

The anti-HIV peptide drug T20 (enfuvirtide) is the only membrane fusion inhibitor available for treatment of viral infection; however, it exhibits relatively weak antiviral activity, short half-life, and a low genetic barrier to inducing drug resistance. Design of lipopeptide-based fusion inhibitors with extremely potent and broad antiviral activities against divergent HIV-1, HIV-2, and SIV isolates have provided drug candidates for clinical development. Here, we have verified a high therapeutic efficacy for the lipopeptide LP-52 in SIVmac239-infected rhesus monkeys. The resistance mutations selected in vivo have also been characterized, providing insights into the mechanism of action of newly designed fusion inhibitors with a membrane-anchoring property. For the first time, the data show that HIV-1 and SIV can share a similar genetic pathway to develop resistance, and that a lipopeptide fusion inhibitor could have a same resistance profile as its template peptide.
KEYWORDS: HIV, SIV, drug resistance, fusion inhibitor, lipopeptide
ABSTRACT
We recently reported a group of lipopeptide-based membrane fusion inhibitors with potent antiviral activities against human immunodeficiency virus type 1 (HIV-1), HIV-2, and simian immunodeficiency virus (SIV). In this study, the in vivo therapeutic efficacy of such a lipopeptide, LP-52, was evaluated in rhesus macaques chronically infected with pathogenic SIVmac239. In a pilot study with one monkey, monotherapy with low-dose LP-52 rapidly reduced the plasma viral loads to below the limit of detection and maintained viral suppression during three rounds of structurally interrupted treatment. The therapeutic efficacy of LP-52 was further verified in four infected monkeys; however, three out of the monkeys had viral rebounds under the LP-52 therapy. We next focused on characterizing SIV mutants responsible for the in vivo resistance. Sequence analyses revealed that a V562A or V562M mutation in the N-terminal heptad repeat (NHR) and a E657G mutation in the C-terminal heptad repeat (CHR) of SIV gp41 conferred high resistance to LP-52 and cross-resistance to the peptide drug T20 and two newly designed lipopeptides (LP-80 and LP-83). Moreover, we showed that the resistance mutations greatly reduced the stability of diverse fusion inhibitors with the NHR site, and V562A or V562M in combination with E657G could significantly impair the functionality of viral envelopes (Envs) to mediate SIVmac239 infection and decrease the thermostability of viral six-helical bundle (6-HB) core structure. In conclusion, the present data have not only facilitated the development of novel anti-HIV drugs that target the membrane fusion step, but also help our understanding of the mechanism of viral evolution to develop drug resistance.
IMPORTANCE The anti-HIV peptide drug T20 (enfuvirtide) is the only membrane fusion inhibitor available for treatment of viral infection; however, it exhibits relatively weak antiviral activity, short half-life, and a low genetic barrier to inducing drug resistance. Design of lipopeptide-based fusion inhibitors with extremely potent and broad antiviral activities against divergent HIV-1, HIV-2, and SIV isolates have provided drug candidates for clinical development. Here, we have verified a high therapeutic efficacy for the lipopeptide LP-52 in SIVmac239-infected rhesus monkeys. The resistance mutations selected in vivo have also been characterized, providing insights into the mechanism of action of newly designed fusion inhibitors with a membrane-anchoring property. For the first time, the data show that HIV-1 and SIV can share a similar genetic pathway to develop resistance, and that a lipopeptide fusion inhibitor could have a same resistance profile as its template peptide.
INTRODUCTION
HIV entry into target cells is mediated by trimeric envelope (Env) glycoproteins composed of the surface subunit gp120 and the noncovalently associated transmembrane subunit gp41 (1, 2). As modeled, binding of gp120 to the cellular receptor CD4 and a coreceptor (CCR5 or CXCR4) triggers dramatic structural changes in the Env complex and activates the fusogenic activity of gp41. First, the N-terminal fusion peptide (FP) of gp41 inserts into the cell membrane to bridge the viral and cellular membranes; then, three C-terminal heptad repeats (CHR) of gp41 pack antiparallelly into three hydrophobic grooves formed by central N-terminal heptad repeat (NHR) coiled coils, resulting in a six-helix bundle (6-HB) structure that pulls two membranes in close apposition for fusion (3–5). CHR- or NHR-derived peptides can bind to the gp41 prehairpin intermediate and block 6-HB formation, and thus possess potent anti-HIV activities. Currently, T20 (enfuvirtide), a 36-mer CHR peptide, is the only fusion inhibitor available for treatment of viral infection, and is effective in combination therapy for HIV-1 infection (6–8). However, T20 has relatively low therapeutic efficacy and a short half-life, and it easily induces drug resistance (9, 10).
Previous studies have demonstrated that chemically or genetically anchoring fusion inhibitor peptides to the cell surface is a vital strategy for improving antiviral activity (11–19). It is thought that the resulting peptides can interact with the cell membranes, thus raising the local concentrations of the inhibitors at the site where viral entry occurs (11, 15). By using this strategy, we previously developed short lipopeptides with a template that mainly targets the gp41 NHR pocket site, which exhibited greatly increased in vitro, ex vivo, and in vivo antiviral activities and stability (13, 14). As illustrated in Fig. 1, we also modified T20 by replacing its C-terminal lipid-binding sequence with a fatty acid group, generating a lipopeptide (LP-40) with a markedly increased potency (20). Astonishingly, the further optimization of LP-40 with different peptide sequences and conjugation strategies rendered a group of lipopeptides (LP-50, LP-51, LP-52, LP-80, and LP-83) with extremely potent and broad antiviral activities (21–24). In the in vitro studies, they inhibited divergent HIV-1, HIV-2, simian immunodeficiency virus (SIV), and T20-resistant strains with mean 50% inhibitory concentrations (IC50s) in the very low picomolar range. Of these lipopeptides, LP-51 and LP-80 exhibited high therapeutic efficacies in chimeric simian-human immunodeficiency virus (SHIV)-infected rhesus macaques (22, 24). In this study, we evaluated the in vivo antiviral activity of the lipopeptide LP-52 in SIV-infected rhesus macaques to assess its potential therapeutic efficacy. SIV mutants selected by LP-52 in treated monkeys were further characterized, providing critical information on the resistance pathway and mechanism of lipopeptide-based viral fusion inhibitors that possess membrane-anchoring features.
FIG 1.

Schematic diagram of SIV/HIV gp41 and peptide derivatives. (A) The sequence structure of gp41 and fusion inhibitor peptides. FP, fusion peptide; NHR, N-terminal heptad repeat; CHR, C-terminal heptad repeat; TM, transmembrane domain; CT, cytoplasmic tail. The positions and sequences corresponding to the T-20-resistance mutation site and the pocket-forming site in the NHR of HIV-1HXB2 reference are marked, and in which the “GIV” motif is colored blue. The pocket-binding domain (PBD) and tryptophan-rich motif in the CHR sequence are colored red and purple, respectively. C16, C18, and Chol in parentheses represent palmitic acid, stearic acid, and cholesterol, respectively; PEG8 represents a flexible linker of 8-unit polyethylene glycol. Engineered amino acids in newly designed lipopeptides are marked in pink. (B) SIVmac239 Env-derived NHR and CHR peptides. The amino acids corresponding to the characterized resistance mutations are marked in blue.
