

Epstein-Barr virus is carried by most humans and can cause life-threatening diseases. Virus-specific T cells have been used in different clinical settings with variable success rates. One way to improve immunotherapy is to better suit T cell generation protocols to viral targets available in different diseases. BNRF1 is present in viral particles and therefore likely available as a target for T cells in diseases with virus amplification. Here, we studied healthy Epstein-Barr virus (EBV) carriers for BNRF1 immunogenicity and report our results indicating BNRF1 to be a dominant target of the EBV-specific CD4+ T cell response. BNRF1-specific CD4+ T cells were found to be cytotoxic and capable of limiting EBV-driven B cell transformation in vitro. The findings of this work contribute to forwarding our understanding of host-virus interactions during health and disease and are expected to find direct application in the generation of specific T cells for immunotherapy.
KEYWORDS: Epstein-Barr virus, T cells, cytotoxic CD4+, virion structure, antigen
ABSTRACT
Cellular immunotherapy is a proven approach against Epstein-Barr virus (EBV)-driven lymphoproliferation in recipients of hematopoietic stem cells. Extending the applicability and improving the response rates of such therapy demands improving the knowledge base. We studied 23 healthy donors for specific CD4+ T cell responses against the viral tegument protein BNRF1 and found such T cells in all seropositive donors, establishing BNRF1 as an important immune target in EBV. We identified 18 novel immune epitopes from BNRF1, all of them generated by natural processing of the full-length protein from virus-transformed lymphoblastoid cell lines (LCL). BNRF1-specific CD4+ T cells were measured directly ex vivo by a cytokine-based method, thus providing a tool to study the interaction between immunity and infection in health and disease. T cells of the cytotoxic Th1 type inhibited the proliferation of autologous LCL as well as virus-driven transformation. We infer that they are important in limiting reactivations to subclinical levels during health and reducing virus propagation during disease. The information obtained from this work will feed into data sets that are indispensable in the design of patient-tailored immunotherapeutic approaches, thereby enabling the stride toward broader application of T cell therapy and improving clinical response rates.
IMPORTANCE Epstein-Barr virus is carried by most humans and can cause life-threatening diseases. Virus-specific T cells have been used in different clinical settings with variable success rates. One way to improve immunotherapy is to better suit T cell generation protocols to viral targets available in different diseases. BNRF1 is present in viral particles and therefore likely available as a target for T cells in diseases with virus amplification. Here, we studied healthy Epstein-Barr virus (EBV) carriers for BNRF1 immunogenicity and report our results indicating BNRF1 to be a dominant target of the EBV-specific CD4+ T cell response. BNRF1-specific CD4+ T cells were found to be cytotoxic and capable of limiting EBV-driven B cell transformation in vitro. The findings of this work contribute to forwarding our understanding of host-virus interactions during health and disease and are expected to find direct application in the generation of specific T cells for immunotherapy.
INTRODUCTION
Epstein-Barr virus (EBV) is a WHO-classified carcinogenic human herpesvirus (1) associated causally or otherwise with several diseases, including cancers such as Burkitt and Hodgkin lymphoma. Following primary infection, EBV elicits a strong cellular immune response that is generally believed to be instrumental in keeping potential virus-related disorders in healthy carriers in check (2). EBV transforms B cells in vitro to generate so-called lymphoblastoid cell lines (LCL) that serve as efficient stimulators to EBV-reactive T cells in the peripheral blood of immune individuals, thereby providing a laboratory model to study immunogenicity of infected B cells and serving as readily available stimulators for selectively activating and expanding virus-specific T cells for clinical use (3).
In vitro EBV-transformed B cells express a maximum of nine so-called latency proteins (4), with a small percentage of cells undergoing spontaneous lytic replication. Early studies on the cellular immune response to EBV focused largely on understanding cytotoxic CD8+ T cell responses to the latency proteins of the virus and yielded several antigenic epitopes in different viral proteins (5). The body of knowledge on the immune responses against the virus has immensely expanded over the years, but CD4+ T cell responses have remained less well studied (6).
The early understanding of the cellular immune responses was successfully translated to clinical application in the form of adoptive transfer of T cells for the prevention as well as treatment of some EBV-associated clinical disorders, most prominently in the context of posttransplant lymphoproliferation disorders (PTLD) in recipients of hematopoietic stem cell transplants (HSCT) (7). The challenge now is to extend and improve the applicability and success of T cell therapy to further clinical disorders such as Hodgkin lymphoma and nasopharyngeal carcinoma (8). Two important factors that are relevant to this pursuit are (i) the application-specific relevance of the antigen specificity of the T cell preparations and (ii) the CD4+ component in clinically used T cell preparations (9).
Improving the response rates to immunotherapy is likely to depend on tailoring immunotherapy to the viral antigen expression context of the disease. The changes in protocols used to prepare T cells for therapy reflect this. Whereas early protocols generally used LCL as stimulators of EBV-specific T cells used for immunotherapy, approaches that are more recent have incorporated the use of antigenic peptides (10, 11). In the face of the rise to prominence and continuing improvements in T cell receptor transfer technologies, the rather laborious and time-consuming protocols using in vitro stimulation of T cells can be expected to be complemented or even replaced by tailored approaches using receptor transfer (12–14). This would be of special value in transplantations involving EBV-negative donors, where the donor lacks in naturally primed EBV-specific T cells (15). A requisite for such advancement is the expansion of our knowledge of T cell epitopes and receptors that target them.
We have observed in the past that B cells can efficiently extract structural proteins from virions following receptor-mediated uptake and present derivative peptides to CD4+ T cells (16). Viral particles contain over 30 different proteins of the virus (17). The immunogenicity of most of them remains largely unexplored. In this background, we systematically studied CD4+ T cell responses against the virion structure protein encoded by the BNRF1 gene of the virus. We chose BNRF1 for several reasons. Number one, in the past we have observed that BNRF1 is an immunodominant target in more than one LCL-stimulated CD4+ T cell line as well as a virus-like particle-stimulated CD4+ T cell line from healthy virus carriers (18, 19). Number two, in a patient with PTLD that had received EBV-derived peptide-specific T cells, we observed that CD4+ T cells targeting a peptide derived from BNRF1 expanded upon transfer, with the peak of the peptide-specific T cell numbers correlating with the drop in viral load (10). Number three, BNRF1 is highly conserved across different strains of EBV, an important aspect to consider given the recent realization that field strains of EBV can carry variant proteins, leading to variability in known T cell epitopes (20). Number four, BNRF1 has been suggested to be expressed in latently infected cells, making it an interesting candidate to target in EBV-driven malignancies (21).
The aim of this study was to assess the breadth of BNRF1-specific CD4+ T cells in healthy virus carriers and, in the process, define clinically relevant target epitopes.
Our study establishes BNRF1 as a common target of CD4+ T cells. We identified 18 novel peptide epitopes of BNRF1 that are immunogenic to CD4+ T cells. Importantly, these epitopes are generated from full-length protein and thus are potentially clinically relevant. We expect the identified peptide epitopes to be useful in future clinical settings and suggest the approach used in this work for future studies into the immunogenicity of further EBV proteins.
RESULTS
CD4+ T cells with BNRF1 reactivity can readily be expanded in vitro from the peripheral blood of EBV-seropositive individuals.
For our search of BNRF1-specific CD4+ T cells in healthy carriers, we used a protein stimulation-based enrichment approach. Recombinant BNRF1 protein was expressed in HEK293T cells with a C-terminal 6× His tag to allow purification using nickel (Ni)-NTA agarose beads and detection using anti-His antibody (Fig. 1A).
FIG 1.

