

Abstract
Conflict resolution in genomic variant interpretation is a critical step towards improving patient care. Evaluating interpretation discrepancies in copy number variants (CNVs) typically involves assessing overlapping genomic content with focus on genes/regions that may be subject to dosage sensitivity (haploinsufficiency (HI) and/or triplosensitivity (TS)). CNVs containing dosage sensitive genes/regions are generally interpreted as “likely pathogenic” (LP) or “pathogenic” (P), and CNVs involving the same known dosage sensitive gene(s) should receive the same clinical interpretation. We compared the Clinical Genome Resource (ClinGen) Dosage Map, a publicly available resource documenting known HI and TS genes/regions, against germline, clinical CNV interpretations within the ClinVar database. We identified 251 CNVs overlapping known dosage sensitive genes/regions but not classified as LP or P; these were sent back to their original submitting laboratories for re-evaluation. Of 246 CNVs re-evaluated, an updated clinical classification was warranted in 157 cases (63.8%); no change was made to the current classification in 79 cases (32.1%); and 10 cases (4.1%) resulted in other types of updates to ClinVar records. This effort will add curated interpretation data into the public domain and allow laboratories to focus attention on more complex discrepancies.
Keywords: CNV discrepancy, dosage sensitivity, variant interpretation, ClinVar, ClinGen
Introduction
Advances in genetic testing technologies have allowed the genomics community to greatly expand its ability to diagnose and care for patients. Historically, genetic diagnoses have been made using a “phenotype-first” approach, where a patient’s clinical features were used to determine a possible clinical diagnosis, and genetic testing was ordered (if available) to confirm. Today, many diagnoses are “genotype-first,” where genome-wide assays, such as chromosomal microarray (CMA), whole exome sequencing (WES), or whole genome sequencing (WGS) are ordered as an initial diagnostic step (Mefford, 2009; Stessman, Bernier, & Eichler, 2014). Variants identified as a result of this testing often lead the clinician to a specific diagnosis, one that may not have been readily apparent given the presenting clinical features, particularly if those features are nonspecific, such as developmental delay. In this “genotype-first” era, the clinical interpretation of genomic test results is of paramount importance, as these results may lead a clinician to confirm or refute a particular diagnosis, and may ultimately have an effect on a given patient’s medical management. Ensuring that laboratories provide accurate and consistent variant interpretations is a critical step toward improving patient care.
The increasing clinical usage of genome-wide assays required laboratories to be prepared to interpret variants that may occur throughout the genome. Interpreting variants in genes or genomic regions with which a laboratory has little to no experience, or about which little is known, is challenging. As clinical genomic testing became more routine, the genomics community recognized that making variant interpretations and the evidence supporting them publicly available could potentially help with these limitations. One early example of a community effort to encourage genomic data sharing was the International Standards for Cytogenomic Arrays (ISCA) Consortium, a group focused on building standards and encouraging collaboration amongst those laboratories performing clinical CMA testing (Miller et al., 2010). The ISCA Consortium was among the first groups to make data obtained from clinical testing publicly available through the National Center for Biotechnology Information’s (NCBI) dbVar database (Kaminsky et al., 2011). This and other shared datasets became essential tools for the clinical interpretation of copy number variants (CNVs) (Coe et al., 2014; Cooper et al., 2011; MacDonald, Ziman, Yuen, Feuk, & Scherer, 2014). As the utility of sharing genomic variants with clinical interpretations became more apparent, NCBI established ClinVar, a publicly available repository of genomic variation and its relationship to human health (Landrum et al., 2014).
As more clinical laboratories began to make their variant interpretation data publicly available through ClinVar, variant interpretation discrepancies became more apparent (Lincoln et al., 2017; Yang et al., 2017). Interpretation discrepancies can arise for a number of reasons, including (but not limited to): time (new evidence may have emerged since a laboratory last evaluated the variant); access to information (one laboratory may have access to information that another may not, such as extensive internal databases, segregation or phenotype information for a particular patient, etc.); opinion (though evaluation guidelines have been published for both sequence (Richards et al., 2015) and copy number variants (Kearney et al., 2011), there is still a level of subjectivity involved when assessing the strength of particular pieces of evidence); and human error (data entry errors, etc.). The transparency provided by ClinVar, however, has encouraged many laboratories to work together to identify the reasons behind these discrepancies and resolve them, a powerful step toward more standardized variant interpretations and ensuring quality within and across laboratories (Garber et al., 2016; Harrison et al., 2017).
Thus far, the majority of reported conflict resolution efforts involving ClinVar data have focused on sequence-level variants, while limited review and re-analysis has been performed for CNV data. The major challenge to identifying and resolving potential CNV interpretation discrepancies has to do with their inherent singular nature. With the exception of recurrent events (such as those mediated by segmental duplications), most CNVs have unique breakpoints. In many cases, other CNVs with matching breakpoints are not available for direct comparison. Even determining when a conflict exists between two or more CNVs is difficult; though they may have areas of overlap, important genomic features may exist within the non-overlapping regions, providing logical reasons for differing classifications. For example, even though two deletions may overlap the same known haploinsufficient gene, one may be interpreted as “pathogenic” due to the fact that it involves most of the gene, while the other may be interpreted as “variant of uncertain significance” or “likely benign” because it overlaps only a non-coding exon, or only exon(s) involved in an isoform not thought to be clinically relevant, etc. Potential CNV conflicts must be evaluated on the basis of overlapping genomic content, with special focus on those genes or genomic regions that may be subject to dosage sensitivity - haploinsufficiency (HI) and/or triplosensitivity (TS).
To facilitate the process of genomic content evaluation and promote interpretation consistency, the ISCA Consortium began systematically evaluating genes and genomic regions for dosage sensitivity in 2011 (Riggs et al., 2012). Though the ISCA Consortium has officially become part of the Clinical Genome Resource (ClinGen), a National Institutes of Health (NIH)-funded effort dedicated to identifying clinically relevant genes and variants for use in precision medicine and research (Rehm et al., 2015), these activities remain ongoing. Dosage evaluations are made publicly available through the ClinGen Dosage Sensitivity Map website (https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/). For each individual gene or genomic region, current medical literature is evaluated for evidence supporting or refuting dosage sensitivity as the mechanism for any associated constitutional disease. Evidence for HI and TS is considered separately, and for each gene and genomic region, both a haploinsufficiency and triplosensitivity score are provided, corresponding to the strength of the available evidence for each. Genes/regions receiving the highest score (3) are considered to have “sufficient” evidence supporting HI and/or TS as a mechanism of disease. In general, CNVs containing genes or genomic regions with an HI or TS score of 3 should be classified as pathogenic (P) or likely pathogenic (LP), unless there is evidence to suggest otherwise (Riggs et al., 2012). For example, a deletion fully encompassing a known HI gene should be interpreted as P/LP, whereas a deletion fully contained within an intron (and unlikely to result in loss of function) of a known HI gene may not. Likewise, a one copy gain that fully contains a known TS gene (whole gene duplication) should be classified as P/LP, whereas a partial gene duplication that contains one or both breakpoints within a gene, and could in fact disrupt gene expression, may not, unless the disrupted gene is also a known HI gene. Other factors to consider when evaluating the clinical significance of a copy number gain include the location and orientation of the additional genomic material.