RESULTS
Therapeutic efficacy of LP-52 in SIV-infected rhesus macaques.
As demonstrated by our previous studies (21), the lipopeptide LP-52 was an extremely potent fusion inhibitor against diverse HIV-1/2 and SIV isolates. To gain insight into its therapeutic potential, we here evaluated LP-52 in SIV-infected rhesus macaques. Because one monkey (designated M1) chronically infected by SIVmac239 over 440 days was available, a pilot experiment was initiated by subcutaneously treating monkey M1 with LP-52 at 3 mg/kg of body weight once daily for 4 weeks. As shown in Fig. 2A, the plasma viral load of monkey M1 decreased from 4.2 log10 RNA copies/ml to below the detection limit (100 copies/ml) at day 3 after the initiation of treatment, which was the first blood sampling time after three drug injections. The viral load retained undetectable during the treatment but it rebounded 14 days after LP-52 was withdrawn. After a 120-day interruption, a second round of treatment was given to monkey M1 with the same dosage of LP-52 (3 mg/kg once daily for 4 weeks). Similarly, the plasma viral load quickly declined from 4.6 log10 RNA copies/ml to an undetectable level after three injections and maintained undetectable during the treatment. After a 120-day break, the monkey was treated with a reduced dosage of LP-52 (1 mg/kg once daily for 4 weeks). Encouragingly, the viral load declined to below the detection limit after treatment for 14 days and was fully controlled during the treatment period.
FIG 2.

Therapeutic efficacy of LP-52 in SIV-infected rhesus macaques. (A) Pilot study of structurally interrupted treatment of a rhesus monkey (M1) chronically infected with SIVmac239 over 440 days. LP-52 was subcutaneously administered at 3 mg/kg for the first and second rounds of treatment and at 1 mg/kg for the third round of treatment. (B to E) Treatment of rhesus monkeys M2 to M5 chronically infected with SIVmac239. After viral intravenous inoculation for 97 days, LP-52 was subcutaneously administered once daily for 4 weeks. The plasma viral loads and blood CD4+ T cells of the monkeys were monitored at different time points.
To further evaluate the therapeutic efficacy of LP-52, four rhesus monkeys (designated M2 to M5) were intravenously infected with SIVmac293 over 3 months, by which time chronic infection had been established with set point viral loads of 4.9 to 6.5 log10 RNA copies/ml. The monkeys were subcutaneously treated with LP-52 at 1 mg/kg once daily for 4 weeks. As shown, the plasma viral load of monkey M2 declined precipitously to below the detection limitation at day 3 after the initiation of treatment and retained undetectable during the treatment (Fig. 2B). The viral load in monkey M3 was also undetectable by day 14, but a viral rebound was observed by day 28 before the last injection was given (Fig. 2C). The viral load of monkey M4 declined from 6.5 to 4.3 log10 RNA copies/ml and then rebounded to a baseline level by day 21 (Fig. 2D). Monkey M5 had a viral load that declined from 5.6 log10 RNA copies/ml to an undetectable level by day 21 and then rebounded under the LP-52 treatment (Fig. 2E). Taken together, these results suggested that LP-52 was effective in treating SIV infection in rhesus monkeys, but a dosage of 1 mg/kg per day might not be enough to efficiently control SIV replication.
In vivo selection of SIV mutants resistant to LP-52.
To understand the mechanism of action of our newly designed lipopeptide fusion inhibitors, we focused on identifying the genetic mutations that mediated SIV rebound during the LP-52 treatment. The entire set of SIV env genes was amplified by PCR from the plasma samples of monkeys M3, M4, and M5 and cloned into an expression vector for DNA sequencing. As shown in Table 1, a total of 27 env clones were obtained from three monkeys and all the clones contained a single V562M or V562A mutation in the gp41 NHR site. In addition, an E567G mutation in the CHR site was frequently identified in the Envs from monkey M3, whereas a S760G mutation in the cytoplasmic tail region of gp41 universally emerged in the Envs cloned from monkey M5. In the gp120 subunit, all the Envs from monkey M5 also possessed a consistent F346L mutation. No consistent substitutions that apparently caused the resistance were localized in the other sites of gp120 or gp41 sequence.
TABLE 1.
Characterization of LP-52-selected mutations in SIVmac239 Env protein
| Monkey ID | SIV env clone | gp120 mutation | gp41 mutation |
|---|---|---|---|
| M3 | m3-1 | V562M, E657G | |
| M3 | m3-2 | E283G | V562M, A836V |
| M3 | m3-3 | V562M, E657G | |
| M3 | m3-4 | V562M, E657G | |
| M3 | m3-5 | V562M, E657G | |
| M3 | m3-6 | Q359R | V562A, K631E |
| M3 | m3-7 | V562M, A836V, D851G | |
| M3 | m3-8 | V562M, E657G | |
| M3 | m3-9 | V562M, E657G | |
| M4 | m4-1 | E66G, M309I, G348K, M404I, I502V | V562M, I710V, I737T, S806T, A807V |
| M4 | m4-2 | T118P, E163G | V562M, R705K, I710V, G754R, W838R, R858K |
| M4 | m4-3 | E66G, M309I, G348K, M404I, I502V | V562M, I710V, I737T, S806T, A807V |
| M4 | m4-4 | V562A | |
| M4 | m4-5 | V562M, L802F | |
| M4 | m4-6 | V562A, A608T, N759D | |
| M4 | m4-7 | I502V | V562M, L802F, H831R |
| M5 | m5-1 | F346L | V562A, S760G |
| M5 | m5-2 | F346L | V562A, S760G |
| M5 | m5-3 | F346L | V562A, S760G, R839K |
| M5 | m5-4 | F346L | V562A, S760G |
| M5 | m5-5 | F346L | V562A, S760G |
| M5 | m5-6 | F346L | V562A, S760G |
| M5 | m5-7 | F346L | V562A, S760G |
| M5 | m5-8 | Y21S, F346L | V562A, S760G |
| M5 | m5-9 | F346L | V562A, S760G |
| M5 | m5-10 | F346L | V562A, S760G |
| M5 | m5-11 | F346L | V562A, S760G |
Resistance profile of SIV and HIV mutants to LP-52.
In order to identify the mutations responsible for LP-52 resistance, we generated a panel of SIVmac239 env mutants that carried a single or combined amino acid substitution as characterized above. The corresponding pseudoviruses were prepared to determine the inhibitory activity of LP-52 by a single-cycle infection assay. As shown in Table 2, a single V562A or V562M mutation in the NHR of gp41 rendered the virus highly resistant to LP-52, with a resistance level of 324.59- or 31.65-fold changes (FC). Interestingly, a single CHR mutation, E657G, also conferred 5.12-fold resistance to LP-52, and it dramatically boosted the resistance levels mediated by V562A and V562M mutations. Specifically, the viruses with double mutations of V562A/E657G and V562M/E657G displayed 4,747.1- and 401.41-fold resistance, respectively. The mutations F346L in gp120 and S760G in the gp41 cytoplasmic site did not contribute to the resistance. We also generated a panel of HIV-1 pseudoviruses with Envs bearing a single V562A/M mutation or in combinations with E657G, which also displayed high levels of resistance to LP-52, verifying the roles of the characterized mutations in the resistance phenotypes.