BNRF1-specific CD4+ T cells are present in the peripheral blood of EBV-seropositive individuals. (A) Expression of recombinant proteins. BZLF1, transaldolase, and BNRF1 were expressed as C-terminal 6 His-tagged proteins in HEK 293T cells and purified using Ni-NTA agarose beads. A sample of the eluate was loaded on a polyacrylamide gel, and Western blotting was performed using the anti-His antibody 3D5. Molecular masses (according to www.uniprot.org) were 142,844 Da for BNRF1, 26,860 Da for BZLF1, and 37,540 Da for transaldolase. (B) BNRF1-stimulated T cell lines from EBV-seropositive and EBV-seronegative donors. CD4+ T cell lines established by restimulations with protein-pulsed PBMC were tested against PBMC, either untreated or pulsed with BNRF1 or with the control protein BZLF1. The IFN-γ indices were calculated as cytokine concentration in supernatants of T cells responding to PBMC pulsed with either BNRF1 (black bars) or control protein BZLF1 (gray bars) divided each by that of T cells incubated with untreated PBMC. Shown are the IFN-γ indices for T cells from different donors after up to seven passages (indicated as D1p1 to D22p7 along the x axis). Representative assays are shown. D21 and D22 represent EBV-seronegative donors, and the remaining donors were seropositive. (C) BNRF1 specificity of T cell lines improves upon restimulation. The T cell lines were restimulated biweekly for at least seven rounds. The IFN-γ indices for BNRF1 are shown for the T cell lines from different donors (D1 to 22). Each data point depicts an IFN-γ index (y axis) for a given donor at a given passage number. The passage numbers are denoted on the x axis (p1 to p7). Representative assays are shown. D21 and D22 represent EBV-seronegative donors, while the remaining donors were seropositive. The T cell line from seropositive donor D1 showed specific responses as passage 1 and was thus not restimulated further. At some passages, some T cell lines could not be tested, due to a lack either of sufficient PBMC or of T cells.
T cell lines were initiated using PBMC pulsed with recombinant BNRF1 protein as stimulators from 22 healthy volunteers, 20 of them EBV seropositive and 2 EBV seronegative. At the end of 2 weeks, the cultures were magnetically sorted using CD4+ microbeads, and the positive fractions were stimulated again with irradiated PBMC pulsed overnight with recombinant BNRF1. Such stimulation was repeated every 2 weeks, and the growing cultures were tested for specific response against BNRF1. As shown in Fig. 1B, 20 of the 22 T cell lines showed specific recognition of BNRF1 (depicted in the figure as a ratio of the interferon gamma [IFN-γ] signal obtained against PBMC pulsed with BNRF1 compared to PBMC pulsed with a recombinant protein expressed similarly and used in similar amounts). The two cases where we found no BNRF1-specific responses were from EBV-negative individuals. These T cell lines were restimulated fortnightly up to nine times, but no consistent selective BNRF1-specific activity could be established.
Where the T cell lines became BNRF1 specific, the number of restimulations that were required for a specific response varied between individuals, perhaps indicating the difference in the precursor frequencies in the peripheral blood. Thus, whereas T cell lines from some donors demonstrated specificity within two rounds of stimulations, others required up to seven rounds. As shown in Fig. 1C, the response of the T cells to BNRF1 compared to the response against PBMC pulsed with an irrelevant protein improved over restimulations. This was not the case with T cell lines from EBV-negative donors D21 and D22.
Breadth of the CD4+ T cell response to BNRF1.
The selective BNRF1 reactivity of the T cell lines from EBV-positive individuals indicated that BNRF1 is immunogenic to CD4+ T cells. The successful establishment of specific T cell lines from all seropositive donors suggested that the immunogenicity of BNRF1 to CD4+ T cells spreads across a broad range of major histocompatibility complex II (MHCII) molecules. The donors were MHC genotyped. In order to further characterize the T cell lines, we established LCL and mini-LCL from available donors by infecting peripheral blood B cells either with wild-type virus or with BZLF1-deficient virus, respectively. The latter efficiently infects B cells, which then transform to mini-LCL that are deficient in lytic replication. Such mini-LCL were used as antigen-presenting cells in some experiments for subsequent T cell characterization, since our previous work has shown that mini-LCL are recognized by BNRF1-specific T cells only after pulsing with antigen.
We tested the T cell lines for responses against BNRF1 peptides. A total of 327 15-mer peptides were synthesized to cover the length of the BNRF1 protein sequence from the B95.8 strain of EBV. The peptides were subdivided into seven pools with similar numbers of peptides. The T cell lines were then tested for responses against the peptide pools. As shown in Fig. 2A, specific T cell lines showed above-background responses against at least one of the pools, suggesting that most of the donors had developed T cell responses against more than one target peptide.
FIG 2.

Identification of peptide epitopes targeted by BNRF1-specific CD4+ T cells. (A) BNRF1-specific T cell lines recognize pools of peptides derived from BNRF1. T cell lines shown here from five donors (D2, D6, D7, D13, and 19) were tested against seven peptide pools (I to VII), pools I to VI each with 47 unique peptides and pool VII with 45 unique peptides drawn out of a library of 327 15-mer synthetic peptides covering the entire amino acid length of BNRF1, with neighboring peptides overlapping by at least 11 amino acids. Autologous PBMC were used as antigen-presenting cells in these studies. Representative responses from five donors are shown. (B) Identification of individual target peptides recognized by BNRF1-specific T cells. A total of 20 peptide subpools (8 row subpools and 12 column subpools) were derived from each 96-well plate of peptides. Each peptide was present in one row subpool and one column subpool. Subpools relevant to the positive pool were tested in further T cell cytokine release assays using PBMC as antigen-presenting cells and responsive T cells as effectors. In the example depicted here, the T cell line from donor D11, which was found to be responsive to pools I and II, was tested against row (r) subpools and column (c) subpools from 96-well plate number 1 (left). Once positive subpools were identified, candidate single peptides were determined and subsequently tested (right), allowing the identification of single target peptides (19 to 95), which were further confirmed. (C) Identification of T cells recognizing adjacent peptides. Some T cell clones (shown for donor D1 clone 4) recognized two pools (pools II and III), which was found to be due to the recognition of two consecutive peptides (numbers 94 and 95) that were the last peptide in pool II and the first peptide in pool III, respectively. Some T cell clones responded against the same pool (lower-left panel, clones 7 and 12 from donor D1 both recognized pool I) but recognized different peptides in the pool. D1 clone 7 recognized peptide 21, and D1 clone 12 recognized peptides 19 and 20.
To identify the target peptide of the T cell lines, subpools were created. Individual peptides that were contained in positive subpools were tested as single peptides. In some cases, two consecutive peptides elicited similar responses from the T cells. Figure 2B and C show representative results of experiments using the described approach.
BNRF1-specific CD4+ T cells can be detected directly ex vivo.
Given the above results, we attempted to quantify BNRF1-specific T cells directly ex vivo. EBV-specific CD4+ T cells have generally been described to be present in numbers around 10 times lower than CD8+ T cells (6). Therefore, CD8-depleted PBMC are generally used in enzyme-linked immunosorbent spot (ELISPOT) assays (22). We exposed CD8-depleted PBMC to recombinant BNRF1 and the control protein transaldolase after washing them following an overnight rest.
After 16 h of incubation with the antigen, the ELISPOT plates were processed, and IFN-γ spot-forming units were counted. The results are shown in Fig. 3. The results indicated that BNRF1-reactive T cells can be detected directly ex vivo by ELISPOT assay, but the frequency of BNRF1-specific CD4+ T cells in peripheral blood differed widely across donors and did not correlate with the number of restimulations required to detect BNRF1-specific reactivity in the T cell lines. EBV-seronegative donors were clearly negative in the ELISPOT assays validating this approach.
FIG 3.

