

Abstract
The molecular mechanisms involved in induced pluripotent stem cells (iPSCs) generation are poorly understood. The cell death machinery of apoptosis-inducing caspases have been shown to facilitate the process of iPSCs reprogramming. However, the effect of other cell death processes, such as programmed necrosis (necroptosis), on iPSCs induction has not been studied. In this study, we investigated the role of receptor-interacting protein kinase 3 (RIP3), an essential regulator of necroptosis, in reprogramming mouse embryonic fibroblast cells (MEFs) into iPSCs. RIP3 was found to be upregulated in iPSCs compared to MEFs. Deletion of RIP3 dramatically suppressed the reprogramming of iPSCs (~82%). RNA-seq analysis and qRT-PCR showed that RIP3 KO MEFs expressed lower levels of genes that control cell cycle progression and cell division and higher levels of extracellular matrix-regulating genes. The growth rate of RIP3 KO MEFs was significantly slower than WT MEFs. These findings can partially explain the inhibitory effects of RIP3 deletion on iPSCs generation and show for the first time that the necroptosis kinase RIP3 plays an important role in iPSC reprogramming. In contrast to RIP3, the kinase and scaffolding functions of RIPK1 appeared to have distinct effects on reprogramming.
Keywords: Reprogramming, Programmed necrosis, Necroptosis, Cell death, RIPK3, RIPK, RIP
1. Introduction
Reprogramming of differentiated cells into induced pluripotent stem cells (iPSCs) can be achieved by ectopic expression of certain combinations of transcription factors, such as Oct4, Sox2, Klf4, and c-Myc, or Oct4, Sox2, Nanog, and Lin28 (Liu et al., 2014;Takahashi and Yamanaka, 2006,2016;Yu et al., 2007;Zhao et al., 2009). Given that iPSCs are molecularly and functionally similar to embryonic stem cells (ESCs) and can be generated from patient somatic cells without destroying human embryos, they hold great promise for the fields of regenerative medicine, disease modeling, and drug discovery (Liu et al., 2014;Takahashi and Yamanaka, 2006,2016;Trounson and DeWitt, 2016;Yu et al., 2007;Zhao et al., 2009). However, the mechanisms underlying transcription factor-mediated reprogramming are still poorly understood (Li et al., 2013;Smith et al., 2016;Takahashi and Yamanaka, 2016).
Multiple factors contribute to the iPSCs reprogramming process (Jaenisch and Young, 2008;Smith et al., 2016;Yamanaka, 2009;Yamanaka and Blau, 2010), which has been modeled as a stochastic process that can be accelerated through both cell-division-rate-dependent and cell-division-rate- independent manners (Hanna et al., 2009;Smith et al., 2016). However, cell death pathways have not been well studied in iPSCs reprogramming process. An interesting, yet surprising, study by Li et al. showed that inhibition of caspase 3 or 8 in human fibroblast cells partially or completely, respectively, prevented the induction of iPSCs, indicating that apoptotic caspases positively regulate iPSCs reprogramming from human fibroblasts (Li et al., 2010a). Besides apoptosis, other cell death processes exist, such as necrosis. Necrosis or necroptosis was considered for years to be an unregulated accidental cell death process (Pasparakis and Vandenabeele, 2015). However, later evidence proved that necrosis is also a regulated and controlled form of cell death (Trichonas et al., 2010; Pasparakis and Vandenabeele, 2015). Receptor interacting protein kinase 3 (RIPK3 or RIP3) is a central mediator of necroptosis and has been shown to be involved in numerous pathological conditions (Galluzzi et al., 2017;Murakami et al., 2012;Orozco and Oberst, 2017;Trichonas et al., 2010;Weinlich et al., 2016). However, its role in iPSC induction has not been studied thus far.
The incomplete understanding of the mechanisms underlying transcription factor-mediated reprogramming limits our ability to further enhance the efficiency of iPSC induction. Since previous work has shown that caspases, which regularly promote cell death in adult cells, actually promote iPSC induction, we wanted to investigate if this can be expanded to other non-apoptotic cell death machinery. For this reason, we investigated the effect of the main necroptosis mediator RIP3 on the process of transcription factor-mediated reprogramming of mouse embryonic fibroblast cells (MEFs) to iPSCs and its possible mechanisms.
2. Methods
Detailed methods are described in the supplementary information.
3. Results
3.1. Expression of RIP3 is upregulated in induced pluripotent stem cells (iPSCs)
To examine the potential role of RIP3 in iPSCs reprogramming process, we first examined the expression of RIP3 at the protein level using immunoblot analysis in mouse iPSCs (miPSCs) generated from MEFs. We observed that the protein level of RIP3 was increased in miPSCs compared to MEFs (Fig. 1A, B). Time course analysis during the reprogramming process showed that the protein level of RIP3 was gradually increased during the second half of the reprogramming process (Fig. 1C, D). This indicates that RIP3, a key regulator of necroptosis, might be involved in regulation of somatic cells reprogramming to iPSCs.