TABLE 2.
Resistance profiles of diverse HIV fusion inhibitorsa
|
Pseudovirus |
LP-52 |
T20 |
LP-80 |
LP-83 |
LP-19 |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (nM) | FC | EC50 (nM) | FC | EC50 (nM) | FC | EC50 (nM) | FC | EC50 (nM) | FC | |
| SIVmac239 | ||||||||||
| WT | 0.09 ± 0.01 | 1 | 394.55 ± 22.56 | 1 | 0.03 ± 0.001 | 1 | 0.002 ± 0.000 | 1 | 0.38 ± 0.03 | 1 |
| F346L | 0.08 ± 0.03 | 0.94 | 400.58 ± 30.16 | 1.02 | 0.03 ± 0.004 | 1.09 | 0.002 ± 0.000 | 1 | 0.38 ± 0.02 | 0.99 |
| V562A | 27.59 ± 3.31 | 324.6 | 23832.66 ± 1590.59 | 60.4 | 1.83 ± 0.43 | 70.19 | 0.025 ± 0.002 | 12.67 | 0.33 ± 0.03 | 0.86 |
| V562M | 2.69 ± 0.41 | 31.65 | 3188.33 ± 775.16 | 8.08 | 0.38 ± 0.05 | 14.46 | 0.013 ± 0.001 | 6.5 | 0.25 ± 0.01 | 0.67 |
| E657G | 0.44 ± 0.2 | 5.12 | 811.77 ± 228.22 | 2.06 | 0.17 ± 0.01 | 6.5 | 0.006 ± 0.001 | 3.17 | 1.35 ± 0.26 | 3.55 |
| S760G | 0.07 ± 0.02 | 0.76 | 339.27 ± 74.99 | 0.86 | 0.03 ± 0.01 | 1.1 | 0.002 ± 0.001 | 0.83 | 0.36 ± 0.03 | 0.94 |
| V562A/E657G | 403.5 ± 76.27 | 4747 | 21548.56 ± 2143.45 | 54.62 | 108.78 ± 6.61 | 4184 | 0.696 ± 0.140 | 348 | 1.24 ± 0.22 | 3.27 |
| V562M/E657G | 34.12 ± 14.1 | 401.4 | 5344.44 ± 872.17 | 13.55 | 3.19 ± 0.49 | 122.5 | 0.09 ± 0.004 | 45.17 | 0.73 ± 0.05 | 1.92 |
| V562A/S760G | 20.93 ± 5.49 | 246.2 | 12122.89 ± 2895.06 | 30.73 | 1.03 ± 0.09 | 39.65 | 0.031 ± 0.006 | 15.33 | 0.36 ± 0.03 | 0.95 |
| V562M/S760G | 3.31 ± 0.89 | 38.94 | 2795.22 ± 370.75 | 7.08 | 0.31 ± 0.05 | 11.76 | 0.014 ± 0.004 | 6.83 | 0.21 ± 0.01 | 0.56 |
| E657G/S760G | 0.38 ± 0.17 | 4.47 | 899.03 ± 92.97 | 2.28 | 0.13 ± 0.03 | 4.82 | 0.006 ± 0.001 | 3 | 1.06 ± 0.09 | 2.78 |
| HIV-1 NL4-3 | ||||||||||
| WT | 0.01 ± 0.002 | 1 | 103.18 ± 2.49 | 1 | 0.004 ± 0.001 | 1 | 0.002 ± 0.000 | 1 | 0.14 ± 0.02 | 1 |
| V547A | 0.27 ± 0.03 | 42.26 | 1577.78 ± 204.37 | 15.29 | 0.25 ± 0.06 | 62 | 0.016 ± 0.001 | 8 | 0.16 ± 0.003 | 1.16 |
| V547M | 0.15 ± 0.02 | 23.16 | 537.26 ± 58.61 | 5.21 | 0.12 ± 0.02 | 31 | 0.008 ± 0.001 | 4 | 0.21 ± 0.01 | 1.49 |
| E646G | 0.01 ± 0.002 | 1.84 | 125.97 ± 9.46 | 1.22 | 0.01 ± 0.004 | 3 | 0.001 ± 0.000 | 0.5 | 0.17 ± 0.02 | 1.24 |
| V547A/E646G | 0.71 ± 0.15 | 111.6 | 1519.33 ± 164.57 | 14.73 | 0.67 ± 0.1 | 167.8 | 0.043 ± 0.016 | 21.5 | 0.17 ± 0.03 | 1.23 |
| V547M/E646G | 0.39 ± 0.04 | 62.11 | 660 ± 56.7 | 6.4 | 0.26 ± 0.08 | 65.75 | 0.011 ± 0.004 | 5.5 | 0.2 ± 0.06 | 1.44 |
Data were derived from three independent experiments and express as means ± standard deviations; FC, fold change; EC50, 50% effective concentration.
Cross-resistance to the peptide drug T20 and newly designed lipopeptides.
We next sought to characterize the cross-resistance profiles of LP-52-induced mutants to T20 and newly designed lipopeptides. As shown in Table 2, SIV mac239 with V562A, V562M, and E657G displayed 60.4-, 8.08-, and 2.06-fold resistance levels to T20, respectively, whereas HIV-1 NL4-3 with V547A, V546M, and E646G conferred 15.29-, 5.21-, and 1.22-fold resistance to T20, respectively. E657G did not show an enhancing effect on the V562A- and V562M-mediated T20 resistance. The V562A, V562M, and E657G mutations also mediated cross-resistance to the lipopeptide inhibitors LP-80 and LP-83, which share the same peptide sequence with LP-52 but possess increased antiviral activities (23, 24). Similar to that of LP-52, both the V562A/M-mediated resistance to LP-80 and LP-83 could be boosted by an E657G mutation. In contrast, V562A and V562M did not render either the SIV or HIV-1 isolates cross-resistant to LP-19, a small lipopeptide inhibitor mainly targeting the NHR pocket site rather than the T20- and LP-52-resistance sites (14). It was also noteworthy that a single E657G mutation remained to mediate mild resistance to LP-19 in SIVmac239 but not HIV-1 NL4-3.
The resistance mutations reduced the binding stability of fusion inhibitors.
To exploit the underlying mechanism of LP-52-selected resistance, we sought to determine the effects of V562A and V562M mutations on the helical interaction and binding stability of fusion inhibitor peptides. To this end, the SIV Env-derived peptide N39 with a wild-type sequence and its mutant peptides with a V562A or V562M substitution were synthesized as target surrogates (see Fig. 1B). As measured by CD spectroscopy, T20 and the lipopeptides LP-52, LP-80, and LP-83 could interact with N39 to form a typical α-helical conformation, as indicated by double minima at 208 and 222 nm; however, the V562A and V562M mutations resulted in markedly decreased α-helicity of the peptide complexes (Fig. 3 and Table 3). Because LP-19 primarily targets the NHR pocket site and does not interact with N39, we thus synthesized wild-type and mutant N36 peptides. Circular dichroism (CD) spectra of the peptide complexes showed that V562A and V562M did not change the α-helical interaction between N36 and LP-19. Defined as the midpoint of thermal unfolding transition (Tm), the thermal stability of peptide complexes demonstrated the binding stability of inhibitors. It was found that V562A and V562M severely attenuated the binding stability of LP-52, LP-80, and LP-83. Specifically, LP-52 interacted with the wild-type N39 with a Tm of 61.1°C, whereas it bound to N39V562A and N39V562M with Tm values of 45.4 and 50.7°C, respectively. The Tm of T20-based complexes could not be defined owing to low α-helical contents, and V562M did not affect the binding of LP-19 to N36 whereas V562A caused a decreased thermostability. Combined, these results suggest that the V562A or V562M mutation in the gp41 NHR helices can dramatically reduce the binding stability of fusion inhibitors, thus critically determining viral resistance and cross-resistance.