BNRF1-specific T cells can be detected ex vivo. CD8-depleted PBMC from 21 different donors (EBV-seropositive except for D21 and D29) were rested overnight, followed by exposure to BNRF1 or to the control protein transaldolase for 16 h on a precoated IFN-γ ELISPOT plate. ELISPOT assays were set up with three to four replicates of each condition. Background-subtracted mean counts of spot-forming units (SFU) per million cells along with the standard deviation (error bars) are shown. The mean of the number of spots in the absence of any antigen was considered background.
Identification of MHCII molecules presenting the epitopes.
For subsequent studies, we performed limiting dilution cloning on the T cell line and tested outgrowing cultures for BNRF1 recognition. Shown in Fig. 4A are results of cytokine secretion assays using outgrowing clones from T cell lines from two donors. Outgrowing T cells included both BNRF1-specific and not BNRF1-specific clones, indicating that T cell lines were not constituted of BNRF1-specific T cells only. We expanded the BNRF1-specific T cells for further characterization, starting with identification of the MHCII restriction element.
FIG 4.

Characterization of polyclonal T cell lines and T cell clones. (A) BNRF1-pulsed PBMC-stimulated T cell lines contain BNRF1-specific as well as nonspecific T cells. Limiting dilution cloning yielded several outgrowing clones. Representative results for nine clones (1 to 9) each obtained from the line from donors D1 (top) and D12 (bottom) were tested in cytokine secretion assays against antigen-presenting cells (mini-LCL), either untreated or pulsed with BNRF1 or the control protein BZLF1. (B) Testing T cells against target cells with partly overlapping MHCII genotype of the donor allows identification or narrowing down of potential antigen-presenting molecules. T cell clones D14 no. 5 and D12 no. 3 were tested in cytokine secretion assays against BNRF1-pulsed mini-LCL from different donors (marked as D14 to D39 and D12 to D44 on the x axis) with known, partially overlapping MHCII profiles, allowing for the identification of the antigen-presenting molecule (top: DRB3*02:02) or for narrowing down the potential antigen-presenting molecules (bottom: DRB1*01:01 and DQB1*05:01). (C) Test for DP molecules as potential antigen-presenting molecules using an anti-DP blocking antibody. T cell clones (named along the x axis) were tested against matched BNRF1-pulsed target cells, either untreated or pretreated with an anti-human DP inhibitory antibody or with an IgG isotype-matched control antibody. Cytokine secretion in response to antibody-treated (anti-DP or control) target cells is shown as a percentage of cytokine secretion in response to untreated targets. (D) Test for antigen-presenting molecule using transfection of MHCII molecules in the transfection-permissive cell line DG75. For some T cell clones, the restriction element was identified by expressing single MHCII molecules using expression plasmids (p). In the example shown here, the EBV-negative Burkitt lymphoma cell line DG75 was transfected with expression plasmids coding for BNRF1 alone or along with another plasmid coding for the MHCII molecules DRB1*1301, DRB1*1501, DRB5*0101, or DQB1*0603. Transfected cells were tested for recognition by the T cell clone D37 no. 1. Mini-LCL, either untreated or pulsed with recombinant BNRF1, served as negative and positive controls. DRB5*0101 was identified as the molecule presenting BNRF1 to D37 no. 1 T cells.
Different approaches were used to identify the restricting MHCII molecules. Where available, we tested BNRF1-pulsed mini-LCL from different allogeneic donors that shared part of the MHCII profile with the T cell donor for recognition by the T cells. This allowed identification of the candidate MHCII molecules that were responsible for presenting the antigen to T cells in some of the cases. Two examples are shown in Fig. 4B for the T cell clones D14 no. 5 recognizing peptide 125 (upper panel) and D12 no. 3 recognizing peptide 281 (lower panel), respectively. For the T cell clone recognizing peptide 125, we identified the restricting MHCII molecule as HLADRB3*0202. By this approach, for the T cell clone recognizing peptide 281, we could only narrow down the candidates to one of either HLADRB1*0101 or HLADQB1*0501.
Another approach involved the blocking of MHCII molecules. Preliminary experiments suggested this approach to work reliably for HLA-DP molecules. Therefore, we applied this approach to test for HLA DP-restriction of some T cell clones. An example is shown in Fig. 4C.
For some T cell clones for which candidate MHCII transient expression plasmids were available, such plasmids were transfected into DG75 cells alone or along with a BNRF1-encoding plasmid and transfected DG75 cells used for identifying the presenting MHCII molecule. An example is depicted in Fig. 4D for the T cell clone 1 from donor D37. The above approaches were combined with using the MHCII binding prediction program NetMHCII (http://www.cbs.dtu.dk/services/NetMHCIIpan/). A combination of the mentioned approaches allowed the identification of single MHCII allotypes as restriction elements for the majority of the BNRF1-derived antigenic peptides recognized by T cell clones from different donors. In one case, the MHCII-presenting molecule could not be identified.
Table 1 presents the BNRF1 peptides and the presenting MHCII molecules, as well as the position of those peptides in the full-length protein sequence. Where two adjacent peptides were recognized as potential targets in T cell cytokine release assays, the combined sequence was used to predict binding, and the core sequence was identified. For such cases, the core sequence plus three upstream and three downstream amino acids are presented as the epitope. Figure 5 shows the amino acid sequence of BNRF1 from the B95.8 viral strain with the identified epitope regions marked. As depicted in Table 1, BNRF1 was presented in a variety of MHCII contexts. Thus, BNRF1 is immunogenic to CD4+ T cells across a broad set of antigen-presenting molecules, with no obvious epitope “hot spots” within the protein (Fig. 5). In the same individual, we often identified more than one BNRF1 peptide recognized by T cells, indicating that BNRF1 is indeed a frequent target for EBV-specific CD4+ T cells.
TABLE 1.
CD4+ T cell epitopes in BNRF1 and their presenting molecules
| Amino acid coordinates in B95.8 protein | Sequence of peptide recognized | No. of responsive T cell lines | Antigen-presenting candidate(s) | Method(s) |
|---|---|---|---|---|
| 37-51 | RLYELLSDPRSALGL | 1 | DRB1*14:01 | Common MHCII between recognized target cells combined with binding prediction |
| 53-67 | PGPLIAENLLLVALR | 1 | DPB1*04:02 | MHCII blocking experiments combined with binding predictions |
| 77-91 | RQERARELALVGILL | 3 | DPB1*104:01 | MHCII blocking experiments combined with common MHCII between recognized target cells |
| 81-95 | ARELALVGILLGNGE | 1 | DRB1*01:02 | Common MHCII between recognized target cells |
| 133-147 | QQFLRLLGATYVLRV | 2 | DRB1*01:02 | Common MHCII between recognized target cells combined with binding prediction |
| 171-185 | NHLVLFDNALRKYDS | 1 | DPB1*05:01 | MHCII blocking experiments combined with binding predictions |
| 277-295 | AAGTIQANCPQLFMRRQHP | 1 | DRB3*0101/DRB5*0202 | Binding prediction |
| 375-389 | LGAIKHQALDTVRYD | 3 | DRB4*01:03 | Common MHCII between recognized target cells |
| 427-441 | LELFSALYPAPCISG | 1 | DRB1*01:01 | Common MHCII between recognized target cells combined with binding predictions |
| 449-463 | SAVIEHLGSLVPKGG | 1 | DRB4*0103 | Common MHCII between recognized target cells |
| 492-506 | MQQFVSSYFLNPACS | 1 | DPB1*04:01 | MHCII blocking experiments combined with common MHCII between recognized target cells |
| 497-511 | SSYFLNPACSNVFIT | 2 | DRB3*02:02 | Common MHCII between recognized target cells |
| 548-562 | LGGLNFVNDLASPVS | 2 | DRB3*01:01 | Common MHCII between recognized target cells |
| 915-929 | LPEMFAEHPGLVFEV | 1 | DRB5*01:02 | Expression of MHCII in trans |
| 983-997 | TWSSFASEQYECLRP | 3 | DPB1*04:01 | Common MHCII between recognized target cells |
| 1006-1020 | VSDYGYNEALAVSPL | 2 | DRB3*02:02 | Expression of MHCII in trans |
| 1121-1135 | LNRPDTFSVALGELG | 1 |
DRB1*01:01/ DQB1*05:01 |
Binding predictions, IEDBa |
| 1238-1252 | TDAWRFAMNYPRNPT | 1 | DRB5*01:01 | Expression of MHCII in trans |
IEDB, Immune Epitope Database (www.iedb.org).
