

Abstract
Stroke affects millions each year. Post-stroke brain edema predicts the severity of eventual stroke damage, yet our concept of how edema develops is incomplete and treatment options remain limited. In early stages, fluid accumulation occurs owing to a net gain of ions, widely thought to enter from the vascular compartment. Here we used magnetic resonance imaging, radiolabeled tracers, and multiphoton imaging in rodents, to show instead that cerebrospinal fluid surrounding the brain enters the tissue within minutes of an ischemic insult along perivascular flow channels. This process was initiated by ischemic spreading depolarizations along with subsequent vasoconstriction, which in turn enlarged the perivascular spaces and doubled glymphatic inflow speeds. Thus, our understanding of post-stroke edema needs to be revised and these findings could provide a conceptual basis for development of alternative treatment strategies.
One Sentence Summary:
Cerebrospinal fluid entry into the brain along the glymphatic pathway is the principal mechanism for early edema formation and ion perturbation during ischemic stroke.
Main text:
Stroke is among the leading causes of death worldwide, affecting 10 million patients annually(1, 2). Recent strides in treatment have improved these statistics, but stroke remains a principal cause of long-term disability(1, 3). A detrimental complication is cerebral edema, the abnormal accumulation of fluid that leads to secondary ischemia, additional tissue loss, and potential death (4, 5). Targeting edema represents a promising therapeutic strategy, because the severity of swelling predicts long-term functional outcomes(6–8). Although edema also develops in a range of other central nervous system (CNS) diseases (e.g., trauma, tumors, and infections), treatment options remain limited, and those available are suboptimal. Traditionally, cerebral edema is divided into two distinct phases: an early cytotoxic phase and a later vasogenic phase(9). Cytotoxic edema occurs within minutes of an ischemic insult and is triggered by spreading depolarizations that result from dysregulated ion homeostasis causing cell swelling (6, 7, 10–14). During the subsequent phase, vasogenic edema, fluid from the blood enters the brain as a result of blood-brain barrier (BBB) breakdown(15). Ions and fluid can cross the BBB via transcellular and paracellular routes(12): the transcellular pathway allows for the early entry of plasma proteins and other osmotically active solutes facilitating fluid entry, while the paracellular pathway takes two days to become active (16, 17). However, edema actually develops several hours before significant BBB dysfunction(18). This terminology used for staging edema was defined in the 1960s, and the conceptual framework for understanding and treating edema is still based on these early observations(8, 9, 19). The term ionic edema was introduced to explain this intermediary phase, during which tissue Na+ content increases(5, 18). This generates an osmotic gradient with plasma allowing water to move into the brain across an intact BBB(20, 21). However, the source of the excess Na+ has been attributed exclusively to influx from the intravascular compartment(5). Yet, the brain is bathed in cerebrospinal fluid (CSF), which has a high Na+ concentration and accounts for almost 10% of intracranial fluid(22). CSF has not been recognized as a source of edema fluid(8), but the notion that perivascular spaces (PVS) provide a conduit for CSF influx prompted us to reassess this idea. Glymphatic exchange of CSF with the interstitial fluid provides a means by which CSF in large quantities may rapidly enter the brain and drive tissue swelling(23, 24). Here, we find that CSF can indeed provide a key source for the initial rise in brain water content in the ischemic brain.
CSF flows into the brain after stroke, driving acute tissue swelling
To determine whether CSF contributes to edema in the early phases of acute ischemic stroke, the middle cerebral artery (MCA) was occluded by injecting macrospheres into the right common carotid artery in wildtype mice (Fig. 1A-B)(25). Embolic MCA occlusion (MCAO) resulted in an immediate, steep reduction in relative cerebral blood flow (rCBF: 92.5±1.2%; Fig. 1C). Mice exhibited severe neurological deficits the following day, and the infarct occupied most of the vascular territory of the MCA (Fig. S1). We used this model to map influx of CSF tagged with a fluorescent tracer immediately after MCAO. The CSF tracer was injected 15min prior to occlusion to ensure that it had distributed along the Circle of Willis, but not yet transported up along the MCA (Fig. 1D). Imaging revealed no difference in CSF tracer entry between ipsilateral and contralateral hemispheres prior to stroke, but MCA occlusion resulted in a sharp 3-fold increase in ipsilateral CSF tracer influx compared with the contralateral hemisphere (Fig. 1E-F). The derivative of the fluorescence curves identified that CSF influx occurred at two separate time points: an early peak (11.4±1.8s) and a second peak (5.24±0.48min) after MCAO (Fig. 1F-G). We next asked whether this CSF influx contributed to edema. The tissue water content of the ischemic cortex increased rapidly 15min after occlusion, while the water content in the non-ischemic hemisphere remained constant (Fig. 1H). To assess the contribution of blood versus CSF to the rapid increase in water content, the two fluid compartments were tagged by either intravenous (i.v.) or intracisternal injection of 22Na and 3H-mannitol. Early ischemic Na+ accumulation is an indirect measure of edema(5, 18, 26); mannitol, on the other hand, is a small BBB-impermeable tracer whose accumulation reflects opening of the barrier (Fig. 1I)(27). 22Na and 3H-mannitol accumulation in the ischemic and non-ischemic hemisphere were directly comparable after i.v. injection, suggesting that the vascular compartment was not the source of edema fluid in early times (Fig. 1J-K). In contrast, both 22Na and 3H-mannitol accumulated to a greater degree in the ischemic hemisphere, when the tracers were delivered to the CSF (Fig. 1L-M). To determine which pool of CSF was the primary source of edema fluid, we performed CSF cisternography using 3D-fast imaging employing steady-state acquisition (3D-FIESTA) magnetic resonance imaging (MRI). The CSF compartment including the lateral, third, and fourth ventricles and the cisterna magna accounted for a total of 13±0.7mm3 (Fig. 2A). Smaller collections of fluid (7.6±1.3mm3) were observed along the PVS of the large arteries of the Circle of Willis, and the rest of the intracranial volume was occupied by the brain tissue including the intravascular compartment (509±11mm3). After MCAO, CSF in the lateral ventricle (Fig. 2B, Movie 1) of the ischemic hemisphere and in the cisterna magna (Fig. 2C) slowly disappeared. Total CSF volume in the ventricular system and cisterna decreased by 9.0±2.5% at 15min and 17±3.0% at 29min post-stroke (Fig. 2D-E). PVS fluid volume began decreasing at around 15 min (Fig. 2F), several minutes after the CSF compartment started shrinking (~5min). Combined, these results add additional support to the notion that CSF is the primary source of early edema fluid after ischemic stroke.
Fig. 1. CSF rapidly enters the brain after stroke resulting in edema.
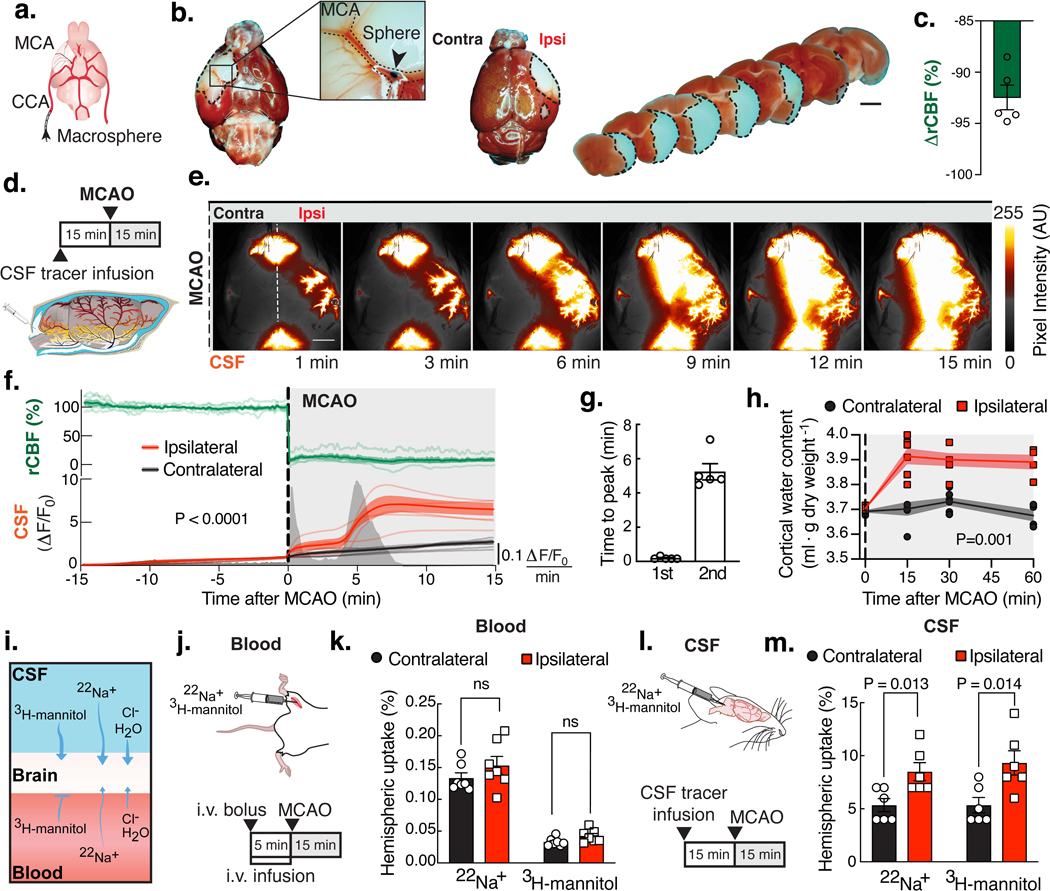
(A) Macrospheres (arrow) were infused into the common carotid artery (CCA), occluding the middle cerebral artery (MCA). (B) TTC staining after MCA occlusion (MCAO). (C) Ipsilateral relative cerebral blood flow (rCBF) after MCAO. (D) Intracisternal BSA-647 was infused 15min prior to MCAO. (E) Ipsilateral (Ipsi) and contralateral (Contra) tracer influx was imaged while measuring rCBF (white line: sagittal suture and fiber optic probe), arbitrary units (AU). (F) Time series of rCBF and fluorescence intensity (ΔF/F0) in each hemisphere. Peaks are mean rate of change in fluorescence intensity over time. Repeated measures two-way ANOVA, interaction P value; n=5 mice. (G) Timing of the 1st and 2nd peak. (H) Water content of the cortex. Mixed-effects repeated measures two-way ANOVA with Sidak’s multiple comparisons test; n=4–7 mice/time point. (I) The source of the edema fluid was identified by labeling either the blood or CSF compartment. (J) 22Na+ and 3H-mannitol were delivered intravenously (i.v.). (K) Percent of the total injected radiation found in each hemisphere after i.v. delivery. T-tests with Holm-Sidak correction; n=7 mice. (L) Isotopes were delivered into CSF. (M) Percent of the total injected radiation in each hemisphere after intracisternal delivery. T-tests with Holm-Sidak correction; n=6 mice. Scale bars: 2mm.
Fig. 2. After MCAO, CSF shifts into other intracranial compartments.