FIG 3.

Effects of the resistance mutations on the binding stability of fusion inhibitors determined by CD spectroscopy. The α-helicity (left panels) and thermostability (right panels) of T20 (A), LP-52 (B), LP-80 (C), LP-83 (D), and LP-19 (E) in complexes with the SIV Env NHR-derived peptide N39 or N36 with a wild-type or mutant sequence were measured with the final concentration of each peptide at 10 μM in PBS. The experiments were performed two times and obtained consistent results, and representative data are shown.
TABLE 3.
Effect of LP-52-selected resistance mutations on the helical interaction and binding stability of diverse fusion inhibitorsa
| Peptide complex | [θ]222 | Helix content (%) | Tm (°C) |
|---|---|---|---|
| T20+N39 | −8201 | 24.9 | NA |
| T20+N39V562A | −4836 | 14.7 | NA |
| T20+N39V562M | −4588 | 13.9 | NA |
| LP-52+N39 | −23162 | 70.2 | 61.1 |
| LP-52+N39V562A | −12143 | 36.8 | 45.4 |
| LP-52+N39V562M | −14978 | 45.4 | 50.7 |
| LP-80+N39 | −20960 | 63.5 | 62.8 |
| LP-80+N39V562A | −11406 | 34.6 | 39.1 |
| LP-80+N39V562M | −13426 | 40.7 | 50.6 |
| LP-83+N39 | −20647 | 62.6 | 68.4 |
| LP-83+N39V562A | −14578 | 44.2 | 58.4 |
| LP-83+N39V562M | −15897 | 48.2 | 58 |
| LP-19+N36 | −23907 | 72.4 | 51.3 |
| LP-19+N36V562A | −23282 | 70.6 | 46.6 |
| LP-19+N36V562M | −22782 | 69 | 51.2 |
| C34+N36 | −16114 | 48.8 | 45.9 |
| C34+N36V562A | −11260 | 34.1 | 39.3 |
| C34+N36V562M | −13230 | 40.1 | 42.1 |
| C34E657G+N36 | −14886 | 45.1 | 37.1 |
| C34E657G+N36V562A | −10618 | 32.2 | 31.7 |
| C34E657G+N36V562M | −9443 | 28.6 | 34.1 |
[θ]222, mean residue ellipticity at 222 nm; Tm, melting point melting temperature defined as the midpoint of the thermal unfolding transition; NA, not applicable.
Effect of the resistance mutations on Env-mediated viral fusion and entry.
To gain more insights into the mechanism of viral resistance to LP-52 and cross-resistance, we next focused on characterizing the effects of the resistance mutations on the functionality of viral Env glycoprotein by two approaches. First, the infectivity of SIVmac239 pseudoviruses bearing single or combined mutations was determined by single-cycle infection assay. As shown in Fig. 4A, the infectivity of the wild-type viruses was normalized to 100% and the relative infectivity of the mutant viruses was accordingly calculated for comparison. None of the single mutations (F346L, V562A, V562M, E657G, or S760G) affected the cell entry efficiency of SIVmac239 pseudoviruses; however, the combinations of V562A/E657G, V562M/E657G, and E657G/S760G resulted in viruses with significantly decreased infectivity. Second, we measured Env-mediated cell-cell fusion by a dual split protein (DSP)-based fusion assay. Surprisingly, none of the mutations significantly affected the functionality of SIV Envs to mediate cell-cell fusion except that a single S760G mutation showed an enhancing activity (Fig. 4B). We further analyzed the expression of viral Envs in transfected cells by Western blotting. As shown in Fig. 4C, all the mutants displayed similar expression levels and processing profiles. Thus, the reduced infectivity of the three mutant pseudoviruses might be attributed to the decreased amounts of Env incorporation into the budding virions.
FIG 4.
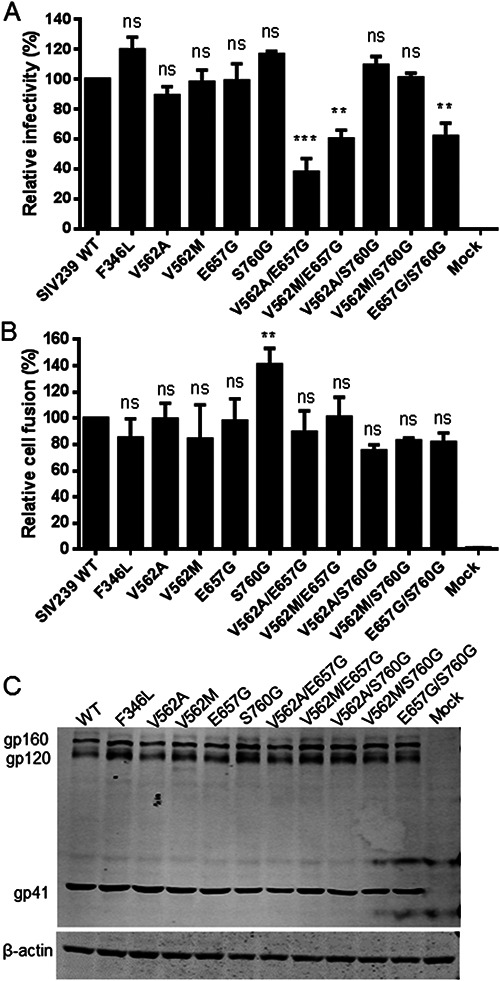
Effects of LP-52-selected mutations on the functionality of SIV Env. (A) Infectivity of the wild-type (WT) and mutant SIVmac239 pseudoviruses in TZM-bl cells. The viral particles were normalized to a fixed amount by p24 antigen and their relative infectivity was determined by a single-cycle infection assay. The luciferase activity of WT SIVmac239 was treated as 100%, and the relative activities of other mutant viruses were calculated accordingly. (B) Relative cell-cell fusion activity of WT and mutant SIVmac239 Envs determined by a dual split-protein (DSP) assay. HEK293T cells expressing viral Env and DSP1-7 were used as effector cells and 293FT cells expressing CXCR4/CCR5 and DSP8-11 were used as target cells. Similarly, the luciferase activity of WT Env was treated as 100%, and the relative fusion activities of other mutant Envs were calculated accordingly. The data were derived from the results of three independent experiments and are expressed as means and standard deviations (SD). Statistical comparisons were conducted by ANOVA (**, P < 0.01; ***, P < 0.001). (C) The expression and processing of SIVmac239 Envs determined by Western blotting. The viral glycoproteins in the lysates of transfected cells were detected with a monkey anti-SIV serum. The bands corresponding to gp160, gp120, and gp41 are respectively marked. The experiments were repeated two times, and representative data are shown.