FIG 5.

Relative positions of CD4+ T cell epitopes in the reference BNRF1 protein sequence from B95.8 virus. The BNRF1 protein contained epitope sequences recognized by T cell lines from only one (red), two (bold green), or three (bold italic print face purple) donors. Where epitopes overlapped, amino acids common to both epitopes are shown in brown bold typeface.
CD4+ T cells against BNRF1 are cytotoxic Th1 type T cells.
Others and we have described EBV-specific cytotoxic CD4+ T cells as cytotoxic (16, 23, 24). In terms of the cytokine phenotype, Th1 as well as Th2 type CD4+ T cells have been described (25). All BNRF1-specific T cells secreted IFN-γ and, as shown in Fig. 6A, secreted perforin as well as granzyme B upon recognition of their targets, thereby establishing themselves as Th1 type cytotoxic T cells. For a more direct demonstration of cytotoxicity, we used Calcein-AM release-based cytotoxicity assay. For such assays, we used autologous mini-LCL as antigen-presenting cells. Prior to labeling with the dye, the antigen-presenting cells were pulsed with either a control peptide or the cognate BNRF1-derived peptide. Peptide-pulsed, dye-labeled cells were incubated with BNRF1-specific T cells. The T cells consistently caused up to 80% specific lysis of targets loaded with the cognate antigenic peptide but not with a control peptide (Fig. 6B).
FIG 6.
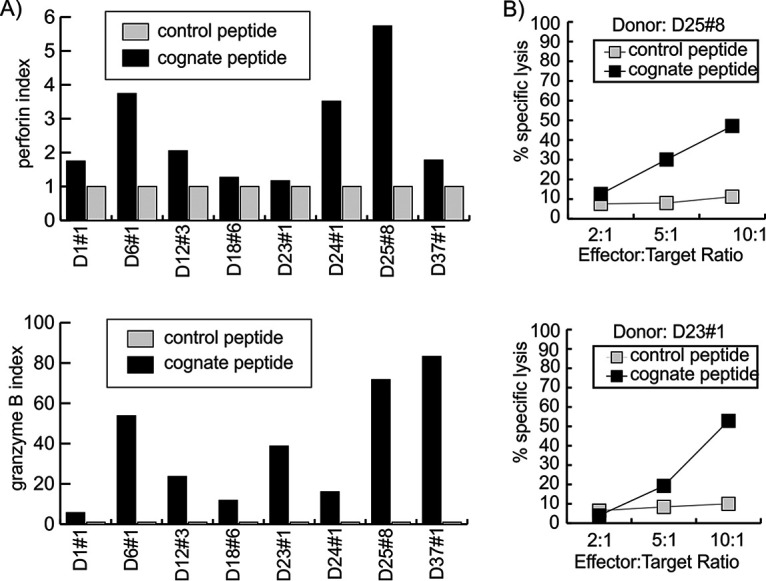
BNRF1-specific CD4+ T cells are cytolytic. (A) Secretion of cytolytic molecules by BNRF1-specific T cells. T cell clones were tested for secretion of perforin (top) and granzyme B (bottom) in response to mini-LCL either unpulsed or pulsed with cognate or control peptide. The perforin (or granzyme B) index represents the perforin (or granzyme B) concentration in supernatants of T cells in response to cognate (black) or control (gray) peptide in relation to that of untreated T cells. The x axis depicts the donor number along with the T cell clone number. (B) Specific lysis of antigen-presenting target cells by BNRF1-specific T cells. T cell clones D25 no. 8 (top) and D23 no. 1 (bottom) were tested for their cytolytic potential in Calcein-AM cytotoxicity assays using mini-LCL pulsed with the cognate peptide or a control peptide as target cells. Peptide-pulsed mini-LCL were labeled with Calcein-AM dye and then brought out with T cells at different effector-to-target ratios as marked on the x axis.
BNRF1-specific T cells recognize, kill, and inhibit the proliferation of lymphoblastoid cell lines capable of lytic replication and counteract EBV-driven primary B cell transformation.
A small percentage of wild-type EBV-transformed B cells undergo lytic viral replication (6) and thereby package BNRF1 into viral particles and release them. We therefore tested all available BNRF1-specific T cell clones for recognition of wild-type EBV-transformed LCL. While lytic replication-deficient mini-LCL were not recognized by the T-cells, lytic replication-competent LCL were readily recognized (Fig. 7A). The degree of recognition of LCL varied from donor to donor but, in general, corresponded to the extent of lytic replication as measured by viral load in the supernatants of the LCL. BNRF1-specific T cells from donor D6 responded to autologous LCL at only background levels, and low viral load in the supernatant supported the finding of the cytokine enzyme-linked immunosorbent assay (ELISA).
FIG 7.

BNRF1-specific CD4+ T cells inhibit EBV-driven proliferation and transformation. (A) BNRF1-specific CD4+ T cells recognize EBV-transformed B cells that are permissive to lytic replication. T cell clones were tested in IFN-γ secretion assays against autologous mini-LCL, mini-LCL pulsed with recombinant BNRF1, or LCL. (B) BNRF1-specific T cells recognize unmanipulated EBV-transformed B cells and can efficiently kill them. (Left) T cell clones (10,000 cells per well) were tested in IFN-γ ELISPOT assays against autologous mini-LCL or LCL brought out in serial dilutions starting at 2,500 target cells per well. (Right) T cells at different T cell-to-target ratios were used to assess cytolytic activity in 4-h cytotoxicity assays using untreated or peptide-pulsed autologous mini-LCL and untreated LCL as targets. (C) BNRF1-specific T cells restrict the proliferation of EBV-transformed LCL but not mini-LCL. Autologous LCL or mini-LCL (target cells) were plated in serial dilutions from 10,000 to 30 cells per well in round-bottom 96-well plates, in four replicas, with or without T cells (10,000 per well). In all cases CD4+ BNRF1-specific T cells (BNRF1 T cells) were used, except for donor D23, where in addition to BNRF1-specific T cells, an influenza M1-specific CD4+ T cell clone (M1 T cells) was available from the same donor and was therefore also included as a control T cell clone. After a month, the plate was inspected for proliferation of mini-LCL or LCL, and wells with target outgrowths in the absence and presence of T cells were noted. The results are expressed as ratios of the number of input target cells that led to outgrowth in the presence of T cells to the number of input target cells that led to outgrowth in the absence of T cells. (D) Donor-derived spontaneous LCL efficiently present antigen to BNRF1-specific CD4+ T cells. BNRF1-specific CD4+ T cell clones from donors D25, D1, and D23 were tested for responses against autologous spontaneous LCL. The corresponding mini-LCL served as controls. (E) BNRF1-specific CD4+ T cells inhibit EBV-driven transformation of primary B cells. Magnetically sorted CD19+ PBMC from three donors (D6, D23, D24) were exposed to B95.8 virus for 2 h and then brought out in serial dilutions from 10,000 to 10 cells per well in 96-well plates in two different conditions, either with or without BNRF1-specific T cells (10,000 per well), with each condition in four replicates. After 4 weeks, the plate was monitored for B cell transformation by microscopy. Shown in the figure are the fold input cell numbers that led to transformation. The number of B cells that led to a transformed proliferating culture when plated without T cells was taken as fold 1.