Fig. 1.

RIP3 is upregulated during the iPSCs reprogramming process. (A) Upper figure: Immunoblot analysis of RIP3 in MEFs and 3 different MEFs-derived iPSCs colonies (miPSCs) generated by the three transcription factors (Oct4, Sox2, Klf4; OSK) combined with the small molecule medium supplements. Ponceau S staining was used to confirm equal protein loading. Lower figure: Immunoblot analysis of RIP3 in the commercially available MEFs (#PMEF-CFL from Millipore) and the commercially available miPSC (#iPS02M from ALSTEM) (B) Density values of RIP3 bands in the upper figure of (A) are graphically expressed. (C) Immunoblot analysis of RIP3 in MEFs transduced with OSK and treated with the small molecule medium supplements. Transduced cells were analyzed for RIP3 at the indicated time points after lentiviral transduction. Ponceau S staining was used to confirm equal protein loading. (D) Density values of RIP3 bands in (C) are graphically expressed. Data are means ± SEM. *P < .05 and ***P < .001 from two-tailed Student’s t-test.
3.2. Deletion of RIP3 reduces iPSCs reprogramming efficiency
To investigate the effect of RIP3 on iPSCs reprogramming, the efficiency of iPSCs colony formation was compared between RIP3 KO and WT MEFs. The same number of MEF cells of each genotype were seeded and then reprogrammed to iPSCs. The resulting iPSC colonies were stained at day 15 with alkaline phosphatase (AP) and quantified for reprogramming efficiency. Deletion of RIP3 dramatically decreased the number of AP-positive iPSC colonies (~ 82% less colonies in RIP3 KO compared to WT MEFs) (Fig. 2B, C), indicating that RIP3 plays a critical role in facilitating the iPSCs reprogramming process. Knockout of RIP3 was verified by western blotting (Fig. 2A) and the pluripotency of the resulting iPSCs was confirmed by immunostaining with pluripotency markers (Fig. 2D).
Fig. 2.
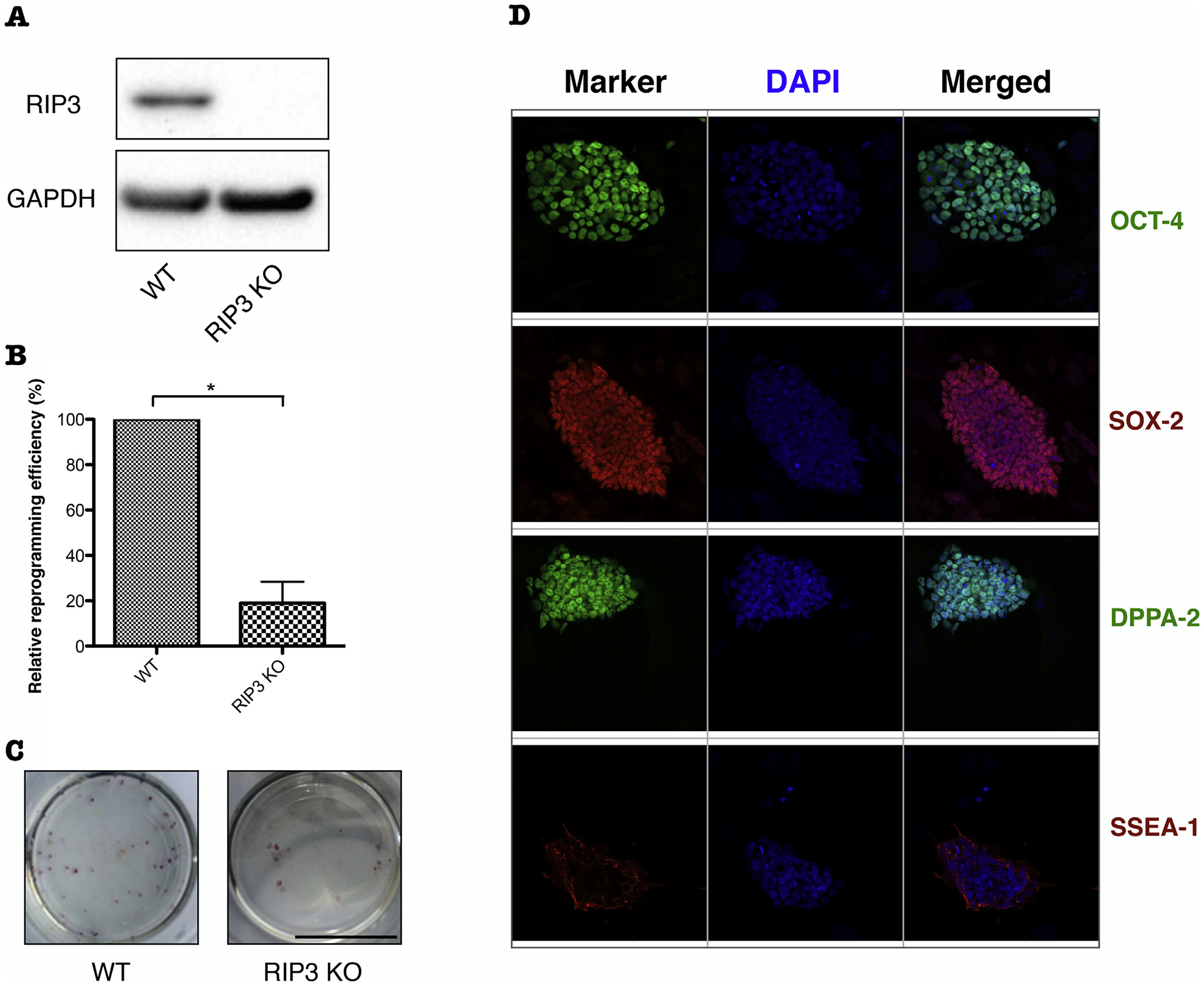
