

Abstract
Vorinostat (suberoylanilide hydroxamic acid) was the first approved histone deacetylase (HDAC) inhibitor in a group of validated cancer therapeutic agents targeting epigenetics. Riluzole is a drug used to treat amyotrophic lateral sclerosis, the antitumor potency of which has been recently revealed. Herein, a novel hybrid of vorinostat and riluzole (compound 1) was rationally designed, synthesized, and evaluated. Compared with vorinostat, compound 1 exhibited superior total HDAC inhibitory activity and similar HDAC isoform selective profiles. The intracellular HDAC inhibition of compound 1 was confirmed by Western blot analysis. Moreover, compound 1 possessed more potent in vitro antiproliferative activity against all tested solid and hematological tumor cell lines than vorinostat. In vitro metabolic stability evaluation of compound 1 revealed better human plasma stability and comparable human liver microsomal stability than vorinostat. Additionally, compound 1 demonstrated more significant in vivo antitumor activity in a MDA-MB-231 xenograft model than vorinostat, which could be attributed to its superior in vitro antiproliferative activity and metabolic stability. Taken together, the results presented here support further research and development of compound 1 as a promising antitumor agent.
Keywords: HDAC, inhibitor, drug design, hybrid molecule, anticancer
Introduction
The acetylation status of lysine residues of nuclear histones, regulated by histone deacetylases (HDACs) and histone acetyl transferases (HATs), is one of the epigenetic mechanisms regulating gene expression (Allis and Jenuwein, 2016). Generally, HDAC overexpression causes a low histone acetylation level, which can downregulate the expression of many genes, including tumor suppressor genes, leading to cancer (Falkenberg and Johnstone, 2014). Therefore, targeting the HDAC family, especially the zinc-dependent HDACs, using small molecular inhibitors became a hot cancer therapeutic strategy, which has been well validated by the approval of five HDAC inhibitors for the treatment of hematological malignancies (Zagni et al., 2017). Vorinostat [suberoylanilide hydroxamic acid (SAHA); Figure 1] is the first approved HDAC inhibitor. Its structure summarizes well the common pharmacophore of most HDAC inhibitors, which contain a zinc binding group (ZBG) that chelates the catalytic zinc ion, a hydrophobic linker that occupies the tunnel of the active site, and a terminal cap that interacts with the amino acid residues around the entrance of the active site (Miller et al., 2003). Structural modification of the terminal cap of vorinostat is a feasible and efficient strategy to develop novel HDAC inhibitors. For example, the introduction of various biologically active fragments, including nitrogen mustard (Xie et al., 2017), proapoptotic stilbene (Giacomini et al., 2014), colchicine (Zhang et al., 2013), and platinum complex (Griffith et al., 2009), to the cap part of vorinostat successfully led to corresponding hybrid molecules with antitumor potency (Figure 1).
FIGURE 1.

Compound design strategy by introducing various biologically active fragments to the terminal cap of vorinostat. The three parts of the HDAC inhibitor pharmacophore are indicated in different colors.
Riluzole is an approved drug for the treatment of amyotrophic lateral sclerosis (ALS; Bellingham, 2011). Many preclinical studies have revealed the anticancer potential of riluzole against breast cancer (Speyer et al., 2012, 2014, 2016; Teh et al., 2015), melanoma (Namkoong et al., 2007; Le et al., 2010; Khan et al., 2011; Lee et al., 2011; Wall et al., 2014; Wen et al., 2014; Rosenberg et al., 2015), glioma (Zhang et al., 2015), and prostate cancer (Akamatsu et al., 2009). Importantly, one preliminary clinical trial of riluzole in patients with resectable stage III or IV melanoma showed promising results (Yip et al., 2009). In addition, one clinical trial evaluating riluzole combined with sorafenib in patients with melanoma or advanced solid tumors is currently active (National Cancer Institute [NCI], 2011). It is worth noting that several analogs of vorinostat with benzothiazole cap groups were previously reported to show potent HDAC inhibitory and antitumor activity, suggesting that the introduction of benzothiazole-based riluzole to the terminal cap group of vorinostat can be tolerated (Tung et al., 2013). In the present study, because of its promising antitumor potency and appropriate physicochemical properties, riluzole was introduced to the terminal cap group of vorinostat, in the hope of obtaining a novel riluzole–vorinostat hybrid with potent HDAC inhibitory and antitumor activity (compound 1; Figure 1).