The recognition of autologous LCL by BNRF1-specific T cell clones from almost all donors raised the questions as to what percentage of cells in an LCL culture could be targeted by the T cells and whether the recognition of LCL could lead to detectable killing. To address this, we tried to enumerate the number of cells in an LCL targeted by the T cells using ELISPOT assays. T cell clones from two donors recognized over 10% of cells in an LCL culture. However, even in the LCL in which around 20% of the cells were recognized by BNRF1-specific T cells, we were unable to detect BNRF1 by Western blot analysis in cell lysates (our unpublished data). BNRF1 could only be detected in lysates of the Burkitt lymphoma cell line Akata in which lytic replication had been induced by anti-IgM treatment (unpublished data). These findings substantiate our previous finding that antigen transfer by virions is the major contributor to MHCII presentation of BNRF1 by LCL (16, 19).
Recognition of LCL led to their killing (Fig. 7B, right). The killing was readily detectable in 4-h cytotoxicity assays, when the T cell-to-target cell ratio was already at 2:1.
We next tested the T cells for inhibition of LCL proliferation. Autologous LCL were incubated in serial dilutions with or without T cells. In the presence of T cells, 3- to 30-fold higher starting cell numbers were required to obtain a proliferating LCL culture, except for donor D6, whose T cells failed to demonstrate a measurable inhibition of LCL proliferation. T cells from none of the donors inhibited the proliferation of mini-LCL, and in one case where T cells against a non-EBV antigen were available, that T cell clone was not able to limit the proliferation of LCL (Fig. 7C), indicating that the inhibition of LCL proliferation by BNRF1-specific CD4+ T cells was target antigen specific.
Whereas B95.8 is a representative laboratory strain, it has been recognized over the last few years that viral strains in healthy carriers and in diseased people can be deviant from the laboratory reference strain. Therefore, we tested spontaneously outgrowing EBV-transformed B cell cultures that were available from three healthy individuals. Although the sample size is small, experiments with these “spontaneous LCL” with field strains of the virus demonstrated that the lytic replication in these spontaneous LCL was enough to sensitize the cells for recognition by the T cells (Fig. 7D).
These findings illustrated that BNRF1-specific T cells can recognize B cells transformed by the laboratory EBV strain B95.8 as well as by the individual field EBV strain. Given the presence of the BNRF1 protein in viral particles in amounts sufficient to trigger recognition by the T cells, we were interested in discovering whether such T cells were also able to inhibit the transformation of B cells by EBV. For these experiments, we enriched B cells in PBMC by positively sorting CD19+ cells using magnetic beads. Sorted B cells were then exposed to wild-type EBV at a multiplicity of infection of 0.1, and after 2 h of incubation B cells were seeded onto 96-well plates in serial dilutions starting at 30,000 in the presence or absence of BNRF1-specific clonal T cells (at a constant number of 10,000). Half the medium in these cocultures was replaced once a week. In order to limit the T cell activity to the initial phase in which virus binds to and enters B cells, no cytokine support was afforded to the T cells. After a month of incubation, the plate was read for EBV-driven transformation. Flow cytometry was performed to confirm the outgrowing cells to be B cells. There was a donor-to-donor difference in the results, but in all donors tested, the presence of BNRF1-specific T cells led to inhibition of EBV-driven transformation, as evidenced by the requirement of higher starting numbers of EBV-exposed primary B cells (Fig. 7E).
DISCUSSION
The optimism generated by the success of EBV T cell therapy in HSC-associated PTLD has driven the exploration of ways to advance adoptive therapy toward improving current response rates and extending its clinical indications (26). Tailoring T cell therapy to suit the need of the patient at hand demands knowledge of the immunogenicity of the disease in question and of the target viral antigens available for presentation, as well as of antigenic epitopes generated by infected cells and presented to T cells in the given MHC context. Findings from in vitro studies and mouse models of PTLD-like tumors, as well as from the clinical application of EBV-specific T cells have established CD4+ T cells as important components of the immune response against EBV-driven PTLD (9, 27, 28). However, studies into the antigen repertoire recognized by EBV-specific CD4+ T cells are still limited. In this work we studied the major EBV tegument protein BNRF1, identified 18 naturally processed antigenic peptide epitopes presented to CD4+ T cells across a number of MHCII molecules, and demonstrated antiproliferative effects of BNRF1-specific CD4+ T cell clones.
We were able to establish BNRF1-specific CD4+ T cell lines from 20 out of 20 EBV-seropositive individuals studied in this work. A study into the CD4+ response against EBV latency proteins in healthy carriers has found a maximum of 75% of donors to respond against EBNA1, the most ubiquitously expressed viral protein (22). A similar study into CD4+ T cells against EBV lytic cycle proteins did not investigate BNRF1, but among three late lytic proteins studied, the best donor response rate was found for BXLF2, CD4+ T cells against which were detected in 10 out of 14 healthy carriers (24). Against this background, our finding of all EBV-positive donors studied having CD4+ T cells against BNRF1, a structural protein of the virus, was unexpected. One possible explanation for our finding is that BNRF1 might also be expressed in viral latency, as has been recently suggested, and that BNRF1 is available for presentation even in the healthy carrier state (21). However, we have consistently observed that mini-LCL, EBV-transformed cells genetically deficient in supporting lytic replication, cannot trigger a response from BNRF1-specific CD4+ T cells, unless exposed to an exogenous source of antigen (19). Therefore, if it is indeed a latency protein, BNRF1 in EBV latency either is expressed in insufficient amounts to trigger cytokine and cytotoxicity responses from CD4+ T cells or is not accessible to the MHCII antigen-presentation pathway in the absence of productive lytic replication.
Our ELISPOT assay results show that using the CD8-depletion approach, BNRF1-reactive CD4+ T cells can be measured directly ex vivo. However, this is not true in all donors, in spite of the fact that all seropositive donors had BNRF1-specific T cells as evidenced by the specificity of the BNRF1-reactive T cell lines. This discrepancy indicates that direct ELISPOT assays ex vivo, as generally employed, may not be sensitive enough to identify all positive individuals and that donor positivity data obtained from such ELISPOT studies may underreport specific T cell frequencies and/or diversities. Indeed, whereas the aforementioned peptide-based ELISPOT approach by Long et al. identified EBNA1-specific CD4+ T cells in only 75% of the donors studied (24), the in vitro stimulation approach by Münz et al. detected EBNA1-specific CD4+ T cells in all of 10 seropositive donors studied (29). These findings together imply that in vitro stimulation approaches might be more reliable in detecting CD4+ T cells against EBV.
It is generally believed that de novo EBV-infected cells are predominantly targeted by CD8+ T cells, with CD4+ T cells being better at recognizing latently infected cells (6). The findings of this work add further evidence to our previous proposition that CD4+ T cells might actually be involved as prominent players during de novo infection too (20). As we have observed in the past for BLLF1 and BALF4, two glycoproteins of the viral envelope, here we demonstrated that another viral structural protein, namely, BNRF1, can mediate the inhibition of B cell transformation and of LCL proliferation by CD4+ T cells.
Individuals latently infected with herpesviruses demonstrate subclinical virus reactivation from latency from time to time (30–33). It has been suggested that chronic virus replication, a common phenomenon in the persistent carrier state, may be essential for the maintenance of latency (31). Such a low-level replication, if not promptly controlled, could amplify to produce subclinical reactivations or even full-fledged clinical disease. It is thus conceivable that virion protein-specific CD4+ T cells deliver a major contribution to keeping the likely baseline reactivation under check and thus maintain a controlled latent infection in healthy carriers. EBV-associated diseases are often associated with high viral loads (34, 35), and viral strains associated with cancers are likely to be associated with enhanced lytic replication (36), suggesting an increased availability of virion proteins like BNRF1. Efficient targeting of EBV-driven diseases likely requires targeting latently infected cells but also cells in lytic replication as well as freshly infected cells. The importance of the immune control of B cells that have picked up virus and are in the process of transformation into continuously proliferating colonies cannot be exaggerated. Our findings support the notion that T cell immunotherapy against EBV-driven diseases could draw benefits from the conscious inclusion of virion-protein-specific CD4+ T cells in T cell preparations. Such CD4+ T cells are likely to be able to play a crucial role during amplification of lytic viral activity, which is a potential key stage in the establishment and or maintenance of virus-driven disease(s) states.