Deletion of RIP3 suppresses iPSCs reprogramming. (A) Knockout (KO) of endogenous RIP3. Immunoblot analysis of RIP3 in WT and RIP3 KO MEFs. GAPDH was detected as a loading control. (B) Frequency of AP-positive iPSCs colonies derived from RIP3 KO relative to those derived from WT MEFs at day 15 after transduction. (C) Representative dishes of AP staining of iPSC colonies derived from MEFs in WT and RIP3 KO genotypes. (D) Characterization of RIP3 KO miPSCs. Immunostaining patterns for OCT-4, SOX-2, DPPA-2, and SSEA-1. Data are means ± SEM. *P < .05 from two-tailed Student’s t-test.
3.3. RIP3 KO MEFs display lower expression of cell cycle genes and a slower proliferation rate compared to WT MEFs
To investigate the molecular mechanisms for the inhibitory effect of RIP3 deletion on iPSCs reprogramming, we performed whole transcriptome expression analysis (RNA-seq) of passage 2 of WT and RIP3 KO MEFs. The results showed that the gene expression profile of RIP3 KO MEFs was distinct from WT cells as expected with several hundred genes changing > 2 folds and 50 genes changing > 5 folds (Fig. 3A). To better understand the RNA-Seq changes, gene ontology (GO) analysis was performed revealing that cell cycle progression and cell division were downregulated in RIP3 KO MEFs compared to WT cells (Fig. 3B, C). We validated this by using quantitative RT-PCR of select genes, such as cyclin-dependent kinase (CDK1), CDK6, cell division cycle 6 (CDC6), and CDC7 (Fig. 3D). In addition, the expression level of protein regulator of cytokinesis 1 (Prc1), epithelial cell transforming 2 (Ect2), and aurora kinase A (Aurka), which function mainly in cell division, was also decreased in RIP3 KO compared to WT MEFs (Fig. 3E).
Fig. 3.
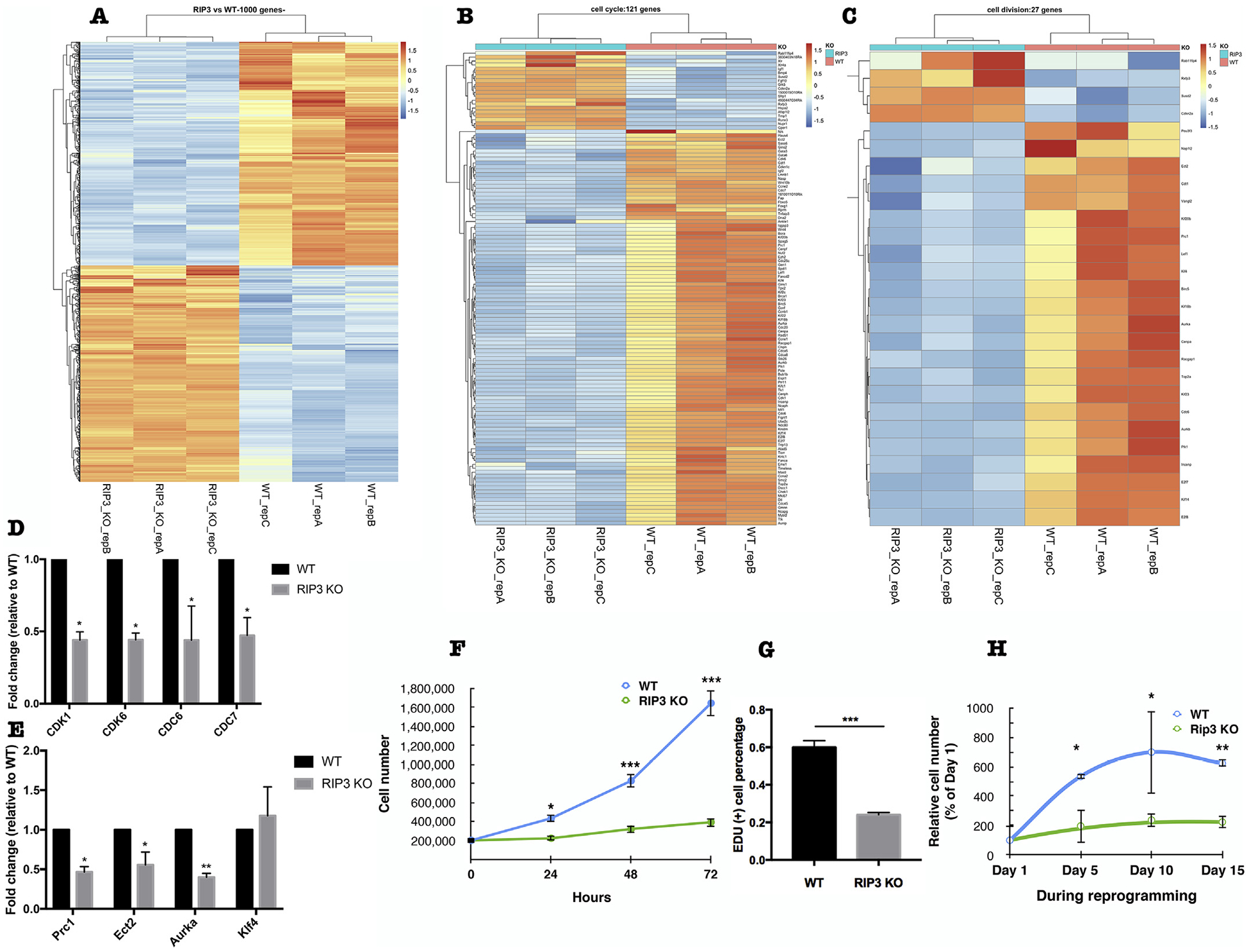
Deletion of RIP3 leads to lower expression of cell cycle and cell division genes and a slower proliferation rate of MEFs. (A) Transcriptome expression analysis (RNA-seq) of WT and RIP3 KO MEF cells. Heatmap displaying the differential gene expression patterns of passage 2 WT and RIP3 KO MEF cells. Genes displayed are the top 1000 genes that showed statistical significant difference based on fold changes of 2 or more and FDR adjusted P value < .05 between WT and RIP3 KO. (B) Heatmap displaying the differential expression patterns of genes related to cell cycle progression and (C) cell