Materials and Methods
Molecular Docking Study
Compound 1 was docked into the active site of HDAC2 (PDB code 4LXZ) using Tripos SYBYL-X 2.0. Before the docking process, the structure of the protein was treated by removing co-crystallized ligands, deleting water molecules, adding hydrogen atoms, and assigning AMBER7 FF99 charges. A 100-step energy minimization was performed to further optimize the protein structure. The molecular structure of compound 1 was generated with the Sybyl/Sketch module. It was optimized using Powell’s method with the TRIPOS force field with the convergence criterion set at 0.005 kcal/(Å mol) and assigned charges with the Gasteiger–Hückel method. Other parameters were set as default values. Molecular docking was carried out via the Sybyl/Surflex-Dock (SFXC) module.
Chemistry
Unless specified otherwise, all starting materials, reagents, and solvents were commercially available. All reactions were monitored by thin-layer chromatography on 0.25 mm silica gel plates (60GF-254) and visualized with ultraviolet light, ferric chloride, or iodine vapor. Nuclear magnetic resonance (NMR) spectra were determined on Varian INOVA spectrometers, with δ in parts per million and J in Hertz, using tetramethylsilane as an internal standard. Measurements were made in dimethyl sulfoxide (DMSO)-d6 solutions. Electrospray ionization–mass spectrometry (ESI-MS) was carried out on an API 4000 spectrometer. High-resolution mass spectroscopy (HRMS) was conducted by the Shandong Analysis and Test Center. Silica gel was used for column chromatography purification. Melting points were determined on an electrothermal melting point apparatus and were uncorrected.
Procedure for the Synthesis of Octandioic Anhydride (3)
A solution of octandioic acid 2 (5.00 g, 28.7 mmol) in acetic anhydride (10 mL) was refluxed for 4 h. Then the mixture was dissolved in acetonitrile (60 mL) and frozen overnight. The resulting precipitate was filtered. The filter residue was dried to give compound 3 (2.78 g, yield 62%) as a light-yellow solid, which was used in the following reaction without further purification. ESI-MS m/z: 167.1 [M + H]+.
Procedure for the Synthesis of 8-oxo-8-((6-(Trifluoromethoxy)benzo[d]thiazol-2-yl) amino)octanoic Acid (4)
To a solution of compound 3 (1.17 g, 7.5 mmol) in tetrahydrofuran (THF; 50 mL) was added riluzole (1.17 g, 5.0 mmol). After reflux for 48 h, the solvent was removed under reduced pressure, followed by addition of ethyl acetate (EtOAc; 50 mL). The EtOAc solution was extracted with 1 M aqueous NaOH (3 × 20 mL). Then the aqueous phase was acidified until no precipitate appeared. The precipitate was filtered and the residue was dried to give compound 4 (1.56 g, yield 80%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 12.45 (s, 1H), 12.00 (s, 1H), 8.11 (d, J = 1.8 Hz, 1H), 7.81 (d, J = 9.0 Hz, 1H), 7.42 (dd, J = 1.8 Hz, 9.0 Hz, 1H), 2.50–2.51 (m, 2H), 2.18–2.22 (m, 2H), 1.60–1.63 (m, 2H), 1.48–1.51 (m, 2H), 1.28–1.31 (m, 4H). HRMS [atmospheric pressure ESI (AP-ESI)] m/z: calculated for C16H18F3N2O4S [M + H]+ 391.0939; experimental 391.0924.