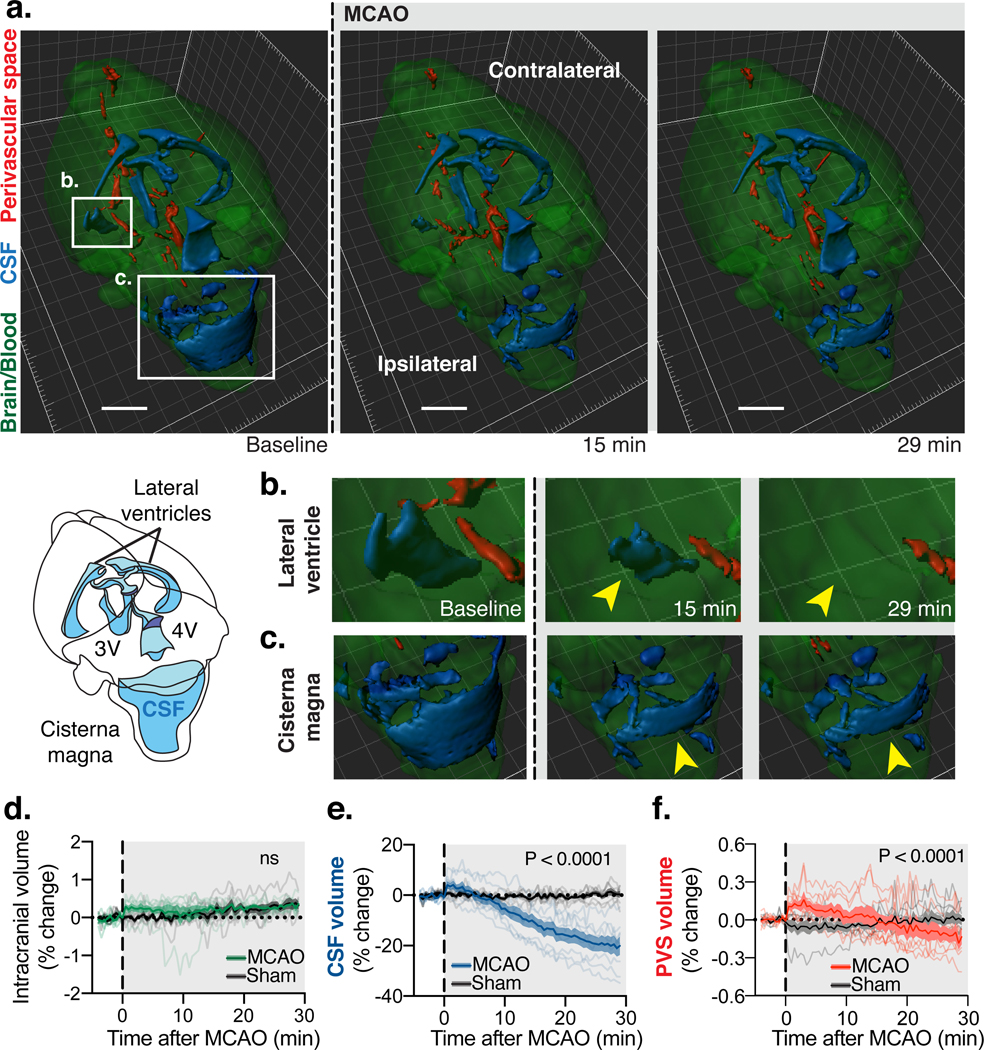
(A) 3D-fast imaging employing steady-state acquisition (FIESTA) shows three main compartments: 1) Intraventricular CSF in the lateral, 3rd (3V) and 4th (4V) ventricles and cisternal CSF in the cisterna magna (blue); 2) free fluid in the perivascular spaces (PVS; red) of the Circle of Willis along the anterior, middle, posterior cerebral, and the basilar arteries; 3) Intracranial content primarily composed of brain tissue and cerebral blood volume (green). 3D-FIESTA at baseline before MCAO and 15 and 29 min later. Scale bar: 2 mm. Insets of (A) of the (B) ventral anterior horn of the ipsilateral lateral ventricle and the (C) cisterna magna demonstrate the loss of free water at 15 min and 29 min after MCAO most notably in the lateral ventricles and the cisternal magna (yellow arrows). (D) Percent change of intracranial volume after MCAO and in sham animals. Repeated measures two-way ANOVA; interaction P value. (E) Percent change of CSF volume after MCAO. Repeated measures two-way ANOVA; interaction P value. (F) Percent change of PVS volume after MCAO. Repeated measures two-way ANOVA; interaction P value; Time-lapse measurements from n= 7 mice in Sham and 8 mice in MCAO group.
Spreading depolarizations trigger CSF flow into the ischemic brain
MRI suggested that the volume of CSF in ventricles rapidly declined upon artery occlusion, so we next employed dynamic contrast-enhanced MRI to determine the most likely location of CSF brain entry (Fig. S2A, Movie 2). MRI analysis confirmed an abrupt increase in ipsilateral perivascular influx of a CSF-delivered contrast agent (gadobutrol) within the first 5min after occlusion (Fig. S2B). Significant contrast enhancement was found at all measured sites along the perivascular trajectory of the ipsilateral MCA: ventral segment of the MCA (ROI1: 2.9-fold compared to the contralateral hemisphere), convexity MCA (ROI2: 4.3-fold), and far from the main MCA trunk in the parenchyma (ROI3: 5.5-fold; Fig. S2C). Tracer signal was seen deep below the cortical surface of the ipsilateral hemisphere, reaching 450±43μm at 14 min and 580±58μm at 28 min after MCAO, indicating that CSF enters the brain tissue (Fig. S3A-E). The entry route was along the glymphatic pathway, with tracer influx occurring primarily along penetrating arterioles (97.7% vs. 2.3% venules; P<0.0001; Fig. S4A-D) (28). Tracers moved along PVSs of the branching vascular network including capillaries and were found deep within parenchyma 30 min after MCAO (Fig. S4B). Thus, several independent sets of analyses point to acceleration of glymphatic influx into the ipsilateral brain in the setting of focal ischemia(6, 8). To identify the driver of CSF influx, we speculated that a loss in blood flow after stroke could cause a hydrostatic pressure gradient that would facilitate CSF flow into the brain. To test this, we measured intracranial pressure (ICP) changes during MCAO (Fig. S5A). ICP dropped from 5.49±0.85mmHg to 4.63±0.88mmHg after MCAO (Fig. S5A-B), but this response was variable between mice, and in some cases, returned to baseline within a few minutes. Thus, the ICP decrease coincided with the first influx peak but could not explain the second, larger peak in CSF influx (Fig. 1F and S5C). We next asked whether this large influx of CSF contributed to the cytotoxic phase of edema. To address this question, cytotoxic edema was detected as a decrease in the apparent diffusion coefficient (ADC) maps using diffusion-weighted MRI(12). A large ADC lesion first appeared in the primary somatosensory cortex after MCA occlusion and then slowly expanded in the form of a spreading wave across the ipsilateral hemisphere (Fig. S2D-E, Movie 3). ADC dropped by 17.5±1.3% within the lesion and spread over 22±2.3% of the brain volume within the first 4.8±0.9min after stroke (Fig. S2F-G). We generated a registered average of the contrast-enhanced and ADC datasets to compare the evolution of cytotoxic edema with CSF influx. (Movie 4). The aligned contrast-enhanced and ADC data and the onset times of both indicated that the ADC changes happened simultaneously with the second peak of CSF entry (P=0.70; Fig. 1F-G). Does the ADC drop trigger the large CSF influx? The wave-like kinetics of the ADC decrease is caused by a spreading depolarization (SD)(29). Ischemic SD consists of waves of sustained, mass depolarizations of cells in the gray matter of the CNS that result from the near complete loss of cellular transmembrane ion gradients(29). Ischemic SD begins in the barrel cortex of the primary somatosensory cortex in rodents (Movie 3)(30) and is thought to trigger cytotoxic edema after ischemia(12). Genetically encoded calcium indicators can be used to visualize the mass depolarization caused by the SD wave(31). To directly test the hypothesis that SD drives glymphatic influx after stroke, we used mice that express GCaMP7 under the Glt1 promoter (GCaMP)(32). GCaMP mice received an intracisternal tracer injection 15min prior to MCAO and were imaged using dual-channel macroscopic imaging (Fig. 3A, Movie 5). Several minutes after MCAO, a spreading wave of GCaMP fluorescence was seen, starting at the primary sensory cortex and travelling slowly over the cortical surface with CSF tracer following closely behind. The area covered by the SD and the expansion kinetics were consistent between mice, but the onset time varied (Fig. 3B). Aligning the data to the peak of GCaMP coverage revealed that the SD preceded a 5.6-fold increase in the area covered by CSF tracer (Fig. 3C). The SD peak coincided with maximal CSF influx, and SD onset time was a near-perfect predictor of the time of CSF influx (R2=0.94; Fig. 3D-E). To gain insight into the spatiotemporal dynamics of the SD wave and CSF influx, we employed a front-tracking algorithm(33). The SD wave propagated at a maximum speed of 36±4.8μm/s, consistent with an isotropic reaction/diffusion mechanism (Fig. 3F-I)(12). The CSF tracer front sped up after the onset of the SD wave but reached a much slower maximum speed of 16±1.0μm/s, indicating that the SD might not directly drive the CSF front. To quantify this delay, we computed the time it took CSF tracer to reach each given pixel after the SD wave had passed and found that 30±6s separated the SD and the tracer fronts (Fig. 3J-K). Thus, the ischemic SD triggers the onset of glymphatic influx after stroke.
Fig. 3. CSF influx is triggered by spreading depolarizations (SD) after focal ischemia.