division in passage 2 WT and RIP3 KO MEF cells. Cell cycle progression and cell division and genes related to them are chosen from the results of GO term analysis of the differentially expressed genes between WT and RIP3 KO MEF cells. (D) Validation of some cell cycle-related genes and (E) cell division-related genes identified in (B) and (C) by quantitative RT-PCR. (F) Growth curve of passage 3 WT and RIP3 KO MEF cells. (G) EdU cell proliferation assay of passage 3 WT and RIP3 KO MEF cells. (H) Growth curve of passage 3 OSK-transduced WT and RIP3 KO MEF cells during the reprogramming process as determined by trypan blue exclusion counting. Data are means ± SEM. *P < .05, **P < .01 from two-tailed Student’s t-test.
Proliferation rate of somatic cells affects the efficiency of their reprogramming to iPSCs, with high proliferation rate being required for reprogramming (Ruiz et al., 2011). Considering the RNA-seq results, we assessed the proliferation of RIP3 KO MEFs. We observed that the growth rate of RIP3 KO MEFs was significantly lower than WT MEFs (Fig. 3F, G). Furthermore, the growth rate of RIP3 KO MEFs during the reprogramming process was also significantly lower than WT MEFs (Fig. 3H).
This link between RIP3 and cell proliferation explain, at least partially, the mechanism by which deletion of RIP3 leads to suppression of iPSCs reprogramming.
3.4. RIP3 KO MEFs express higher levels of extracellular matrix (ECM) genes
Prior studies have suggested that the ECM is a barrier for iPSCs reprogramming (Jiao et al., 2013). Interestingly, GO analysis of the differentially expressed genes from the RNA-seq showed that genes whose function is related to ECM were upregulated in RIP3 KO compared to WT MEFs (Fig. 4A). qRT-PCR validation showed that the expression level of collagen type III alpha 1 chain (Col3a1), Col8a1, Col12a1, and hyaluronan and proteoglycan link protein 1 (Hapln1), whose functions are related to ECM, were increased in RIP3 KO compared to WT MEFs (Fig. 4B). This novel link between RIP3 and ECM could also partially explain the inhibitory effects of deleting RIP3 on iPSCs reprogramming.
Fig. 4.