Procedure for the Synthesis of N1-Hydroxy-N8- (6-(Trifluoromethoxy)benzo[d]thiazol-2-yl) Octanediamide (1)
To a solution of compound 4 (0.78 g, 2.0 mmol) in THF (40 mL), triethylamine (Et3N) (0.22 g, 2.2 mmol) was added. Isobutyl chloroformate (0.30 g, 2.2 mmol) dissolved in THF (5 mL) was added to the reaction mixture in an ice bath, and the mixture was stirred for 1 h in an ice bath. A mixture of hydroxylamine hydrochloride (0.15 g, 2.2 mmol) and Et3N (0.22 g, 2.2 mmol) in methanol (5 mL) was stirred for 5 min then poured directly into the reaction mixture. The reaction continued for 4 h at room temperature, then the solvent was removed under reduced pressure followed by the addition of 30 mL of water. Then, 1 M HCl was used to adjust the pH to 6. The resulting precipitate was filtered and washed with water to obtain the crude product, which was purified by recrystallization to afford compound 1 (0.36 g, yield 45%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 12.44 (s, 1H), 10.33 (s, 1H), 8.66 (s, 1H), 8.11 (d, J = 1.2 Hz, 1H), 7.80 (d, J = 9.0 Hz, 1H), 7.41 (dd, J = 1.2 Hz, 9.0 Hz, 1H), 2.50–2.51 (m, 2H), 1.93–1.95 (m, 2H), 1.59–1.64 (m, 2H), 1.47–1.51 (m, 2H), 1.26–1.30 (m, 4H). HRMS (AP-ESI) m/z: calculated for C16H19F3N3O4S [M + H]+ 406.1048; experimental 406.1055.
Biology
In vitro HDAC Inhibition Fluorescent Assay
An aliquot of 10 μL of enzyme solution (HeLa cell nuclear extract, HDAC2, HDAC6, or HDAC8) was mixed with different concentrations of test compound (50 μL). The mixture was incubated at 37°C for 5 min, followed by the addition of 40 μL of fluorogenic substrate tert-butyl (S)-(6-acetamido- 1-((4-methyl-2-oxo-2H-chromen-7-yl)amino)-1-oxohexan-2-yl) carbamate (Boc-Lys(acetyl)-AMC) for HeLa cell nuclear extracts, HDAC2, and HDAC6; tert-butyl (S)-(1-((4-methyl- 2-oxo-2H-chromen-7-yl)amino)-1-oxo-6-(2,2,2-trifluoroacetam ido)hexan-2-yl)carbamate (Boc-Lys(trifluoroacetyl)-AMC) for HDAC8). After incubation at 37°C for 30 min, the mixture was quenched by the addition of 100 μL of developer containing trypsin and trichostatin A. Following incubation at 37°C for 20 min, the fluorescence intensity was measured using a microplate reader at excitation and emission wavelengths of 390 and 460 nm, respectively. The inhibition ratios were calculated from the fluorescence intensity readout of tested wells relative to those of control wells, and the half maximal inhibitory concentration (IC50) values were calculated using the prism non-linear curve-fitting method.
Western Blot Analysis
The MDA-MB-231 cells were treated with compounds or DMSO for a specified period of time. Then the cells were washed twice with cold phosphate-buffered saline (PBS) and lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer. Lysates were cleared by centrifugation. Protein concentrations were determined using the bicinchoninic acid assay. Equal amounts of cell extracts were then resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and probed with acetyl-histone H4 antibody (intracellular substrate of HDAC1 and HDAC2), acetyl-α-tubulin antibody (intracellular substrate of HDAC6), and β-actin antibody (used as a loading control), respectively. Blots were detected using an enhanced chemiluminescence system.
In vitro Antiproliferative Assay
All cell lines were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine serum at 37°C in a 5% CO2 humidified incubator. The cell proliferation assay was determined by the MTT (3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide) method. Briefly, cells were passaged the day before dosing into a 96-well plate, allowed to grow for 12 h, and then treated with different concentrations of compound for 72 h. A 0.5% MTT solution was added to each well. After incubation for another 4 h, formazan formed from MTT was extracted by adding 200 μL of DMSO. Absorbance was then determined using a microplate reader at 570 nm.
In vitro Metabolic Stability Assay in Human Plasma
Human plasma samples containing compound 1 were incubated at 37°C. At the specific time points, samples were added to acetonitrile to terminate the reaction, then subjected to vortex mixing for 5 min and stored in a freezer at –80°C. Before analysis, the samples were centrifuged. The remainder of 1 in the supernatants was analyzed by liquid chromatography with tandem mass spectrometry (LC-MS/MS). The t1/2(half–time) values were calculated using the equation t1/2(half–time) = −0.693/k, where k is the slope found in the linear fit of the natural logarithm of the fraction remaining of compound 1 versus incubation time.
