

Herpes simplex virus 1 (HSV-1) establishes a lifelong infection in neuronal cells. The virus periodically reactivates and causes recurrent disease. Strains of HSV-1 vary greatly in their virulence and potential to reactivate in animal models. Although these differences are phenotypically well defined, factors contributing to the strains’ abilities to reactivate are largely unknown. We utilized a human neuronal cell line model of HSV latency and reactivation (LUHMES) to characterize the latent infection of two HSV-1 wild-type strains. We find that strain-specific differences in reactivation are recapitulated in LUHMES. Additionally, these differences correlate with the degree of heterochromatinization of the latent genomes. Our data suggest that the epigenetic state of the viral genome is an important determinant of reactivation that varies in a strain-specific manner. This work also shows the first evidence of strain-specific differences in reactivation outside the context of the whole animal at a human neuronal cell level.
KEYWORDS: herpes simplex virus, latency, neurotropism, neurovirulence, reactivation
ABSTRACT
Herpes simplex virus 1 (HSV-1) establishes a lifelong latent infection in peripheral nerve ganglia. Periodically, the virus reactivates from this latent reservoir and is transported to the original site of infection. Strains of HSV-1 have been noted to vary greatly in their virulence and reactivation efficiencies in animal models. While HSV-1 strain 17syn+ can be readily reactivated, strain KOS(M) shows little to no reactivation in the mouse and rabbit models of induced reactivation. Additionally, 17syn+ is markedly more virulent in vivo than KOS. This has raised questions regarding potential strain-specific differences in neuroinvasion and neurovirulence and their contribution to differences in the establishment of latency (or ability to spread back to the periphery) and to the reactivation phenotype. To determine if any difference in the ability to reactivate between strains 17syn+ and KOS(M) is manifest at the level of neurons, we utilized a recently characterized human neuronal cell line model of HSV latency and reactivation (LUHMES). We found that KOS(M) established latency with a higher number of viral genomes than strain 17syn+. Strikingly, we show that the KOS(M) viral genomes have a higher burden of heterochromatin marks than strain 17syn+. The increased heterochromatin profile for KOS(M) correlates with the reduced expression of viral lytic transcripts during latency and impaired induced reactivation compared to that of 17syn+. These results suggest that genomes entering neurons from HSV-1 infections with strain KOS(M) are more prone to rapid heterochromatinization than those of 17syn+ and that this results in a reduced ability to reactivate from latency.
IMPORTANCE Herpes simplex virus 1 (HSV-1) establishes a lifelong infection in neuronal cells. The virus periodically reactivates and causes recurrent disease. Strains of HSV-1 vary greatly in their virulence and potential to reactivate in animal models. Although these differences are phenotypically well defined, factors contributing to the strains’ abilities to reactivate are largely unknown. We utilized a human neuronal cell line model of HSV latency and reactivation (LUHMES) to characterize the latent infection of two HSV-1 wild-type strains. We find that strain-specific differences in reactivation are recapitulated in LUHMES. Additionally, these differences correlate with the degree of heterochromatinization of the latent genomes. Our data suggest that the epigenetic state of the viral genome is an important determinant of reactivation that varies in a strain-specific manner. This work also shows the first evidence of strain-specific differences in reactivation outside the context of the whole animal at a human neuronal cell level.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is an important human pathogen that is estimated to infect two-thirds of the world’s adult population (1). One of the key features of HSV-1 is the virus’s ability to establish a lifelong latent infection within peripheral nerve ganglia. Periodically, the virus reactivates from this latent reservoir and is transported anterogradely back to the original site of infection, typically the mucosal epithelia of the oral cavity or the corneal epithelium. The capacity of the latent virus to reactivate may be essential for reseeding of the host, as well as for dissemination to additional hosts. Therefore, the molecular understanding of the events that control reactivation is key to developing improved therapeutic approaches to reduce recurrent episodes as well as the spread of the virus through the population.
Current understanding of HSV-1 latency and reactivation has been gained by utilizing a variety of well-characterized wild-type laboratory strains. Among the most commonly used wild-type strains, 17syn+ and KOS(M), there are key differences reported in the frequency and extent of reactivation in vivo (2–7). While HSV-1 strain 17syn+ has been described as a high reactivator, strain KOS(M) displays little to no reactivation when induced to reactivate in vivo (2). Previous work investigating factors that influence the difference observed in reactivation found that HSV-1 strain KOS(M) establishes latency with a lower viral genome copy number than 17syn+ in vivo (6). It was suggested that this difference in latent viral genome load between the two virus strains explains the reduced reactivation frequency of KOS(M) compared to 17syn+ (6). While this conclusion correlates well with the reduced reactivation phenotype of KOS(M), it has been difficult to prove, since KOS(M) also exhibits a reduced neuroinvasion and neurovirulence phenotype (8). Therefore, the question remains whether strain 17syn+ is inherently more efficient at reactivating in neurons or whether a decreased establishment of latency or decreased ability to spread following reactivation was responsible for the reduced ability to reactivate in animal models for KOS(M).
HSV-1 latency and reactivation have been studied primarily in rabbit and mouse models, which has made dissociating reactivation from the neuroinvasion phenotypes challenging (9). Recent advances have made available neuronal cell models that support the latent phase of HSV-1 infections (10–15). These models offer the opportunity to study latency and reactivation at a neuronal cell level independent of outside factors, such as the immune system and spread between the neuronal and epithelial compartments. In this study, we examined the establishment and reactivation efficiency of HSV-1 strains 17syn+ and KOS(M) in the Lund human mesencephalic neuronal cell line (LUHMES) (10). We observe that HSV-1 strain KOS(M) establishes latency with a higher number of viral genomes per cell than strain 17syn+. We also show that these viral genomes have a higher burden of the heterochromatin marker H3K27me3 on their histones during both latency and upon induced reactivation than strain 17syn+. Furthermore, this increased heterochromatin profile for KOS(M) correlates with reduced viral transcription and infectious virus release following chemically induced reactivation from latency compared to that of 17syn+. Our findings suggest that the increased heterochromatinization of KOS(M) latent genomes accounts for the reduced reactivation phenotype compared to that of strain 17syn+.