Deletion of RIP3 leads to higher expression of extracellular matrix (ECM) genes in MEFs. (A) Heatmap displaying the differential expression patterns of genes related to extracellular matrix in passage 2 WT and RIP3 KO MEF cells. Extracellular matrix and genes related to it are chosen from the results of GO term analysis of the differentially expressed genes between WT and RIP3 KO MEF cells. (B) Validation of some extracellular matrix-related genes identified in (A) by quantitative RTPCR. Data are means ± SEM. *P < .05, ***P < .001 from two-tailed Student’s t-test.
Due to technical difficulties, we were not able to overexpress RIP3 in MEFs before and during reprogramming to assess whether this could reverse the effects observed in MEFs that are deficient in RIP3.
4. Discussion
In this study, necroptotic RIP3 kinase is shown to play an important role in reprogramming of somatic cells to iPSCs. RIP3 was upregulated during iPSC induction and deletion of RIP3 led to a dramatic suppression of iPSCs induction. Mechanistically this could be explained, at least partially, by the fact that RIP3 KO MEFs had lower expression levels of cell cycle and cell division related genes, slower growth rate, and higher expression of ECM related genes.
Cell death is a substantial component of homeostasis in multicellular organisms. It is generally thought that apoptosis has a major physiologic role during embryonic development, while necroptosis is thought to be mainly involved in pathologic states (Berger et al., 2014;Grootjans et al., 2017;Newton et al., 2004). Decreasing cell death and increasing cell survival is known to enhance iPSCs reprogramming (Jiang et al., 2016). Surprisingly, however, inhibition of caspase 3 and caspase 8 in human fibroblasts has been shown to partially or completely, respectively, prevent the induction of iPSCs (Li et al., 2010a). This effect was mediated through targeting of the retinoblastoma susceptibility (Rb) gene, which is a tumor suppressor gene that regulates cell cycle progression, by caspase 3 and caspase 8 during reprogramming (Li et al., 2010a).
Our study shows for the first time that RIP3, a main regulator of the other major cell death pathway (necroptosis), also affects reprogramming efficiency likely through effects on genes that regulate cell cycle progression and ECM. Few prior reports have looked at RIP3 effects on cellular proliferation. Elevated expression of RIP3 suppresses the growth of vascular smooth muscle cells through inhibiting phosphoinositide 3-kinase-Akt axis, while silencing RIP3 gene enhances serum- and platelet-derived growth factor-induced cell proliferation (Li et al., 2010b). Overexpression of RIP3 has been shown to inhibit the growth of intrahepatic and lung tumors in vivo, although it did not affect the cell proliferation rate (Vucur et al., 2013;Yang et al., 2017). Similarly, deleting RIP3 in acute myeloid leukemia (AML) cells did not significantly affect the cell proliferation rate, but led to partial cellular differentiation and decreased the colony formation ability of the cells (Xin et al., 2017). Although opposite to some published reports in other cell types, our findings that RIP3 deletion leads to slower MEFs growth rate further confirms the potential role of RIP3 as a cell growth regulator. This role, however, seems to be cell type- and condition-dependent. A proteome-wide analysis of RIP3-regulated phosphorylation sites in macrophages and MEFs isolated from wild type and Rip3 KO mice found that a large number of the identified phosphopeptides were exclusive to macrophages or to MEFs, indicating that cell type-specific function of RIP3 exists (Wu et al., 2012). Interestingly, many of the RIP3-regulated phosphoproteins were functionally associated with the cell cycle (Wu et al., 2012).
Stem cell and cell cycle machineries are intertwined and mechanisms controlling the cell cycle are an integral mechanistic part of the pluripotent state of stem cells (Boward et al., 2016;Kareta et al., 2015b). Cell cycle regulation is also critical for establishment as well as maintenance of iPSCs (Boward et al., 2016;Ruiz et al., 2011), as evident by multiple factors. First, there is a strong link between the proliferative capacity of somatic cells and their ability to be reprogrammed (Boward et al., 2016;Utikal et al., 2009). Rapid proliferation of the starting cell population enhances the reprogramming and enriching for proliferating cells increases reprogramming efficiency (Ghule et al., 2011;Kareta et al., 2015b;Roccio et al., 2013;Ruiz et al., 2011). Second, cell cycle genes are upregulated and cell cycle remodeling occurs early during reprogramming (Boward et al., 2016;Cacchiarelli et al., 2015;Kareta et al., 2015b;Ruiz et al., 2011). Third, cells with CDK inhibition become refractive to reprogramming (Kawamura et al., 2009;Li et al., 2009;Marión et al., 2009;Zhu et al., 2016) and ectopic expression of CDK-cyclin complexes increases the reprogramming efficiency (Boward et al., 2016;Ruiz et al., 2011). Lastly, reducing factors that antagonizes cell division, such as Rb, Cyclin-dependent kinase inhibitors (CDKIs), and p53 enhances reprogramming (Kareta et al., 2015a,b;Li et al., 2012;Lin et al., 2014). Therefore, the reduced growth rate of RIP3 KO MEFs and the downregulation of many cell cycle and cell division genes, including CDKs, CDCs, and Aurka, in our study can largely explain the inhibitory effect of RIP3 deletion on iPSCs reprogramming. Furthermore, this novel link between RIP3 and cell cycle progression sheds light on an important mechanism that can explain the effect of RIP3 on the growth of various cell types, as previously mentioned, and might have other implications that require further studies.