In vitro Metabolic Stability Assay in Human Liver Microsomes
Human liver microsomes containing compound 1 were incubated with NADPH at 37°C. At the specific time points, samples were added to acetonitrile to terminate the reaction, then subjected to vortex mixing for 5 min and stored in a freezer at –80°C. Before analysis, the samples were centrifuged. The remainder of 1 in the supernatants was analyzed by LC-MS/MS. The t1/2(half–time) values were calculated using the equation t1/2 = −0.693/k, where k is the slope found in the linear fit of the natural logarithm of the fraction remaining of compound 1 versus incubation time.
In vivo Antitumor Experiment Against MDA-MB-231 Xenograft
In vivo human tumor xenograft models were established as previously described (Zang et al., 2018; Liang et al., 2019). For the in vivo antitumor efficacy study, 5 × 106 human breast cancer cells (MDA-MB-231) were inoculated subcutaneously in the right flank of female athymic nude mice (BALB/c-nu, 5–6 weeks old; Beijing HFK Bioscience Co., Ltd.). Ten days after injection, tumors were palpable and mice were randomized into treatment and control groups (six mice per group). The treatment groups received compound 1 or vorinostat by oral administration (30 mg/kg/day), and the blank control group received oral administration of an equal volume of PBS (5% DMSO). Subcutaneous tumors were measured with a vernier caliper every 3 days. Tumor volumes (V) were estimated using the equation (V = ab2/2, where a and b are the longest and shortest diameter, respectively). The body weight of the mice was also monitored regularly. At the end of the experimental period, mice were sacrificed and the tumor tissues were dissected and weighed. Tumor growth inhibition (TGI) and the relative increment ratio (T/C) were calculated at the end of treatment to reveal the antitumor effects in tumor weight and tumor volume, respectively.
TGI = (the mean tumor weight of control group – the mean tumor weight of treated group)/the mean tumor weight of control group.
T/C = the mean RTV of treated group (T)/the mean RTV of blank control group (C).
where the relative tumor volume (RTV) is Vt/V0 (Vt is the tumor volume measured at the end of treatment; V0 is the tumor volume measured at the beginning of the treatment).
All the obtained data were used to evaluate the antitumor potency and toxicity of compounds. Data were analyzed by Student’s one-tailed t-test. A p-value of <0.05 was considered statistically significant.
Results and Discussion
Proposed Binding Mode of Compound 1 in HDAC2
Before synthesis, a molecular docking study was performed to elucidate the potential binding mode of compound 1 in the active site of HDAC2. The results in Figure 2 indicate that compound 1 fitted well in the active site of HDAC2. In detail, the hydroxamic acid group of Compound 1 form four hydrogen bonds with His145, His146, and Tyr308, respectively. The aliphatic chain occupied the hydrophobic channel of the active site and the riluzole-based cap group occupied a shallow pocket around the entrance of the active site. The design strategy of compound 1 is rationalized via this binding mode.
FIGURE 2.

Proposed binding mode of compound 1 (green) with HDAC2 (PDB code 4LXZ). (A) Surface of HDAC2 with compound 1. (B) Detailed interactions between HDAC2 and compound 1. Yellow dashed lines represent the hydrogen bonds. Oxygen, nitrogen, fluorine, sulfur, and polar hydrogen atoms are shown in red, blue, pale cyan, bright orange, and white, respectively. The Zn2+ is shown as a magenta sphere. The figure was generated using PyMol (http://www.pymol.org/).
Synthesis
The hybrid compound 1 was synthesized according to the procedures described in Scheme 1. The starting material, octandioic acid 2, was refluxed in acetic anhydride to get the anhydride 3, which reacted with riluzole to get the carboxylic acid 4. Compound 4 was then condensed with hydroxylamine to get the target compound 1.
SCHEME 1.
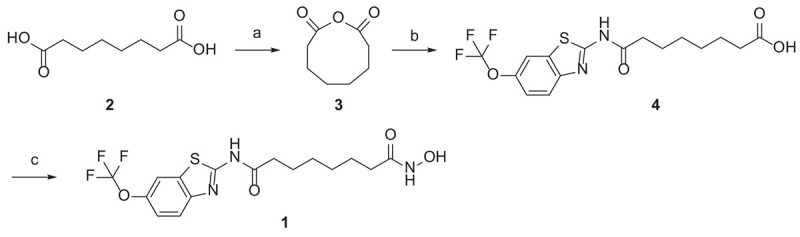
Reagents and conditions: (a) acetic anhydride, reflux; (b) riluzole, THF, reflux; (c) ClCOOi-Bu, TEA, NH2OH.HCl, THF, r.t.