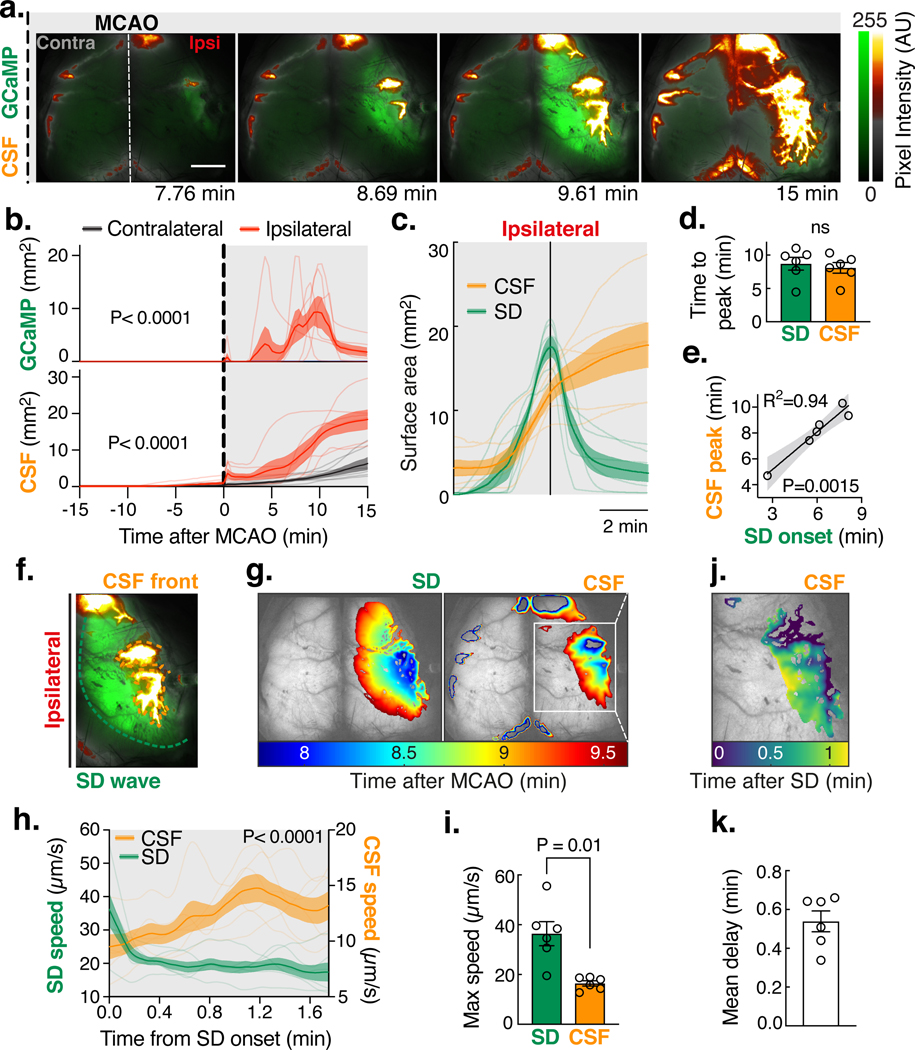
(A) A fluorescent CSF tracer (BSA-594) was delivered into the cisterna magna of Glt1-GCaMP7 mice 15 min before MCAO and imaged using a dual channel macroscope for 15 min as done in Fig. 1d. Pixel intensity in arbitrary units (AU). Scale bar: 2 mm. (B) Area covered by GCaMP and CSF tracer fluorescence. Repeated measures two-way ANOVA with Sidak’s multiple comparisons test; interaction P value; n=6 mice. (C) CSF tracer surface area aligned to the SD (GCaMP) peak. (D) Time to peak influx rate for the SD (GCaMP) and the CSF tracer. Paired t-test. (E) Linear regression of CSF peak influx and SD onset time with 95% confidence intervals. (F) The fronts of the ipsilateral SD wave and the CSF tracer were tracked. (G) Area covered by the SD (left panel) and the CSF tracer (right panel) over time. (H) Front speed of the SD wave and the CSF front. Repeated measures two-way ANOVA with Sidak’s multiple comparisons test; interaction P value. (I) Maximum speed of the SD and CSF front. Paired t-test. (J) Delay time between the SD wave and the CSF tracer front. (K) Mean delay between the SD and CSF front.
Spreading ischemia drives a rapid increase in perivascular CSF flow
A phenomenon termed spreading ischemia has been shown to take place 10–30s after SD, which would temporally coincide with glymphatic influx(34–36). Pronounced constriction of parenchymal and pial arterioles occurs following SD, owing to release of K+ and other vasoactive substances, and the depletion of nitric oxide in the endothelium(35, 37). CSF flow within PVSs is highly dependent on arterial diameter changes (38, 39). Could vasoconstriction drive the increase in CSF entry? To address the role of spreading ischemia on CSF flow, we used two-photon (2P) microscopy (Fig. 4A, Movie 6)(23, 24). Immediately after occlusion, there was a transient loss of intravascular flow as rCBF dropped (Fig. 4B-C). Despite intravascular dye returning a few seconds later owing to collateral flow, the hemisphere remained severely hypoperfused (Fig. 1F). Several minutes after MCAO, a SD wave propagated across the imaging field, followed by a potent vasoconstriction of the pial and penetrating arterioles, culminating in an intense increase in CSF tracer signal (Fig. 4D-F, Movie 7). The propagation speed of the SD wave was comparable to that seen in macroscopic experiments (42±1.7μm/s), and the delay between SD and the maximal vasoconstriction (30±2.4s) was temporally consistent with the delay between the SD wave and the CSF tracer front from the previous analysis (P = 0.395; Fig. 4G-I). The vasoconstrictive response was variable between mice and arteriolar location, with penetrating arterioles constricting more (79±10%) and earlier than pial arterioles (45±10%; Fig. S6A-D). The variability of this vasomotor response has been attributed to differences in the intravascular perfusion pressure and the location of vessels relative to the infarct core(35, 37). Constriction of both the penetrating and pial arterioles was followed by a parallel increase in the space occupied by the PVS tracer (Fig. 4F and S6B, E-F)(39). PVSs are believed to form an interconnected hydraulic network, where flow is transported via perivascular pumping generated by the cardiac cycle(38). However, we hypothesized that an increase in the size of the distal PVSs owing to vasoconstriction would create a pressure gradient that could effectively speed up perivascular flow. To test the hypothesis that diameter changes in the perivascular network could drive flow, we simulated the event using a topological network from mice pial MCAs (Fig. 5A-B, Movie 8)(40). The simulation showed that as a result of the SD, the constricting arteries increased the PVS size, drawing fluid into the tissue owing to reduced pressure in the expanding PVS. This inrush of fluid resulted in a 2.2-fold increase in baseline flow speed at the MCA, which eventually decreased (Fig 5C). To determine whether we could detect a similar increase in CSF flow speed at the inlet of the network, we performed particle tracking velocimetry(38). CSF flow speed was analyzed for changes in pulsatile velocity (vpulsatile) or net flow velocity (vdownstream) to determine if flow speed variations were caused by changes in pulsatility or by an overall pressure gradient (Fig. S7A-B, Movie 9). After MCAO, there was a large, instantaneous increase in vdownstream that quickly returned to baseline. But vpulsatile never recovered to baseline owing to the occlusion. This first component was consistent with the first peak of CSF influx. At the onset of SD (3.79±0.44min after MCAO), the large pial MCA also constricted, albeit to a lesser degree (27±6.1%) and was accompanied by a ~2.5-fold increase in vdownstream, while vpulsatile remained below baseline (Fig. S7C-D). The degree of vasoconstriction was a significant predictor of vdownstream, with every 10% decrease in diameter causing a 2-fold increase in flow, but no such correlation was seen with vpulsatile (Fig. S7E). Thus, post-ischemic CSF influx is not driven by changes in pulsatility, but instead by an overall pressure gradient caused by vasoconstriction. The simulation also supported the notion that vasoconstriction is a sufficient mechanism to explain water accumulation in the tissue. However, this does not exclude the possibility that ionic or osmotic gradients secondary to the SD might also play a role in driving flow. In conclusion, both simulations and experimental evidence indicate that spreading ischemia after SD abruptly accelerates perivascular CSF influx into the brain after stroke.
Fig. 4. Spreading ischemia after spreading depolarization drives perivascular CSF influx.
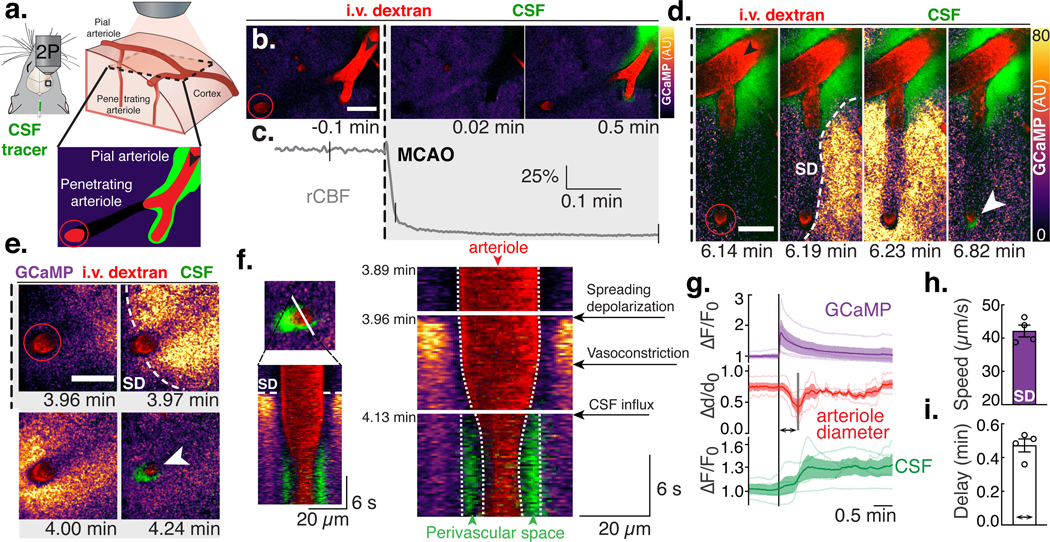
(A) Pial (black arrows) and penetrating arterioles (red circles, 40–50 μm below surface), branches of the ipsilateral MCA were imaged using two-photon (2P) microscopy. (B) Pial and penetrating arteriole (i.v. dextran) in a Glt1-GCaMP7 mouse after receiving an intracisternal tracer injection (BSA-647) during MCAO. Scale bars (B-E): 50 μm. (C) Relative cerebral blood flow (rCBF) measurements; ticks align with images in (B). (D) Spreading depolarization (SD) after MCAO. Normalized GCaMP fluorescence (ΔF-Faverage) is color-coded for pixel intensity and displayed in arbitrary units (AU). (E) Constriction of penetrating arteries after SD causes tracer influx into brain (white arrow). (F) Line scan over the penetrating arteriole in (E) depicting the appearance of the SD, the subsequent vasoconstriction and the CSF tracer influx filling the perivascular space left by the constricted arteriole. (G) Quantification of GCaMP and CSF tracer fluorescence (ΔF/F0) and arteriole diameter (Δd/d0) aligned to the onset of the SD; n=4 mice. (H) SD wave speed. (I) Delay time between SD onset and minimum arteriolar diameter.
Fig. 5. Topological glymphatic network model of perivascular spaces around the mouse middle cerebral artery (MCA).
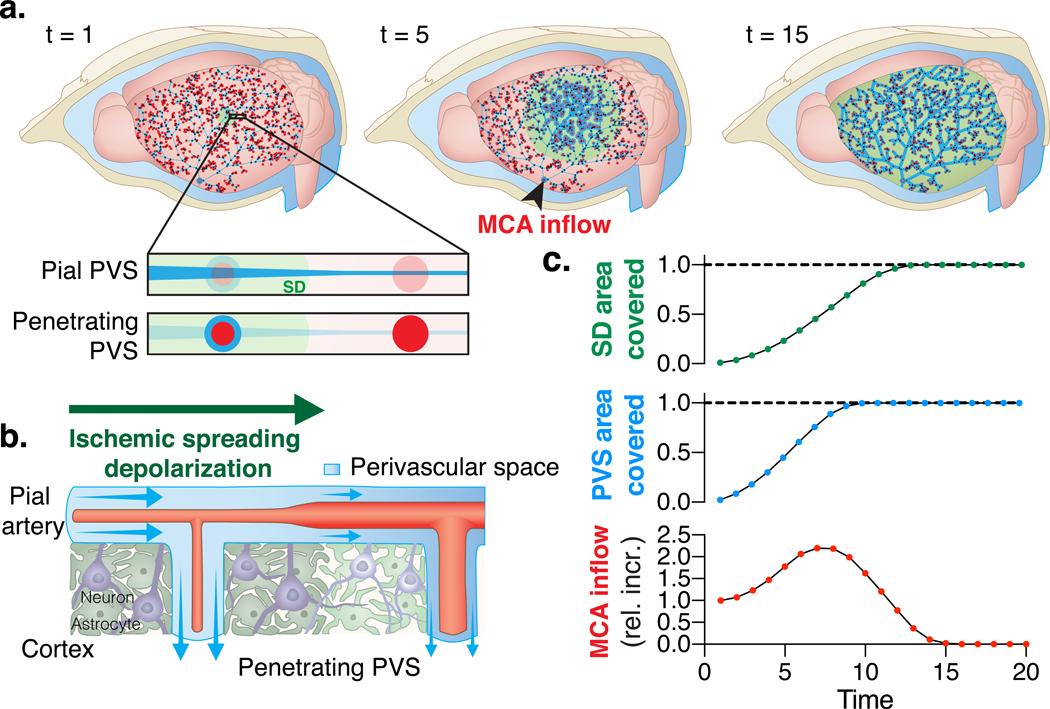
(A) Network model representing a system of interconnected perivascular spaces (PVS) surrounding the mouse pial MCA at different time points (t) during an ischemic spreading depolarization (SD; green). Pial PVS are depicted as blue lines that get wider during spreading ischemia. Penetrating arteries are depicted as red circles and the blue circle surrounding it signifies an increase in the area of the penetrating PVS as the arteriole constricts after SD. The simulation evaluated the relative increase (rel. incr.) in baseline flow at the inlet of the MCA (MCA inflow; black arrow). (B) Pial and penetrating PVS area increases as the arteries constrict due to the passage of a SD, thus increasing the fluid volume in the network. Conservation of mass controls the resulting MCA inflow, (C) resulting in a net increase in fluid volume in the network. Dashed line represents the tissue border of the cortical surface. The SD travels over the entire cortex spanning an area larger than that covered by the MCA network.