RESULTS
Differences between HSV-1 strains 17syn+ and KOS in LUHMES cells during acute infection.
The LUHMES cell line is an engineered human neuronal precursor line that expresses the v-myc gene under the control of a TET-off promoter (16). The cells can be efficiently differentiated into postmitotic neurons by the addition of tetracycline, dibutyryl-cyclic AMP, and glial cell-derived neurotrophic factor (GDNF) to the growth medium and express markers of mature (dopaminergically fated) neurons within 5 days of differentiation (17). We have previously demonstrated that HSV-1 can establish a latent infection in LUHMES cells, and reactivation can be induced using an inhibitor of the phosphoinositide 3-kinase (PI3K) pathway (10). In the present study, we investigated potential strain-specific differences between HSV-1 strains 17syn+ and KOS(M) both during the acute infection and then during PI3K inhibitor-induced reactivation from a latent state in LUHMES neurons.
LUHMES cells were infected with 17syn+ or KOS(M) at a multiplicity of infection (MOI) of 5, and quantitative PCR (qPCR) was performed to monitor viral genome accumulation during the course of the acute infection. The experimental design, including the differentiation of the LUHMES cells and the acute infection time points that were examined, are illustrated in Fig. 1A. Surprisingly, immediately following the 1-h adsorption of virus with the cells (t = 0), 10-fold higher genome copies were measured for KOS(M)-infected LUHMES cells than to 17syn+-infected cells (Fig. 2A). Additionally, this 10-fold difference in genome copies between KOS(M) and 17syn+ was consistent across all time points during 24 h of the acute infection (Fig. 2A). Interestingly, these findings suggest that KOS(M) has a higher particle-to-PFU ratio, with an incoming genomic burden around 10-fold higher than that of 17syn+. We also found that 17syn+ genome copies do not reach KOS(M) genome levels during the first 24 h of an acute infection.
FIG 1.

Experimental timeline of neuronal differentiation, infection, latency, and reactivation. (A) Experimental timeline of acute infection. LUHMES neuronal cells were plated and allowed to proliferate for 3 days, followed by 5 days of differentiation, as described in Materials and Methods. Once differentiated, cells were infected with either 17syn+ or KOS(M) at an MOI of 5 for 1 h, with harvesting of DNA/RNA at the indicated times. (B) Experimental timeline of latency and reactivation. LUHMES neuronal cells were plated and allowed to proliferate for 3 days, followed by 5 days of differentiation. Once differentiated, cells were pretreated for 2 h in medium containing 50 μM acyclovir (ACV). Following pretreatment with ACV, cells were infected with either 17syn+ or KOS(M) at an MOI of 5 in medium containing 50 μM ACV. Forty-eight hours later, the medium was changed to medium without ACV and the infection was allowed to proceed, with harvesting at the latent time point 8 days postinfection (labeled 0 hours postreactivation). Reactivation was induced at day 8 postinfection (labeled as day 16) with 10 μM final concentration of Ly294002, and reactivation samples were harvested at the indicated times.
FIG 2.

HSV-1 genome copies during acute infection. (A) HSV-1 strain KOS(M) delivers and maintains significantly higher genome copies than 17syn+ during the acute infection of LUHMES neurons. HSV-1 genome copy numbers were tracked by qPCR using primers and probes designed against UL30 over a 24-h time course of infection of LUHMES cells with either 17syn+ or KOS(M) at an MOI of 5. (B) HSV-1 strain 17syn+ genome copies were determined as described above. A significant difference in genome copies was seen 12 and 24 h postinfection compared to levels at 0 h. Interestingly, at 12 h postinfection, genomic copies were reduced compared to those at 0 h, and input genomes appear to have replicated by 24 h postinfection. (C) HSV-1 strain KOS(M) input genomes undergo statistically signification replication by 24 h postinfection compared to levels at 0 h. For each time point, significance was determined by ordinary two-way ANOVA with Sidak’s multiple-comparison test across 6 biological replicates (*, P < 0.05; ***, P < 0.0001) (n = 6).
When we examined genome copies during the acute infection compared to that at 0 h postinoculation, we found additional differences between strains 17syn+ and KOS(M) (Fig. 2B and C). At 12 h postinfection, a 2-fold decrease in 17syn+ genomes was seen, which was not observed for KOS(M) (Fig. 2B and C). Additionally, at 24 h postinfection, while an increase in genomes was seen for both strains 17syn+ and KOS(M), 17syn+ had a 4-fold induction, while KOS(M) had a 2-fold induction (Fig. 2B and C). These results suggest that during an acute infection in LUHMES cells, the replication kinetics of HSV-1 genomes is restricted in general but is more pronounced in the case of strain KOS(M) compared to the fold increase of total genomes 24 h postinfection. Furthermore, these results reveal notable strain-specific differences in genome copy number between 17syn+ and KOS(M) and point to a potential genome dose-dependent effect on replication at later time points.
To further understand the biological implications of viral genome copy number during an acute infection of LUHMES neurons, we quantified viral RNA for select genes and analyzed the relationship between genomic copies and the relative gene expression during acute infection. RNA was reversed transcribed, and primers and probes specific for the viral targets ICP4, TK, and the LAT intron were assayed by qPCR (see Table 2). We chose to present the data as transcripts per genome to accurately reflect transcripts made from an equal number of genomes. Analysis of relative gene expression of the essential immediate-early gene ICP4 per genome reveals higher ICP4 messages being made per genome for 17syn+ than KOS(M) (Fig. 3A). Interestingly, we also find that the expression of the early gene TK relative to viral genomic content in the neurons is also strain dependent, as we measured higher TK expression for 17syn+ than for KOS(M) at 3 and 24 h postinfection (Fig. 3B). Lastly, when we look at relative LAT Intron expression, we saw higher expression per vial genome at 0, 6, and 24 h postinfection with 17syn+ (Fig. 3C). These results indicate that during an acute infection, HSV-1 transcription is more restricted in the case of KOS(M) than for 17syn+.