HDAC Inhibition and Isoform Selectivity
The total HDAC inhibitory activity of compound 1 was evaluated against the HeLa cell nuclear extract. The results listed in Table 1 show that compound 1 (IC50 = 0.12 μM) was more potent than the approved drug vorinostat (IC50 = 0.25 μM). To profile the HDAC isoform selectivity, compound 1 was further tested against HDAC2, HDAC6, and HDAC8 with vorinostat as the reference compound. The overall selectivity profile of compound 1 was similar to that of vorinostat (Table 1). Note that the HDAC6 inhibitory activity of 1 (IC50 = 0.012 μM) was more than sevenfold higher than that of vorinostat (IC50 = 0.091 μM).
TABLE 1.
HDAC inhibition and isoform selectivity of compounds 1 and vorinostat.
| Compound |
IC50 (μ M)a |
|||
| HeLa nuclear extract | HDAC2 | HDAC6 | HDAC8 | |
| 1 | 0.12 ± 0.01 | 0.33 ± 0.04 | 0.012 ± 0.002 | 3.3 ± 0.2 |
| Vorinostat | 0.25 ± 0.02 | 0.23 ± 0.02 | 0.091 ± 0.004 | >5 |
aAll assays were replicated (n = 3). The IC50 values are shown as mean ± SD.
Western blot analysis was performed to verify the intracellular target engagement of compound 1. The results in Figure 3 show that both compound 1 and vorinostat could dramatically increase the levels of acetyl-histone H4 (intracellular substrate of HDAC1 and HDAC2) and acetyl-α-tubulin (intracellular substrate of HDAC6) in the MDA-MB-231 cell line. It is worth noting that, at the same concentration of 0.5 μM, the effect of 1 on acetyl-α-tubulin was superior to that of vorinostat, which is in line with the more potent HDAC6 inhibition of 1 compared with vorinostat, as shown in Table 1.
FIGURE 3.

MDA-MB-231 cells were treated with dimethyl sulfoxide (DMSO) or compounds for 3 h. The levels of acetyl-α-tubulin (Ac-Tub) and acetyl-histone H4 (Ac-HH4) were determined by immunoblotting. β-Actin was used as a loading control. The result is a representative of three independent experiments.
In vitro Antiproliferative Activity
Because of its HDAC inhibitory potency, compound 1 was progressed to an in vitro antiproliferative assay against human tumor cells, including breast cancer cell lines MDA-MB-231 and MCF-7, prostate adenocarcinoma cell line PC-3, neuroblastoma cell line SK-N-BE(2), acute myelogenous leukemia cell line KG-1, acute lymphoblastic leukemia cell line MOLT-4, and erythroleukemia cell line HEL. Remarkably, compound 1 with IC50 values ranging from 0.14 to 2.74 μM was more potent against all tested human cancer cell lines than the approved drug vorinostat (Table 2). Note that riluzole showed less than 50% growth inhibition at 10 μM against the tested tumor cell lines, which was consistent with previous studies (Namkoong et al., 2007; Zhang et al., 2015; Speyer et al., 2016).
TABLE 2.
In vitro antiproliferative activity of compound 1, vorinostat, and riluzole.
| Compound |
IC50 (μ M)a |
||||||
| MDA-MB-231 | MCF-7 | PC-3 | SK-N-BE(2) | KG-1 | MOLT-4 | HEL | |
| 1 | 0.77 ± 0.02 | 2.74 ± 0.06 | 2.52 ± 0.33 | 0.29 ± 0.007 | 0.65 ± 0.02 | 0.17 ± 0.007 | 0.14 ± 0.007 |
| Vorinostat | 1.58 ± 0.06 | 5.62 ± 0.06 | 9.21 ± 0.37 | 1.16 ± 0.27 | 1.59 ± 0.14 | 0.36 ± 0.049 | 0.21 ± 0.028 |
| Riluzole | >10 | NDb | >10 | NDb | >10 | NDb | >10 |
aAll assays were replicated (n = 3). The IC50 values are shown as mean ± SD. bNot determined.