The resistance mutations impaired the stability of viral 6-HB core structure.
Our previous studies demonstrated that the resistance mutations to HIV-1 fusion inhibitors can enhance or reduce the interaction of viral 6-HB core structure, thus contributing to the resistance phenotypes. Here, we were interested to know whether the same case occurred in SIV Env. To this aim, we synthesized the SIV Env-derived peptide C34 that has been considered a core sequence of viral 6-HB conformation. A C34 peptide carrying an E657G substitution was also generated to examine the effect of such a CHR mutation. As shown in Fig. 5 and Table 3, both the V562A and V562M mutations impaired the α-helicity and thermostability of the N36/C34-based 6-HB, and the E657G mutation also reduced the thermostability significantly. When V562A or V562M and E657G were combined, they displayed synergistic effects to reduce the α-helical content and Tm of the peptide complexes. Therefore, both the NHR and CHR mutations might contribute to the resistance in a coordinated manner.
FIG 5.

Effects of the resistance mutations on the helical interaction and stability of 6-HB structure modeled by the NHR and CHR peptides. The α-helicity and thermostability of 6-HBs formed between SIV-derived peptides C34 and N36 or N36 with a V562A or V562M substitution (A and B) and between C34 with an E657G substitution and N36 or its mutants (C and D) were determined by CD spectroscopy. The final concentration of each peptide in PBS was 10 μM. The experiments were performed two times and obtained consistent results, and representative data are shown.
DISCUSSION
In this study, we first set out to assess the in vivo therapeutic efficacy of LP-52, a lipopeptide-based fusion inhibitor with a highly potent and broad-spectrum antiviral activity. In a pilot study with rhesus monkey M1, who was chronically infected with SIVmac239 over 440 days, we were surprised to observe that a monotherapy with low-dose LP-52 dramatically reduced the plasma viral load to below the detection limit and maintained viral suppression during the 4-week treatment. In order to gain more insights into the therapeutic efficacy of LP-52 in SIV-infected monkeys, we thus decided to treat the monkey M1 with a structurally interrupted treatment protocol. The second round of treatment used the same dosage of LP-52, i.e., 3 mg/kg once daily for 4 weeks, which also achieved viral suppression efficiently. Therefore, we reduced the dose of LP-52 to 1 mg/kg of body weight and administered the monkey once daily for 4 weeks in the third round of treatment. Similarly, the viral load precipitated below the detection limit and retained undetectable until LP-52 was withdrawn. Encouraged by the results from the monkey M1, we thus applied a low-dose LP-52 for treating four monkeys with newly established chronic infections. The plasma viral loads in three of the monkeys (M2, M3, and M5) reduced to undetectable levels but rebounded in two monkeys. LP-52 decreased the viral load over 2 log RNA copies/ml in the monkey M4. Regardless, the present results still suggested that LP-52 is a highly active inhibitor in vivo, especially where it is tested with an SHIV-infected monkey model. We would thus like to speculate that LP-52 can fully control SIV replication if an increased dosage is rationally applied.
Since T20 was discovered in the early 1990s, its resistance pathway was extensively characterized by in vitro selection and in treated patients (9, 10, 25–27). The resistance mutations to T20 were primarily mapped to the peptide-binding sites on the NHR of HIV-1 gp41, and in particular the amino acid stretch 547GIVQQQNNLL556 (the sequence numbering of HIV-1HXB2), corresponding to the 560GIVQQQQQLL569 of SIVmac239, which served as a key determinant to the resistance phenotype. In addition to the primary NHR mutations, some mutations in the gp41 CHR site, such as N637K, E647G, E648K, and S649A, were identified as secondary mutations that compensated for the fusion kinetics of resistant viruses (28–40). In the past decades, the resistance profiles of a panel of new CHR-based HIV-1 fusion inhibitors, including C34 (32), T1249 (28), T2635 (30), and SC34EK (29), were finely characterized by selecting resistant HIV-1 mutants, which revealed the critical amino acid mutations responsible for the resistance. In our studies, we have not only dedicated to develop viral inhibitors that block the membrane fusion step, but also focused on exploiting the resistance mechanism for various fusion inhibitor peptides, such as sifuvirtide (31, 41), SC22EK (42), MTSC22EK (43), and SC29EK (44). Recently, we have been interested to determine how HIV-1 mutants are escaping the newly designed lipopeptide inhibitors that display dramatically increased antiviral potency and in vivo stability, as well as membrane-anchoring characteristics. In light of our failure to select resistant HIV-1 mutants in vitro, the viral rebounds emerging under the LP-52 treatment in SIVmac239-infected monkeys offered the opportunity to study this resistance. On the basis of our results, we would here like to address several points that surprised us during the resistance analysis. First, a single mutation of V562A or V562M (corresponding to the primary mutation V549A/M in HIV-1HXB2) or its combination with a CHR-based E657G mutation (corresponding to the secondary mutation E648G in HIV-1HXB2) was observed to confer high levels of resistance, which could imply a relatively low genetic resistance barrier for LP-52, at least in the treatment of SIV isolates. Second, it was found for the first time that SIV and HIV-1 can share a similar genetic pathway to develop resistance. Third, an extremely potent lipopeptide fusion inhibitor with membrane-anchoring features also shares a similar resistance profile with its original template peptide.
Eggink et al. (45) previously described several pathways underlying the mechanism of HIV-1 resistance to fusion inhibitors, including small amino acid-mediated reduced contact, large amino acid-mediated steric obstruction, acidic amino acid-mediated electrostatic repulsion, and basic amino acid-mediated electrostatic attraction (45) We further proposed that the disruption of hydrogen bonds and hydrophobic contacts would severely impair the binding of inhibitors, thus determining the resistance (46). Indeed, our previous studies repeatedly showed that both the primary NHR mutations and secondary CHR mutations could reduce the binding stability of fusion inhibitors, impair the functionality of viral Env, and the conformation and stability of viral 6-HB core structure (41–44). Again, our present studies verified that the V562A, V562M, and E657G mutations behaved similarly. As this study was the first time assessing a viral fusion inhibitor in SIV-infected monkey models, we therefore infer that SIV and HIV-1 can share a similar molecular mode to develop resistance to diverse peptide- and lipopeptide-based membrane fusion inhibitors. In our future projects, we will definitely continue to investigate the genetic pathways and mechanisms of HIV-1 and SIV resistances to other newly developed lipopeptides that possess more potent antiviral activities than LP-52, such as LP-80 and LP-83 (23, 24).
MATERIALS AND METHODS
Synthesis of peptides and lipopeptides.