FIG 3.

More transcription is occurring per genome for strain 17syn+ than KOS(M) during acute infection of LUHMES. Shown are the relative gene expression profiles for HSV-1 ICP4 (A), TK (B), and LAT intron (C) expressed as copies per genome following a 24-h time course of infection of LUHMES with 17syn+ or KOS(M) at an MOI of 5. RT-qPCR was performed with primers and probes against the indicated genes (Table 2). Interestingly, on a per-genome basis, higher gene expression is seen for all three transcripts measured when LUHMES cells are infected with 17syn+ versus KOS(M). For each time point, significance was determined by ordinary two-way ANOVA with Sidak’s multiple-comparison test across 6 biological replicates (*, P < 0.05; ***, P < 0.0001) (n = 6).
Establishment of HSV-1 latency by strains 17syn+ and KOS in LUHMES cells.
HSV-1 strains 17syn+ and KOS(M) have been shown to vary in their respective rate of viral shedding during the establishment of a latent infection in vivo in the rabbit ocular model (2). In addition, murine trigeminal ganglia neurons latently infected with strain KOS in vivo have been shown to contain fewer HSV genomes than latently infected 17syn+ neurons (6). These differences in the establishment of latency were noted in both the mouse and rabbit models; therefore, differences in viral replication and efficiency of neuronal invasion to the sensory ganglia could play a role in the efficiency of the establishment of latency. In light of these earlier in vivo findings, we wanted to determine if we could detect strain-specific differences in either the shedding/production of infectious virus during the establishment of latency or in the genomic load during latency and if this is recapitulated in human LUHMES neuronal cells. This would allow us to assess the establishment of latency within neurons directly, as opposed to indirectly through infection of the neurons from the periphery, as in the case of the mouse and rabbit models.
To assess differences in the shedding/production of infectious virus between strains 17syn+ and KOS(M) during the establishment of latency, we assayed cell supernatants for infectious virus during the establishment period. In an effort to increase the sensitivity of the traditional HSV plaque assay, we incubated the entire cell culture supernatant from infected LUHMES cells with rabbit skin cell monolayers and did not remove the inoculum following the adsorption period. We detected fewer infectious particles in the supernatant for KOS(M) than for 17syn+ at 4 days postinfection (Fig. 4). Additionally, following 4 days postinfection, we did not detect infectious virus for strain KOS(M) compared to 17syn+, in which we see low levels of spontaneous reactivation.
FIG 4.
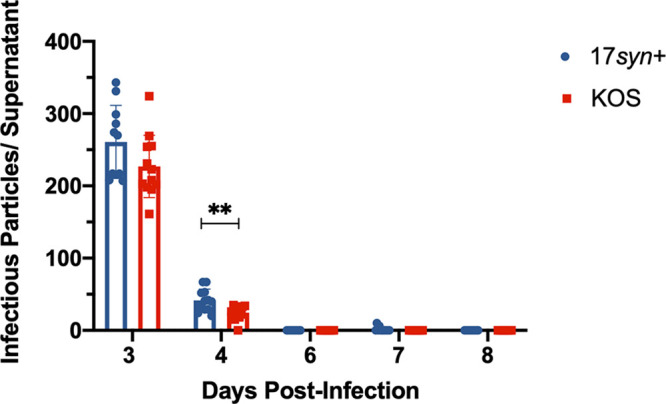
Time course plaque assay during establishment of latent infection. KOS(M) appears to enter latency earlier than 17syn+, as measured by less infectious virus production during the course of latency establishment. The data are represented as the number of infectious particles per LUHMES culture containing 150,000 cells. For each time point, significance was determined by ordinary two-way ANOVA with Sidak’s multiple-comparison test across 12 biological replicates (**, P < 0.01) (n = 12).
These data suggest that either KOS(M) establishes latency more quickly than 17syn+ or that it establishes latency less efficiently than 17syn+. To differentiate between the two possibilities, PCR to assess HSV-1 genomes was performed. As indicated in Fig. 5A (0-h time point), KOS(M) established latency as efficiently as 17syn+, and the latent KOS(M) cultures contained >2-fold more genomes than the 17syn+ latent cultures. These results suggest that KOS(M) establishes latency more efficiently than strain 17syn+.
FIG 5.

KOS(M) genomes are maintained at higher copies overall during latency and reactivation than with 17syn+ but are not significantly replicated following PI3K inhibitor-induced reactivation. (A) HSV-1 strain KOS(M) maintains significantly higher genome copies than 17syn+ during latency as well as during PI3K inhibitor-induced reactivation in LUHMES. Reactivation was induced at day 8 postinfection (0 h) with 10 μM Ly294002, and cultures were harvested for qPCR at the indicated times using primers and probes against UL30. (B) 17syn+ genome copies have been significantly replicated by 12 h postreactivation (compared to 0 h). (C) HSV-1 strain KOS(M) genome copies were determined as described above. We found no significant difference in genomes copy number during reactivation compared to that at 0 h, implying fewer genomes were replicated. For each time point, significance was determined by ordinary two-way ANOVA with Sidak’s multiple-comparison test across 6 biological replicates (*, P < 0.05; ***, P < 0.0001) (n = 6).
HSV-1 strain KOS(M) displays reduced reactivation compared to that of strain 17syn+.