In vitro Metabolic Stability
One drawback of hydroxamate-based compounds, including vorinostat, is their poor metabolic stability. Therefore, the in vitro metabolic stability of compound 1 in human plasma and liver microsomes was assessed and compared with the reported data of vorinostat. The results in Table 3 show that, although compound 1 possessed similar stability in human liver microsomes to vorinostat (t1/2 = 56 min vs t1/2 = 60 min), its stability in human plasma was much better than that of vorinostat (t1/2 > 120 min vs t1/2 = 75 min).
TABLE 3.
In vitro metabolic stability of compound 1 and vorinostat.
| Compound |
t1/2 (min) |
|
| Human plasma | Human liver microsomes | |
| 1 | >120a | 56a |
| Vorinostat | 75b | 60c |
aAll assays were replicated (n ≥ 2). bData from Konsoula and Jung (2008). cData from Venkatesh et al. (2007).
In vivo Antitumor Activity Assay
Based on the promising in vitro antiproliferative activity and metabolic stability of compound 1, an MDA-MB-231 xenograft model was used to further evaluate the in vivo antitumor potency of compound 1. After 21 consecutive days of treatment (30 mg/kg/day), TGI and T/C were calculated. As shown in Table 4, compound 1 demonstrated significantly better in vivo efficacy than vorinostat. The tumor growth curve and the final tumor tissue size are shown in Figures 4, 5, respectively, which explicitly demonstrate the potent antitumor activity of compound 1 in vivo. Moreover, the mouse body weights in Figure 6 indicate the high tolerability and low toxicity of compound 1.
TABLE 4.
In vivo antitumor activity in the MDA-MB-231 xenograft modela.
| Compound | TGI (%) | T/C (%) |
| 1 | 59 | 55 |
| Vorinostat | 33 | 78 |
aCompared with the control group, compound 1 showed statistically significant (p < 0.05) tumor growth inhibition (TGI) and relative increment ratio (T/C) by Student’s one-tailed t-test, and vorinostat showed no statistically significant (p > 0.05) TGI and T/C by Student’s one-tailed t-test. The TGI values are based on tumor weight.
FIGURE 4.

Growth curve of implanted MDA-MB-231 xenografts in nude mice. Data are expressed as the mean ± standard deviation.
FIGURE 5.

Dissected MDA-MB-231 tumor tissues.
FIGURE 6.

Animal body weight. Data are expressed as the mean ± standard deviation.
Conclusion
In the present work, based on the molecular hybridization strategy, a novel riluzole–vorinostat hybrid 1 was rationally designed, synthesized, and evaluated. Compared with vorinostat, compound 1 exhibited superior total HDAC inhibitory activity and similar HDAC isoform selective profiles, which was confirmed by Western blot analysis. Remarkably, compound 1 exhibited superior in vitro antitumor activity and metabolic stability to vorinostat, which contributed to its promising in vivo antitumor activity in the MDA-MB-231 xenograft model. In summary, our results support further mechanism studies and preclinical evaluation of compound 1 as a novel antitumor agent.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Ethics Statement
The animal study was reviewed and approved by Shandong University Laboratory Animal Center Ethics Committee.
Author Contributions
QX and CL contributed equally to research investigation, data analysis, manuscript writing, and organizing. JZ, CC, and SG performed the data validation and manuscript review. YZ contributed to conceptualization, supervision, and funding acquisition. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the Natural Science Foundation of Shandong Province (Grant No. ZR2018QH007), the Key Research and Development Program of Shandong Province (2017CXGC1401), and the Young Scholars Program of Shandong University (YSPSDU, 2016WLJH33).