BNRF1-specific T cells were found to recognize wild-type virus-transformed B cell lines to variable degrees that correlated with the rate of lytic replication in the LCL. The nonrecognition of mini-LCL but recognition of LCL even though BNRF1 could not be detected by immunoblot analysis in mini-LCL or in LCL could be explained by our previous observation that viral particles released from cells undergoing lysis can serve as sources of antigen for neighboring cells (19). In agreement with this, ELISPOT assays revealed the number of LCL recognized by T cells to be more than the generally low, limited lytic replication in LCL (6, 37). The previous indication that mini-LCL can present BNRF1 to CD8+ T cells (21) raises the question of whether there is a preferential presentation of intracellular BNRF1 to CD8+ T cells and, if yes, whether the differential presentation can be attributed to differences in levels of expression. In order to address this further, we are in the process of establishing a system to express BNRF1 intracellularly in an inducible manner. Such a system is expected to also allow the assessment of the number of cells expressing BNRF1 intracellularly that can actually be targeted by CD4+ and CD8+ T cells as well.
The lytic replication-dependent recognition of LCL suggests that one approach to optimize EBV-specific T cell therapy might be the induction of lytic replication to aid the recognition of tumor cells by EBV-specific CD4+ T cells, such as those against BNRF1. Induction of lytic replication has been an avenue that has been under investigation as a way to make infected cells susceptible to antivirals such as ganciclovir (38). Our findings with virion-specific CD4+ T cells raise the intriguing question of whether controlled induction of lytic replication can aid immunotherapy by increasing the availability of target antigens.
This work has identified 18 previously unknown target peptide epitopes recognized by BNRF1-specific CD4+ T cells and has established an approach to quantifying BNRF1-specific CD4+ T cells directly ex vivo by ELISPOT. This ELISPOT approach needs to be optimized to improve sensitivity so as to determine true precursor frequencies of such T cells in the peripheral blood. Using peptides instead of full-length recombinant protein might be one way of optimization. Peptides are purer than recombinant protein in terms of BNRF1 and can be expected to reduce the background signals, but full-length protein-elicited spots are probably more meaningful since they indicate appropriate processing of full-length BNRF1 and presentation to T cells. Therefore, these two aspects need to be weighed against each other. In PBMC from two seropositive donors, ELISPOT assays performed with individual peptides or a combination of peptides recognized by their T cell lines have shown promising results (data not shown). Such an approach could be used for assessing virus-specific CD4+ T cells in health and disease.
Healthy virus carriers may be different in terms of the basal activation rate, as has been suggested by previous studies (32). This may contribute to different degrees of activation of BNRF1-specific CD4+ T cells in different individuals, thereby producing highly variable precursor frequencies when measured in cross-section studies like that done in this work. If BNRF1-specific T cells were stimulated to a different extent in different individuals due to different rates of basal viral replication, the T cells might differ in their differentiation phenotype. With the knowledge of a considerable number of target peptides presented on common MHCII molecules, the phenotype question might be systematically addressed with MHCII multimer technology as has been employed in the past by Long and colleagues for several MHCII target epitopes in other EBV proteins (39). An understanding of the effector-memory phenotype of the T cells will likely contribute to understanding virus reactivation during health.
Our findings suggest that cytotoxic CD4+ BNRF1-specific T cells can efficiently recognize and eliminate lytically infected B cells. It was indeed noted nearly 40 years ago that the suppression of EBV infection in vitro takes place after infection but before transformation and that viral determinants left on the surface are potential triggers of the T cell response (40). Our results support these early observations and add BNRF1 to other viral targets with high clinical impact in the immune control of viral spreading and EBV-associated cell proliferation.
MATERIALS AND METHODS
Donors and primary blood cells.
This work used peripheral blood mononuclear cells (PBMC) and derivatives thereof. PBMC were purified using standard procedures from peripheral blood obtained from healthy volunteer donors. The use of material of human origin in this work was approved by the ethics committee of the Technical University of Munich (approvals 934/03 and 1872/07) and was in accordance with the Declaration of Helsinki of the World Medical Association (last amended in 2013).
The donors were assigned donor numbers starting with D1. All donors from whom T cell lines and clones were raised as well as those from whom LCLs and mini-LCLs were established were HLA-typed at the Laboratory of Immunogenetics at the University Hospital of the Ludwig Maximilians University, Munich, Germany. PBMC or derivatives thereof were not available from all donors for all experiments. Therefore, based on availability, material derived from different sets of donors was used for different experiments.
Protein preparation.
Protein preparations that were used as antigens were expressed in human embryonic kidney cells (HEK 293T) (ATCC CRL3216) from a cytomegalovirus (CMV) promoter (pCMV)-driven plasmid construct as described before (41). The cloning allowed proteins to be expressed with a C-terminal 6× histidine-tag and purified out of the lysates of transfected cells using Ni-NTA agarose beads (Qiagen).
We used wild-type EBV DNA of B95.8 viral strain origin (GenBank accession number AJ507799.2) as the template for PCRs to clone the EBV protein-coding genes BNRF1 and BZLF1. The human protein transaldolase (UniProt accession number P37837) used as a control in ELISPOT assays was similarly expressed as EBV proteins following cloning from cDNA derived from HEK 293T cells.
In order to ensure consistent antigen delivery, we pooled multiple preparations of recombinant proteins, measured the protein concentration, and froze appropriate aliquots.
Protein concentration, polyacrylamide gel electrophoresis, and Western blot analysis.
Protein concentration was measured by spectrophotometry at a wavelength of 595 nm using the Bradford reagent and standard protocols. Western blot analysis of 6× histidine (His)-tagged protein preparations was performed using the mouse monoclonal anti-His6 antibody 3D5 (kindly provided by the Monoclonal Antibody Facility, Helmholtz Centre Munich, Germany; https://www.helmholtz-muenchen.de/mab).
T cell lines and clones.
T cells were cultured in AIM-V medium (Invitrogen) supplemented with antibiotics, l-glutamine, and 10% heat-inactivated pooled human serum obtained from healthy donors following informed consent.
Polyclonal T cell lines were established by exposing PBMC to recombinant BNRF1 overnight followed by irradiation of the cells and culture with nonirradiated PBMC. Then, 1 to 1.5 μg of recombinant BNRF1 was used for 2 million PBMC in 2 ml medium in a well of a 24-well plate. Cultures started for T cell lines were treated with recombinant human interleukin-2 (IL-2) (50 U/ml) 24 to 48 h after initiation of culture. Depending on proliferation, the cultures were expanded using medium supplemented with IL-2. Two weeks after the initiation of T cell lines, the cells were harvested and the CD4+ fraction obtained by magnetic sorting stimulated again by coculture with irradiated PBMC pulsed with recombinant BNRF1 protein. This mode of stimulation was repeated fortnightly. Clonal T cell lines were obtained by dilution culture of T cell lines and cultures as described before (16). T cell clones were assigned numbers that follow the donor numbers.
B cell lines.
LCL were established by infection of peripheral blood B cells using either the wild-type B95.8 strain of EBV or laboratory strains derived from B95.8 and maintained in culture as previously described (41). For simplicity’s sake, LCL established by infection with B95.8 are called LCL, whereas those established by infection with laboratory viral strains deficient in lytic replication are called mini-LCL. The laboratory strain used to generate mini-LCL is genetically deficient in the Epstein-Barr virus lytic cycle switch gene BZLF1. It was kindly provided by Henri-Jacques Delecluse, at the German Cancer Research Center, Heidelberg. The same culture conditions were used for LCL and mini-LCL as well as for the EBV-negative Burkitt lymphoma cell line DG-75 (ATCC CRL-2625). The transfection of DG-75 cells for their use in antigen presentation experiments has been described (42).