HSV-1 strains 17syn+ and KOS(M) have been shown to differ in their reactivation frequency during in vivo infections of mice and rabbits (2–6). To determine if there were strain-specific differences in the efficiency of reactivation, we established a latent infection in LUHMES cells with 17syn+ and KOS(M). Following establishment of a latent infection, we chemically induced reactivation using a 10 μM final concentration of the PI3K inhibitor Ly294002 and assayed cell supernatant for evidence of entry into the productive viral lytic phase by plaque assay. We found that following the induction of reactivation, strain KOS(M) did not productively reactivate, as measured by plaque production, compared to strain 17syn+ (Table 1). Importantly, during all time points of reactivation studied, no infectious virus was detected for strain KOS(M) compared to various levels of lytic reactivation for 17syn+ (Table 1). These results show an impaired reactivation phenotype for KOS(M) compared with that of 17syn+ in human LUHMES neurons.
TABLE 1.
HSV-1 strain KOS(M) is not competent for full reactivation from latencya
| Time (h) postreactivation | Cultures positive for plaques [% (no. positive/total no.)] |
|
|---|---|---|
| 17syn+ | KOS(M) | |
| 0 | 8.3 (1/12) | 0 (0/12) |
| 3 | 0 (0/12) | 0 (0/12) |
| 6 | 50 (6/12) | 0 (0/12) |
| 12 | 66 (8/12) | 0 (0/12) |
| 18 | 16 (2/12) | 0 (0/12) |
| 24 | 42 (5/12) | 0 (0/12) |
These data demonstrate a pronounced difference in phenotypic reactivation for two different strains of HSV-1. Remarkably, KOS(M) fails to produce infectious virus upon PI3K inhibitor-induced reactivation. 17syn+, in contrast, readily reactivates under the same conditions. We see spontaneous reactivation in latent cultures of 17syn+ LUHMES of around 4 to 6% (unpublished data). For these experiments, around 8% of the latent cultures (0 h) were also able to enter the lytic phase. LUHMES culture supernatants were harvested at the indicated time points following reactivation with Ly294002. The latent time point is 8 days postinfection (labeled 0 h postreactivation). Shown are the percentages of each LUHMES culture (150,000 cells) for a total population of 1.8 × 106 cells that were positive for plaques during reactivation.
The striking difference in reactivation between these strains led us to query whether the inability of KOS(M) to reactivate was due to a reduced induction of lytic gene transcription and/or genome replication compared to those of strain 17syn+. DNA and RNA were extracted following induced reactivation, as depicted in Fig. 1B. An analysis of genome copies upon the induction of reactivation revealed roughly 3-fold higher numbers of genome copies for KOS(M) than for strain 17syn+ at all time points studied (Fig. 5A). When comparing genome copies during reactivation to our latent time point (0 h), we find that a significant increase in genomes for 17syn+ occurs at 12 h following inhibition of PI3K, which was not observed for strain KOS(M). The amplification of genomes and production of infectious virus was only observed for 17syn+ following induced reactivation, implying that strain 17syn+ reactivates more efficiently than KOS(M).
We then quantified viral transcript levels following Ly294002-induced reactivation. We chose to look at a gene from each kinetic class, as well as LAT intron levels during reactivation, to determine if differences in gene expression between the two strains correlate with the reactivation phenotypes described thus far. The immediate-early transcript ICP4 was detected at higher relative expression levels for strain 17syn+ than for KOS(M) at early time points (Fig. 6A). The same trend is seen for the early gene TK and the late gene US3 (Fig. 6B and D). Interestingly, higher levels of LAT intron expression were seen for strain KOS(M) than for strain 17syn+ (Fig. 6C). Overall, we find that the treatment of latently infected LUHMES cells with a PI3K inhibitor to induce reactivation results in more robust expression of lytic transcripts for 17syn+ than is seen for KOS(M) (Fig. 6). It is not clear to what degree the increased expression of the LAT is contributing to the inability of KOS(M) to reactivate from latency, but this is clearly an area of great interest and is currently being investigated in the LUHMES neuronal model.
FIG 6.

Transcripts produced per genome during the latent infection and following PI3K inhibitor-induced reactivation differ between 17syn+ and KOS(M). Relative ICP4 (A), TK (B), LAT intron (C), and US3 (D) expression calculated during latency and following reactivation are expressed on a per-genome basis. Reactivation was induced at day 8 postinfection with 10 μM Ly294002, and reactivation samples were harvested at the indicated times. RT-qPCR was performed as described in Materials and Methods and Table 2. For each time point, significance was determined by ordinary two-way ANOVA with Sidak’s multiple-comparison test across 6 to 12 biological replicates (*, P < 0.05; **, P < 0.001; ***, P < 0.0001) (n = 6 to 12).
The structure and conformation of the HSV-1 chromatin in LUHMES cells have not yet been determined. The differences we see in relative expression levels of HSV-1 transcripts between the two strains indicate that a reactivation phenotype is tied to chromatin modifications affecting the accessibility of viral genomes. To answer this question, we assayed facultative heterochromatin marks on the genomes of these two strains. Following reactivation with 10 μM Ly294002, we performed chromatin immunoprecipitation (ChIP) reverse transcription-qPCR (RT-qPCR) on the facultative heterochromatin mark H3K27me3 at various time points (Fig. 1B). H3K27me3 was chosen because it occupies the HSV-1 genome during latency, and removal of this mark has been found to be key to the ability of the virus to reactivate (18–20). We found that, both during latency and following induced reactivation, KOS(M) genomes were more enriched for H3K27me3 than 17syn+ genomes (Fig. 7). Collectively, these data point to a more heterochromatinized genome state for KOS(M) than for 17syn+. This chromatin inaccessibility for KOS(M) likely leads to the reduced reactivation phenotype we demonstrate here.
FIG 7.