Extracellular matrix (ECM) plays a role in iPSCs reprogramming. Collagen has been shown to be a barrier for reprogramming MEFs to iPSCs and downregulation of collagen gene expression can significantly improve the reprogramming efficiency (Jiao et al., 2013). The relationship between RIP3 and extracellular matrix has not been widely studied. One study showed that deletion of RIP3 reduces the levels of fibrosis and Col1a1 in the liver of a non-alcoholic steatohepatitis mouse model (Gautheron et al., 2014). Our novel finding that RIP3 KO MEFs express higher levels of ECM regulating genes than WT MEFs may, at least partially, explain the results of reduced iPSCs reprogramming efficiency and provides a new insight regarding other functions of RIP3. Importantly, previous studies have shown that ECM collagen inhibits the growth and alter the cell cycle progression of various cancer cell lines and skin fibroblasts (Ivanov et al., 2007;Koohestani et al., 2013;Rhudy and McPherson, 1988). This suggests that the increased expression of ECM genes in RIP3 KO MEFs could reduce iPSCs reprogramming efficiency through affecting the cell cycle progression and cell proliferation, as we show in our results, rather than by a completely independent mechanism.
Besides RIP3, Receptor Interacting Protein kinase 1 (RIP1) is another key regulator of necroptosis, that can also promote cell survival depending on the situation (Christofferson et al., 2014). Similar to its complex role in cell survival and death, it also appears to play a complicated role in iPSCs induction. We observed that the protein level of RIP1 was reduced in iPSCs compared to MEFs (S Fig. 1A, B), but during reprogramming it mildly increased at day 4 after induction followed by a gradual decrease as reprogramming progressed (S Fig. 1C, D). Deletion of RIP1 showed that reprogramming efficiency of RIP1 KO MEFs was similar to WT MEFs in half of the experiments and higher in another half, leading to an overall statistically insignificant increase (S Fig. 2). Furthermore, reprogramming efficiency of kinase dead RIP1 MEFs or RIP1 kinase inhibitor-treated WT MEFs was lower than WT MEFs (S Fig. 1D, E). These results indicate that RIP1 plays a role in iPSCs generation, but kinase and scaffolding functions of RIP1 have distinct and maybe opposing effects on this process. This complex result on the role of RIP1 in iPSC reprogramming is not unlike the effects seen in cell death and survival (Christofferson et al., 2014), and further studies are needed to completely understand the effect of RIP1 vs RIP3 on iPSCs reprogramming.
In summary, our results shed light on unknown functions of RIP3 and show for the first time that the necroptosis-mediator RIP3 Kinase is important for iPSCs reprogramming through, at least in part, cell cycle and ECM regulation. This adds to our knowledge about the role of cell death regulator in iPSC induction and improves our understanding of this important and complicated process.
Supplementary Material
Acknowledgments
We thank: Dr. Gustavo Mostoslavsky from Center for Regenerative Medicine, BUSM for his technical support in this project; Dr. Michelle Kelliher from UMass Medical School and Dr. Douglas Green from St. Jude Children’s Research Hospital for providing us with cell lines; Biostatistics and Bioinformatics Core at HSPH for the analysis of the RNA-seq data. Grant supportNEIR21EY023079-01A1,R01-EY025362-01;Yeatts Family Foundation;Loefers Family Fund;Macula Society Research Grant award; a Physician Scientist Award fromRPBand theAlcon Research Institute Young Investigator Award, an unrestricted grant from theResearch to Prevent Blindness FoundationandNEIgrantEY014104.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.scr.2019.101387.
