

SUMMARY
Amyloid β-protein (Aβ) toxicity is hypothesized to play a seminal role in Alzheimer’s disease (AD) pathogenesis. However, it remains unclear how Aβ causes synaptic dysfunction and synapse loss. We hypothesize that one mechanism of Aβ-induced synaptic injury is related to the cleavage of amyloid β precursor protein (APP) at position D664 by caspases that release the putatively cytotoxic C31 peptide. In organotypic slice cultures derived from mice with a knock-in mutation in the APP gene (APP D664A) to inhibit caspase cleavage, Aβ-induced synaptic injury is markedly reduced in two models of Aβ toxicity. Loss of dendritic spines is also attenuated in mice treated with caspase inhibitors. Importantly, the time-dependent dendritic spine loss is correlated with localized activation of caspase-3but is absent in APP D664A cultures. We propose that the APP cytosolic domain plays an essential role in Aβ-induced synaptic damage in the injury pathway mediated by localized caspase activation.
Graphical Abstract
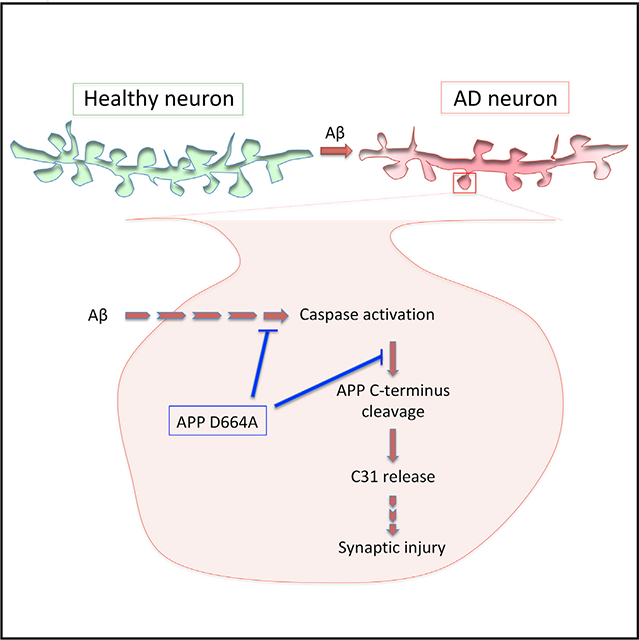
In Brief
Park et al. show that inhibition of amyloid precursor protein (APP) C terminus cleavage by D664A mutation prevents Aβ-induced local caspase activation, leading to localized synaptic injury.
INTRODUCTION
The pathological hallmarks of Alzheimer’s disease (AD), the most common age-related neurodegenerative disease, are extracellular deposition of amyloid β-protein (Aβ) in senile plaques in brain and intracellular accumulation of tau protein in neurofibrillary tangles. In addition, although their presence is not specific to AD, neuronal death and synapse loss invariably accompany the two characteristic hallmarks. The molecularly complex and physically small synapses play an indispensable role in neuronal network function via the release and uptake of neuro-transmitters between neurons (Gillingwater and Wishart, 2013). Accordingly, dysfunction or loss of synapses and dendritic spines are believed to be a major contributor to the cognitive impairments seen in AD and represent early changes in disease pathophysiology.
The amyloid cascade hypothesis posits that the accumulation of Aβ in brain is one of the initiators of the disease cascade, but the mechanisms by which Aβ is cytotoxic in vivo are still unclear. Aβ is derived from sequential cleavages of the amyloid precursor protein (APP) by β- and γ-secretases and released primarily from neurons in an activity-dependent manner (Bero et al., 2011; Cirrito et al., 2005; Kamenetz et al., 2003). In addition to secretases, APP can be cleaved by other proteases, including caspases or caspase-like proteases in the C-terminal cytosolic region after the aspartate residue at position 664 (APP695 numbering; Dyrks et al., 1988; Gervais et al., 1999; Weidemann et al., 1989; Wertkin et al., 1993), leading to the release of a C-terminal-derived peptide of 31 amino acids in length, coined C31 (Lu et al., 2000). In cultured neurons, C31 has been demonstrated to exhibit cytotoxicity, and interestingly, this toxicity can be attenuated by the co-expression of an APP mutant construct encoding an aspartate to alanine substitution at position 664 (D664A) to abrogate caspase-mediated cleavage (Lu et al., 2003a, 2003b). Similarly, cytotoxicity induced by Aβ treatment is diminished in the presence of APP D664A mutant, presumably due to inhibition of this cleavage (Lu et al., 2003a, 2003b). In this context, Aβ toxicity in cultured neurons and in vivo has been shown to be in part accompanied by caspase activation (Guillot-Sestier et al., 2012; Harada and Sugimoto, 1999), and further, there is evidence of caspase-mediated cleavage in brains of APP-expressing transgenic mice (D’Amelio et al., 2011; Higuchi et al., 2005; Rohn et al., 2008). Furthermore, in AD patients, caspase-3 activation has been documented in postsynaptic fractions (Banwait et al., 2008; Louneva et al., 2008). However, the relationship among caspase-mediated APP cleavage, Aβ-induced synapse loss, and caspase activation has not been directly tested.