HSV-1 strain KOS has increased H3K27me3 marks compared to those of strain 17syn+ during latency and PI3K inhibitor-induced reactivation in LUHMES. Chromatin immunoprecipitation was performed to measure heterochromatin formation on HSV-1 genomes from cultures of LUHMES cells infected with 17syn+ or KOS(M) at 8 days postinfection (latency) or following Ly294002-induced reactivation from latency. Shown are three regions of the HSV-1 genome queried by qPCR following ChIP. (A to C) ICP4 (A), the LAT promoter (B), and the late gene, gC (C). Heterochromatin was assessed by immunoprecipitation with an antibody recognizing H3K27me3. ChIP-qPCR data were normalized to percent input over total H3. Reactivation was induced at day 8 postinfection with 10 μM Ly294002, and reactivation samples were harvested at the indicated times. These data point to a significant reason why KOS(M) fails to reactivate. As shown here, 17syn+ genomes are associated with fewer heterochromatin marks than KOS(M), indicating less accessibility of the KOS genome for replication and transcriptional activities. For each time point, significance was determined by ordinary two-way ANOVA with Sidak’s multiple-comparison test across 3 biological replicates (*, P < 0.05; **, P < 0.001; ***, P < 0.0001) (n = 3).
DISCUSSION
HSV-1 strains 17syn+ and KOS(M) are known to have key differences in the frequency and magnitude of reactivation in vivo (2–7). The reduced neuroinvasive and neurovirulence phenotype of KOS(M) compared to that of 17syn+ has made it difficult to determine whether strain-specific differences in reactivation occur at a neuronal cell level or are mediated by indirect effects of the neuroinvasion and neurovirulence that reduce the efficiency of amplification and spread of the virus after it reactivates (8). In this study, we demonstrate that the ability of HSV-1 to reactivate is strain dependent in human neurons. We found that HSV-1 strain KOS(M) has a reduced reactivation phenotype compared to that of strain 17syn+ in LUHMES cells. Additionally, while KOS(M) actually establishes latency with a greater number of genomes than 17syn+, the KOS(M) genomes are more epigenetically silenced and have lower expression of lytic transcripts than 17syn+. These results suggest that the reduced reactivation phenotype of KOS(M) in vivo is due to a more epigenetically silenced genome that does not reactivate efficiently at a neuronal cell level.
Unexpectedly, we found that at the same multiplicity of infection (MOI) for strains 17syn+ and KOS(M), there was higher incoming genomic burden for KOS(M) than 17syn+ (Fig. 2). Interestingly, previous data have shown that immediately following intracerebral inoculation of mice with either KOS(M) or 17syn+ at the same MOI, there was an increase in viral yield for KOS(M) compared to 17syn+ at 0 h postinfection (8, 21). HSV-1 particle-to-PFU ratios have been known to differ between cell types and strains (22). These results suggest that KOS(M) and 17syn+ may have different particle-to-PFU ratios, which results in the increased genomic load for KOS(M). Another interpretation of the data suggests that KOS(M) enters LUHMES cells more efficiently than 17syn+. One explanation for the difference in entry could be the difference in the structure of gB between strains 17syn+ and KOS(M) (23). Studies have found that gB is one of the key glycoproteins thought to mediate cell fusion (24). If the fusion rates of HSV-1 strains 17syn+ and KOS(M) are different, this could explain the increased genomic burden at 1 h postinfection. Further studies are needed to determine if differences in genomic burden at the same MOI in LUHMES cells are mediated by differences in the particle/PFU ratio, gB-mediated fusion, or other mechanisms and whether this difference may be manifested in a neuron-specific manner.
Here, we show that the higher genomic burden for KOS(M) than 17syn+ is seen not only during acute infection but also during latency (Fig. 5). In striking contrast to our observations, it was previously reported that 17syn+ has a higher genomic burden than KOS(M) in mouse trigeminal nerve ganglion (6). However, one of the key differences between that study and the current study was that it was performed in vivo. Since KOS(M) is known to exhibit reduced neuroinvasion and neurovirulence in vivo, it has been difficult to tease apart the factors that lead to the impaired reactivation phenotype described thus far for this strain (8). For the first time, we can compare the two strains in an in vitro latency model, independent of nonneuronal host factors, which may contribute to a difference in establishment between the strains. Using this model, we find that KOS(M) establishes latency equal to, if not better than, that of strain 17syn+, meaning that another factor besides genome load likely influences the efficiency of reactivation. In addition, the accumulation of heterochromatic marks occurs earlier for KOS(M) than 17syn+, suggesting that KOS(M) establishes a latent state earlier.
Our data show that HSV-1 strain KOS(M) reactivates less frequently and with less infectious virus release, which is consistent with what has been seen in animal model data (2–7). Specifically, our data tracks most closely with models where reactivation is induced in vivo. In both the rabbit eye model, where reactivation is adrenergically induced, and in the murine ocular model, where reactivation is induced by hyperthermic stress, the reduced KOS(M) reactivation phenotype observed is consistent with the data we report here from human LUHMES neurons (2, 6). Interestingly, when we examine data from explant-induced reactivation models of murine sensory nerve ganglia latently infected with KOS(M) and 17syn+, KOS(M) still reactivates less efficiently than 17syn+; however, there is a much smaller difference between the strains than that seen in the LUHMES or the in vivo models (21, 25). One thing to note when comparing these data is the various methods of inducing reactivation and the methods of detection. It remains unknown whether there are differences in efficiencies between different inducers of reactivation (26, 27). For example, adrenergic or hyperthermic stress may activate different pathways than the stressors that induce reactivation in explanted ganglia. An additional caveat to consider when comparing these data sets is that, in the mouse and rabbit ocular models, reactivation is measured by productive virus shed rather than molecular reactivation. All these variables make it difficult to directly compare one system to another. However, here we were able to control some of these variables through the use of a human neuronal cell culture model that allowed us to measure reactivation at the cellular level, independent of the host.
From the studies we presented here, the number of incoming viral genomes during infection of the LUHMES cells does not appear to dictate the ability of HSV-1 to establish and maintain latency, and it does not appear to correlate with reactivation potential. It is, however, possible that the regulation of latency is, in fact, influenced during the establishment phase by a genome dose-dependent mechanism. It is conceivable that a higher incoming genomic burden leads to more tightly repressed genomes and, therefore, a lower frequency of reactivation. One could envision that a higher level of genomic burden could heighten host antiviral responses, which could result in a more transcriptionally repressed chromatin state. Additionally, it is possible that incoming defective genomes compete for host factors needed for reactivation. Further work needs to be done to characterize the effects of incoming genomic load on the outcome of infection in human neurons, and it will be interesting to juxtapose these data with effective incoming genome burdens that one sees during an in vivo infection.