Peptides (T20, N39 and its mutants, and C34 and its mutants) and lipopeptides (LP-19, LP-52, LP-80, and LP-83) were synthesized using a standard solid-phase 9-flurorenylmethoxycarbonyl (Fmoc) method as described previously (23). Fatty acid-conjugated peptides were prepared with a template peptide containing a C-terminal lysine residue with a 1-(4,4-dimethyl-2,6-dioxocyclohexylidene) ethyl (Dde) side chain-protecting group, which requires a deprotection step in a solution of 2% hydrazinehydrate-N,N-dimethylformamide. Cholesterylated peptide LP-83 was prepared by chemoselective thioether conjugation between a template peptide containing a C-terminal cysteine residue and bromoacetic acid cholesterol. All peptides were acetylated at the N terminus and amidated at the C terminus. They were purified by reverse-phase high-performance liquid chromatography (HPLC) and characterized by mass spectrometry. Concentrations of the peptides were measured by UV absorbance and a theoretically calculated molar extinction coefficient based on the tryptophan and tyrosine residues.
Treatment of SIV-infected rhesus macaques with LP-52.
Five adult Chinese rhesus macaques (Macaca mulatta) screened to be negative for SIV, herpes B virus, and simian T-lymphotropic virus were used for evaluating the in vivo therapeutic efficacy of LP-52. Monkey M1 chronically infected by pathogenic SIVmac239 over 440 days was enrolled in this study. Four monkeys (M2, M3, M4, and M5) were intravenously inoculated once with 100 TCID50 of SIVmac239 and chronic infection was established over 3 months. The monkeys were treated with LP-52 at 1 or 3 mg/kg of body weight once daily. Peripheral blood was periodically collected during the whole observation. Plasma viral loads of SIVmac239 were detected by a quantitative real-time reverse transcription-PCR (qRT-PCR) assay. Briefly, viral RNA was isolated from cell-free plasma using a QIAamp Viral RNA minikit (Qiagen, Valencia, CA) and cDNA samples were prepared with SuperScript III first-strand synthesis system for RT-PCR kit (Invitrogen, Carlsbad, California). RT-PCRs were carried out on an ABI 9700 real-time PCR system (Applied Biosystems), in which samples were processed in duplicate using the following cycling protocol: 48°C for 30 min, 95°C for 10 min, followed by 40 cycles at 95°C for 15 s, and 60°C for 1 min. The sequences of forward primer (5′-GCAGAGGAGGAAATTACCCAGTAC-3′), reverse primer (5′-CAATTTTACCCAGGCATTTAATGTT-3′), and a probe used for viral RNA were targeted against the conserved region of SIVmac239 gag protein. The limit of detection was 100 copy equivalents of RNA per ml of plasma. To measure the kinetics of CD4+ T cells, ethylenediaminetetraacetic acid (EDTA)-anticoagulated whole blood was stained with monoclonal antibodies CD3-PE/Cy7 (SP34-2), CD4-Percp/Cy5.5 (L200), and CD8-APC/Cy7 (RPA-T8), and CD4 T cell counts were determined with BD truecount tubes according to the manufacturer’s instructions (BD Biosciences, San Jose, CA). All the samples were acquired and analyzed on a FACS Aria II instrument (BD).
Amplification and sequencing of SIVmac239 env.
Viral RNA and cDNA samples from LP-52-treatted monkeys were prepared as described above. An endpoint limiting-dilution nested-PCR assay was applied to amplify the full-length env gene of SIVmac239. The outer forward primer was 5′-TCCTCTCTCAGCTATACCGCCCTC-3′ and reverse primer was 5′-ACTTTTGGCCTCACTGATACCCCTA-3′, and the inner primer pair was 5′-CGGGATCCTGAGCAGTCACGAAAGAGAAGAAGAACTC-3′ and 5′-GCTCTAGAACTGCCCCTGATTGTATTTCTGTCCCTC-3′. The PCR was carried out as follows: 94°C for 10 min, followed by 30 cycles at 94°C for 30 s, 60°C for 30 s, and 72°C for 3 min, and then a final extension at 72°C for 10 min. The PCR products were purified and subjected to DNA sequencing (SinoGenoMax Co., Beijing, China).
Site-directed mutagenesis.
The selected resistance mutations were introduced into the Env glycoprotein of SIVmac239 or HIV-1 NL4-3 by QuickMutation site-directed mutagenesis kit (Beyotime Biotechnology, Shanghai, China). Two primers were designed to contain specific mutations and occupied the same starting and ending positions on the opposite strands of an env-expressing plasmid. DNA synthesis was conducted by PCR in a 25-μl reaction volume using 50 ng of template plasmid. PCR amplification was performed for one cycle of denaturation at 95°C for 1 min, followed by 20 cycles of 95°C for 40 s, 60°C for 1 min, and 68°C for 8 min, with a final extension at 72°C for 10 min. The amplicons were treated with restriction enzyme DpnI for 1 h at 37°C, and DpnI-resistant molecules were recovered by transforming Escherichia coli strain DH5α with antibiotic resistance. The required mutations were confirmed by DNA sequencing.
Single-cycle infection assay.
The infectivity of SIVmac239 or HIV-1 NL4-3 and their inhibitions were determined by a single-cycle infection assay as described previously (47). In brief, pseudoviruses were generated by cotransfecting HEK293T cells with an Env-encoding plasmid and a viral backbone plasmid pSG3△env. Cell culture supernatants containing the packaged pseudoviruses were harvested 48 h posttransfection. The same amounts of pseudovirus particles were normalized by p24 antigen and their infectivity was determined in TZM-bl cells. To measure the inhibitory activity of fusion inhibitors, peptides were prepared in 3-fold dilutions, mixed with 100 TCID50 of viruses. After incubation for 1 h at room temperature, the mixture was added to TZM-bl cells (104 cells/well) and then incubated for 48 h at 37°C. Luciferase activity was determined using luciferase assay reagents and a luminescence counter (Promega, Madison, WI, USA). Percent inhibition of the pseudovirus and 50% inhibitory concentration (IC50) of an inhibitor were calculated using GraphPad Prism software (GraphPad Software Inc., San Diego, CA).
Cell-cell fusion assay.
A dual split protein (DSP)-based fusion cell-cell assay was used to measure SIV Env-mediated cell-cell fusion activity as described previously (47). Briefly, a total of 1.5 × 104 HEK293T cells (effector cells) were seeded on a 96-well plate and incubated overnight, and they were then transfected with a mixture of an Env-expressing plasmid and a DSP1-7 plasmid. After 24 h, 3 × 104 293FT cells expressing CXCR4/CCR5 and DSP8-11 (target cells) were resuspended in prewarmed culture medium that contained EnduRen live-cell substrate (Promega) at a final concentration of 17 ng/μl and then transferred to the effector cell wells at equal volumes. The mixed cells were spun down to maximize cell-cell contact, and the luciferase activity was measured as described above.
Circular dichroism spectroscopy.
Circular dichroism (CD) spectroscopy was performed to determine the α-helicity and thermostability of the peptide complexes as described previously (41). Briefly, the SIV-derived NHR peptide N39 or N36 was incubated with an equal molar concentration of a fusion inhibitor peptide at 37°C for 30 min in phosphate-buffered saline (PBS, pH 7.2). CD spectra were acquired on a Jasco spectropolarimeter (model J-815) using a 1-nm bandwidth with a 1-nm step resolution from 195 to 260 nm at 20°C. Spectra were corrected by subtraction of a solvent blank. The α-helical content was calculated from the CD signal by dividing the mean residue ellipticity ([θ]) at 222 nm by the value expected for 100% helix formation (−33,000 degrees cm2 dmol−1). Thermal denaturation was conducted by monitoring the ellipticity change at 222 nm from 20 to 98°C at a rate of 2°C/min using a temperature controller, and melting temperature (Tm) was defined as the midpoint of the thermal unfolding transition.