As of late 2017, ClinVar contained over 19,000 CNVs; approximately 17% of these (3164) were deposited as the result of the initial efforts of the ISCA Consortium, and have not been updated since they were initially made publicly available in 2011 (Kaminsky et al., 2011). In an effort to increase the quality of CNV interpretations available to the genomics community through ClinVar, we used evidence scores from the ClinGen Dosage Sensitivity Map to identify CNVs with interpretations that appear to be in conflict with current understanding of the genes and/or genomic regions they overlap. As evidence supporting dosage sensitivity of genes or genomic regions included within a particular CNV may have emerged since these CNVs were last evaluated, the original submitting laboratories were contacted to re-evaluate these CNVs with currently available evidence. This effort represents an important first step in establishing a CNV conflict resolution process that may be utilized beyond resolution of conflicts in ClinVar. In addition, this work paves the way for the identification and re-evaluation of other CNV classification conflict types, including inter- and intra-laboratory CNV conflicts, conflicts with other evidence-based scoring and/or predictive dosage sensitive metrics, and conflicts with sources of CNV data in the general population (such as the Database of Genomic Variants).
Methods
The ClinGen Dosage Sensitivity Map
As of August 2017, the ClinGen Dosage Sensitivity Map included dosage sensitivity evaluation on 1303 single genes and genomic regions (both recurrent, such as the 16p11.2 region associated with neurodevelopmental disorders [MIM:611913, 614671] and non-recurrent, such as the 4p13.6 region associated with Wolf-Hirschhorn syndrome [MIM:194190]). At that time, there were 257 genes and 38 genomic regions reaching the threshold of “known” dosage sensitive (HI and/or TS score of 3) (see Supp. Table S1 for a full list). This list of 295 known dosage sensitive genes/regions was downloaded from the ClinGen Dosage Sensitivity Map website (ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/clingen) and used to compare against CNVs in ClinVar.
Identification of Potential Conflicts in ClinVar
Variant and clinical significance data were imported into a local database (Neo4j, Malmo, Sweden) from NCBI’s ClinVar database (XML full release, Aug. 2017). From the ClinVar XML, we selected variants with type “copy_number_gain” or “copy_number_loss.” Variants that were not mapped to GRCh38 were mapped using the NCBI remap tool. The GRCh38 coordinates of each CNV were then compared for overlap with all exons of genes in the dosage map using an algorithm written in the Clojure language. Detected overlaps of exons and variants were stored back in the Neo4j database. This database was then queried for potential conflicts of interpretation; deletions overlapping a HI gene and interpreted as benign (B), likely benign (LB), or variant of uncertain significance (VUS) were identified as potential conflicts (Figure 1A). As intragenic duplications could potentially result in loss of function, only duplications completely encompassing a TS gene and interpreted as B, LB, or VUS were considered potential conflicts.
Figure 1: Schematic Representation of the CNV Discrepancy Identification and Resolution Process.
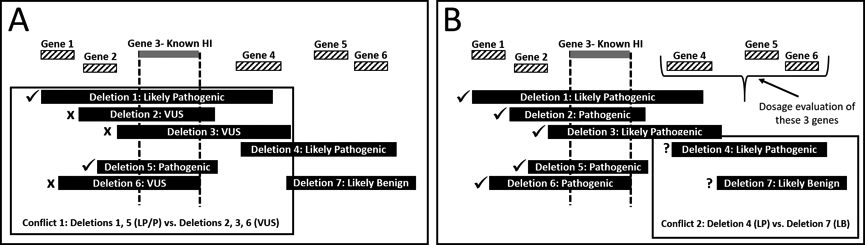
In this generic region of the genome, there are at least two potential conflicts involving CNVs overlapping by ≥50% with different clinical interpretations. A) Conflict 1 involves known haploinsufficient (HI) Gene 3. This information can be used to mediate the conflict resolution process. Likely pathogenic (LP)/pathogenic (P) interpretations are expected for deletions fully encompassing this gene, such as Deletions 1 and 5. Deletions 2, 3, and 6 also fully encompass this gene, but are interpreted as variants of uncertain significance (VUS). These cases would be flagged for reevaluation by the submitting laboratory. B) Assuming Conflict 1 is resolved by the reevaluation process, Conflict 2 can be assessed. To mediate this process, Genes 4, 5, and 6 would be triaged for dosage sensitivity evaluation. The dosage evaluation process could resolve the conflict in and of itself; for example, if Gene 4 were found to be a known HI gene, Deletions 4 and 7 would no longer be in conflict, as Deletion 7 does not include Gene 4.
A different process was used to detect potential conflicts with dosage sensitive genomic regions annotated in the dosage map. Several of our dosage sensitive genomic regions are non-recurrent; to annotate these within the dosage map, coordinates are manually selected by the ClinGen Dosage Sensitivity working group, typically based on the established critical region or smallest genomic range reported in the literature to be associated with the clinical phenotype. A description of how the coordinates were determined is included in each region entry (for example, see https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/clingen_region.cgi?id=ISCA-37434). Historically, these manually curated coordinates have been recorded using build GRCh37; updating these coordinates to GRCh38 will require manual review to ensure intended genes/regions are included, a process currently underway within the ClinGen Dosage Sensitivity working group. Since GRCh38 coordinates were not available for all dosage sensitive genomic regions in August 2017, potential conflicts were manually identified using the UCSC Genome Browser. The browser was configured to show only variants in the publicly available “ClinGen CNVs” track with “benign”, “likely benign”, or “uncertain” clinical significance interpretations. Each nstd45 region was then viewed in the genome browser on the GRCh37 build, and all variants that overlapped with at least 50% of a given region and had a type/interpretation that conflicted with the region were recorded. For instance, if a variant was a “benign” copy number loss covering at least 50% of a haploinsufficient region, it would be recorded as a conflict.
In total, we initially identified 284 potential conflicts (Figure 2). Eighteen CNVs that were identified as part of research testing and/or from somatic tissue were excluded. An additional 15 CNVs appeared to have problems with remapping to GRCh38 (for example, a dosage sensitive gene was contained within the remapped coordinates but not the original submitted coordinates, or vice versa). These CNVS were also excluded from further analysis.