References
- Akamatsu K., Shibata M.-A., Ito Y., Sohma Y., Azuma H., Otsuki Y. (2009). Riluzole induces apoptotic cell death in human prostate cancer cells via endoplasmic reticulum stress. Anticancer. Res. 29 2195–2204. [PubMed] [Google Scholar]
- Allis C., Jenuwein T. (2016). The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17 487–500. 10.1038/nrg.2016.59 [DOI] [PubMed] [Google Scholar]
- Bellingham M. C. (2011). A Review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci. Ther. 17 4–31. 10.1111/j.1755-5949.2009.00116.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg K., Johnstone R. (2014). Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 13 673–691. 10.1038/nrd4360 [DOI] [PubMed] [Google Scholar]
- Giacomini E., Nebbioso A., Ciotta A., Ianni C., Falchi F., Roberti M., et al. (2014). Novel antiproliferative chimeric compounds with marked histone deacetylase inhibitory activity. ACS Med. Chem. Lett. 5 973–978. 10.1021/ml5000959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith D., Morganb M. P., Marmion C. J. (2009). A novel anti-cancer bifunctional platinum drug candidate with dual DNA binding and histone deacetylase inhibitory activity. Chem. Commun. 47 6735–6737. 10.1039/B916715C [DOI] [PubMed] [Google Scholar]
- Khan A. J., Wall B., Ahlawat S., Green C., Schiff D., Mehnert J. M., et al. (2011). Riluzole enhances ionizing radiation–induced Cytotoxicity in human melanoma cells that ectopically express metabotropic glutamate Receptor 1 In Vitro and In Vivo. Clin. Cancer Res. 17 1807–1814. 10.1158/1078-0432.CCR-10-1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konsoula R., Jung M. (2008). In vitro plasma stability, permeability and solubility of mercaptoacetamide histone deacetylase inhibitors. Int. J. Pharm. 361 19–25. 10.1016/j.ijpharm.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le M. N., Chan J. L.-K., Rosenberg S. A., Nabatian A. S., Merrigan K. T., Cohen-Solal K. A., et al. (2010). The glutamate release inhibitor riluzole decreases migration, invasion, and proliferation of melanoma cells. J. Invest. Dermatol. 130 2240–2249. 10.1038/jid.2010.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. J., Wall B. A., Wangari-Talbot J., Shin S. S., Rosenberg S., Chan J. L., et al. (2011). Glutamatergic pathway targeting in melanoma: single-agent and combinatorial therapies. Clin. Cancer Res. 17 7080–7092. 10.1158/1078-0432.CCR-11-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X., Zang J., Li X., Tang S., Huang M., Geng M., et al. (2019). Discovery of Novel Janus Kinase (JAK) and Histone Deacetylase (HDAC) dual inhibitors for the treatment of hematological malignancies. J. Med. Chem. 62 3898–3923. 10.1021/acs.jmedchem.8b01597 [DOI] [PubMed] [Google Scholar]
- Miller T. A., Witter D. J., Belvedere S. (2003). Histone deacetylase inhibitors. J. Med. Chem 46 5097–5116. 10.1021/jm0303094 [DOI] [PubMed] [Google Scholar]
- Namkoong J., Shin S.-S., Lee H. J., Marin Y. E., Wall B. A., Goydos J. S., et al. (2007). Metabotropic Glutamate Receptor 1 and glutamate signaling in human melanoma. Cancer Res. 67 2298–2305. 10.1158/0008-5472.CAN-06-3665 [DOI] [PubMed] [Google Scholar]
- National Cancer Institute [NCI] (2011). Riluzole and Sorafenib Tosylate in Treating Patients With Advanced Solid Tumors or Melanoma. ClinicalTrials.gov Identifier: NCT01303341. Bethesda, MD: NCI. [Google Scholar]
- Rosenberg S. A., Niglio S. A., Salehomoum N., Chan J. L., Jeong B. S, Wen Y., et al. (2015). Targeting Glutamatergic Signaling and the PI3 kinase pathway to halt melanoma progression. Trans. Oncol 8 1–9. 10.1016/j.tranon.2014.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speyer C. L., Hachem A. H., Assi A. A., Johnson J. S., DeVries J. A., Gorski D. H. (2014). Metabotropic Glutamate Receptor-1 as a novel target for the antiangiogenic treatment of breast cancer. PLoS One 9:e88830. 10.1371/journal.pone.0088830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speyer C. L., Nassar M. A., Hachem A. H., Bukhsh M. A., Jafry W. S., Khansa R. M., et al. (2016). Riluzole mediates anti-tumor properties in breast cancer cells independent of metabotropic glutamate receptor-1. Breast Cancer Res. Treat. 157 217–228. 10.1007/s10549-016-3816-x [DOI] [PubMed] [Google Scholar]