Accordingly, to test this hypothesis that caspase activation triggered by Aβ is associated with synapse loss in an APP-dependent manner via cleavage of the APP C terminus, we generated an APP D664A knock-in (KI) mouse line, hereon designated as APP D664A KI, where Asp (D) to Ala (A) substitution at the 664 position was introduced into the endogenous mouse APP gene. This APP D664A KI transgenic mouse line therefore presented an opportunity to determine whether caspase-mediated cleavage of APP contributes to Aβ-induced synaptic toxicity. Here, we showed that APP D664A KI mice were protected from Aβ-induced synaptic toxicity. Hippocampal organotypic slice cultures (OTSCs) incubated with Aβ-containing media or virally infected with a truncated APP construct (C99ΔC) as a localized source of Aβ induced local spine loss in wild-type (WT) but not in APP D664A KI mice. In addition, there was evidence of localized caspase-3 activation within dendritic spines following expression of C99ΔC. This time-dependent activation of caspase-3 was negatively correlated with dendritic spine loss. These results together indicate that the APP D664 residue in the C terminus plays an important role in Aβ-induced synaptic injury that involves both caspase activation and likely cleavage of APP.
RESULTS
APP D664A KI Mice Exhibit Normal Dendritic Spine Density
To investigate the roles of APP caspase cleavage in synaptic injury in vivo, we generated an APP D664A KI mouse line by homologous recombination with an Asp (D) to Ala (A) substitution (mutation of nucleotide sequence from GAC to GCT) at position 664 in the N terminus of exon 18, the last exon in the APP cDNA (Figure 1A). Genetic modification in embryonic stem cells was confirmed by southern blotting and PCR genotyping analysis (data not shown). The homozygous KI mice did not exhibit any overt phenotypes up to more than 1 year of age. Morphological analyses demonstrated normal brain anatomy and cytoarchitecture, while biochemical analyses revealed comparable levels of full-length APP and Aβ between D664A KI and WT littermate control mice (Figure S1). APP D664A KI mice crossed with the GFP-M line of transgenic mice showed that the density of apical and basal dendritic spines of CA1 neurons were similar between D664A KI and WT littermate mice at 6 (Figure 1B) and 12 months of age (data not shown). Finally, hippocampal OTSCs grown for 5 or 10 days also showed no difference in the spine density in CA1 pyramidal neurons between D664A KI and littermate WT control mice (Figure 1C). Thus, mice carrying the D664A point mutation of the APP gene in the homozygous state did not result in any overt toxicity or perturbations as assessed by APP processing or measurements of dendritic spine density.
Figure 1. APP D664A KI Mice Show Normal Characteristics of Synapse.