Spreading edema depends on aquaporin-4 expression
We next asked whether blocking the SD using a glutamate N-methyl-D-aspartate (NMDA) receptor antagonist (MK-801) or inhibiting spreading ischemia (SI) using a cocktail of vasodilators (nimodipine, papaverine, and S-nitroso-N-acetylpenicillamine) would prevent the rapid entry of CSF (Fig. S8). Both approaches have been shown to either modulate the spatiotemporal dynamics of SD or the severity of SI, but neither has been shown to inhibit completely the effects of the first depolarization after an ischemic insult(35, 36, 41–44). In our model, pre-treatment with MK-801 or infusion of a vasodilator cocktail were ineffective in blocking the SD or SI (Fig. S8A-J). As before, MCAO was followed by a 3 to 4-fold increase in CSF influx in all groups (Fig. S8K-N, P=0.39). However, residual CBF after SI was a significant predictor of the degree of CSF influx, suggesting that targeting reperfusion and SI might be a more feasible approach than trying to block the SD (Fig. S8O). Alternatively, CSF transport into the brain has been shown to be facilitated by aquaporin-4 (AQP4) water channels expressed on the endfeet of astrocytes that form the outer wall of the penetrating PVS (23, 45). Because AQP4 facilitates the transport of CSF out of the PVS, we hypothesized that AQP4–9 null mice (Aqp4−/−) might have reduced CSF influx after MCAO (Fig. 6). This is in line with evidence demonstrating that knockout animals and wildtypes treated with AQP4 inhibitors are protected from edema after stroke(46–49). Hence, we tested the effect of AQP4-knockout on CSF tracer entry after MCAO using transcranial macroscopic imaging (Fig. 6A-B). As with wildtypes (Aqp4+/+), Aqp4−/− mice had a SD followed by SI after occlusion (Fig. 6C). Likewise, CSF tracer also distributed in the ipsilateral cortex but to a reduced degree in Aqp4−/− mice compared to wildtype (P=0.018; Fig. 6D). Could the absence of AQP4 at the perivascular endfoot reduce the entry of CSF to the tissue? To test this, we acquired two-photon imaging after intracisternal infusion of a small CSF tracer (3kDa dextran; Fig. 6E). During SI, tracer entered the penetrating PVS in large quantities reaching depths of up to 200 μm below the cortical surface and distributing into the interstitial fluid in wildtype (ISF; Fig. 6F). Aqp4−/− mice have been shown to have abnormal SD kinetics, so we first determined whether differences in the vasodynamics of penetrating arterioles during SI could be responsible for the differential response in CSF influx. Knockout animals exhibited slower arteriolar constrictions but to a similar degree as Aqp4+/+ animals, which would not explain the observed results (Fig.6G-J). We then looked to see how much traced CSF actually reached the penetrating PVS and how much of it exchanged with the ISF. Less tracer entered the penetrating PVSs of Aqp4−/− mice compared to their wildtype counterparts (P<0.0001; Fig.6K). At the same time, less of the CSF tracer could be found in the neighboring ISF of the penetrating PVS in the knockouts, as in previous reports (Fig.6L)(23). Global tracer quantification in ex vivo brain sections confirmed a significant reduction in fluorescence intensity in the ipsilateral and contralateral hemisphere of Aqp4−/− mice (P=0.005 and 0.001, respectively; Fig. 6M-O). In line with these results, AQP4 knockout animals did not develop edema within the first 15 min after MCAO, as opposed to Aqp4+/+ mice (Fig.6P, 1H). Thus, deletion of AQP4 significantly suppresses ischemia-induced CSF tracer influx.
Fig. 6. CSF-mediated edema after MCAO is dependent on aquaporin-4 (AQP4) expression.
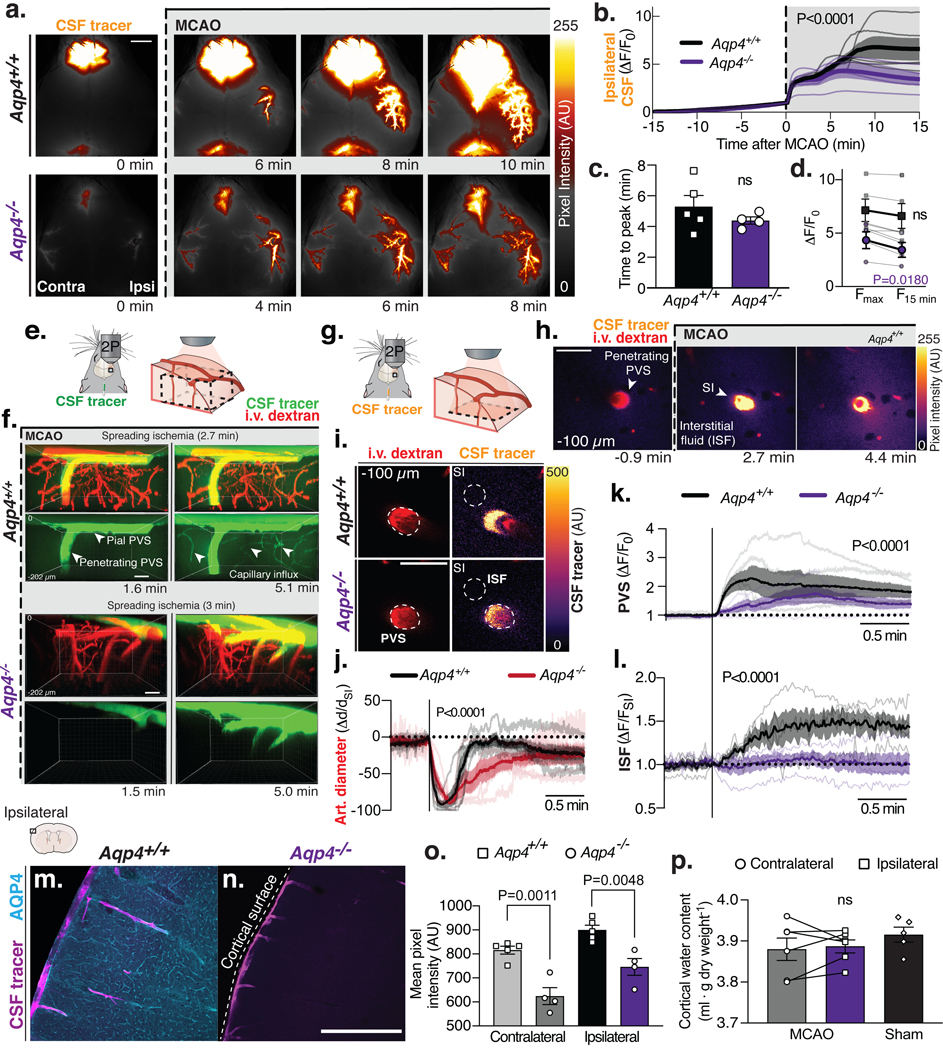
(A) Transcranial imaging after intracisternal tracer injection (BSA-647) in Aqp4+/+ and Aqp4−/− mice. (B) Quantification of ipsilateral CSF tracer influx (ΔF/F0). Two-way repeated measures ANOVA, P value from interaction term; n=4–5 mice/genotype. (C) Time of CSF influx peak. Unpaired t-test. (D) Change in maximum ΔF/F0 (Fmax) to 15 min after MCAO (F15 min). Paired t-test. (E-F) Volumetric two-photon imaging shows CSF tracer (3kDa dextran) entering brain via surface and penetrating perivascular spaces (PVS) in Aqp4+/+ and Aqp4−/− mice. After spreading ischemia (SI) the tracers were also found surrounding capillaries only in Aqp4+/+. (G) Time-lapse 2-photon imaging of a penetrating arteriole 100 μm below the cortical surface. (H) Tracer enters the interstitial fluid (ISF) after SI. (I) CSF tracer entered the penetrating PVS of both Aqp4+/+ and Aqp4−/− mice. (J) Penetrating arteriolar (art.) diameter changes after MCAO. Tracer influx was quantified in the (K) penetrating PVS and the (L) ISF neighboring the same PVS; 1 PVS/mouse; n=4–5 mice/genotype. (M) Immunohistochemical labeling for AQP4 from ipsilateral dorsal cortex of Aqp4+/+ and (N) Aqp4−/− after CSF tracer injection. (O) Mean pixel intensity from 6 coronal sections from 4–5 mice/genotype. (P) Cortical water content 15 min after MCAO or bilateral cortices from sham-treated Aqp4−/− mice; paired t-test; n=5–6 mice/group; ns: not significant. Scale bars: (A) 2mm, (F-I) 50μm, (M-N) 500μm.
Discussion
Historically, brain swelling has been assumed to develop entirely through the accumulation of fluid from the intravascular compartment(15). The present study demonstrates that CSF influx, initiated by ischemic SD, plays an important role in driving acute tissue swelling (Movie 10)(16, 50–52). This new finding is further supported by a detailed examination of edema formation at CSF-ISF exchange zones, which we observed in cortex adjacent to leptomeningeal vessels as well as in periventricular zones 30 min after MCAO in mice and during the acute phase of human cerebral ischemic infarction (Fig. S9, Table S1). Edema was primarily seen in brain regions that are known flow pathways for CSF-ISF rather than in deeper structures such as striatum that are farther away. Is glymphatic edema a previously unrecognized contributor to secondary ischemic injury? The entry of CSF through PVSs is likely to facilitate swelling of astrocytic endfeet, and together with pericyte constriction would further reduce blood flow and cause infarct expansion(53, 54). However, this study does not exclude the possibility that vascular fluid is a significant source of swelling in later stages after stroke, because transvascular ion and fluid influx are increasingly important at 24h after MCAO when the BBB is already open (Fig. S10A-D). Several hours after CSF influx, de novo expression of the nonselective monovalent cation channel, SUR1-TRPM4, in brain endothelial cells contributes to additional Na+ influx, and blockade of these transporters with glibenclamide reduces edema(5, 55). Nonetheless, CSF production continues to be a significant source of Na+ and water after stroke (Fig. S10E-H). Our results posit that CSF is the earliest source of Na+ and fluid, driving tissue swelling. This early phase is likely to play an additive role to the later stages of edema formation. Perhaps even more important is the fact that spreading depolarizations continue for several days after stroke and are also observed in other acute conditions, including subarachnoid hemorrhage, intracerebral hemorrhage, and traumatic brain injury(29). Our study predicts that SDs will worsen edema and possibly explains why the frequency of depolarizations correlates with secondary injury and poor outcome(29, 39, 56–58). Thus, targeting glymphatic edema may offer a therapeutic strategy for treatment of a broad range of acute brain pathologies.