Western blotting assays.
The expression profile of SIVmac239 Env was examined by Western blotting assay as described previously (47). In brief, HEK293T cells were transfected with an Env-expressing plasmid and cultured for 48 h. Then, the lysates of transfected cells were centrifuged at 20,000 × g at 4°C for 15 min to remove insoluble materials. Equal amounts of total proteins were separated by SDS-PAGE and then transferred to a nitrocellulose membrane. After blocking with 5% nonfat dry milk solution in Tris-buffered saline (TBS, pH 7.4) at room temperature for 1 h, the membrane was incubated with a 1:200 diluted monkey serum derived from the SIVmac239-infected rhesus monkey overnight at 4°C. After washing three times with TBS-Tween 20, the membrane was incubated with horseradish peroxidase (HRP)-mouse-anti-monkey IgG. The β-actin protein was detected as an internal control with a mouse anti-β-actin monoclonal antibody (Sigma) and IRDye 680LT donkey-anti-mouse IgG. The membrane was then scanned using the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA).
Ethics statement.
Protocols for the use of animals were approved by the Institutional Animal Care and Use Committee (IACUC) at the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (No. ILAS-VL-2015-004). To ensure personnel safety and animal welfare, the study of animals was conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Science and the recommendations of the Weatherall report for the use of nonhuman primates in research (http://www.acmedsci.ac.uk/more/news/the-use-of-non-human-primates-in-research/). All monkeys were housed and fed in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facility. The experiments were performed in a bio-safety level 3 laboratory.
Data availability.
All data are fully available without restriction.
ACKNOWLEDGEMET
We thank Zene Matsuda at the Institute of Medical Science, University of Tokyo, for providing plasmids and cells for the DSP-based cell-cell fusion assay.
This work was supported by grants from the National Science and Technology Major Project of China (2018ZX10301103, 2017ZX10304402-001-011, 2017ZX10202102-001-003), and CAMS Innovation Fund for Medical Sciences (2017-I2M-1-014).
REFERENCES
- 1.Eckert DM, Kim PS. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem 70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 2.Colman PM, Lawrence MC. 2003. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol 4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 3.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 4.Tan K, Liu J, Wang J, Shen S, Lu M. 1997. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A 94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 6.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci U S A 91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lalezari JP, Henry K, O’Hearn M, Montaner JS, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ Jr, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo M, Group TS. 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med 348:2175–2185. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 8.Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med 4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 9.Rimsky LT, Shugars DC, Matthews TJ. 1998. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J Virol 72:986–993. doi: 10.1128/JVI.72.2.986-993.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenberg ML, Cammack N. 2004. Resistance to enfuvirtide, the first HIV fusion inhibitor. J Antimicrob Chemother 54:333–340. doi: 10.1093/jac/dkh330. [DOI] [PubMed] [Google Scholar]
- 11.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. 2009. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A 106:5801–5806. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Augusto MT, Hollmann A, Castanho MA, Porotto M, Pessi A, Santos NC. 2014. Improvement of HIV fusion inhibitor C34 efficacy by membrane anchoring and enhanced exposure. J Antimicrob Chemother 69:1286–1297. doi: 10.1093/jac/dkt529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chong H, Wu X, Su Y, He Y. 2016. Development of potent and long-acting HIV-1 fusion inhibitors. AIDS 30:1187–1196. doi: 10.1097/QAD.0000000000001073. [DOI] [PubMed] [Google Scholar]
- 14.Chong H, Xue J, Xiong S, Cong Z, Ding X, Zhu Y, Liu Z, Chen T, Feng Y, He L, Guo Y, Wei Q, Zhou Y, Qin C, He Y. 2017. A lipopeptide HIV-1/2 fusion inhibitor with highly potent in vitro, ex vivo, and in vivo antiviral activity. J Virol 91:e00288-17. doi: 10.1128/JVI.00288-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashkenazi A, Viard M, Unger L, Blumenthal R, Shai Y. 2012. Sphingopeptides: dihydrosphingosine-based fusion inhibitors against wild-type and enfuvirtide-resistant HIV-1. FASEB J 26:4628–4636. doi: 10.1096/fj.12-215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wexler-Cohen Y, Shai Y. 2009. Membrane-anchored HIV-1 N-heptad repeat peptides are highly potent cell fusion inhibitors via an altered mode of action. PLoS Pathog 5:e1000509. doi: 10.1371/journal.ppat.1000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang X, Jin H, Chen Y, Li L, Zhu Y, Chong H, He Y. 2019. A membrane-anchored short-peptide fusion inhibitor fully protects target cells from infections of HIV-1, HIV-2, and simian immunodeficiency virus. J Virol 93:e01177-19. doi: 10.1128/JVI.01177-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hildinger M, Dittmar MT, Schult-Dietrich P, Fehse B, Schnierle BS, Thaler S, Stiegler G, Welker R, von Laer D. 2001. Membrane-anchored peptide inhibits human immunodeficiency virus entry. J Virol 75:3038–3042. doi: 10.1128/JVI.75.6.3038-3042.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egelhofer M, Brandenburg G, Martinius H, Schult-Dietrich P, Melikyan G, Kunert R, Baum C, Choi I, Alexandrov A, von Laer D. 2004. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J Virol 78:568–575. doi: 10.1128/jvi.78.2.568-575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding X, Zhang X, Chong H, Zhu Y, Wei H, Wu X, He J, Wang X, He Y. 2017. Enfuvirtide (T20)-based lipopeptide is a potent HIV-1 cell fusion inhibitor: implication for viral entry and inhibition. J Virol 91:e00831-17. doi: 10.1128/JVI.00831-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chong H, Zhu Y, Yu D, He Y. 2018. Structural and functional characterization of membrane fusion inhibitors with extremely potent activity against HIV-1, HIV-2, and simian immunodeficiency virus. J Virol 92:e01088-18. doi: 10.1128/JVI.01088-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chong H, Xue J, Zhu Y, Cong Z, Chen T, Guo Y, Wei Q, Zhou Y, Qin C, He Y. 2018. Design of novel HIV-1/2 fusion inhibitors with high therapeutic efficacy in rhesus monkey models. J Virol 92:e00775-18. doi: 10.1128/JVI.00775-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Chong H, Yu D, Guo Y, Zhou Y, He Y. 2019. Design and characterization of cholesterylated peptide HIV-1/2 fusion inhibitors with extremely potent and long-Lasting antiviral activity. J Virol 93:e02312-18. doi: 10.1128/JVI.02312-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chong H, Xue J, Zhu Y, Cong Z, Chen T, Wei Q, Qin C, He Y. 2019. Monotherapy with a low-dose lipopeptide HIV fusion inhibitor maintains long-term viral suppression in rhesus macaques. PLoS Pathog 15:e1007552. doi: 10.1371/journal.ppat.1007552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sista PR, Melby T, Davison D, Jin L, Mosier S, Mink M, Nelson EL, DeMasi R, Cammack N, Salgo MP, Matthews TJ, Greenberg ML. 2004. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. AIDS 18:1787–1794. doi: 10.1097/00002030-200409030-00007. [DOI] [PubMed] [Google Scholar]