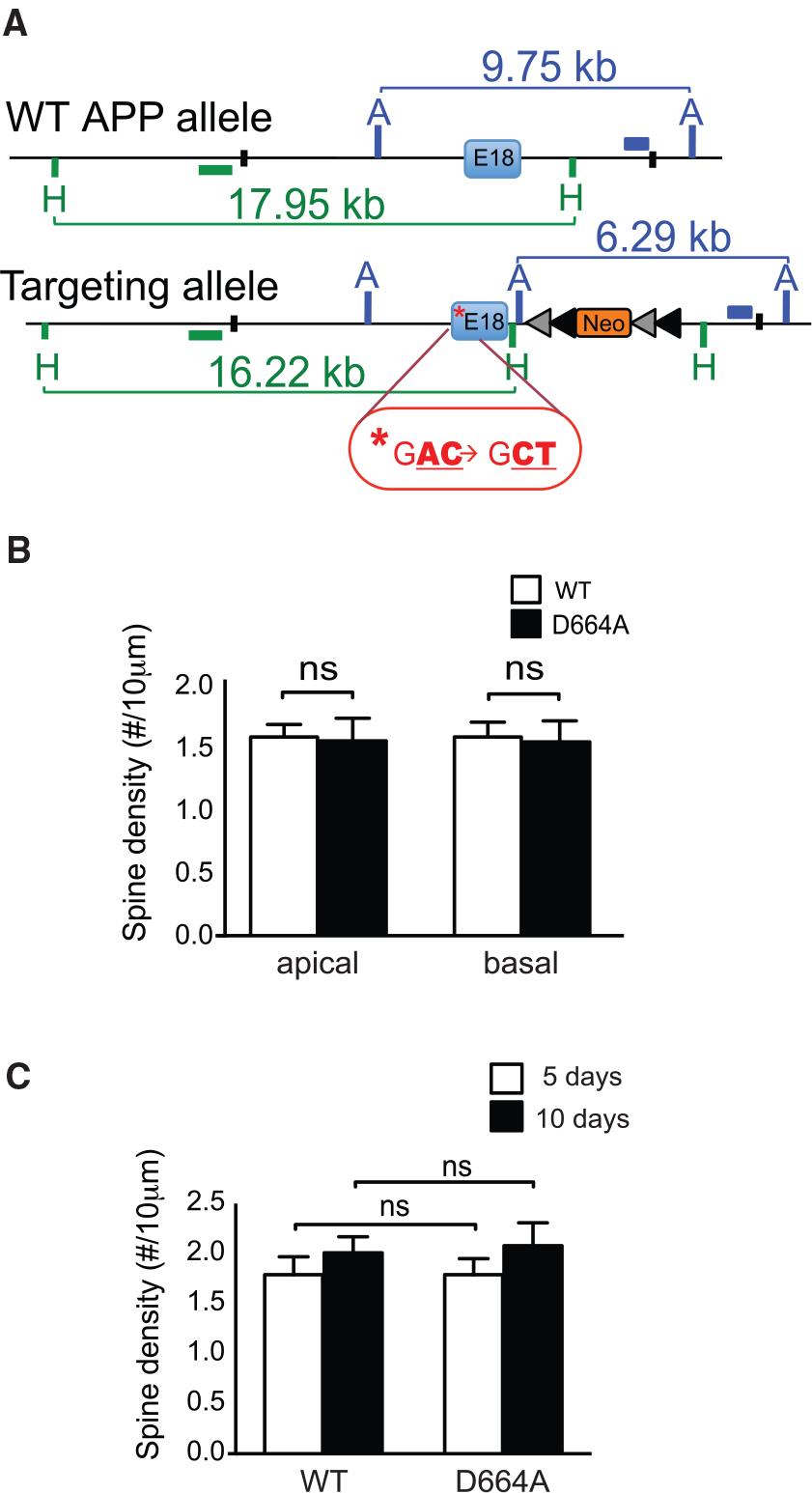
(A) Illustration showing scheme to generate the APP D664A KI allele. Following successful homologous recombination and germline transmission, APP D664A KI mice were identified for targeted integration and confirmed by southern blot and PCR analyses. Green and blue horizontal bars indicate the location of internal probes for southern blot analysis. Asterisk indicates the mutated nucleotide sequence in exon 18. The neomycin cassette (neo) is flanked by the loxP (gray) and frt (black) sequences. The position of the restriction sites, AflIII (A) and HpaI (H), are indicated in the schematic. (B) Analysis of apical and basal spine densities of GFP-M mice crossed with APP D664A KI mice as compared to control GFP-M mice. Dendritic segments were selected from hippocampal CA1 region of 6-month-old mice. There were no differences in dendritic spine numbers between APP D664A KI and WT littermate controls in either apical or basal dendrites. n = 48 neurons from 8 APP D664A KI mice and n = 54 neurons from 9 WT control mice. NS, not significant by two-tailed Student’s t test. (C) Analysis of dendritic spine density of CA1 neurons in hippocampal OTSCs from WT and APP D664A KI mice. Neurons were infected with GFP-expressing Sindbis virus and cultured for 5 or 10 days in CHO media. n = 50 neurons (5 days) and n = 10 neurons (10 days) from 12 APP D664A KI mice. n = 43 neurons (5 days) and n = 10 neurons (10 days) from 11 WT littermate controls. NS, not significant by two-tailed Student’s t test. Scale bar, 10 μm.