Figure 2:
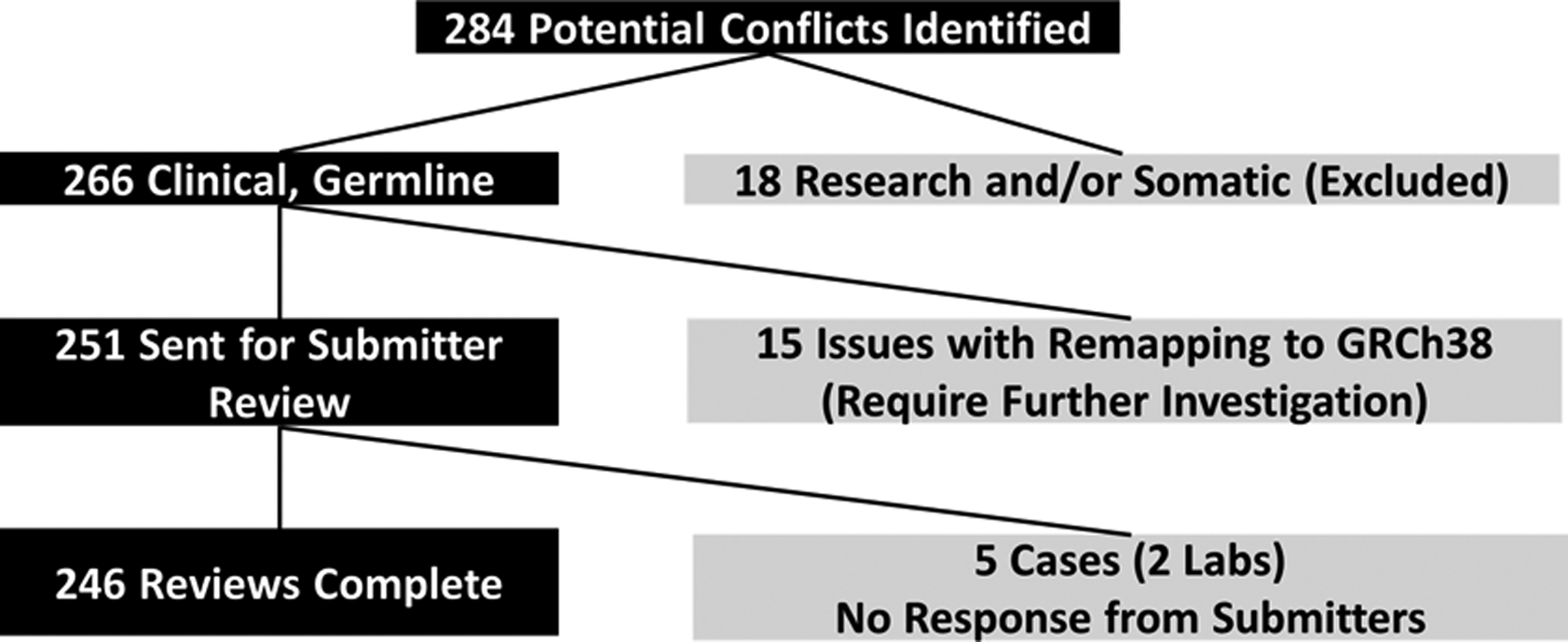
Number of potential conflicts identified and sent for re-evaluation by original submitting laboratory.
Conflict Resolution
Between August-September 2017 and January-February 2018, two rounds of CNV conflict resolution activities were performed. A total of 251 potential conflicts were sent to 14 different original submitting laboratories for re-evaluation (see Supplemental Information for the full list). For each CNV, the submitting laboratory received a summary of the originally submitted information (variant coordinates; original submitted interpretation; associated sex, phenotype, inheritance information, if available; ClinVar and dbVar identifiers). The specific dosage sensitive gene/genomic region that triggered each conflict was also provided. Laboratories were asked to re-evaluate and, if warranted, re-classify the CNV in light of currently available evidence. Participating laboratories were asked to return a new classification (if applicable) and a free-text rationale for their decision. The free text rationales were reviewed and grouped into general categories for analysis.
Results
Twelve of 14 laboratories re-evaluated 246 of the 251 potential conflicts, for a response rate of 98%; two laboratories representing 5 potential conflicts did not respond to requests for participation (a complete list of all re-evaluated cases is available in the supplemental material). Of the 246 re-evaluated CNVs, 125 (50.8%) overlapped a known dosage sensitive gene (121 deletions overlapping HI genes and 4 duplications fully encompassing TS genes), and 121 (49.2%) represented CNVs overlapping a known dosage sensitive region (34 deletions overlapping HI regions, 87 duplications overlapping TS regions). As suspected, many of these cases had not been evaluated in several years; 74.3% (183) of the cases had last been evaluated five or more years ago (see Supp. Table S2 for all original dates of evaluation). In all, 155 (63.0%) of the re-evaluated potential conflicts resulted in updated classifications; 81 (32.9%) resulted in no change to the original classification; and 10 (4.1%) resulted in some “other” type of update to the ClinVar record, discussed in further detail below (Figure 3). Of the 236 cases where a re-classification decision (yes or no) was made, 78.4% (n=185) were returned with a free-text description of the rationale supporting their decision, whereas 21.6% (n=51) of the CNVs were returned without a corresponding decision rationale.