References
- Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, Harris PA, Kaiser WJ, Mocarski ES, Bertin J, Gough PJ, 2014. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol 192, 5476–5480. 10.4049/jimmunol.1400499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boward B, Wu T, Dalton S, 2016. Concise review: control of cell fate through cell cycle and pluripotency networks. Stem Cells 34, 1427–1436. 10.1038/ncomms1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacchiarelli D, Trapnell C, Ziller MJ, Soumillon M, Cesana M, Karnik R, Donaghey J, Smith ZD, Ratanasirintrawoot S, Zhang X, Ho Sui SJ, Wu Z, Akopian V, Gifford CA, Doench J, Rinn JL, Daley GQ, Meissner A, Lander ES, Mikkelsen TS, 2015. Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell 162, 412–424. 10.1016/j.cell.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofferson DE, Li Y, Yuan J, 2014. Control of life-or-death decisions by RIP1 kinase. Annu. Rev. Physiol 76, 129–150. 10.1146/annurev-physiol-021113-170259. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Chan FK-M, Kroemer G, 2017. Necroptosis: mechanisms and relevance to disease. Annu. Rev. Pathol. Mech. Dis 12, 103–130. 10.1146/annurev-pathol-052016-100247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, Kreggenwinkel K, Schneider AT, Bartneck M, Neumann UP, Canbay A, Reeves HL, Luedde M, Tacke F, Trautwein C, Heikenwalder M, Luedde T, 2014. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med 6, 1062–1074. 10.15252/emmm.201403856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghule PN, Medina R, Lengner CJ, Mandeville M, Qiao M, Dominski Z, Lian JB, Stein JL, van Wijnen AJ, Stein GS, 2011. Reprogramming the pluripotent cell cycle: restoration of an abbreviated G1 phase in human induced pluripotent stem (iPS) cells. J. Cell. Physiol 226, 1149–1156. 10.1002/jcp.22440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootjans S, Vanden Berghe T, Vandenabeele P, 2017. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 24, 1184–1195. 10.1038/cdd.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, Creyghton MP, van Oudenaarden A, Jaenisch R, 2009. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 462, 595–601. 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov V, Ivanova S, Roomi MW, Kalinovsky T, Niedzwiecki A, Rath M, 2007. Naturally produced extracellular matrix inhibits growth rate and invasiveness of human osteosarcoma cancer cells. Med. Oncol 24, 209–217. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Young R, 2008. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 132, 567–582. 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Du M, Wu M, Zhu Y, Zhao X, Cao X, Li X, Long P, Li W, Hu B, 2016. Phosphatidic acid improves reprogramming to pluripotency by reducing apoptosis. Stem Cells Dev. 25, 43–54. 10.1089/scd.2015.0159. [DOI] [PubMed] [Google Scholar]
- Jiao J, Dang Y, Yang Y, Gao R, Zhang Y, Kou Z, Sun X-F, Gao S, 2013. Promoting reprogramming by FGF2 reveals that the extracellular matrix is a barrier for reprogramming fibroblasts to pluripotency. Stem Cells 31, 729–740. 10.1002/stem.1318. [DOI] [PubMed] [Google Scholar]
- Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos A-F, Cecchini MJ, Spacek D, Batista LFZ, O’Brien M, Ng Y-H, Ang CE, Vaka D, Artandi SE, Dick FA, Brunet A, Sage J, Wernig M, 2015a. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 16, 39–50. 10.1016/j.stem.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kareta MS, Sage J, Wernig M, 2015b. Crosstalk between stem cell and cell cycle machineries. Curr. Opin. Cell Biol 37, 68–74. 10.1016/j.ceb.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Izpisua Belmonte JC, 2009. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460, 1140–1144. 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koohestani F, Braundmeier AG, Mahdian A, Seo J, Bi J, Nowak RA, 2013. Extracellular matrix collagen alters cell proliferation and cell cycle progression of human uterine leiomyoma smooth muscle cells. PLoS One 8, e75844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Collado M, Villasante A, Strati K, Ortega S, Canamero M, Blasco MA, Serrano M, 2009. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460, 1136–1139. 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, He Z, Shen J, Huang Q, Li W, Liu X, He Y, Wolf F, Li C-Y, 2010a. Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Stem Cells 7, 508–520. 10.1016/j.stem.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Li G, Lan X, Zheng M, Chen K-H, Cao C-M, Xiao R-P, 2010b. Receptor interacting protein 3 suppresses vascular smooth muscle cell growth by inhibition of the phosphoinositide 3-kinase-akt axis. J. Biol. Chem 285, 9535–9544. 10.1161/01.CIR.98.16.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Collado M, Villasante A, Matheu A, Lynch CJ, Canamero M, Rizzoti K, Carneiro C, Martinez G, Vidal A, Lovell-Badge R, Serrano M, 2012. p27(Kip1) directly represses Sox2 during embryonic stem cell differentiation. Cell Stem Cell 11, 845–852. 10.1016/j.stem.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Feng H, Gu H, Lewis DW, Yuan Y, Zhang L, Yu H, Zhang P, Cheng H, Miao W, Yuan W, Cheng S-Y, Gollin SM, Cheng T, 2013. The p53–PUMA axis suppresses iPSC generation. Nat. Commun 4, 1–9. 