APP D664A KI Mice Are Neuroprotective against Exogenous Aβ Toxicity
We next asked whether mice carrying the D664A substitution would confer any neuroprotection to Aβ-induced synaptotoxicity. Accordingly, OTSCs from APP WT and D664A KI mice were incubated in conditioned media from CHO (Chinese Hamster Ovary) cells stably transfected with full-length APP carrying the V717F mutation (“7PA2” cells) that have been shown to produce Aβ oligomers (Walsh et al., 2002) or media from control cells. OTSCs were treated with 7PA2 condition media at 150 or 250 pM of Aβ42, which was previously determined to be synaptotoxic in OTSC pyramidal neurons, but not to hippocampal neurons for 5 or 10 days following published protocol (Li et al., 2009; Shankar et al., 2008). Spine density in hippocampal neurons within the OTSCs was reduced by about 30% in WT neurons treated with 150 pM of Aβ42 as compared to CHO-treated control cultures. Interestingly, there was no reduction in spines in OTSCs derived from D664A KI mice. Thus, at 150 pM Aβ42, there was a ~30% reduction in spines in WT versus D664A OTSCs. At 250 pM Aβ42, there was a ~20% non-significant reduction in spines in WT versus D664A neurons. Immunodepletion of Aβ from 7PA2 media resulted in a loss of the synaptic toxicity (Figures 2B and S2A), as reported previously (Nieweg et al., 2015; Walsh et al., 2002; Welzel et al., 2014). Consistent with the morphological observations, there was similar protection against Aβ-induced impairment in synaptic plasticity as measured by long-term potentiation (LTP) in acute hippocampal slices. The depression in LTP seen with 7PA2 media, as noted by other reports previously (Lei et al., 