Materials and Methods
Animals.
All experiments were approved by the University Committee on Animal Resources of the University of Rochester and the Danish Animal Experiments Inspectorate. Efforts were taken to minimize the number of animals used. Male C57BL/6 mice (Charles River) or Glt1-GCaMP7 mice (RIKEN BioResource Research Center, Japan) on a C57BL/6 background 8 weeks of age were used for all the experiments. Glt1-GCaMP7 mice express GCaMP7 in 95.2% of cortical astrocytes with minimal expression in other glial cell types and about 50% of cortical neurons in L4 and L6(32). Male eight-week-old NG2-dsRed (Jackson Laboratory, Stock No. 008241) mice were used to identify perivascular inflow routes. AQP4-knockout (Aqp4−/−) mice on a C57BL/6 background between 8 and 16 weeks old were used(45). Aqp4−/− mice have been shown to have slower SD, delayed [K]e increase and uptake kinetics(59). The knockouts also exhibit shorter extracellular glutamate elevations compared to wildtype(60). Some of these studies have attributed this difference to the larger extracellular space volume observed in these mice, causing a slower increase in [K]e during depolarization (59). Other studies have instead credited it to AQP4-dependent changes in volume-regulated anion channels (60). Aqp4−/− mice also show significant differences in O2 diffusion, especially to remote areas away from microvessels (61). All experiments performed in this study were done on mice anesthetized with ketamine/xylazine (100/10 mg/kg, i.p.). The vasomotor response to spreading depolarizations in naïve cortex is different in mice compared to other species(29). However, in ischemic cortex as seen in our model of MCAO, spreading ischemia is the predominant vasomotor response after SD across a variety of diseases and animal models (i.e. mice(35), rats(62), cats(34), swine(63), and humans(64–67)).
Intracisternal injections.
Animals were fixed in a stereotaxic frame. A 30G needle was connected to PE-10 tubing filled with artificial (aCSF) into the cisterna magna as described here(68). For intracisternal injections, 10 μl of CSF tracer was injected at a rate of 2 μl/min over 5 min with a syringe pump (Harvard Apparatus).
CSF tracers.
AlexaFluor647 or AlexaFluor594-conjugated bovine serum albumin (BSA-647 or BSA-594, 66 kDa, Invitrogen) and Cascade Blue-conjugated 3 kDa dextran (Invitrogen) were diluted in artificial CSF (0.5% or 1% m/v) and used as a fluorescent CSF tracer. Radio-labeled 22Na (0.5 μCi, Perkin Elmer) and 3H-mannitol (1 μCi, American Radiolabeled Chemicals) were dissolved in artificial CSF. For particle tracking velocimetry experiments, red fluorescent polystyrene microspheres (FluoSpheres™ 1.0 μm, 580/605 nm, 0.25% solids in aCSF, Invitrogen) were briefly sonicated and infused as before(38).
Drugs.
MK801 (5mg/kg in 0.9% NaCl; Tocris) was administered via intraperitoneal injection 15 min prior to MCAO. The vasodilator cocktail contained nimodipine (2μg/kg/min; Tocris), papaverine (3mg/kg/min; Sigma Aldrich) and S-nitroso-N-acetylpenicillamine (3μmol/kg/min in 0.9% NaCl; Sigma-Aldrich) and was delivered intravenously through a PE-10 tubing catheter in the femoral vein using an infusion pump (Harvard Apparatus).
Physiological recordings.
Heart rate and respiratory rate were acquired using an animal physiological monitoring device (Harvard Apparatus). Intracranial pressure (ICP) was measured through a 30G needle connected to a PE-10 catheter filled with aCSF into the cisterna magna. The line was connected to a pressure transducer and monitor (World Precision Instruments). Relative cerebral blood flow (rCBF) was measured using laser Doppler flowmetry (PF5010 Laser Doppler Perfusion Module, PR 418–1, Perimed). The fiber optic probe (MT500–0, Perimed) was fixed onto the skull above the MCA vascular territory (5mm lateral and 1mm posterior to bregma) on the right hemisphere with cyanoacrylate glue. For two-photon experiments, rCBF was measured at the lateral aspect of the skull over the right temporal bone in order to place the fiber optic probe below the headplate and shield it from laser irradiation. All the signals were collected using a 1440A digitizer and AxoScope software (Axon Instruments) and analyzed using Matlab.
Middle cerebral artery occlusion.
The anesthetized animal was laid on its back on a heating pad. A surgical midline incision was made from the clavicle to the chin to expose the right common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA). Then the distal side of ECA and proximal side of CCA were ligated by 7–0 silk suture. The distal side of the ICA was clamped using a micro clamp (B-1, Fine Science Tools). A small incision was subsequently made in the proximal portion of the CCA. A PE-10 polyethylene tube (PE-10, Beckton-Dickinson) filled with heparinized saline and six macrospheres, 200 μm in diameter, was inserted into the CCA and the clamp was subsequently removed. The tubing was secured in position by 7–0 silk suture. The macrospheres were advanced into the ICA via an injection of 0.02 ml heparinized saline. Ischemia was confirmed by laser Doppler flowmetry. The incision was closed with a 5–0 suture. All mice were monitored using laser Doppler flowmetry during the macrosphere infusion. If rCBF did not decrease >70% of baseline or rCBF returned to baseline the animal was excluded.
Behavior testing.
Twenty-four hours following MCAO or sham, mice were placed in a 45cm x 61cm open field apparatus. Movement of the mice was recorded for 30 min and then analyzed on ANY-maze video tracking software. MCAO and sham mice were placed on an accelerating rota-rod (Ugo Basile) that accelerated from 5 rpm to 40 rpm. The mice were allowed to remain on the rota-rod until they could no longer continue, and a final time was recorded once they fell off the rotating cylinder. MCAO and sham mice were allowed to grasp a wire bar suspended 43 cm above the surface with only their forepaws before being released. Each mouse was tested for three 20-second trials and given a score from 0 to 3 depending on how well it was able to stay on the string. A score of 3 was given to mice who would immediately fall from the string when released; a 2 if the mouse was able to hang on the string and attempted to climb; a 1 was given to mice that were able to put both forepaws and one or both hind paws onto the string; if the mouse placed all four paws and the tail onto the string and made lateral movement, a score of 0 was given(69).
Transcranial optical imaging.
Mice were fixed to either a MAG-1 or MAG-2 stereotaxic holding device (Narishige) and injection, via a cisterna magna cannula, of 10 μl of bovine serum albumin (BSA)-conjugated Alexa fluor-594 or 647 (Invitrogen) in aCSF, occurred over a period of five minutes using a syringe pump (Harvard Apparatus). Whole cortical macroscopic imaging was performed using either an Olympus MVX10 with a PRIOR Lumen LED and Hamamatsu ORCA-Flash4.0 V2 Digital CMOS camera using Metamorph software or a Leica M205 FA fluorescence stereomicroscope equipped with an Xcite 200DC light source and A12801–01 W-View GEMINI (Hamamatsu) for simultaneous GFP (excitation 480 nm, emission 510 nm, Leica) and Cy3 (excitation 560 nm, emission 630 nm, Leica) acquisition. Images were acquired with a Hamamatsu ORCA-Flash4.0 V2 Digital CMOS camera using LAS X Leica software, with a 512×512, 16-bit resolution, at 20Hz.
MRI scans.
RI was performed using a 9.4 Tesla animal scanner (BioSpec 94/30 USR, Bruker BioSpin, Ettlingen, Germany) equipped with a cryogenically-cooled quadrature-resonator (CryoProbe, Bruker BioSpin, Ettlingen, Germany). Mice were placed on an MR-compatible stereotactic holder with ear bars to minimize head movement during scanning. Body temperature was maintained at 37°C and monitored along with the breathing rate by a remote monitoring system (SA Instruments, NY, USA). All required adjustments and T2-weighted scans (2D TurboRARE: TR/TE: 8000/36 ms, Matrix 384 × 256, FOV 19.2 × 12.8 mm, NEX 1, 48 horizontal slices, slice thickness = 0.2 mm) were performed within 15 min after CCA catheter placement for visualization of the geometry before the baseline DCE, DWI, or cisternography scans. Ischemia was confirmed by flow-compensated 2D time-of-flight MR angiography scans (2D-GEFC: TR/TE 10/2.4 ms, Matrix 192 × 128, FA 80, FOV 19.2 × 12.8 mm, NEX 1, 10 horizontal slices, slice thickness = 0.3 mm) 30 seconds immediately after macrosphere infusion: if angiography did not show occlusion of MCA the animal was excluded.
Dynamic contrast enhancement MRI (DCE-MRI).
Pre and post contrast T1-weighted (T1W) images were collected with 3D-FLASH sequence (TR/TE 17.7/3.1 ms, FA=15°, Matrix 192 × 128 × 96, FOV 19.2 × 12.8 × 9.6 mm, NEX 1). T1W of the entire mouse brain were performed in 2 min at an isotropic spatial resolution of 100 μm. As a T1-enhancing contrast agent, gadobutrol (Gadovist, Bayer Pharma AG, Leverkusen, Germany) was injected into cisterna magna. The time series T1W scanning protocol was comprised of three baseline scans (6 min) followed by intracisternal infusion of 12.5 mM gadobutrol at a constant rate of 1.0 μL/min for 10 min. Considering the dead space within the catheter, the actual infused volume of the contrast was 9 μL. Scans continued over 50 measurements, and MCAO started at 24 min after the first T1W scan.
Diffusion-weighted imaging (DWI).
DWI was performed with 2D single-shot echo-planar DWI sequence with parameters set as follow: TR/TE 2500/20 ms, NEX 1, FOV 18 × 15 mm, Matrix 108 × 96, 8 axial slices, slice thickness 0.8 mm. Z-direction motion probing gradient (MPG) was acquired with multiple b values (0, 100, 400, and 1000 s/mm2). For the DWI study, 30 baseline images before MCAO were acquired, followed by a series of images acquired over time after MCAO. Scans continued over 360 measurements with each measurement lasting 10 seconds. Maps of the mean diffusivity (ADC-map) were derived using the standard algorithm of Paravision 6.0.1 software (Bruker).
MR CSF cisternography.
CSF cisternography was performed with 3D-fast imaging employing steady-state acquisition (3D-FIESTA) sequence (TR/TE 17.7/3.1 ms, FA=15°, Matrix 192 × 128 × 96, FOV 19.2 × 12.8 × 9.6 mm, NEX 1). For CSF volumetric measurements, 10 baseline images were acquired before MCAO, followed by a series of images acquired over time after MCAO. Scans continued over 60 measurements with each measurement lasting 30 seconds. Ventricular compartments were segmented, and temporal volume change was measured by IMARIS (v. 9.2.1, Bitplane, Concord, MA, USA).
In vivo two-photon laser scanning microscopy.