Figure 3:
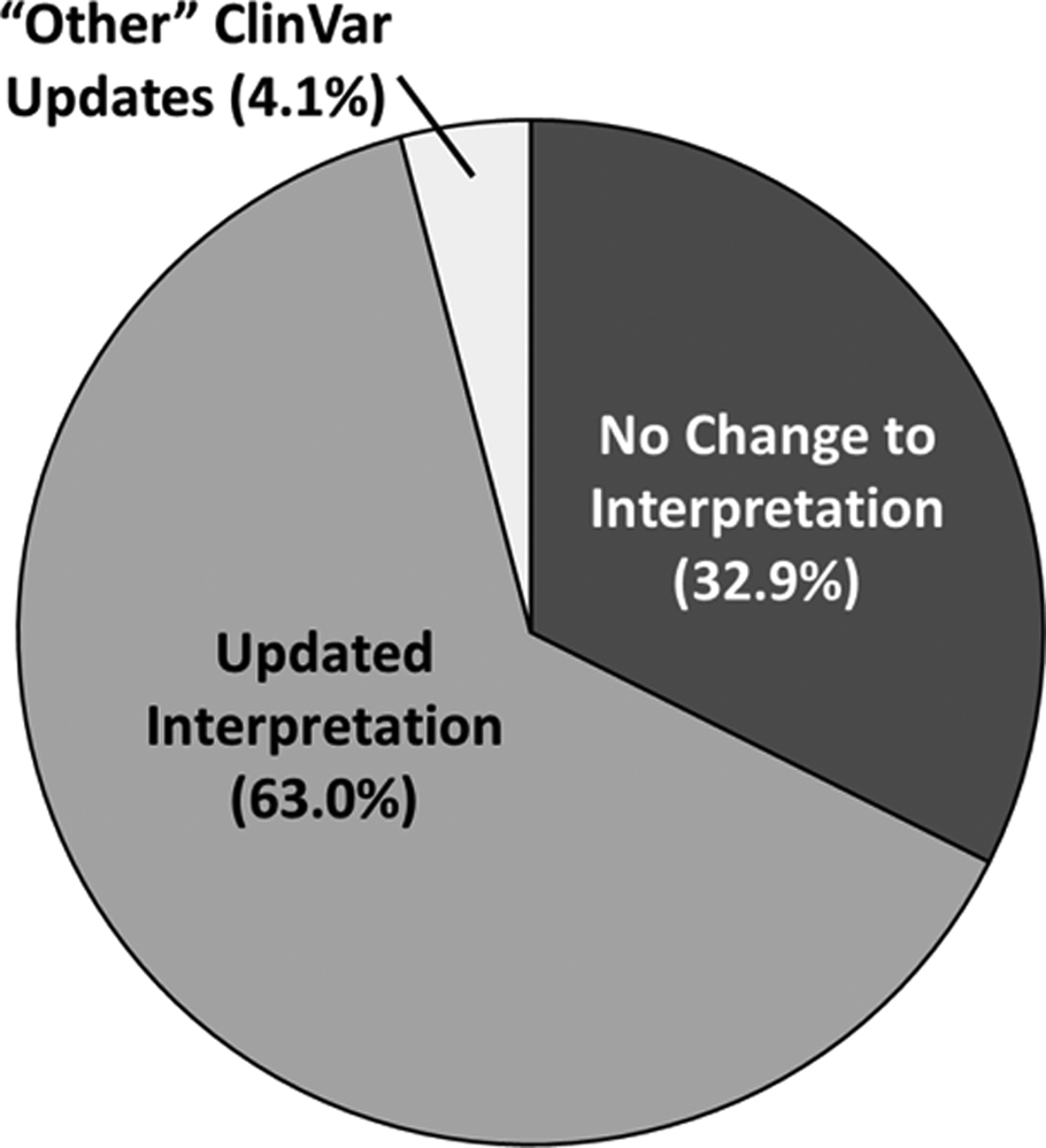
Percentage of cases re-evaluated by the original submitting laboratory (n=246) resulting in updates to the ClinVar record.
Updated Classifications
After re-evaluation by the original submitting laboratory, 63.0% (n=155) of the potential conflicts resulted in updated classifications. Potential conflicts involving dosage sensitive genomic regions received updated classifications more frequently than those involving dosage sensitive genes (86.0% (104/121) vs. 40.8% (51/125), respectively). Perhaps not surprisingly (based on the selection criteria), most potential conflicts that did result in updated classifications were updated to P/LP (94.8%, n=147) (Figure 4). Most of the updated P/LP CNVs were originally classified as VUS (89.0%), likely reflecting emergence of new data.
Figure 4: Classification Updates.
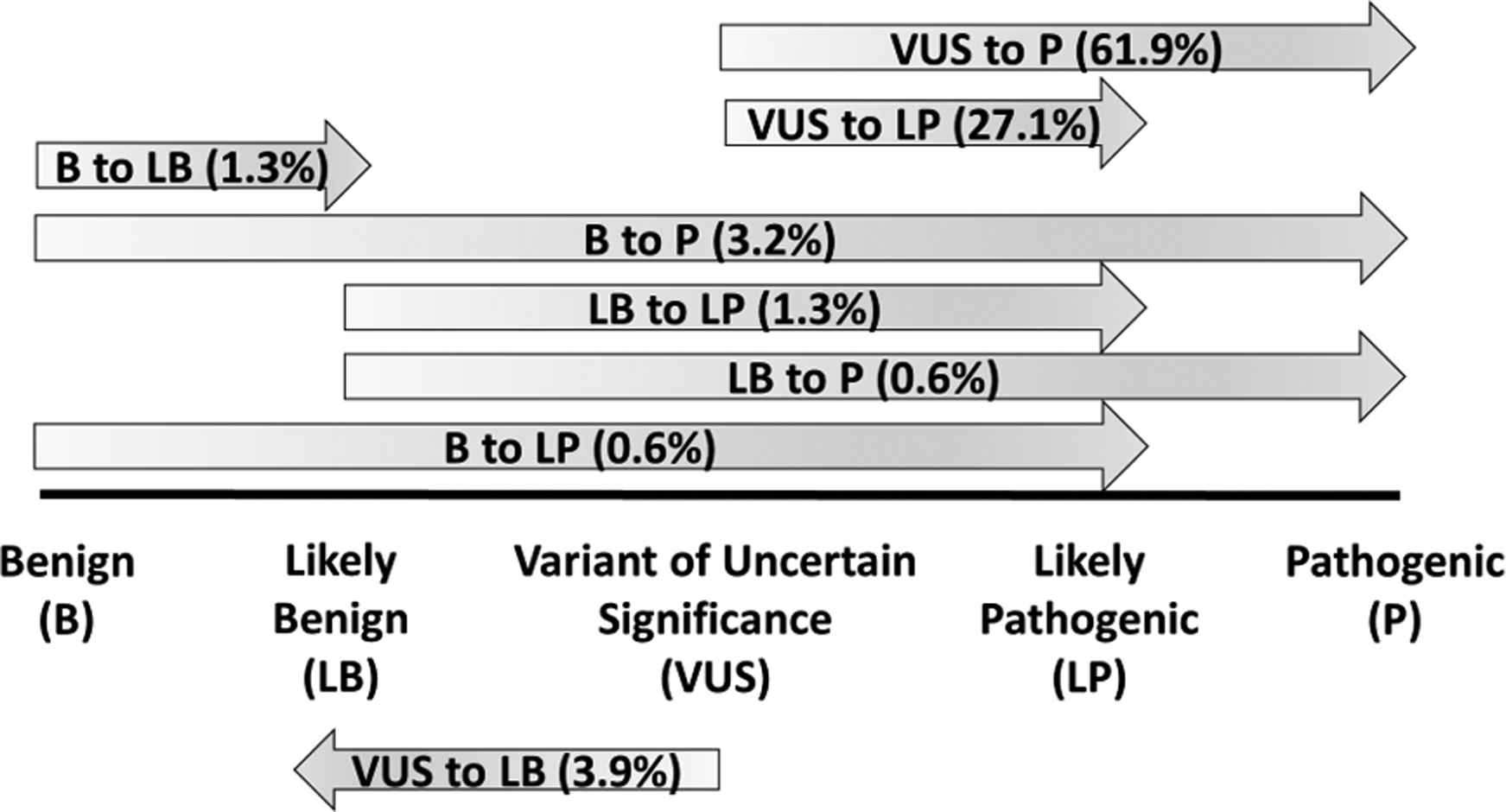
Laboratory re-evaluation resulted in updated classification in 155 cases; the exact classification changes are shown here. Overall, 94.8% of cases with updated classifications were changed to likely pathogenic (LP) or pathogenic (P).