2016; Li et al., 2009), was absent in slices from APP D664A mice (Figure 2C). In short, these results demonstrated that this D664A substitution in the cytosolic tail of the endogenous APP gene protected neurons in OTSCs and acute hippocampal slices against Aβ-induced synaptic toxicity.
Figure 2. The D664A Point Mutation Rescues Aβ-Mediated Synaptic Dysfunction.
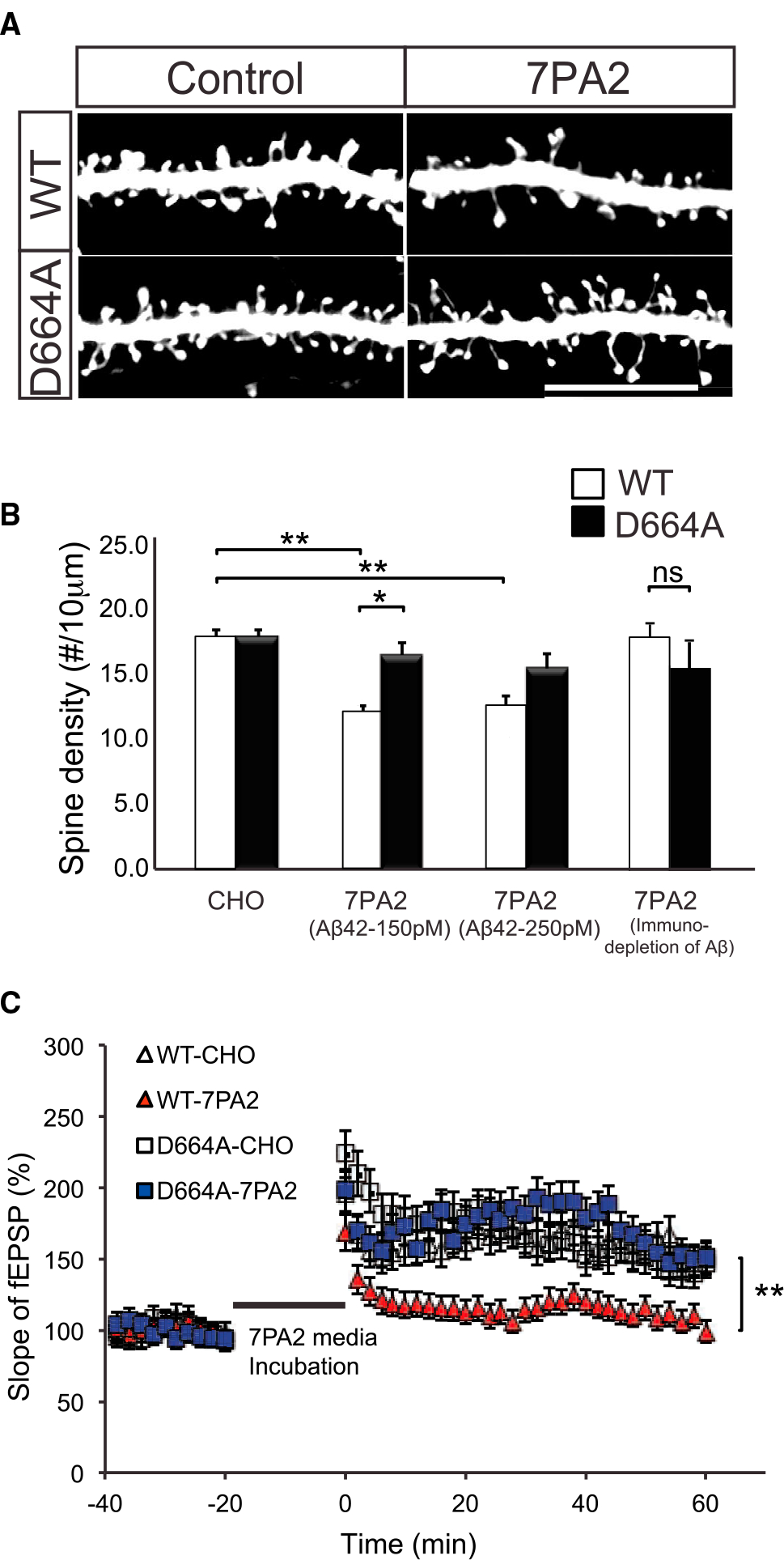
(A) Representative dendritic spine images of hippocampal OTSCs from APP WT and D664A KI mice. The OTSCs were incubated in 7PA2-conditioned media containing 150 pM Aβ42 for 5 days in vitro. GFP-expressing Sindbis virus was injected 1 day prior to experiment endpoint. Scale bar, 20 μm. (B) Quantification of dendritic spine densities in OTSCs shown in (A). 7PA2 containing 150 pM or 250 pM Aβ42 significantly reduced spine density; however, spine density was unchanged in D664A KI mice. n = 50 neurons (CHO media), n = 37 neurons (150 Aβ42), n = 30 neurons (250 Aβ42) from 9 APP D664AKI mice; n = 43 neurons (CHO media), n = 40 neurons (150 pM Aβ42), n = 31 neurons (250 pM Aβ42) from 10 WT littermate control mice. *p ≤ 0.05, **p ≤ 0.01 by two-way ANOVA followed by Tukey’s multiple comparisons test. Aβ-immunodepleted 7PA2-conditioned medium showed no significant reduction in spine density. n = 10 neurons from 4 APP D664A KI mice and n = 10 neurons from 4 WT littermate controls. NS, not significant by Student’s t test. (C) 7PA2-conditioned media depressed LTP as compared to untransfected CHO media. This impairment was rescued in APP D664A KI mice. n = 11 slices for each of four conditions. **p ≤ 0.01 by two-tailed Student’s t test comparing WT-7PA2 versus D664A-7PA2.