- Speyer C. L., Smith J. S., Banda M., DeVries J. A., Mekani T., Gorski D. H. (2012). Metabotropic glutamate receptor-1: a potential therapeutic target for the treatment of breast cancer. Breast Cancer Res. Treat. 132 565–573. 10.1007/s10549-011-1624-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh J. L. F., Shah R., La Cava S., Dolfi S. C., Mehta M. S., Kongara S., et al. (2015). Metabotropic glutamate receptor 1 disrupts mammary acinar architecture and initiates malignant transformation of mammary epithelial cells. Breast Cancer Res. Treat. 151 57–73. 10.1007/s10549-015-3365-3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung T. T., Oanh D. T., Dung P. T., Hue V. T., Park S. H., Han B. W., et al. (2013). New benzothiazole/thiazole-containing hydroxamic acids as potent histone deacetylase inhibitors and antitumor agents. Med. Chem. 9 1051–1057. 10.2174/15734064113099990027 [DOI] [PubMed] [Google Scholar]
- Venkatesh P. R., Evelyn G. E., Zeng P. (2007). In Vitro Phase I Cytochrome P450 Metabolism, Permeability and Pharmacokinetics of SB639, a Novel Histone Deacetylase inhibitor in preclinical species. Biol. Pharm. Bull. 30 1021–1024. 10.1248/bpb.30.1021 [DOI] [PubMed] [Google Scholar]
- Wall B. A., Yu J. L., Khan A., Haffty B., Goydos J. S., Chen S. (2014). Riluzole is a radio-sensitizing agent in an in vivo model of brain metastasis derived from GRM1 expressing human melanoma cells. Pigment Cell Melanoma Res. 28 105–109. 10.1111/pcmr.12327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y., Li J., Koo J., Shin S.-S., Lin Y., Jeong B. S., et al. (2014). Activation of the Glutamate Receptor GRM1 Enhances Angiogenic Signaling to Drive Melanoma Progression. Cancer Res. 74 2499–2509. 10.1158/0008-5472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R., Li Y., Tang P., Yuan Q. (2017). Rational design, synthesis and preliminary antitumor activity evaluation of a chlorambucil derivative with potent DNA/HDAC dual-targeting inhibitory activity. Bioorg. Med. Chem. Lett. 27 4415–4420. 10.1016/j.bmcl.2017.08.011 [DOI] [PubMed] [Google Scholar]
- Yip D., Le M. N., Chan J. L., Lee J. H., Mehnert J. A., Yudd A., et al. (2009). A Phase 0 Trial of Riluzole in Patients with Resectable Stage III and IV Melanoma. Clin. Cancer Res. 15 3896–3902. 10.1158/1078-0432.CCR-08-3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagni C., Floresta G., Monciino G., Rescifina A. (2017). The search for potent, small-molecule HDACIs in cancer treatment: a decade after Vorinostat. Med. Res. Rev. 37 1373–1428. 10.1002/med.21437 [DOI] [PubMed] [Google Scholar]
- Zang J., Liang X., Huang Y., Jia Y., Li X., Xu W., et al. (2018). Discovery of Novel Pazopanib-Based HDAC and VEGFR dual inhibitors targeting cancer epigenetics and angiogenesis simultaneously. J. Med. Chem. 61 5304–5322. 10.1021/acs.jmedchem.8b00384 [DOI] [PubMed] [Google Scholar]
- Zhang C., Yuan X., Li H., Zhao Z., Liao Y., Wang X., et al. (2015). Anti-Cancer Effect of Metabotropic Glutamate Receptor 1 Inhibition in Human Glioma U87 Cells: involvement of PI3K/Akt/mTOR pathway. Cell Physiol. Biochem. 35 419–432. 10.1159/000369707 [DOI] [PubMed] [Google Scholar]
- Zhang X., Zhang J., Tong L., Luo Y., Su M., Zang Y., et al. (2013). The discovery of colchicine-SAHA hybrids as a new class of antitumor agents. Bioorg. Med. Chem. 21 3240–3244. 10.1016/j.bmc.2013.03.049 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.