Spontaneous LCL were established by setting up PBMC culture as for the establishment of LCL, with cyclosporine A as described before (41) but without any exogenous virus. Outgrowing cultures were treated similarly as LCL.
Magnetic sorting.
Magnetic sorting was performed employing the MACS sorting technology (Miltenyi) following published protocols of the manufacturer. Magnetic microbeads against human CD4, CD8, and CD19 were used.
Cytokine ELISA and ELISPOT.
For characterization of T cell cytokine secretion, appropriate cell culture supernatants were used for measurement of cytokine content using commercial kits and accompanying protocols. The following kits (all from Mabtech) were used: human interferon gamma (IFN-γ), granzyme B, and perforin.
Major histocompatibility complex class II HLA-DP inhibition experiments were performed using clone B7/21 (Abcam) against HLA-DP. IFN-γ cytokine ELISPOT assays were performed using kits from Mabtech according to the accompanying protocol, and cell numbers were adjusted as described in the main text. We performed quantification of ELISPOT using a scanner/analyzer from C.T.L. (Cellular Technology Ltd.) using the Immunospot 5.0 software.
Cytotoxicity assays.
The T cell cytotoxicity assays performed in this work were Calcein-AM-based cytotoxicity assays (43). Briefly, cells to be used as targets were labeled with Calcein-AM and cell-permeant dye (Thermo Fischer) followed by coculture with T cells. The number of target cells used was 5,000 per well, and the number of T cells depended upon the desired T cell to antigen-presenting cell (APC) ratio. After 4 h of coculture to allow for cytolysis by the T cells, we measured absorbance at 485 nm as a proxy of Calcein-AM released into the supernatant with an Infinite F 200 PRO microplate reader (Tecan).
We recorded the reading for target cells exposed to a detergent as the maximum and the reading from supernatants where the target cells were left untreated as the minimum. The following formula was used to calculate cytotoxicity: [(reading in the presence of T cells – minimum)/(maximum – minimum)] × 100. The result is presented as percent specific lysis.
Peptides.
A total of 327 15-amino acid peptides, each 15-amino acids long, were synthesized (JPT Peptide Technologies) covering the whole length of BNRF1. Two adjacent peptides overlapped by 11 amino acids, with one exception—the last two peptides at the C-terminal end of BNRF1 overlapped by 12 amino acids.
ACKNOWLEDGMENTS
D.A. designed and performed experiments, analyzed and interpreted experimental data, and wrote the initial manuscript draft. J.D. performed and analyzed experiments and contributed to the initial manuscript draft. J.M. designed and performed experiments, analyzed and interpreted experimental data, and finalized the manuscript. U.B. designed experiments, interpreted experimental data, and finalized the manuscript.
REFERENCES
- 1.IARC. 2012. A review of human carcinogens. Part B: biological agents, vol 100 B, p 475 International Agency for Research on Cancer, Lyon, France. [Google Scholar]
- 2.Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. 2015. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol 33:787–821. doi: 10.1146/annurev-immunol-032414-112326. [DOI] [PubMed] [Google Scholar]
- 3.Rooney CM, Smith CA, Ng CY, Loftin S, Li C, Krance RA, Brenner MK, Heslop HE. 1995. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 345:9–13. doi: 10.1016/s0140-6736(95)91150-2. [DOI] [PubMed] [Google Scholar]
- 4.Kang MS, Kieff E. 2015. Epstein-Barr virus latent genes. Exp Mol Med 47:e131. doi: 10.1038/emm.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rickinson AB, Moss DJ. 1997. Human cytotoxic T lymphocyte responses to Epstein-Barr virus infection. Annu Rev Immunol 15:405–431. doi: 10.1146/annurev.immunol.15.1.405. [DOI] [PubMed] [Google Scholar]
- 6.Longnecker RM, Kieff E, Cohen JI. 2013. Epstein-Barr virus, p 1898–1959. In Knipe DM, Howley PM (ed), Field’s virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 7.Bollard CM, Rooney CM, Heslop HE. 2012. T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nat Rev Clin Oncol 9:510–519. doi: 10.1038/nrclinonc.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shum T, Kruse RL, Rooney CM. 2018. Strategies for enhancing adoptive T-cell immunotherapy against solid tumors using engineered cytokine signaling and other modalities. Expert Opin Biol Ther 18:653–664. doi: 10.1080/14712598.2018.1473368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haque T, Wilkie GM, Jones MM, Higgins CD, Urquhart G, Wingate P, Burns D, McAulay K, Turner M, Bellamy C, Amlot PL, Kelly D, MacGilchrist A, Gandhi MK, Swerdlow AJ, Crawford DH. 2007. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood 110:1123–1131. doi: 10.1182/blood-2006-12-063008. [DOI] [PubMed] [Google Scholar]
- 10.Moosmann A, Bigalke I, Tischer J, Schirrmann L, Kasten J, Tippmer S, Leeping M, Prevalsek D, Jaeger G, Ledderose G, Mautner J, Hammerschmidt W, Schendel DJ, Kolb HJ. 2010. Effective and long-term control of EBV PTLD after transfer of peptide-selected T cells. Blood 115:2960–2970. doi: 10.1182/blood-2009-08-236356. [DOI] [PubMed] [Google Scholar]
- 11.Papadopoulou A, Gerdemann U, Katari UL, Tzannou I, Liu H, Martinez C, Leung K, Carrum G, Gee AP, Vera JF, Krance RA, Brenner MK, Rooney CM, Heslop HE, Leen AM. 2014. Activity of broad-spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci Transl Med 6:242ra83. doi: 10.1126/scitranslmed.3008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orentas RJ, Roskopf SJ, Nolan GP, Nishimura MI. 2001. Retroviral transduction of a T cell receptor specific for an Epstein-Barr virus-encoded peptide. Clin Immunol 98:220–228. doi: 10.1006/clim.2000.4977. [DOI] [PubMed] [Google Scholar]
- 13.Schaft N, Lankiewicz B, Drexhage J, Berrevoets C, Moss DJ, Levitsky V, Bonneville M, Lee SP, McMichael AJ, Gratama JW, Bolhuis RL, Willemsen R, Debets R. 2006. T cell re-targeting to EBV antigens following TCR gene transfer: CD28-containing receptors mediate enhanced antigen-specific IFNgamma production. Int Immunol 18:591–601. doi: 10.1093/intimm/dxh401. [DOI] [PubMed] [Google Scholar]
- 14.Frumento G, Zheng Y, Aubert G, Raeiszadeh M, Lansdorp PM, Moss P, Lee SP, Chen FE. 2013. Cord blood T cells retain early differentiation phenotype suitable for immunotherapy after TCR gene transfer to confer EBV specificity. Am J Transplant 13:45–55. doi: 10.1111/j.1600-6143.2012.04286.x. [DOI] [PubMed] [Google Scholar]