C99ΔC-Induced Spine Change Is Relative to Time-Dependent Aβ Production but Not in APP D664A KI Mice
Previous studies have shown that overexpression of the D664A mutation protected against cell death in cultured systems (Lu et al., 2000, 2003b). The preceding observations now demonstrated that Aβ-induced synaptic dysfunction and synaptic injury could also be prevented or attenuated in cultures derived from APP D664A KI mice. To define the synaptic changes at higher resolution, we adapted an approach pioneered by the Malinow laboratory, where sparse infection of OTSCs with a Sindbis virus expressing the APP “C99” construct (i.e., C-terminal fragment [CTF] generated after β-secretase cleavage) enabled electrophysiological and morphological characterization of Aβ toxicity in single neurons that expressed this APP CTF (Kamenetz et al., 2003). Though C99 is a rich source of Aβ, it encompasses the entire cytosolic region, which includes both the caspase cleavage sequences and the putatively toxic C31 peptide. Accordingly, we tested whether Sindbis directed expression of a C-terminally truncated C99 fragment deleted of C31 region (C99ΔC) can be utilized as a source of neuronal Aβ at a single-cell level to induce synaptic toxicity. The deletion of C31 (i.e., the residual fragment after the caspase cleavage site) should negate any perturbations resulting from this cleavage event (Figure S3). To test this C99ΔC APP fragment, the expression of the various C99 constructs and their cleavage products were confirmed in vitro with purified active caspase-3 (Figure S3C). As anticipated, Sindbis-mediated expression of C99ΔC in OTSCs resulted in the release of Aβ into the culture medium that increased with time in the culture (Figure S3D). Importantly, the levels of Aβ released were comparable to between the C99ΔC and WT C99 fragment (Figure S3E), as would be expected, given previous reports that Aβ levels from the APP D664A mutation were similar to WT APP (Soriano et al., 2001). As Aβ is released from C99 or C99ΔC after γ-secretase cleavage, the levels of Aβ measured by ELISA were markedly reduced after treatment with a γ-secretase inhibitor (GSI) (Figure S3E). When co-expressed with tdTomato fluorescent marker appended behind an IRES (internal ribosome entry site) sequence, individual neurons expressing C99ΔC could be readily identified, and importantly, dendritic spines were clearly delineated in the infected cells (Figures 3 and S4). Following Sindbis virus expression of C99ΔC in OTSCs, there was a time-dependent reduction in the density of dendritic spines in infected neurons, down