10.1038/ncomms3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-C, Murayama Y, Hashimoto K, Nakamura Y, Lin C-S, Yokoyama KK, Saito S, 2014. Role of tumor suppressor genes in the cancer-associated reprogramming of human induced pluripotent stem cells. Stem Cell Res Ther 5, 58. 10.1186/scrt447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Song Y, Yu H, Zhao T, 2014. Understanding the roadmaps to induced pluripotency. Cell Death Dis. 5 10.1038/cddis.2014.205. e1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, Blasco MA, 2009. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460, 1149–1153. 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Matsumoto H, Roh M, Suzuki J, Hisatomi T, Ikeda Y, Miller JW, Vavvas DG, 2012. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc. Natl. Acad. Sci. U. S. A 109, 14598–14603. 10.1073/pnas.1206937109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Sun X, Dixit VM, 2004. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol 24, 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco S, Oberst A, 2017. RIPK3 in cell death and inflammation: the good, the bad, and the ugly. Immunol. Rev 277, 102–112. 10.1038/icb.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Vandenabeele P, 2015. Necroptosis and its role in inflammation. Nature 517, 311–320. 10.1084/jem.20120189. [DOI] [PubMed] [Google Scholar]
- Rhudy RW, McPherson JM, 1988. Influence of the extracellular matrix on the proliferative response of human skin fibroblasts to serum and purified platelet-derived growth factor. J. Cell. Physiol 137, 185–191. 10.1002/jcp.1041370123. [DOI] [PubMed] [Google Scholar]
- Roccio M, Schmitter D, Knobloch M, Okawa Y, Sage D, Lutolf MP, 2013. Predicting stem cell fate changes by differential cell cycle progression patterns. Development 140, 459–470. 10.1242/dev.086215. [DOI] [PubMed] [Google Scholar]
- Ruiz S, Panopoulos AD, Herrerías A, Bissig K-D, Lutz M, Berggren WT, Verma IM, Belmonte JCI, 2011. A high proliferation rate is requiredfor cell reprogramming and maintenance of human embryonic stem cell identity. Curr. Biol 21, 45–52. 10.1016/j.cub.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Sindhu C, Meissner A, 2016. Molecular features of cellularreprogramming and development. Nat. Rev. Mol. Cell Biol 1–16. 10.1038/nrm.2016.6. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S, 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S, 2016. Perspectives. Nat. Rev. Mol. Cell Biol 1–11. doi: 10.1038/nrm.2016.8. [DOI] [PubMed] [Google Scholar]
- Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW, Vavvas DG, 2010. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc. Natl. Acad. Sci. U. S. A 107, 21695–21700. 10.1073/pnas.1009179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trounson A, DeWitt ND, 2016. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol 17, 194–200. 10.1038/nrm.2016.10. [DOI] [PubMed] [Google Scholar]
- Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, Khalil A, Rheinwald JG, Hochedlinger K, 2009. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460, 1145–1148. 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucur M, Reisinger F, Gautheron J, Janssen J, Roderburg C, Cardenas DV, Kreggenwinkel K, Koppe C, Hammerich L, Hakem R, Unger K, Weber A, Gassler N, Luedde M, Frey N, Neumann UP, Tacke F, Trautwein C, Heikenwalder M, Luedde T, 2013. RIP3 Inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Rep. 4, 776–790. 10.1016/j.celrep.2013.07.035. [DOI] [PubMed] [Google Scholar]
- Weinlich R, Oberst A, Beere HM, Green DR, 2016. Perspectives. Nat. Rev. Mol. CellBiol 18, 127–136. 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- Wu X, Tian L, Li J, Zhang Y, Han V, Li Y, Xu X, Li H, Chen X, Chen J, Jin W, Xie Y, Han J, Zhong C-Q, 2012. Investigation of receptor interacting protein (RIP3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol. Cell. Proteomics 11, 1640–1651. 10.1074/mcp.M112.019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin J, You D, Breslin P, Li J, Zhang J, Wei W, Cannova J, Volk A, Gutierrez R, Xiao Y, Ni A, Ng G, Schmidt R, Xia Z, Pan J, Chen H, Patel MM, Kuo PC, Nand S, Kini AR, Chen J, Zhu J, 2017. Sensitizing acute myeloid leukemia cells to induced differentiation by inhibiting the RIP1/RIP3 pathway. Leukemia 31, 1154–1165. 10.1038/leu.2016.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S, 2009. A fresh look at iPS cells. Cell 137, 13–17. 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- Yamanaka S, Blau HM, 2010. Nuclear reprogramming to a pluripotent state by three approaches. Nature 465, 704–712. 10.1038/nature09229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Li J, Yu L, Zhang Z, Xu F, Jiang L, Zhou X, He S, 2017. Regulation of RIP3 by the transcription factor Sp1 and the epigenetic regulator UHRF1 modulates cancer cell necroptosis. Cell Death Dis. 8 10.1038/cddis.2017.483. e3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA, 2007. Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920. 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zhao X-Y, Li W, Lv Z, Liu L, Tong M, Hai T, Hao J, Guo C-L, Ma Q-W, Wang L, Zeng F, Zhou Q, 2009. iPS cells produce viable mice through tetraploid complementation. Nature 461, 86–90. 10.1038/nature08267. [DOI] [PubMed] [Google Scholar]
- Zhu S, Cao J, Sun H, Liu K, Li Y, Zhao T, 2016. p18 inhibits reprogramming-through inactivation of Cdk4/6. Sci. Rep 1–8. 10.1038/srep31085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.