Approximately 5.2% of re-evaluated CNVs receiving updated classifications (n=8) were not upgraded to P/LP: 2 cases were upgraded by one classification “step,” from B to LB (1.3%), while 6 cases were downgraded from VUS to LB (3.9%). All of these CNVs were deletions involving the same gene, NRXN1; haploinsufficiency of this gene has been associated with developmental brain disorders such as autism, intellectual disability, and schizophrenia (Autism Genome Project Consortium et al., 2007; Bucan et al., 2009; Ching et al., 2010; Gauthier et al., 2011; Lowther et al., 2017; Walsh et al., 2008). Investigation into their genomic content revealed that none of these deletions involved coding sequence, and were last evaluated between 2010–2012. At this time, there have been several NRXN1 intronic deletion variants observed in normal populations cataloged in the Database of Genomic Variants (DGV) Gold Standard Dataset (Zarrei, MacDonald, Merico, & Scherer, 2015), though most are observed at less than the 1% frequency threshold typically used to describe a variant as a polymorphism. This information supports the re-classification of these variants to LB.
In total, 9 potential conflicts (5.7%) underwent greater than two-step re-classification, changing from either B or LB to P or LP. In two of these cases (a deletion involving the PMS2 gene associated with Lynch syndrome [MIM:614337] and a 16p11.2 deletion [MIM:611913]), the laboratories indicated that changing knowledge over time played a role in their updated interpretation. In the other 7 cases, the laboratory indicated that the original submitted classifications were in error, though it was unclear where the error occurred (during the laboratory reporting process, during the data submission process, etc.).
The remaining 138 (89.0%) cases with updated classifications changed from VUS to LP or P. While most who provided a rationale (n=106) cited updated information emerging over time as the reason for the change (86.7%, n=92)), there were 8 cases from this group that also specifically noted an error in submission (the submitted interpretation was not the reported interpretation) as the reason for the change. From the data obtained as part of this study, it is unclear where the error occurred.
The genomic region that generated the most potential conflicts with overlapping CNVs was the proximal, recurrent 16p11.2 region (MIM:614337, 614671). Deletions and duplications of this region are now known to be involved in neurocognitive phenotypes, such as autism, and known to exhibit variable expressivity and reduced penetrance (Bernier et al., 2017; D’Angelo et al., 2016; Fernandez et al., 2010; Kumar et al., 2008; Steinman et al., 2016; Weiss et al., 2008). The clinical significance of this region was typically interpreted as uncertain when laboratories first started performing clinical microarray testing. Variants at this region, particularly the duplications, were frequently observed in reportedly normal parents (who may not have had detailed, neurocognitive phenotyping), contributing to the misconception that they were not clinically relevant. The clinical effects of this region are now better understood and, as such, the region has been evaluated as a known haploinsufficient and triplosensitive region according to the ClinGen Dosage Sensitivity Map. Fifty-one cases were identified as being in potential conflict with this region (3 deletions, 48 duplications); most of these were originally interpreted as VUS (n=49), though one case each was interpreted as LB or B. After re-evaluation, 50 of these cases were reclassified to LP/P; in one case, the submitting laboratory opted to keep the interpretation as VUS because the variant was observed in a mosaic state.
No Change to the Original Classification
Of the 246 cases that were re-evaluated, 32.9% (n=81) resulted in no change to the original interpretation. This decision was made more frequently for cases flagged as potential conflicts due to overlap with a dosage sensitive gene (51.2%, 64/125 potential gene conflicts) than for those flagged due to overlap with a dosage sensitive genomic region (14.0%, 17/121 potential region conflicts). Of those cases opting not to change their classification that provided a rationale for their decision (n=64), the most commonly cited reason for not changing the interpretation was because the case involved a dosage sensitive gene on the X chromosome, and the patient was a female (43.8%, n=28). These cases (female carriers of variants that most likely would have been interpreted pathogenic in males) were instead interpreted as either LB or VUS.
Other reasons for deciding not to change the original classification involved the genomic context of the particular variant. Among cases overlapping dosage sensitive genes, there were cases where the variant was completely intronic (n=6), involved only non-coding exons (n=6), or involved only the last exon (and not expected to result in nonsense-mediated decay) (n=1). There were three cases involving deletions of NRXN1 where the laboratory noted that these three cases were observed several years ago on a lower-resolution array platform; they could not be certain whether the variants actually overlapped with any coding sequence of NRXN1, so they opted to keep the classification as VUS. Among those cases overlapping dosage sensitive regions, the laboratory opted not to change the interpretation because the variants were smaller than the regions as defined by the ClinGen Dosage Sensitivity map (n=4). These 4 cases involved two genomic regions that are not recurrent, segmental-duplication regions, are known to vary in size, and do not have a well-established critical region or causative gene (deletions of 4p16.3, associated with Wolf-Hirschhorn syndrome [MIM:194190], and deletions of 2q37.3, associated with a brachydactyly-intellectual disability phenotype [MIM:600430]).
In several cases, the laboratory opted not to change their original classification because they did not agree with the ClinGen Dosage Sensitivity designations (i.e., they did not feel that there was strong enough evidence to support the dosage sensitivity scores for these genes/regions) (n=9). These 9 cases came from a single laboratory, and involved duplications of the 17q11.2 region (including NF1) (n=2); and duplications of the distal 22q11.2 region (LCR22-D to LCR22-E or -F) (n=7). Each of these duplications have been reported in association with varying neurodevelopmental phenotypes and reduced penetrance/variable expressivity. The clinical significance of these types of events has historically been difficult to determine, given their nonspecific phenotype and presence in reportedly “normal” parents. However, recent literature has shown that, when carefully phenotyped, “normal” carriers of certain CNVs do show subtle neurodevelopmental deficits (Kendall et al., 2017; Mannik et al., 2015; Stefansson et al., 2014). Given the laboratory’s concerns over the evidence supporting triplosensitivity of the 17q11.2 region (including NF1) and the distal 22q11.2 region (LCR22-D to LCR22-E), these regions will be re-evaluated by the ClinGen Dosage Sensitivity working group.