- 15.Roddie C, Peggs KS. 2017. Immunotherapy for transplantation-associated viral infections. J Clin Invest 127:2513–2522. doi: 10.1172/JCI90599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adhikary D, Behrends U, Moosmann A, Witter K, Bornkamm GW, Mautner J. 2006. Control of Epstein-Barr virus infection in vitro by T helper cells specific for virion glycoproteins. J Exp Med 203:995–1006. doi: 10.1084/jem.20051287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A 101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adhikary D, Behrends U, Boerschmann H, Pfunder A, Burdach S, Moosmann A, Witter K, Bornkamm GW, Mautner J. 2007. Immunodominance of lytic cycle antigens in Epstein-Barr virus-specific CD4+ T cell preparations for therapy. PLoS One 2:e583. doi: 10.1371/journal.pone.0000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adhikary D, Behrends U, Feederle R, Delecluse HJ, Mautner J. 2008. Standardized and highly efficient expansion of Epstein-Barr virus-specific CD4+ T cells by using virus-like particles. J Virol 82:3903–3911. doi: 10.1128/JVI.02227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cirac A, Stutzle S, Dieckmeyer M, Adhikary D, Moosmann A, Korber N, Bauer T, Witter K, Delecluse HJ, Behrends U, Mautner J. 2018. Epstein-Barr virus strain heterogeneity impairs human T-cell immunity. Cancer Immunol Immunother 67:663–674. doi: 10.1007/s00262-018-2118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abbott RJ, Quinn LL, Leese AM, Scholes HM, Pachnio A, Rickinson AB. 2013. CD8+ T cell responses to lytic EBV infection: late antigen specificities as subdominant components of the total response. J Immunol 191:5398–5409. doi: 10.4049/jimmunol.1301629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long HM, Haigh TA, Gudgeon NH, Leen AM, Tsang CW, Brooks J, Landais E, Houssaint E, Lee SP, Rickinson AB, Taylor GS. 2005. CD4+ T-cell responses to Epstein-Barr virus (EBV) latent-cycle antigens and the recognition of EBV-transformed lymphoblastoid cell lines. J Virol 79:4896–4907. doi: 10.1128/JVI.79.8.4896-4907.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nikiforow S, Bottomly K, Miller G, Munz C. 2003. Cytolytic CD4(+)-T-cell clones reactive to EBNA1 inhibit Epstein-Barr virus-induced B-cell proliferation. J Virol 77:12088–12104. doi: 10.1128/jvi.77.22.12088-12104.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long HM, Leese AM, Chagoury OL, Connerty SR, Quarcoopome J, Quinn LL, Shannon-Lowe C, Rickinson AB. 2011. Cytotoxic CD4+ T cell responses to EBV contrast with CD8 responses in breadth of lytic cycle antigen choice and in lytic cycle recognition. J Immunol 187:92–101. doi: 10.4049/jimmunol.1100590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steigerwald-Mullen P, Kurilla MG, Braciale TJ. 2000. Type 2 cytokines predominate in the human CD4(+) T-lymphocyte response to Epstein-Barr virus nuclear antigen 1. J Virol 74:6748–6759. doi: 10.1128/jvi.74.15.6748-6759.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gottschalk S, Rooney CM. 2015. Adoptive T-cell immunotherapy. Curr Top Microbiol Immunol 391:427–454. doi: 10.1007/978-3-319-22834-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merlo A, Turrini R, Bobisse S, Zamarchi R, Alaggio R, Dolcetti R, Mautner J, Zanovello P, Amadori A, Rosato A. 2010. Virus-specific cytotoxic CD4+ T cells for the treatment of EBV-related tumors. J Immunol 184:5895–5902. doi: 10.4049/jimmunol.0902850. [DOI] [PubMed] [Google Scholar]
- 28.Linnerbauer S, Behrends U, Adhikary D, Witter K, Bornkamm GW, Mautner J. 2014. Virus and autoantigen-specific CD4+ T cells are key effectors in a SCID mouse model of EBV-associated post-transplant lymphoproliferative disorders. PLoS Pathog 10:e1004068. doi: 10.1371/journal.ppat.1004068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Münz C, Bickham KL, Subklewe M, Tsang ML, Chahroudi A, Kurilla MG, Zhang D, O’Donnell M, Steinman RM. 2000. Human CD4(+) T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J Exp Med 191:1649–1660. doi: 10.1084/jem.191.10.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao QY, Rickinson AB, Epstein MA. 1985. Oropharyngeal shedding of infectious Epstein-Barr virus in healthy virus-immune donors. A prospective study. Chin Med J (Engl) 98:191–196. [PubMed] [Google Scholar]
- 31.Yao QY, Rickinson AB, Epstein MA. 1985. A re-examination of the Epstein-Barr virus carrier state in healthy seropositive individuals. Int J Cancer 35:35–42. doi: 10.1002/ijc.2910350107. [DOI] [PubMed] [Google Scholar]
- 32.Ling PD, Lednicky JA, Keitel WA, Poston DG, White ZS, Peng R, Liu Z, Mehta SK, Pierson DL, Rooney CM, Vilchez RA, Smith EO, Butel JS. 2003. The dynamics of herpesvirus and polyomavirus reactivation and shedding in healthy adults: a 14-month longitudinal study. J Infect Dis 187:1571–1580. doi: 10.1086/374739. [DOI] [PubMed] [Google Scholar]
- 33.Johnson KH, Webb CH, Schmeling DO, Brundage RC, Balfour HH Jr. 2016. Epstein-Barr virus dynamics in asymptomatic immunocompetent adults: an intensive 6-month study. Clin Trans Immunol 5:e81. doi: 10.1038/cti.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tay JK, Chan SH, Lim CM, Siow CH, Goh HL, Loh KS. 2016. The role of Epstein-Barr virus DNA load and serology as screening tools for nasopharyngeal carcinoma. Otolaryngol Head Neck Surg 155:274–280. doi: 10.1177/0194599816641038. [DOI] [PubMed] [Google Scholar]
- 35.Kimura H, Kwong YL. 2019. EBV viral loads in diagnosis, monitoring, and response assessment. Front Oncol 9:62. doi: 10.3389/fonc.2019.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai MH, Raykova A, Klinke O, Bernhardt K, Gartner K, Leung CS, Geletneky K, Sertel S, Munz C, Feederle R, Delecluse HJ. 2013. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep 5:458–470. doi: 10.1016/j.celrep.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 37.Kieff E, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2603–2654. In Fields BN, Knipe DM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 38.Feng WH, Hong G, Delecluse HJ, Kenney SC. 2004. Lytic induction therapy for Epstein-Barr virus-positive B-cell lymphomas. J Virol 78:1893–1902. doi: 10.1128/jvi.78.4.1893-1902.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long HM, Chagoury OL, Leese AM, Ryan GB, James E, Morton LT, Abbott RJ, Sabbah S, Kwok W, Rickinson AB. 2013. MHC II tetramers visualize human CD4+ T cell responses to Epstein-Barr virus infection and demonstrate atypical kinetics of the nuclear antigen EBNA1 response. J Exp Med 210:933–949. doi: 10.1084/jem.20121437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thorley-Lawson DA. 1980. The suppression of Epstein-Barr virus infection in vitro occurs after infection but before transformation of the cell. J Immunol 124:745–751. [PubMed] [Google Scholar]
- 41.Mautner J, Pich D, Nimmerjahn F, Milosevic S, Adhikary D, Christoph H, Witter K, Bornkamm GW, Hammerschmidt W, Behrends U. 2004. Epstein-Barr virus nuclear antigen 1 evades direct immune recognition by CD4+ T helper cells. Eur J Immunol 34:2500–2509. doi: 10.1002/eji.200324794. [DOI] [PubMed] [Google Scholar]
- 42.Fiebiger BM, Pfister H, Behrends U, Mautner J. 2015. Polyubiquitination of lysine-48 is an essential but indirect signal for MHC class I antigen processing. Eur J Immunol 45:716–727. doi: 10.1002/eji.201444830. [DOI] [PubMed] [Google Scholar]
- 43.Neri S, Mariani E, Meneghetti A, Cattini L, Facchini A. 2001. Calcein-acetyoxymethyl cytotoxicity assay: standardization of a method allowing additional analyses on recovered effector cells and supernatants. Clin Diagn Lab Immunol 8:1131–1135. doi: 10.1128/CDLI.8.6.1131-1135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]