze RD, et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat Genet. 2015;47:1047–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, et al. Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell. 2018;172:205–.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yizhak K, Aguet F, Kim J, Hess JM, Kübler K, Grimsby J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364 10.1126/science.aaw0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin J-R, Izar B, Wang S, Yapp C, Mei S, Shah PM, et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife. 2018;7:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci USA. 2013;110:11982–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. 2014;20:436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11:417–22. [DOI] [PubMed] [Google Scholar]
- 44.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. [DOI] [PubMed] [Google Scholar]
- 45.Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep. 2017;19:203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, et al. An immune atlas of clear cell renal cell carcinoma. Cell. 2017;169:736–.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016;13:273–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baker A-M, Huang W, Wang X-MM, Jansen M, Ma X-J, Kim J, et al. Robust RNA-based in situ mutation detection delineates colorectal cancer subclonal evolution. Nat Commun. 2017;8:1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343:1360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016; 353:78–82. [DOI] [PubMed] [Google Scholar]
- 51.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstråhle J, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun. 2018;9:2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014;4:650–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dawson S-J, Tsui DWY, Murtaza M, Biggs H, Rueda OM, Chin S-F, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–209. [DOI] [PubMed] [Google Scholar]
- 55.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014; 158:1110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller AM, Shah RH, Pentsova EI, Pourmaleki M, Briggs S, Distefano N, et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature. 2019;565:654–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352:169–75. [DOI] [PubMed] [Google Scholar]
- 60.Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015;5:1164–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. 2017;32:169–.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siegel MB, He X, Hoadley KA, Hoyle A, Pearce JB, Garrett AL, et al. Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer. J Clin Investig. 2018;128:1371–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514:54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Janiszewska M, Tabassum DP, Castaño Z, Cristea S, Yamamoto KN, Kingston NL, et al. Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nat Cell Biol. 2019;21:879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reeves MQ, Kandyba E, Harris S, Del Rosario R, Balmain A. Multicolour lineage tracing reveals clonal dynamics of squamous carcinoma evolution from initiation to metastasis. Nat Cell Biol. 2018;20:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bhang H-EC, Ruddy DA, Krishnamurthy Radhakrishna V, Caushi JX, Zhao R, Hims MM, et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med. 2015;21:440–8. [DOI] [PubMed] [Google Scholar]
- 67.Hinohara K, Wu H-J, Vigneau Sébastien, McDonald TO, Igarashi KJ, Yamamoto KN, et al. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell. 2019;35:330–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lan X, Jörg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature. 2017;549:227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 2018;560:325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]