Other types of updates
Ten re-evaluations resulted in “other” types of updates to the existing ClinVar record. All 10 cases were flagged as potential conflicts due to overlap with a known dosage sensitive gene; there were no cases overlapping genomic regions in this category. Upon re-evaluation, six cases were identified by the submitting laboratories as either artifacts of testing or false positive calls on array. These cases were originally submitted as part of the original ISCA Consortium pilot data set (Kaminsky et al., 2011), and were observed when the laboratories first started performing clinical CMA testing. After several additional years of experience, the laboratories are now easily able to identify issues such as false positive calls due to poorly performing probes on certain array platforms. These 6 CNVs will be removed from ClinVar.
Three additional CNVs came from a single laboratory that uses a third-party system to submit their data to ClinVar; in these three cases, this third-party process resulted in the inadvertent submission of variants that were identified but not originally classified or reported with “VUS” interpretations in ClinVar. These CNVs will also be removed from ClinVar. The tenth case was flagged as a potential conflict because it appeared to be a deletion involving the EHMT1 gene; haploinsufficiency of this gene has been associated with Kleefstra syndrome (MIM:610253). Upon re-evaluation, the submitting laboratory noted that the observed case was actually a duplication; the case was mistakenly submitted to ClinVar as a deletion. The copy number on this particular case will be corrected in ClinVar, and the original interpretation (VUS) will remain the same.
Inconsistencies in interpretation of CNVs on the X chromosome
As a result of the re-evaluation process, we identified an area of inconsistency among laboratories when interpreting CNVs involving the X chromosome. Of all 246 re-evaluated CNVs, 48 were flagged as potential conflicts due to overlap with dosage sensitive gene on the X chromosome. Of these, 41 had copy numbers of 1 or 3, implying that they were observed in females (sex of the tested individual is not consistently available in ClinVar records). In 10 out of these 41 potential conflicts involving X-linked genes in females (24.4%), the laboratories did opt to change their classification from the original VUS to LP/P. In the remaining 31 cases (68.9%), the laboratories did not opt to change their classifications from their original VUS (n=16), LB (n=14), or B (n=1). Historically, CNV interpretation has been done in the context of the presenting individual - if the observed CNV was not believed to be related to the reason for testing, it may not have been interpreted as pathogenic. For example, CNVs on the X chromosome in females, typically representing a carrier state, may receive classifications other than LP or P, to reflect the fact that these findings are likely not the cause of the individual’s reason for testing. More recently, there has been a movement towards ensuring that variant pathogenicity is assessed independently of the presenting patient’s reason for referral, and that a variant should receive the same interpretation (on the basis of supporting evidence), regardless of the clinical context in which it is observed (Richards et al., 2015). For example, CNVs involving known dosage sensitive genes on the X chromosome should be interpreted as LP/P, regardless of whether they are observed in a male or a female; caveats regarding the clinical significance of this finding for the tested individual should be explained in the body of the report. Our work shows that laboratories are currently utilizing both approaches, resulting in inconsistency in the way X-chromosome CNVs are being classified for males and females. Note that scenarios where the clinical significance for an individual patient may differ based upon their sex are not limited to variants on the X-chromosome; this issue may also arise with autosomal variants involving imprinted genes/regions, or sex-limited phenotypes determined by autosomal loci. Updated CNV interpretation guidelines recommending that variant interpretation be uncoupled from clinical significance for a given individual should make these interpretations more consistent in the future.
Discussion
Publicly available databases containing genomic variants and their clinical interpretations (such as ClinVar) represent an incredible resource for clinical laboratories, clinicians, and researchers; knowing that another group has observed a given variant, how they interpreted it, and the evidence they used to arrive at that conclusion can help shape one’s own evaluation of that variant. In addition, by making their variant interpretations publicly available, laboratories are now more readily able to appreciate when their interpretations are in conflict with others. This process has prompted collaborations between laboratories to resolve interpretation discrepancies, mainly among sequence-level variants (Garber et al., 2016; Harrison et al., 2017).
Interpretation discrepancies among CNVs have been appreciated for some time (Tsuchiya et al., 2009), however, to our knowledge, this study represents the first organized, multi-laboratory effort to resolve them. A conflict for a sequence variant is identified when different clinical interpretations exist for the same exact variant; resolving them involves reviewing currently available evidence for that single, well-defined variant. Because the majority of CNVs have unique breakpoints, our group has needed to take a different approach to discrepancy identification and resolution. Since there are often no other CNVs available with the exact same breakpoints for comparison, CNVs conflicts needed to be identified based on copy number, degree of overlap, and shared genomic content. For this initial effort, we wanted to identify those CNVs most likely to warrant an interpretation update, and therefore focused on those CNVs that overlapped by at least 50% with a known dosage sensitive gene or genomic region, as defined by the ClinGen Dosage Sensitivity Map. Indeed, 63.0% of those cases sent for re-evaluation ultimately received updated interpretations, and 94.8% of those with updated classifications represented changes that were medically relevant (i.e., changes from B, LB, or VUS to LP/P). These data suggest that this approach did effectively identify cases that were not aligned with current understanding of the genes or genomic regions involved.
Since this was a pilot effort, we chose to use the evidence-based ClinGen Dosage Sensitivity Map as our standard for “known” dosage sensitive genes and genomic regions; these genes and genomic regions are designated as dosage sensitive after careful review and consideration of literature-based evidence that loss or gain of these genes/regions causes human disease. Other methods have been developed to predict which genes may be haploinsufficient based on biologically relevant evidence (expression patterns, number of observed vs. expected loss of function (LOF) variants in the general population, etc.) and objective, statistical analysis (Huang, Lee, Marcotte, & Hurles, 2010; Lek et al., 2016; Petrovski, Wang, Heinzen, Allen, & Goldstein, 2013; Uddin et al., 2014; Uddin et al., 2016). As the results of these metrics are computationally derived, they are able to annotate many more genes as potentially haploinsufficient than the manual ClinGen Dosage Sensitivity evaluation process, which can be extremely beneficial for hypothesis exploration in the research setting. However, computational predictors also have limitations, and one must be aware of these when considering incorporating them into clinical use. For example, metrics that account for differences between observed vs. expected loss of function variation data in the general population may not predict haploinsufficiency for genes in which LOF variants are known to cause adult-onset disorders that do not affect reproductive fitness, such as BRCA1. We believe there is a role for both types of methods (manual vs. computational) to evaluate dosage sensitivity; predictors could be used to triage the genes that the ClinGen Dosage Sensitivity team evaluates in the future. Additionally, the 50% overlap threshold was chosen as a conservative measure to ensure that flagged cases had a sufficient degree of overlap with the dosage sensitive gene/region to justify asking the laboratory to re-evaluate. It could be argued that any degree of overlap with a known dosage sensitive gene/region should trigger a potential conflict reevaluation, however, efforts were made to strike a balance between identifying all possible conflicts and overloading participating laboratories with reevaluations unlikely to result in change. Future studies will focus on identifying the ideal overlap threshold for these conflict resolution exercises.