A cranial window was prepared over the right anterolateral parietal bone above the MCA vascular territory. The dura mater was left intact and, to prevent intracranial depressurization, the window was sealed with agarose (1.1% at 37°C) and a glass coverslip (8 mm diameter). Unsealed craniotomies have been shown to affect glymphatic function(45). Two-photon imaging was performed using a resonant scanner Bergamo scope (Thorlabs) and a Chameleon Ultra II laser (Coherent) with a water-immersion 20× objective (1.0 NA, Olympus). Intravascular FITC-dextran (2,000 kDa, 2.5%, Sigma-Aldrich) and either red microspheres or BSA-647 were excited at an 820 nm wavelength. In GCaMP mice, vessels were labeled with TRITC– dextran (2,000 kDa) and CSF with BSA-647 and excited at 860nm. Emission was filtered at 525, 607, and 647nm. Images were acquired using ThorImage software and synchronized with physiological recordings (3 kHz, ThorSync software).
Quantification of CSF production rate.
CSF production rate was quantified as previously described in rats(70, 71). Anesthetized mice were fixed on a stereotactic apparatus and a 0.5mm burr hole was made over the left lateral ventricle (AP=−0.1mm, ML= −0.85mm). A 30-gauge needle connected to PE-10 tubing was lowered through the burr hole to −2.00 mm DV. The mouse then received a MCAO as before. The rCBF drop was confirmed with laser Doppler flowmetry for five minutes and then the mouse was placed back in a stereotactic head frame and the cisterna magna was surgically exposed with the mouse’s neck flexed at 90 degrees. A 30-gauge needle connected to PE10 tubing filled with mineral oil (Sigma Aldrich, M5904) was inserted into the cisterna magna and advanced gently 2mm through the foramen of Magendie into the 4th ventricle. One microliter of mineral oil was infused at a rate of 1μl/min for 1 min with a syringe pump (Harvard Apparatus) to block outflow from the ventricular system. The location of CSF within the intraventricular PE-10 tubing was marked at 10 min intervals. The volume of CSF was calculated as: CSF volume=π x (internal radius: 0.14mm)2 x length. The rate of CSF production (μl/min) was calculated as the slope of the linear regression from each mouse.
Brain water content measurements.
Anesthetized animals were decapitated immediately, and the cortex was dissected and weighed (Wwet; g). The tissue was dried at 65°C until it reached a constant weight (~48 hours) and brains were re-weighed (Wdry). For calculations of the tissue water content (ml/g dry weight), the following formula was used: (Wwet - Wdry)/Wdry.
Radioisotope influx.
To evaluate CSF influx into the brain, radiolabeled tracers 22Na (0.5 μCi, Perkin Elmer) and 3H-mannitol (1 μCi, Perkin Elmer) were injected into the cisterna magna at 2 μl/min for 5 min in aCSF and allowed to circulate for 15 min prior to MCAO. After 15 min, the animals were rapidly decapitated, the skull and dura were removed, and the brain was harvested. Brains were cut into six sections for processing. In a separate set of experiments, the radiolabeled tracers were injected intravenously first as a 2μl bolus and then a 2 μl/min infusion for 5 min. Immediately after the end of the infusion, MCAO was induced and 15 min later animals were rapidly decapitated, the skull and dura were removed, and the brain was harvested. All brain tissue was weighed and solubilized in 0.5 ml tissue solubilizer (Solvable, PerkinElmer) overnight. Upon solubilization, 5 ml scintillation cocktail was added (Ultima Gold, PerkinElmer). The injectate controls were treated in the same way as the tissue samples. All samples were analyzed by liquid scintillation spectrometry using a scintillation counter (LS 6500 Multipurpose Scintillation Counter, Beckman Coulter). The radioactivity (counts per minute) remaining in the brain after injection (percentage of injected dose) was determined as Rb/Ri × 100, where Rb is the radioactivity remaining in the brain at the end of the experiment and Ri is the radioactivity in the injectate controls for each experiment. Only cerebrum was considered, and cerebellum was excluded from analysis since ischemia does not reach this region. All samples were background subtracted to blank samples with 0.5 ml tissue solubilizer and 5 ml scintillation cocktail.
Triphenyltetrazolium chloride (TTC) staining.
TTC was prepared at 2% in phosphate-buffered saline (PBS) and kept in a 37°C water bath. MCAO and sham mice were anesthetized and perfused with 5–10mL of ice-cold PBS. Mice were immediately decapitated, and the brains were extracted and put into TTC. Brains remained in the TTC solution for 15 min and then imaged under an Olympus SZX12 bright-field microscope. In a separate experiment, the brains were sectioned 1mm-thick on a vibratome (Leica VT1200S) in ice-cold PBS. The coronal slices were placed into TTC for 15 min and then 4% paraformaldehyde (PFA) for an additional 30 min. The coronal sections were then imaged under the microscope for infarct volume quantification. Area of the infarct volume was calculated in each slice, by tracing the infarct core using the polygon selection tool in Fiji. Area (mm2) of the infarct was then converted to volume (mm3) accounting for the 1mm slice thickness. The sum of all the coronal infarct volumes was calculated to give whole brain infarct volume. The edema-corrected infarct volume was calculated as previously described(72) and expressed as a percentage of the contralateral hemisphere.
Arteriole/venule labeling experiments.
NG2-dsRed mice were anesthetized and received an intravenous injection of 0.2ml of 1mg/ml wheat-germ agglutinin lectin (Invitrogen Alexa Fluor 647 conjugated) into the saphenous vein. Next, a fluorescent CSF tracer (0.5% 3kDa dextran Cascade Blue; Invitrogen) was infused into the cisterna magna and allowed to circulate for 15 minutes prior to MCAO. Thirty minutes later, the mouse was perfusion-fixed with PBS (100 μg lectin) followed by 4% PFA and then the brains were harvested. The following day, the brains were sectioned into 100μm slices using a vibratome (Leica VT1200S) and mounted onto slides with ProLong Gold antifade mounting medium (Invitrogen). Images were acquired using a Leica TCS SP8 confocal system. A total of 8–10 z-stacks at 20x magnification from cortex were taken from 4–6 coronal sections from each mouse. The percentage of tracer-positive arterioles and venules was calculated from the total number of blood vessels with CSF tracer labeling (42–49 vessels/mouse).
Tissue histology.
Mouse brains were obtained after sacrifice, following sham or MCAO and fixed in 4% PFA. Patients who died with acute cerebral ischemia documented between January 2018 and December 2018, and who underwent autopsy, were identified retrospectively by reviewing the autopsy records of the Department of Pathology at the University of Rochester Medical Center. With these constraints, 5 focal cerebral infarct specimens from patients were identified. As controls, brain samples were also evaluated from adult patients with documented absence of an ischemic brain lesion who died from acute cardiorespiratory collapse. In all cases, the presence or absence of an ischemic brain lesion was confirmed by a neuropathologist. Standard tissue fixation protocols (7–10 days in formalin) were applied. Paraffin-embedded tissue blocks from frontal cortex, basal ganglia, and periventricular regions were obtained in each case by an expert neuropathologist (RIM). Blocks were sectioned at 6 μm thickness, stained with hematoxylin and eosin (HE), and imaged at 1000x (oil immersion) using a Nikon NiU Microscope and Nikon Microscope Solutions Imaging Software (NIS-Elements AR Version 4.30.01). Edema area was quantified by a blinded rater using Fiji. HE images were automatically thresholded to include the highest intensity pixels (Otsu’s method), which consisted of the white pixels of the fluid accumulation. The edema area was measured as a percentage of the total tissue area from a representative image from each animal/subject. In the case of mouse cortex (Fig. S9B), each biological replicate was an average from 4 separate images from the same mouse. For coronal sections in Fig. 6M-O, brains were sliced into 100μm sections using a vibratome (Leica VT1200S) and mounted onto slides with ProLong Gold antifade mounting medium (Invitrogen). Images were acquired with an Olympus MVX10 stereomacroscope and a PRIOR Lumen LED and Hamamatsu ORCA-Flash4.0 V2 Digital CMOS camera using Metamorph software. Images were analyzed as previously described(33). Mean pixel intensity for six sections starting at the anterior aspect of the corpus callosum and skipping 400 μm intervals moving posteriorly was quantified using ROIs for the ipsilateral and contralateral hemispheres in Fiji. An average of the six coronal sections was computed for each mouse.
Transcranial optical imaging analysis.
A region of interest (ROI) was manually drawn around the skull and fluorescence intensity of CSF tracer influx was quantified using Fiji software(33). Mean fluorescence intensity was normalized to the time of MCAO (F0) and the derivative of the curve was computed to calculate the mean rate of change in fluorescence intensity over time. The time at which a peak in the rate of change occurred was determined to calculate time to peak influx. To extract more quantitative information we tracked fronts using an automated Matlab algorithm previously developed at the University of Rochester(33, 73). Fronts are curves that separate bright regions from dark regions in videos of murine brains. Local front speeds quantify the local speeds of CSF influx and spreading depolarization. We tracked fronts only within the brain hemispheres, sometimes treating the ipsilateral and contralateral hemispheres separately. We located those hemispheres by calculating the time-averaged GCaMP brightness of each video, then finding the two largest bright regions. Front tracking requires choosing a brightness threshold: each front separates a region brighter than the threshold from a region dimmer than the threshold. To calculate thresholds, we calculated the mean brightness of each channel in each hemisphere, varying over time. We chose the thresholds as the brightness values halfway between the initial, dim values and the final, saturated values. From the thresholded dataset we calculated the surface area in mm2 over time in an ipsilateral and contralateral hemisphere ROI. The surface area data for the CSF tracer and the GCaMP channels were aligned in time using the maximum area of the GCaMP channel. We next calculated the mean rate of change in the surface area covered by CSF tracer over time. The maximal rate of change was used to compute the time to CSF influx peak and was compared to the time at which maximal GCaMP surface area occurred. SD onset time was measured as the first non-zero value of the thresholded GCaMP channel after MCAO. We next measured front speeds in a smaller ROI over the MCA territory. The average front speed for all pixels was plotted over time. To reduce noise caused by outlier pixels, we smoothed front speeds with a sliding line fit and a 20 second smoothing window. The maximum value of front speeds after the onset of the SD was considered the max speed. To determine the delay time between the SD and the arrival of CSF tracer we evaluated the time elapsed between a pixel having GCaMP fluorescence and CSF tracer fluorescence; an average of all the pixels in the ROI was calculated to obtain mean delay time.
MRI post-processing and analysis.
Both dynamic contrast-enhanced (DCE) and apparent diffusion coefficient (ADC)-map time series data were motion corrected using Advanced Normalization Tools (ANTs) normalization software(74, 75). DCE-MRI time series were motion corrected and converted into percent change from baseline-time series, calculated as the percent signal change from the averaged signal of baseline images. To avoid biases in tracer calculations from availability of tracer to the glymphatic system, we then normalized percent change maps to the peak average. A population-based average of the baseline scans was created by an iterative registration process with 2 rigid, 2 affine and 5 non-linear registration-and-averaging steps. To allow unbiased ipsilateral-contralateral comparisons, original and left-right-flipped images were both used in the template creation. Registration was carried out using Advanced Normalization Tools v. 2.1.0. The average template was skull-stripped manually using ITK-SNAP and was then segmented into brain structures by registering the Waxholm Space Atlas of the C57BL/6J Mouse Brain(76) to the template. Normalized percent change maps were finally transformed to the average space for analysis. We the drew lines extending from the MCA at the level of the temporal ridge and 1 mm orthogonal to the brain surface in the coronal slice. Tracer penetration depth was defined as the distance from the MCA to the deepest voxel with normalized tracer signal > 1. For DWI analysis, edema was defined as the ADC value in a pixel dropping 3 standard deviations below baseline. Analysis of images was performed in ITK-SNAP (version 3.6.0, http://www.itksnap.org), ImageJ (version 2.00, NIH, Bethesda, MD, USA), and Matlab (version 9.3.0, MathWorks, Natick, MA, USA). Circular regions of interests (ROIs) were drawn in the images both for the DCE enhancement ratio calculations and ADC calculations. 3D maximum intensity projection (MIP) and multiplanar reformation (MPR) for DCE data visualization was generated using IMARIS (v. 9.2.1, Bitplane, Concord, MA, USA). To create the registered average of the DCE and ADC time series, the time series were registered to the average template described above, and from those, different slices of the Volumes were processed into videos using Python 3.6.