In summary, we report the differences in the establishment and reactivation of HSV-1 strains KOS(M) and 17syn+ in a human neuronal cell culture system. Overall, we find that although KOS(M) establishes a higher genomic burden than strain 17syn+, those genomes are more epigenetically repressed and do not reactivate to the same extent as 17syn+. An association between the increased facultative heterochromatin histone marks and the decrease in reactivation seen for strain KOS(M) suggests that the epigenetic state of the latent genomes plays a role in overall reactivation. Since H3K9me3 has also been shown to play a role in reactivation, it will be interesting to examine the levels of H3K9me3 on strains KOS(M) and 17syn+ in future studies to see if it has an effect on reactivation efficiency (28–30). Further investigation is necessary to determine the mechanism of the increased heterochromatin profile on KOS(M) and how that contributes to reactivation efficiency.
MATERIALS AND METHODS
Viruses and cells.
HSV-1 viral strains 17syn+ and KOS(M) were used in this study, and they were previously plaque purified 3 times. Low-passage-number stocks of these viruses were obtained from J. Stevens and propagated in rabbit skin (RS) cells. RS cells, obtained from B. Roizman, were maintained in minimum essential medium (MEM; no. 11095-098; Gibco) supplemented with 5% bovine serum (no. 26170-043; Gibco) and 1% PSG (100 U penicillin, 100 mg/ml streptomycin, 0.292 mg/ml l-glutamine; no. SV30082.01; HyClone) at 37°C, 5% CO2. For plaque assays, whole supernatants were harvested at the corresponding time points and added to 6-well plates of RS cells. Following 48 h of incubation, cells were fixed and stained with crystal violet solution (8 mg/ml crystal violet [no. 18640; Sigma], 20.9% final concentration of ethanol [no. 2801G; Decon Labs], 79.1% final concentration of distilled H2O). Lund human mesencephalic (LUHMES) cells were obtained from the ATCC (no. CRL-2927) and were cultured as described previously (10). All experiments performed in this study utilized LUHMES cells at passages 3 to 5 from the original ATCC stock. Briefly, LUHMES cells were cultured in flasks, on plates, and on dishes, which were coated with poly-l-ornithine hydrobromide (no. P3655; Sigma) overnight at room temperature followed by fibronectin (no. F2006; Sigma) overnight at 37°C. Flasks, plates, and dishes then were rinsed with phosphate-buffered saline (no. SH30256.01; HyClone) and allowed to dry overnight. For proliferation, LUHMES cells were propagated in Dulbecco’s modified Eagle’s medium/Ham’s F12 (DMEM/F12; no. 12-719F; Lonza) supplemented with 1% N2 supplement (no. 10378016; ThermoFisher Scientific), 1% PSG, and a 40-ng/ml final concentration of recombinant human fibroblast growth factor (FGF)-basic (no. 100-18B; PeproTech) added fresh to medium before use. Cells were switched to DMEM/F12 supplemented with 1% N2 supplement (no. 10378016; ThermoFisher Scientific), 1% PSG, a 1-μg/ml final concentration of tetracycline hydrochloride (no. T7660; Sigma), 1 mM final concentration N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt (no. D0627; Sigma), and a 2-ng/ml final concentration recombinant human glial cell-derived neurotrophic factor (GDNF) (no. 212-GD-010; R&D Systems) at 70% confluence to induce differentiation as previously described (10).
LUHMES cell infections.
LUHMES cells were grown and differentiated on poly-l-ornithine/fibronectin-coated coverslips as previously described (10). The timeline of acute, latent, and reactivation experiments is described in Fig. 1. Briefly, LUHMES cells were plated at 25,000 cells per well (24-well plates) or 2 × 106 cells (100-mm dishes) and allowed to proliferate for a period of 3 days, followed by 5 days of differentiation. For the acute infection, the postmitotic neurons were infected at a multiplicity of infection (MOI) of 5 with either HSV-1 viral strain 17syn+ or KOS(M) in complete medium containing 50 μM (ACV; no. PHR1254; Sigma) for 1 h. Following the 1-h inoculation, cells were rinsed with phosphate-buffered saline (no. SH30256.01; HyClone) and overlaid with differentiation medium (see “Viruses and cells,” above). Cultures were harvested at the time points described for Fig. 1A. For the latent and reactivation infection, the postmitotic neurons were pretreated with 50 μM ACV for 2 h and infected with HSV-1 viral strain 17syn+ or KOS(M) at an MOI of 5. After 48 h, medium was removed and replaced with differentiation medium without ACV. Supernatants were harvested starting at 3 days postinfection until reactivation, as described for Fig. 1B. Harvesting was done at the latent time point 8 days postinfection, and reactivation was induced with differentiation medium containing 10 μM Ly294002 (no. 9901S; Cell Signaling Technology). Samples were harvested at the time points described for Fig. 1B.
DNA/RNA extraction, reverse transcription, and qPCR.
Genomic DNA and RNA were extracted using a Quick-DNA/RNA miniprep kit (no. D7001; Zymo Research) by following the manufacturer’s recommended instructions. To remove DNA contamination, RNA samples were DNase treated with a TURBO DNA-free kit (no. AM2239; ThermoFisher). Reverse transcription was performed using a high-capacity cDNA reverse transcription kit (no. 4368813; ThermoFisher) with the addition of 250 ng of RNA per reaction. Genomic DNA/cDNA was then subjected to qPCR (StepOnePlus real-time PCR system; no. 4376600; ThermoFisher/Applied Biosystems) with TaqMan fast universal PCR master mix (2×) (no. 4352042; ThermoFisher/Applied Biosystems) and custom primer probes (Table 2). Controls for RT-qPCRs included no-template and no-reverse transcriptase wells for each plate for each gene analyzed.