Due to the unique nature of most of the CNVs involved, instead of working with each other (a model frequently used by sequence variant conflict resolution groups), each laboratory was asked to re-evaluate their cases on their own, using currently available information. By leveraging the existing Dosage Sensitivity Map resource for the evaluation of genomic content, we were able to present participating laboratories with a summary of relevant information in an attempt to streamline their re-evaluation process. The study is limited by the fact that we did not explicitly ask participants how useful they felt this was, however we are encouraged by our high participation and completion rate - 12 of 14 laboratories approached opted to participate, and all 12 labs completed 100% of their assigned re-evaluations. It is possible that having an identified reason for re-evaluation - the fact that a case overlaps a specific gene/genomic region - makes the process of CNV conflict resolution more straightforward and manageable. Given the success of this initial effort, we intend to apply this model to other, potentially more complex CNV discrepancies (Figure 1B). Genes involved in other CNV discrepancies that have not been previously evaluated for dosage sensitivity can be triaged for evaluation by ClinGen, and, pending the results of the evaluation, could contribute to the resolution of the conflict.
The ultimate intent of this effort was to increase the quality of clinically interpreted CNVs available for community use in ClinVar, and we feel this was accomplished in several different ways. The first was facilitating the update of interpretations that were not in line with current understanding of dosage sensitive genes and genomic regions; however, not all cases re-evaluated resulted in an updated clinical interpretation. This was expected; even if a CNV overlaps a dosage sensitive gene/genomic region by 50%, it still may not overlap critical exons/regions. However, the evaluation process as a whole, regardless of whether or not the interpretation was updated, provided valuable information that can be used to update the existing ClinVar record, another way in which the quality of these particular cases was increased. For all cases re-evaluated (n=246), the “Date Last Evaluated” field will be updated. Approximately 74.4% of these cases had not been evaluated in five years or more; once this information is updated in ClinVar, users can be confident that the interpretations have been recently reviewed by the laboratory and are current. Additionally, now that processes for potential conflict identification and re-review are in place, these conflict resolution exercises will take place more frequently. Many re-evaluations (n=185 or 75.2%) included a rationale for the laboratory’s decision to update the classification or not, and this information can also be added to the ClinVar record to make users aware of what considerations went in to the laboratories’ interpretations. The re-evaluation process can result in richer information being added to the ClinVar record, outside of any potential interpretation change.
Finally, identifying and correcting errors in ClinVar CNV submissions increases data quality. We identified several cases (n=22, 8.9% of all cases evaluated) that represented errors - from cases that should not be represented in ClinVar at all (false positives, etc.) to cases that had some kind of incorrect attribute (copy number, clinical interpretation, etc.). For many laboratories, the ClinVar submission process involves at least some degree of manual data manipulation - for example, combining data that may exist in a variant calling system with data that may exist in a completely separate laboratory information management system, getting data to match with ClinVar’s controlled vocabulary when it may be different from one’s own, making sure potentially identifiable information is removed, etc. These processes may introduce errors into the submission. The ClinVar staff perform some “logic” checks on data as they are received; for example, making sure the coordinates listed for a given CNV are not larger than the chromosome it is on, etc. Additional checks, such as comparing data to the ClinGen Dosage Map prior to submission to ensure that cases that overlap with known dosage sensitive genes or genomic regions have been interpreted as the laboratory intended, may prevent some of these errors from making it into the database.
The process of CNV interpretation conflict identification and resolution is perpetual; new information regarding the dosage effects of genes and genomic regions is always being uncovered, and CNV interpretations will change accordingly. Community curation efforts such as the ClinGen Dosage Sensitivity Map are also constantly updated to reflect this knowledge. Future CNV conflict resolution efforts will continue to use the Dosage Sensitivity Map to identify and mediate conflicts by checking CNVs submitted to ClinVar for overlap with current known dosage sensitive genes and genomic regions, as well as by triaging those genes involved in other, inter-laboratory conflicts that have not been previously evaluated for dosage sensitivity (Figure 1B). The Dosage Map can serve as a valuable resource to identify those CNVs that require re-evaluation to align with current knowledge and provide laboratories with up-to-date dosage sensitivity information during the reassessment process with the ultimate goal of improving patient care.
Supplementary Material
Acknowledgments:
This work has been supported by the National Human Genome Research Institute (NHGRI) through grant U41HG006834. Many of the authors are clinical service providers employed by fee-for-service testing laboratories. Employment is noted in the author affiliations. There are no other conflicts to disclose.
This work is supported by the National Human Genome Research Institute (NHGRI) through grant U41HG006834.
Footnotes
Conflict of Interest:
Many of the authors are clinical service providers employed by fee-for-service testing laboratories. Employment is noted in the author affiliations. There are no other conflicts to disclose.