- 26.Baldwin CE, Sanders RW, Deng Y, Jurriaans S, Lange JM, Lu M, Berkhout B. 2004. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J Virol 78:12428–12437. doi: 10.1128/JVI.78.22.12428-12437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/aac.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eggink D, Baldwin CE, Deng Y, Langedijk JP, Lu M, Sanders RW, Berkhout B. 2008. Selection of T1249-resistant human immunodeficiency virus type 1 variants. J Virol 82:6678–6688. doi: 10.1128/JVI.00352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimura K, Nameki D, Kajiwara K, Watanabe K, Sakagami Y, Oishi S, Fujii N, Matsuoka M, Sarafianos SG, Kodama EN. 2010. Resistance profiles of novel electrostatically constrained HIV-1 fusion inhibitors. J Biol Chem 285:39471–39480. doi: 10.1074/jbc.M110.145789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eggink D, Bontjer I, Langedijk JP, Berkhout B, Sanders RW. 2011. Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J Virol 85:10785–10797. doi: 10.1128/JVI.05331-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Z, Shan M, Li L, Lu L, Meng S, Chen C, He Y, Jiang S, Zhang L. 2011. In vitro selection and characterization of HIV-1 variants with increased resistance to sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem 286:3277–3287. doi: 10.1074/jbc.M110.199323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nameki D, Kodama E, Ikeuchi M, Mabuchi N, Otaka A, Tamamura H, Ohno M, Fujii N, Matsuoka M. 2005. Mutations conferring resistance to human immunodeficiency virus type 1 fusion inhibitors are restricted by gp41 and Rev-responsive element functions. J Virol 79:764–770. doi: 10.1128/JVI.79.2.764-770.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lohrengel S, Hermann F, Hagmann I, Oberwinkler H, Scrivano L, Hoffmann C, von Laer D, Dittmar MT. 2005. Determinants of human immunodeficiency virus type 1 resistance to membrane-anchored gp41-derived peptides. J Virol 79:10237–10246. doi: 10.1128/JVI.79.16.10237-10246.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cabrera C, Marfil S, Garcia E, Martinez-Picado J, Bonjoch A, Bofill M, Moreno S, Ribera E, Domingo P, Clotet B, Ruiz L. 2006. Genetic evolution of gp41 reveals a highly exclusive relationship between codons 36, 38 and 43 in gp41 under long-term enfuvirtide-containing salvage regimen. AIDS 20:2075–2080. doi: 10.1097/QAD.0b013e3280102377. [DOI] [PubMed] [Google Scholar]
- 35.Xu L, Pozniak A, Wildfire A, Stanfield-Oakley SA, Mosier SM, Ratcliffe D, Workman J, Joall A, Myers R, Smit E, Cane PA, Greenberg ML, Pillay D. 2005. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob Agents Chemother 49:1113–1119. doi: 10.1128/AAC.49.3.1113-1119.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loutfy MR, Raboud JM, Montaner JS, Antoniou T, Wynhoven B, Smaill F, Rouleau D, Gill J, Schlech W, Brumme ZL, Mo T, Gough K, Rachlis A, Harrigan PR, Walmsley SL. 2007. Assay of HIV gp41 amino acid sequence to identify baseline variation and mutation development in patients with virologic failure on enfuvirtide. Antiviral Res 75:58–63. doi: 10.1016/j.antiviral.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 37.Poveda E, Rodes B, Labernardiere JL, Benito JM, Toro C, Gonzalez-Lahoz J, Faudon JL, Clavel F, Schapiro J, Soriano V. 2004. Evolution of genotypic and phenotypic resistance to Enfuvirtide in HIV-infected patients experiencing prolonged virologic failure. J Med Virol 74:21–28. doi: 10.1002/jmv.20141. [DOI] [PubMed] [Google Scholar]
- 38.Ray N, Blackburn LA, Doms RW. 2009. HR-2 mutations in human immunodeficiency virus type 1 gp41 restore fusion kinetics delayed by HR-1 mutations that cause clinical resistance to enfuvirtide. J Virol 83:2989–2995. doi: 10.1128/JVI.02496-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray N, Harrison JE, Blackburn LA, Martin JN, Deeks SG, Doms RW. 2007. Clinical resistance to enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J Virol 81:3240–3250. doi: 10.1128/JVI.02413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svicher V, Aquaro S, D'Arrigo R, Artese A, Dimonte S, Alcaro S, Santoro MM, Di Perri G, Caputo SL, Bellagamba R, Zaccarelli M, Visco-Comandini U, Antinori A, Narciso P, Ceccherini-Silberstein F, Perno C-F. 2008. Specific enfuvirtide-associated mutational pathways in HIV-1 Gp41 are significantly correlated with an increase in CD4(+) cell count, despite virological failure. J Infect Dis 197:1408–1418. doi: 10.1086/587693. [DOI] [PubMed] [Google Scholar]
- 41.Yu D, Ding X, Liu Z, Wu X, Zhu Y, Wei H, Chong H, Cui S, He Y. 2018. Molecular mechanism of HIV-1 resistance to sifuvirtide, a clinical trial-approved membrane fusion inhibitor. J Biol Chem 293:12703–12718. doi: 10.1074/jbc.RA118.003538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su Y, Chong H, Qiu Z, Xiong S, He Y. 2015. Mechanism of HIV-1 resistance to short-peptide fusion inhibitors targeting the gp41 pocket. J Virol 89:5801–5811. doi: 10.1128/JVI.00373-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su Y, Chong H, Xiong S, Qiao Y, Qiu Z, He Y. 2015. Genetic pathway of HIV-1 resistance to novel fusion inhibitors targeting the gp41 pocket. J Virol 89:12467–12479. doi: 10.1128/JVI.01741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu X, Liu Z, Ding X, Yu D, Wei H, Qin B, Zhu Y, Chong H, Cui S, He Y. 2018. Mechanism of HIV-1 resistance to an electronically constrained alpha-helical peptide membrane fusion inhibitor. J Virol 92:e02044-17. doi: 10.1128/JVI.02044-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eggink D, Langedijk JP, Bonvin AM, Deng Y, Lu M, Berkhout B, Sanders RW. 2009. Detailed mechanistic insights into HIV-1 sensitivity to three generations of fusion inhibitors. J Biol Chem 284:26941–26950. doi: 10.1074/jbc.M109.004416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao X, Chong H, Zhang C, Waltersperger S, Wang M, Cui S, He Y. 2012. Broad antiviral activity and crystal structure of HIV-1 fusion inhibitor sifuvirtide. J Biol Chem 287:6788–6796. doi: 10.1074/jbc.M111.317883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geng X, Liu Z, Yu D, Qin B, Zhu Y, Cui S, Chong H, He Y. 2019. Conserved residue Asn-145 in the C-terminal heptad repeat region of HIV-1 gp41 is critical for viral fusion and regulates the antiviral activity of fusion inhibitors. Viruses 11:609. doi: 10.3390/v11070609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are fully available without restriction.