TABLE 2.
Custom TaqMan primer/probe sequences
| Transcript | Sequencea
|
||
|---|---|---|---|
| Forward primer | Reverse primer | Probe | |
| ICP4 | CACGGGCCGCTTCAC | GCGATAGCGCGCGTAGA | CCGACGCGACCTCC |
| TK | CACGCTACTGCGGGTTTATATAGAC | GGCTCGGGTACGTAGACGATAT | CACCACGCAACTGC |
| LAT intron | CGCCCCAGAGGCTAAGG | GGGCTGGTGTGCTGTAACA | CCACGCCACTCGCG |
| UL30 | AGAGGGACATCCAGGACTTTGT | CAGGCGCTTGTTGGTGTAC | ACCGCCGAACTGAGCA |
| gC | CCTCCACGCCCAAAAGC | GGTGGTGTTGTTCTTGGGTTTG | CCCCACGTCCACCCC |
| US3 | GTATACCACGACCGTCGACAT | ACGGCAGTCTCGAAGATCAC | CAGACCGGCGCTCCAA |
| LAT promoter | CAATAACAACCCCAACGGAAAGC | TCCACTTCCCGTCCTTCCAT | TCCCCTCGGTTGTTCC |
The primer and probe sequences are in the 5′-to-3′ orientation.
ChIP-qPCR.
Chromatin immunoprecipitations (ChIP) were performed as previously described, with the following modifications (31). Briefly, approximately 1 × 107 LUHMES cells were infected, latency was established, and cells were reactivated with Ly294002 (see “LUHMES cell infection,” above). Cells were fixed by adding formaldehyde (no. BP531-500; ThermoFisher) to the culture medium at a final concentration of 1%. After 10 min, the reaction was quenched with 1.15 M glycine (G8898; Sigma) for 5 min at room temperature. Chromatin from fixed cells was sonicated using a Bioruptor Twin (no. UDC-400; Diagenode) with 30-s on and 30-s off cycles on high to produce chromatin fragments between 200 and 500 bp. Immunoprecipitations were performed with 2.5 μg of chromatin per IP reaction at 4°C overnight with shaking. Anti-triMe H3K27 (39155; Active Motif) and anti-histone H3 (no. AB12079; Abcam) antibodies were used at a concentration of 1 μg per sample. For immunocapture, protein A/G magnetic beads (no. 26162; ThermoFisher) were added to the samples and incubated for 4 h at 4°C. The beads then were washed in a series of salt buffers before elution (31). Samples were de-cross-linked and digested with RNase A (no. EN0531; ThermoFisher) and proteinase K (no. P4850; Sigma). DNA was purified according to the manufacturer’s recommendations using ChIP DNA Clean & Concentrator (no. D5201; Zymo). All ChIPs were also conducted using a negative-control antibody (anti-rabbit IgG; no. Ab46540; Abcam) to assess background levels of pulldown. qPCR was performed (see “DNA/RNA extraction, reverse transcription, and qPCR,” above), and samples were analyzed as percent input over total histone H3. Samples were determined for significance using a two-way analysis of variance (ANOVA).
ACKNOWLEDGMENTS
This work was supported by grant R01 AI48633 from the NIH.
We acknowledge E. Barrozo, J. Singh, W. Canty, A. Adolphson, S. Lee, and Z. Scherzer for critical discussions and helpful comments in preparing the manuscript.
REFERENCES
- 1.Looker KJ, Magaret AS, May MT, Turner KME, Vickerman P, Gottlieb SL, Newman LM. 2015. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS One 10:e0140765. doi: 10.1371/journal.pone.0140765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hill JM, Field MAR, Haruta Y. 1987. Strain specificity of spontaneous and adrenergically induced HSV-1 ocular reactivation in latently infected rabbits. Curr Eye Res 6:91–97. doi: 10.3109/02713688709020074. [DOI] [PubMed] [Google Scholar]
- 3.Bloom DC, Hill JM, Devi-Rao G, Wagner EK, Feldman LT, Stevens JG. 1996. A 348-base-pair region in the latency-associated transcript facilitates herpes simplex virus type 1 reactivation. J Virol 70:2449–2459. doi: 10.1128/JVI.70.4.2449-2459.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hill JM, Garza HH Jr, Su YH, Meegalla R, Hanna LA, Loutsch JM, Thompson HW, Varnell ED, Bloom DC, Block TM. 1997. A 437-base-pair deletion at the beginning of the latency-associated transcript promoter significantly reduced adrenergically induced herpes simplex virus type 1 ocular reactivation in latently infected rabbits. J Virol 71:6555–6559. doi: 10.1128/JVI.71.9.6555-6559.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill JM, Maggioncalda JB, Garza HH Jr, Su YH, Fraser NW, Block TM. 1996. In vivo epinephrine reactivation of ocular herpes simplex virus type 1 in the rabbit is correlated to a 370-base-pair region located between the promoter and the 5’ end of the 2.0 kilobase latency-associated transcript. J Virol 70:7270–7274. doi: 10.1128/JVI.70.10.7270-7274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawtell NM, Poon DK, Tansky CS, Thompson RL. 1998. The latent herpes simplex virus type 1 genome copy number in individual neurons is virus strain specific and correlates with reactivation. J Virol 72:5343–5350. doi: 10.1128/JVI.72.7.5343-5350.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strelow LI, Laycock KA, Jun PY, Rader KA, Brady RH, Miller JK, Pepose JS, Leib DA. 1994. A structural and functional comparison of the latency-associated transcript promoters of herpes simplex virus type 1 strains KOS and McKrae. J Gen Virol 75:2475–2480. doi: 10.1099/0022-1317-75-9-2475. [DOI] [PubMed] [Google Scholar]
- 8.Sedarati F, Stevens J. 1987. Biological basis for virulence of three strains of herpes simplex virus type 1. J Gen Virol 68:2389–2395. doi: 10.1099/0022-1317-68-9-2389. [DOI] [PubMed] [Google Scholar]