Two-photon imaging analysis.
Images obtained were 16-bit with either two or three channels depending on the experiment, each with spatial dimensions of 512 × 512 and an average frame rate of 15Hz. The 525 nm channel captured the vascular FITC-dextran or GCaMP7 fluorescence, the 607 nm the vascular Texas Red–dextran or the red microspheres in CSF for particle tracking, while the 647 nm channel captured the fluorescent BSA CSF tracer. Images were analyzed using Fiji and Matlab. The GCaMP channel was processed by converting it to 8-bit and generating a Z-projected average intensity of the entire imaging session and subtracting the average image from each frame (ΔF-Faverage). A Gaussian blur filter with a sigma of 1 was applied to the resulting stack and was subsequently color-coded using the lookup table mpl-inferno. A mask of the CSF tracer and vascular channel was applied, and then all channels were merged. Linescans of the XYT data were generated using Fiji. For quantification purposes, GCaMP and CSF tracer fluorescence were quantified as ΔF/F0, F0 is the first frame of the experiment prior to MCAO. Wave speed was calculated by drawing two ROIs 100 μm apart, along the direction of wave propagation. The time between the GCaMP fluorescence appearing in the first and second ROI was considered the speed of propagation. Perivascular spaces (PVS) were defined as any fluid compartment following a blood vessel. Pial PVS followed leptomeningeal vessels at the brain surface and penetrating PVS dove into cortical parenchyma with penetrating arterioles. PVS area was compared to arteriolar area for both pial and penetrating PVS. Images for both the CSF and i.v. dextran channel were thresholded using an automated method (Otsu) and the diameter of the vessel was measured. In early timepoints after intracisternal infusion, tracers had not reached the penetrating arteriole PVS, so CSF tracer area was normalized to the penetrating arteriolar area (ΔA/Aart). Tracer was found in the pial PVS starting at 15 min after infusion and was therefore normalized to the area covered by tracer prior to MCAO before normalizing to the pial arteriolar area (ΔAnorm/Aart). For experiments in Fig. 6E-G, time-lapse Z-stacks were acquired and processed using IMARIS (v. 9.2.1, Bitplane, Concord, MA, USA). To quantify the arteriolar diameter changes in Aqp4−/− and Aqp4+/+ mice, the diameter was normalized to the beginning of the spreading ischemia-induced constriction (Δd/dSI). An ROI was placed over the penetrating arteriole and the surrounding perivascular space (PVS) and fluorescence intensity was normalized to the background intensity before MCAO (ΔF/F0). For ISF tracer quantification, an ROI was placed next to the PVS making sure to avoid any surrounding capillaries or the overlying pial arteriole. The same size ROI was used for all animals. To determine the amount of PVS tracer that entered the ISF, fluorescence intensity was normalized to the time of onset of the SI (ΔF/FSI).
Particle tracking velocimetry.
To directly measure the speed of cerebrospinal fluid flowing through perivascular spaces we performed particle tracking velocimetry (PTV) using time series of images obtained from in vivo two-photon laser scanning microscopy. Our procedure is similar to one described previously(38). The PTV analysis is performed using automated Matlab software(77, 78) in which tens of thousands of microspheres are tracked with subpixel accuracy through the time series of images to obtain spatially and temporally resolved velocities. We use an improved algorithm in which dynamic masking is performed to remove stagnant particles from the measurements. Specifically, before identifying and tracking particles, we first subtract a unique background image from each frame which is obtained by averaging the nearest 300 frames (150 before and after). To calculate heat maps of average flow speed (Fig. S7A), the spatial domain was divided into 5 pixel x 5 pixel bins, the velocities were time-averaged in each bin using measurements from 1 min or less (centered on the indicated time), and the speed was computed from the average velocity. To calculate the time series of vdownstream first the downstream velocity component of each particle in each frame is computed as where u is the instantaneous particle velocity and is a field of unit vectors computed from time-averaging the velocity field over the entire time series. We then obtain vdownstream by calculating the spatial average of all the downstream velocity components in 1 second bins and normalize the time series by the baseline value. The baseline value is the average value of vdownstream over the initial 2 min of the experiment before the MCA occlusion occurs. To calculate vpulsatile, we first obtain the time series of vmean by computing the spatially averaged speed for each frame. We then define the time series of vpulsatile as max(vmean)—min(vmean) over each cardiac cycle, divided by the baseline value of this quantity. The cardiac cycle is defined using sequential peaks of the R wave in synchronized ECG measurements. The normalized artery diameter time series was measured using custom software developed in Matlab. The measurements were performed by subsampling the time series of images obtained from in vivo two-photon laser scanning microscopy by a factor of ten and analyzing a user-defined ROI centered on a straight segment of the artery in each frame using an automated algorithm. The algorithm first separates the artery segment from the background by binarizing the ROI based on a user-defined threshold. The major and minor axes of the artery segment are then measured using the built-in Matlab function “regionprops.” By ensuring that the segment length is longer than the artery diameter, the major and minor axes then correspond to the segment length and artery diameter, respectively. The normalized artery diameter time series then corresponds to the length of the minor axis in each frame, divided by the baseline value. The baseline value is the average artery diameter over the initial 2 min of the experiment before the MCA occlusion occurs.
Glymphatic network model.
Transport of water follows a network composed of perivascular spaces (PVS), bifurcations and terminal nodes (penetrating PVS). In mathematical language, we call the pial PVS the edges along the network, collected in the set E, and the terminals and bifurcations are collected in the set of nodes, N. Thus, the network is given by nodes ) i ∈ N ( and edges (i, j) ∈ E. The edges of the network have the property of an inverse resistance, or conductance Cij. To simplify the model, we assume that each PVS is a straight pipe segment of length Lij with cross-sectional area Aij. The conductance is then
| (1) |
We assume that the spreading depolarization (SD) wave propagates from (x0, y0) across a two-dimensional domain on the cortical surface with radius R(t) growing at a constant celerity c, so that R(t) = ct. The presence of the wave at node ) with coordinates (xi, yi) is then defined as w(δ) = 1 for δ ≤ 1 or w(δ) = 0 otherwise, where the relative distance from node to the wave’s origin is or from farthest end of an edge to the wave’s origin, δij = max(δi, δj).
The arrival of the SD wave at a penetrating arteriole located at site i ∈ N triggers a dilation process,
| (2) |
Where are the initial and final PVS volumes. The wave covering pial surface vessels (i, j) ∈ E triggers an analogous dilation response,
| (3) |
leading to an increase of the PVS’s conductance are the initial and final cross-sectional PVS conductances. Mass balance demands
| (4) |
where ρ is the mass density, qi is the flow rate in or out of a terminal node, Qij is the flow rate along an edge (i, j) and is the volume of the PVS connected to the bifurcating node i. Summation of (4) over all nodes ) yields the following relation, in which the sum over the edge flows cancels (i.e., over forward and backwards directions). Furthermore, the pial surface volume is defined as is defined as the subset of nodes corresponding to penetrating arterioles which we use to split up the sum over terminal nodes into inflows (QMCA) and outflows. Thus, we can compute the inflow at the MCA,
| (5) |
To determine this flow rate, we first need to compute To obtain an expression for the rate of change of the cross-sectional area Aij, we simply differentiate (1), resulting in where we in the last step again use (1) or Second, we need to express the terminal flow rates qi(t). The following relation holds: Hence, we can fully determine the value of (5) through computation of (3) and (2). We set the initial values consistent with a constant downstream flow (in-/outflow of the network) that terminates at the time of occlusion. For the network graph we used data from(40) and data to reflect realistic vessel diameters, adapted algorithmically to optimize power dissipation losses inside the network(79). The algorithm was implemented in Matlab using a simple Euler forward method.
Estimated diffusion calculations:
The diffusion of tracer presented in Fig. S3E was estimated as previously described(80, 81). To our knowledge, the diffusion coefficient of gadobutrol in live rodent brain has not been reported in the literature, so we estimated the degree of penetration of a similar sized tracer (3kDa dextran). Calculations were done using the error function solution for plane diffusion into a half-space: C = C0erfc(x/2sqrt(D*t)) and an effective diffusivity D*= 5.36 X 10−7 cm2/s(82). Since the D* value was calculated for the cortex of live normoxic rats, we also used a value D*= 0.284×10−7 cm2/s after 1 min of terminal ischemia induced by intracardiac 1M KCl to better reflect the reduced extracellular space after the spreading depolarization as seen in Fig. S2D-F.
Statistical analysis.
All statistical analyses were done in GraphPad Prism 8. Data in all graphs are plotted as mean ± standard error of the mean (SEM) over the individual data points/lines from each mouse. Parametric and nonparametric tests were selected based on normality testing and are reported in the figure legends. Normality tests were chosen depending on the sample size (D’Agostino Pearson omnibus test where possible and Shapiro-Wilk if the n was too small). Sphericity was not assumed in all repeated measures two-way ANOVAs and a Geisser-Greenhouse correction was performed. All hypothesis testing was two-tailed, and significance was determined at an α = 0.05.
Supplementary Material
Acknowledgments:
We would like to thank Dan Xue for assistance with the illustrations.
Funding: National Institute of Neurological Disorders and Stroke and the National Institute on Aging (US National Institutes of Health: R01NS100366 to MN; RF1AG057575 to MN, DHK, JHT; and K08NS089830 to RIM), the US Army Research Office (grant no. MURI W911NF1910280 to MN, DHK, and JHT), Fondation Leducq Transatlantic Networks of Excellence Program, Novo Nordisk and Lundbeck Foundations, and the EU Horizon 2020 research and innovation programme (grant no. 666881; SVDs@target). The views and conclusions contained in this manuscript are solely those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the National Institutes of Health, Army Research Office, or the US Government. The US Government is authorized to reproduce and distribute reprints for Government purposes notwithstanding any copyright notation herein.
Footnotes
Competing interests: Authors declare no competing interests; and
Data and materials availability: All data are available in the manuscript or the supplementary material. Glt1-GCaMP7 mice (No. RBRC09650) are available through the RIKEN BioResource Center repository under a material transfer agreement with RIKEN BRC.