References
- Autism Genome Project Consortium, Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, … Meyer KJ (2007). Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nature Genetics, 39(3), 319–328. doi:ng1985 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier R, Hudac CM, Chen Q, Zeng C, Wallace AS, Gerdts J, … Simons VIP consortium. (2017). Developmental trajectories for young children with 16p11.2 copy number variation. American Journal of Medical Genetics.Part B, Neuropsychiatric Genetics : The Official Publication of the International Society of Psychiatric Genetics, 174(4), 367–380. doi: 10.1002/ajmg.b.32525 [doi] [DOI] [PubMed] [Google Scholar]
- Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, … Hakonarson H (2009). Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genetics, 5(6), e1000536. doi: 10.1371/journal.pgen.1000536 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching MS, Shen Y, Tan WH, Jeste SS, Morrow EM, Chen X, … Children’s Hospital Boston Genotype Phenotype Study Group. (2010). Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. American Journal of Medical Genetics.Part B, Neuropsychiatric Genetics : The Official Publication of the International Society of Psychiatric Genetics, 153B(4), 937–947. doi: 10.1002/ajmg.b.31063 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, … Eichler EE (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nature Genetics, 46(10), 1063–1071. doi: 10.1038/ng.3092 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, … Eichler EE (2011). A copy number variation morbidity map of developmental delay. Nature Genetics, 43(9), 838–846. doi: 10.1038/ng.909 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo D, Lebon S, Chen Q, Martin-Brevet S, Snyder LG, Hippolyte L, … Simons Variation in Individuals Project (VIP) Consortium. (2016). Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry, 73(1), 20–30. doi: 10.1001/jamapsychiatry.2015.2123 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, … Scherer SW (2010). Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. Journal of Medical Genetics, 47(3), 195–203. doi: 10.1136/jmg.2009.069369 [doi] [DOI] [PubMed] [Google Scholar]
- Garber KB, Vincent LM, Alexander JJ, Bean LJH, Bale S, & Hegde M (2016). Reassessment of genomic sequence variation to harmonize interpretation for personalized medicine. American Journal of Human Genetics, 99(5), 1140–1149. doi:S0002-9297(16)30396-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Siddiqui TJ, Huashan P, Yokomaku D, Hamdan FF, Champagne N, … Rouleau GA (2011). Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Human Genetics, 130(4), 563–573. doi: 10.1007/s00439-011-0975-z [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SM, Dolinsky JS, Knight Johnson AE, Pesaran T, Azzariti DR, Bale S, … Rehm HL (2017). Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 19(10), 1096–1104. doi: 10.1038/gim.2017.14 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, & Hurles ME (2010). Characterising and predicting haploinsufficiency in the human genome. PLoS Genetics, 6(10), e1001154. doi: 10.1371/journal.pgen.1001154 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, … Martin CL (2011). An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 13(9), 777–784. doi: 10.1097/GIM.0b013e31822c79f9 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, & Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. (2011). American college of medical genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 13(7), 680–685. doi: 10.1097/GIM.0b013e3182217a3a [doi] [DOI] [PubMed] [Google Scholar]
- Kendall KM, Rees E, Escott-Price V, Einon M, Thomas R, Hewitt J, … Kirov G (2017). Cognitive performance among carriers of pathogenic copy number variants: Analysis of 152,000 UK biobank subjects. Biological Psychiatry, 82(2), 103–110. doi:S0006-3223(16)32711-1 [pii] [DOI] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, … Christian SL (2008). Recurrent 16p11.2 microdeletions in autism. Human Molecular Genetics, 17(4), 628–638. doi:ddm376 [pii] [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, & Maglott DR (2014). ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Research, 42(Database issue), D980–5. doi: 10.1093/nar/gkt1113 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … Exome Aggregation Consortium. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. doi: 10.1038/nature19057 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln SE, Yang S, Cline MS, Kobayashi Y, Zhang C, Topper S, … Nussbaum RL (2017). Consistency of BRCA1 and BRCA2 variant classifications among clinical diagnostic laboratories. JCO Precision Oncology , 1 doi: 10.1200/PO.16.00020 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther C, Speevak M, Armour CM, Goh ES, Graham GE, Li C, … Bassett AS (2017). Molecular characterization of NRXN1 deletions from 19,263 clinical microarray cases identifies exons important for neurodevelopmental disease expression. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 19(1), 53–61. doi: 10.1038/gim.2016.54 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, & Scherer SW (2014). The database of genomic variants: A curated collection of structural variation in the human genome. Nucleic Acids Research, 42(Database issue), D986–92. doi: 10.1093/nar/gkt958 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannik K, Magi R, Mace A, Cole B, Guyatt AL, Shihab HA, … Reymond A (2015). Copy number variations and cognitive phenotypes in unselected populations. Jama, 313(20), 2044–2054. doi: 10.1001/jama.2015.4845 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC (2009). Genotype to phenotype-discovery and characterization of novel genomic disorders in a “genotype-first” era. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 11(12), 836–842. doi: 10.1097/GIM.0b013e3181c175d2 [doi] [DOI] [PubMed] [Google Scholar]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, … Ledbetter DH (2010). Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American Journal of Human Genetics, 86(5), 749–764. doi: 10.1016/j.ajhg.2010.04.006 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, & Goldstein DB (2013). Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genetics, 9(8), e1003709. doi: 10.1371/journal.pgen.1003709 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, … ClinGen. (2015). ClinGen--the clinical genome resource. The New England Journal of Medicine, 372(23), 2235–2242. doi: 10.1056/NEJMsr1406261 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … ACMG Laboratory Quality Assurance Committee. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 17(5), 405–424. doi: 10.1038/gim.2015.30 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs ER, Church DM, Hanson K, Horner VL, Kaminsky EB, Kuhn RM, … Martin CL (2012). Towards an evidence-based process for the clinical interpretation of copy number variation. Clinical Genetics, 81(5), 403–412. doi: 10.1111/j.1399-0004.2011.01818.x [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, … Stefansson K (2014). CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature, 505(7483), 361–366. doi: 10.1038/nature12818 [doi] [DOI] [PubMed] [Google Scholar]
- Steinman KJ, Spence SJ, Ramocki MB, Proud MB, Kessler SK, Marco EJ, … Simons VIP Consortium. (2016). 16p11.2 deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort. American Journal of Medical Genetics.Part A, 170(11), 2943–2955. doi: 10.1002/ajmg.a.37820 [doi] [DOI] [PubMed] [Google Scholar]
- Stessman HA, Bernier R, & Eichler EE (2014). A genotype-first approach to defining the subtypes of a complex disease. Cell, 156(5), 872–877. doi: 10.1016/j.cell.2014.02.002 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya KD, Shaffer LG, Aradhya S, Gastier-Foster JM, Patel A, Rudd MK, … Brothman AR (2009). Variability in interpreting and reporting copy number changes detected by array-based technology in clinical laboratories. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 11(12), 866–873. doi: 10.1097/GIM.0b013e3181c0c3b0 [doi] [DOI] [PubMed] [Google Scholar]
- Uddin M, Pellecchia G, Thiruvahindrapuram B, D’Abate L, Merico D, Chan A, … Scherer SW (2016). Indexing effects of copy number variation on genes involved in developmental delay. Scientific Reports, 6, 28663. doi: 10.1038/srep28663 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M, Tammimies K, Pellecchia G, Alipanahi B, Hu P, Wang Z, … Scherer SW (2014). Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nature Genetics, 46(7), 742–747. doi: 10.1038/ng.2980 [doi] [DOI] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, … Sebat J (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science (New York, N.Y.), 320(5875), 539–543. doi: 10.1126/science.1155174 [doi] [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, … Autism Consortium. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. The New England Journal of Medicine, 358(7), 667–675. doi: 10.1056/NEJMoa075974 [doi] [DOI] [PubMed] [Google Scholar]
- Yang S, Lincoln SE, Kobayashi Y, Nykamp K, Nussbaum RL, & Topper S (2017). Sources of discordance among germ-line variant classifications in ClinVar. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 19(10), 1118–1126. doi: 10.1038/gim.2017.60 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrei M, MacDonald JR, Merico D, & Scherer SW (2015). A copy number variation map of the human genome. Nature Reviews.Genetics, 16(3), 172–183. doi: 10.1038/nrg3871 [doi] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.