- 9.Wagner EK, Bloom DC. 1997. Experimental investigation of herpes simplex virus latency. Clin Microbiol Rev 10:419–443. doi: 10.1128/CMR.10.3.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards TG, Bloom DC. 2019. Lund human mesencephalic (LUHMES) neuronal cell line supports herpes simplex virus 1 latency in vitro. J Virol 93:e02210-18. doi: 10.1128/JVI.02210-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Aiuto L, Bloom DC, Naciri JN, Smith A, Edwards TG, McClain L, Callio JA, Jessup M, Wood J, Chowdari K, Demers M, Abrahamson EE, Ikonomovic MD, Viggiano L, De Zio R, Watkins S, Kinchington PR, Nimgaonkar VL. 2019. Modeling herpes simplex virus 1 infections in human central nervous system neuronal cells using two- and three-dimensional cultures derived from induced pluripotent stem cells. J Virol 93:e00111-19. doi: 10.1128/JVI.00111-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pourchet A, Modrek AS, Placantonakis DG, Mohr I, Wilson AC. 2017. Modeling HSV-1 latency in human embryonic stem cell-derived neurons. Pathogens 6:24. doi: 10.3390/pathogens6020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai G-Y, Pizer LI, Levin MJ. 2002. Fractionation of neurons and satellite cells from human sensory ganglia in order to study herpesvirus latency. J Virol Methods 104:21–32. doi: 10.1016/s0166-0934(02)00032-0. [DOI] [PubMed] [Google Scholar]
- 14.Thellman NM, Botting C, Madaj Z, Triezenberg SJ. 2017. An immortalized human dorsal root ganglion cell line provides a novel context to study herpes simplex virus 1 latency and reactivation. J Virol 91:e00080-17. doi: 10.1128/JVI.00080-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shipley MM, Mangold CA, Szpara ML. 2016. Differentiation of the SH-SY5Y human neuroblastoma cell line. J Vis Exp 108:e53193. doi: 10.3791/53193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P. 2002. Effect of mutant α-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem 277:38884–38894. doi: 10.1074/jbc.M205518200. [DOI] [PubMed] [Google Scholar]
- 17.Scholz D, Pöltl D, Genewsky A, Weng M, Waldmann T, Schildknecht S, Leist M. 2011. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J Neurochem 119:957–971. doi: 10.1111/j.1471-4159.2011.07255.x. [DOI] [PubMed] [Google Scholar]
- 18.Messer HG, Jacobs D, Dhummakupt A, Bloom DC. 2015. Inhibition of H3K27me3-specific histone demethylases JMJD3 and UTX blocks reactivation of herpes simplex virus 1 in trigeminal ganglion neurons. J Virol 89:3417–3420. doi: 10.1128/JVI.03052-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwiatkowski DL, Thompson HW, Bloom DC. 2009. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83:8173–8181. doi: 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devi-Rao GB, Bloom DC, Stevens JG, Wagner EK. 1994. Herpes simplex virus type 1 DNA replication and gene expression during explant-induced reactivation of latently infected murine sensory ganglia. J Virol 68:1271–1282. doi: 10.1128/JVI.68.3.1271-1282.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Everett RD. 1989. Construction and characterization of herpes simplex virus type 1 mutants with defined lesions in immediate early gene 1. J Gen Virol 70:1185–1202. doi: 10.1099/0022-1317-70-5-1185. [DOI] [PubMed] [Google Scholar]
- 23.Colgrove RC, Liu X, Griffiths A, Raja P, Deluca NA, Newman RM, Coen DM, Knipe DM. 2016. History and genomic sequence analysis of the herpes simplex virus 1 KOS and KOS1.1 sub-strains. Virology 487:215–221. doi: 10.1016/j.virol.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foster TP, Melancon JM, Kousoulas KG. 2001. An α-helical domain within the carboxyl terminus of herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) is associated with cell fusion and resistance to heparin inhibition of cell fusion. Virology 287:18–29. doi: 10.1006/viro.2001.1004. [DOI] [PubMed] [Google Scholar]
- 25.Bloom DC, Devi-Rao GB, Hill JM, Stevens JG, Wagner EK. 1994. Molecular analysis of herpes simplex virus type 1 during epinephrine-induced reactivation of latently infected rabbits in vivo. J Virol 68:1283–1292. doi: 10.1128/JVI.68.3.1283-1292.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roizman B, Whitley RJ. 2013. An inquiry into the molecular basis of HSV latency and reactivation. Annu Rev Microbiol 67:355–374. doi: 10.1146/annurev-micro-092412-155654. [DOI] [PubMed] [Google Scholar]
- 27.Suzich JB, Cliffe AR. 2018. Strength in diversity: understanding the pathways to herpes simplex virus reactivation. Virology 522:81–91. doi: 10.1016/j.virol.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, Kristie TM. 2013. Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Sci Transl Med 5:167ra5. doi: 10.1126/scitranslmed.3005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang Y, Quenelle D, Vogel JL, Mascaro C, Ortega A, Kristie TM. 2013. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. mBio 4:e00558-12. doi: 10.1128/mBio.00558-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hill JM, Quenelle DC, Cardin RD, Vogel JL, Clement C, Bravo FJ, Foster TP, Bosch-Marce M, Raja P, Lee JS, Bernstein DI, Krause PR, Knipe DM, Kristie TM. 2014. Inhibition of LSD1 reduces herpesvirus infection, shedding, and recurrence by promoting epigenetic suppression of viral genomes. Sci Transl Med 6:265ra169. doi: 10.1126/scitranslmed.3010643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pierce SE, Tyson T, Booms A, Prahl J, Coetzee GA. 2018. Parkinson’s disease genetic risk in a midbrain neuronal cell line. Neurobiol Dis 114:53–64. doi: 10.1016/j.nbd.2018.02.007. [DOI] [PubMed] [Google Scholar]