References & Notes:
- 1.Benjamin EJ et al. , Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 5; 139, e56–e528 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Feigin VL et al. , Update on the Global Burden of Ischemic and Hemorrhagic Stroke in 1990–2013: The GBD 2013 Study. Neuroepidemiology 45, 161–176 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iadecola C, Anrather J, Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 14, 1363–1368 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rungta RL et al. , The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell 161, 610–621 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V, Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol 6, 258–268 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang D, Bhatta S, Gerzanich V, Simard JM, Cytotoxic edema: mechanisms of pathological cell swelling. Neurosurgical focus 22, E2 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen AJ, Nedergaard M, Brain ion homeostasis in cerebral ischemia. Neurochemical pathology 9, 195–209 (1988). [DOI] [PubMed] [Google Scholar]
- 8.Stokum JA, Gerzanich V, Simard JM, Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab 36, 513–538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klatzo I, Presidental address. Neuropathological aspects of brain edema. J Neuropathol Exp Neurol 26, 1–14 (1967). [DOI] [PubMed] [Google Scholar]
- 10.Van Harreveld A, Changes in the diameter of apical dendrites during spreading depression. Am J Physiol 192, 457–463 (1958). [DOI] [PubMed] [Google Scholar]
- 11.Ochs S, Van Harreveld A, Cerebral impedance changes after circulatory arrest. Am J Physiol 187, 180–192 (1956). [DOI] [PubMed] [Google Scholar]
- 12.Dreier JP, Lemale CL, Kola V, Friedman A, Schoknecht K, Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology 134, 189–207 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Dreier JP, The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17, 439–447 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Nedergaard M, Astrup J, Infarct rim: effect of hyperglycemia on direct current potential and [14C]2-deoxyglucose phosphorylation. J Cereb Blood Flow Metab 6, 607–615 (1986). [DOI] [PubMed] [Google Scholar]
- 15.Klatzo I, Pathophysiological aspects of brain edema. Acta Neuropathol 72, 236–239 (1987). [DOI] [PubMed] [Google Scholar]
- 16.Knowland D et al. , Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 82, 603–617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang EJ et al. , Blood-brain barrier opening to large molecules does not imply blood-brain barrier opening to small ions. Neurobiol Dis 52, 204–218 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Young W, Rappaport ZH, Chalif DJ, Flamm ES, Regional brain sodium, potassium, and water changes in the rat middle cerebral artery occlusion model of ischemia. Stroke 18, 751–759 (1987). [DOI] [PubMed] [Google Scholar]
- 19.Battey TW et al. , Brain edema predicts outcome after nonlacunar ischemic stroke. Stroke 45, 3643–3648 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatashita S, Hoff JT, Salamat SM, Ischemic brain edema and the osmotic gradient between blood and brain. J Cereb Blood Flow Metab 8, 552–559 (1988). [DOI] [PubMed] [Google Scholar]
- 21.Hatashita S, Hoff JT, Brain edema and cerebrovascular permeability during cerebral ischemia in rats. Stroke 21, 582–588 (1990). [DOI] [PubMed] [Google Scholar]
- 22.Thrane AS, Rangroo Thrane V, Nedergaard M, Drowning stars: reassessing the role of astrocytes in brain edema. Trends Neurosci 37, 620–628 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iliff JJ et al. , A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4, 147ra111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie L et al. , Sleep drives metabolite clearance from the adult brain. Science 342, 373–377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inoue Y et al. , Detection of necrotic neural response in super-acute cerebral ischemia using activity-induced manganese-enhanced (AIM) MRI. NMR Biomed 23, 304–312 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Gotoh O, Asano T, Koide T, Takakura K, Ischemic brain edema following occlusion of the middle cerebral artery in the rat. I: The time courses of the brain water, sodium and potassium contents and blood-brain barrier permeability to 125I-albumin. Stroke 16, 101–109 (1985). [DOI] [PubMed] [Google Scholar]
- 27.Sisson WB, Oldendorf WH, Brain distribution spaces of mannitol-3H, inulin-14C, and dextran-14C in the rat. Am J Physiol 221, 214–217 (1971). [DOI] [PubMed] [Google Scholar]
- 28.Iliff JJ et al. , Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest 123, 1299–1309 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dreier JP, Reiffurth C, The stroke-migraine depolarization continuum. Neuron 86, 902–922 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Bogdanov VB et al. , Susceptibility of Primary Sensory Cortex to Spreading Depolarizations. J Neurosci 36, 4733–4743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enger R et al. , Dynamics of Ionic Shifts in Cortical Spreading Depression. Cereb Cortex 25, 4469–4476 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monai H et al. , Calcium imaging reveals glial involvement in transcranial direct current stimulation-induced plasticity in mouse brain. Nat Commun 7, 11100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plog BA et al. , Transcranial optical imaging reveals a pathway for optimizing the delivery of immunotherapeutics to the brain. JCI Insight 3, 120922 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strong AJ et al. , Peri-infarct depolarizations lead to loss of perfusion in ischaemic gyrencephalic cerebral cortex. Brain 130, 995–1008 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Shin HK et al. , Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab 26, 1018–1030 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Dreier JP et al. , Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab 18, 978–990 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Chuquet J, Hollender L, Nimchinsky EA, High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci 27, 4036–4044 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mestre H et al. , Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat Commun 9, 4878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schain AJ, Melo-Carrillo A, Strassman AM, Burstein R, Cortical Spreading Depression Closes Paravascular Space and Impairs Glymphatic Flow: Implications for Migraine Headache. J Neurosci 37, 2904–2915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blinder P, Shih AY, Rafie C, Kleinfeld D, Topological basis for the robust distribution of blood to rodent neocortex. Proc Natl Acad Sci U S A 107, 12670–12675 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dreier JP et al. , Ischaemia triggered by spreading neuronal activation is inhibited by vasodilators in rats. J Physiol 531, 515–526 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iijima T, Mies G, Hossmann KA, Repeated negative DC deflections in rat cortex following middle cerebral artery occlusion are abolished by MK-801: effect on volume of ischemic injury. J Cereb Blood Flow Metab 12, 727–733 (1992). [DOI] [PubMed] [Google Scholar]
- 43.Gill R, Andine P, Hillered L, Persson L, Hagberg H, The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischaemia in the rat. J Cereb Blood Flow Metab 12, 371–379 (1992). [DOI] [PubMed] [Google Scholar]
- 44.Lauritzen M, Hansen AJ, The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J Cereb Blood Flow Metab 12, 223–229 (1992). [DOI] [PubMed] [Google Scholar]
- 45.Mestre H et al. , Aquaporin-4-dependent glymphatic solute transport in the rodent brain. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao X, Derugin N, Manley GT, Verkman AS, Reduced brain edema and infarct volume in aquaporin-4 deficient mice after transient focal cerebral ischemia. Neurosci Lett 584, 368–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manley GT et al. , Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6, 159–163 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Igarashi H, Huber VJ, Tsujita M, Nakada T, Pretreatment with a novel aquaporin 4 inhibitor, TGN-020, significantly reduces ischemic cerebral edema. Neurol Sci 32, 113–116 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pirici I et al. , Inhibition of Aquaporin-4 Improves the Outcome of Ischaemic Stroke and Modulates Brain Paravascular Drainage Pathways. Int J Mol Sci 19, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Betz AL, Keep RF, Beer ME, Ren XD, Blood-brain barrier permeability and brain concentration of sodium, potassium, and chloride during focal ischemia. J Cereb Blood Flow Metab 14, 29–37 (1994). [DOI] [PubMed] [Google Scholar]
- 51.Ishimaru S, Hossmann KA, Relationship between cerebral blood flow and blood-brain barrier permeability of sodium and albumin in cerebral infarcts of rats. Acta Neurochir Suppl (Wien) 51, 216–219 (1990). [DOI] [PubMed] [Google Scholar]
- 52.Lo WD, Betz AL, Schielke GP, Hoff JT, Transport of sodium from blood to brain in ischemic brain edema. Stroke 18, 150–157 (1987). [DOI] [PubMed] [Google Scholar]
- 53.Ito U, Hakamata Y, Kawakami E, Oyanagi K, Temporary [corrected] cerebral ischemia results in swollen astrocytic end-feet that compress microvessels and lead to delayed [corrected] focal cortical infarction. J Cereb Blood Flow Metab 31, 328–338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khennouf L et al. , Active role of capillary pericytes during stimulation-induced activity and spreading depolarization. Brain 141, 2032–2046 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simard JM et al. , Glibenclamide in cerebral ischemia and stroke. Neurocritical care 20, 319–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gaberel T et al. , Impaired glymphatic perfusion after strokes revealed by contrast-enhanced MRI: a new target for fibrinolysis? Stroke 45, 3092–3096 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Luckl J et al. , Peri-infarct flow transients predict outcome in rat focal brain ischemia. Neuroscience 226, 197–207 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hartings JA et al. , The continuum of spreading depolarizations in acute cortical lesion development: Examining Leao’s legacy. J Cereb Blood Flow Metab 37, 1571–1594 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yao X et al. , Aquaporin-4 regulates the velocity and frequency of cortical spreading depression in mice. Glia 63, 1860–1869 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Enger R et al. , Deletion of Aquaporin-4 Curtails Extracellular Glutamate Elevation in Cortical Spreading Depression in Awake Mice. Cereb Cortex 27, 24–33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thrane AS et al. , In vivo NADH fluorescence imaging indicates effect of aquaporin-4 deletion on oxygen microdistribution in cortical spreading depression. J Cereb Blood Flow Metab 33, 996–999 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bere Z, Obrenovitch TP, Bari F, Farkas E, Ischemia-induced depolarizations and associated hemodynamic responses in incomplete global forebrain ischemia in rats. Neuroscience 260, 217–226 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Hartings JA et al. , Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction. Brain 140, 2673–2690 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Woitzik J et al. , Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology 80, 1095–1102 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Hinzman JM et al. , Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain 137, 2960–2972 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Dreier JP et al. , Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 132, 1866–1881 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luckl J et al. , The negative ultraslow potential, electrophysiological correlate of infarction in the human cortex. Brain 141, 1734–1752 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xavier ALR et al. , Cannula Implantation into the Cisterna Magna of Rodents. J Vis Exp, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Albayram O et al. , Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun 8, 1000 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karimy JK et al. , Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat Med 23, 997–1003 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Karimy JK et al. , A novel method to study cerebrospinal fluid dynamics in rats. J Neurosci Methods 241, 78–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McBride DW, Klebe D, Tang J, Zhang JH, Correcting for Brain Swelling’s Effects on Infarct Volume Calculation After Middle Cerebral Artery Occlusion in Rats. Transl Stroke Res 6, 323–338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nevins TD, Kelley DH, Front tracking for quantifying advection-reaction-diffusion. Chaos 27, 043105 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Avants BB et al. , A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage 54, 2033–2044 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Avants B et al. , Multivariate analysis of structural and diffusion imaging in traumatic brain injury. Acad Radiol 15, 1360–1375 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Johnson GA et al. , Waxholm space: an image-based reference for coordinating mouse brain research. Neuroimage 53, 365–372 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ouellette NT, Xu HT, Bodenschatz E, A quantitative study of three-dimensional Lagrangian particle tracking algorithms. Exp Fluids 40, 301–313 (2006). [Google Scholar]
- 78.Kelley DH, Ouellette NT, Using particle tracking to measure flow instabilities in an undergraduate laboratory experiment. Am J Phys 79, 267–273 (2011). [Google Scholar]
- 79.Martens EA, Klemm K, Transitions from Trees to Cycles in Adaptive Flow Networks. Frontiers in Physics 5, (2017). [Google Scholar]
- 80.Sykova E, Nicholson C, Diffusion in brain extracellular space. Physiol Rev 88, 1277–1340 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ringstad G et al. , Brain-wide glymphatic enhancement and clearance in humans assessed with MRI. JCI Insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thorne RG, Nicholson C, In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci U S A 103, 5567–5572 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.