to ~50% of control neurons after 24 h. This loss of dendritic spines was attenuated by treatment with the GSI added at 12 or 18 h after virus infection, thus confirming the role of Aβ in initiating this dendritic spine loss (Figure 3). Interestingly, while the addition of the GSI at 12 h returned spine density to control levels, the GSI added later, at 18 h, only partially attenuated the spine loss (Figure 3B), suggesting that given sufficient time, halting further Aβ production and release can either prevent or possibly restore dendritic spine loss. Surviving spines also showed morphological changes, as mushroom spine heads were reduced, while long, thin spines were much more abundant (Figure S4E). In sum, C99ΔC was able to induce synaptic degeneration in APP WT neurons in an Aβ-dependent manner.
Figure 3. C99ΔC-Induced Synaptotoxicity Is Dependent on Aβ Production.
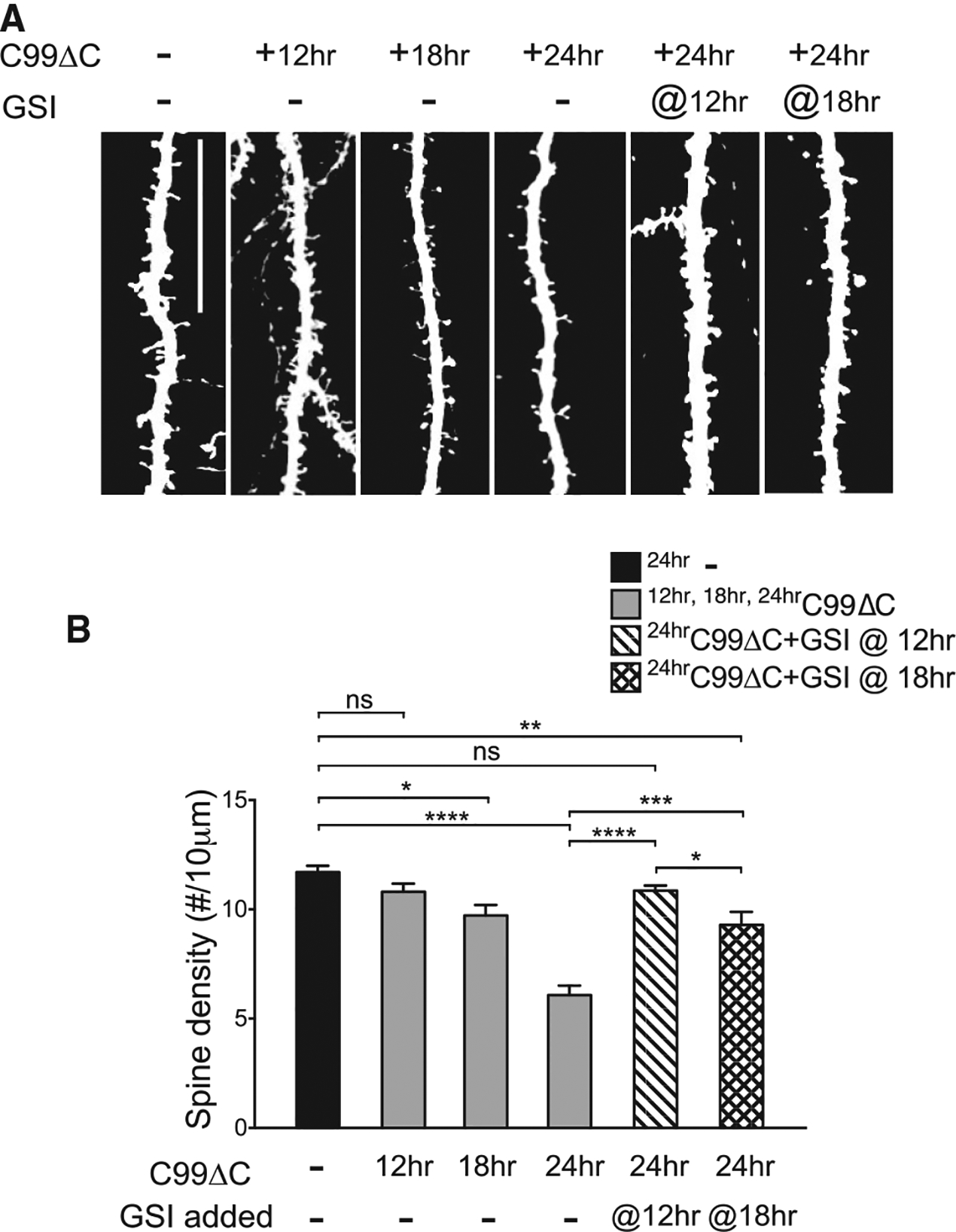
(A) Representative images from Sindbis-mediated viral expression of C99ΔC in CA1 neurons in OTSCs. The time-dependent dendritic spine loss can be blocked by the addition of 10 μM γ-secretase inhibitor (GSI) to inhibit Aβ production. Scale bar, 20 μm. (B) Quantification of spine density from (A) showed significant spine loss starting at 18 h post-infection and decreased to a maximum of ~50% at 24 h post-infection. Addition of GSI at 12 h and 18 h post-infection attenuated synapse loss, with the greatest effect when treatment was started at 12 h, where spine numbers were comparable to control slices not infected with C99ΔC. control tdTomato-infected culture: n = 15 neurons; C99ΔC-infected cultures: 15 neurons (12 h), 8 neurons (18 h), 14 neurons (24 h); GSI-treated cultures: n = 9 neurons (12 h), 11 neurons (18 h) from a total of 21 mice for all experiments in aggregate NS, not significant, *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001 by two-way ANOVA followed by Tukey’s multiple comparisons test.
We next tested whether the APP D664A KI mice exhibited any protection against C99ΔC-induced synaptotoxicity. Accordingly, Sindbis virus expressing C99ΔC was introduced into OTSCs derived from APP D664A KI or WT littermate control mice using the same procedure as before. Twenty-four hours following injection of Sindbis virus into the hippocampus CA1, spine densities in neurons from APP D664A KI mice were similar to untreated cultures, while neurons from control WT mice demonstrated the ~50% reduction spine numbers as seen previously (Figure 4). Therefore, using Sindbis virus to target single neurons for expression of C99ΔC to generate Aβ, OTSCs from APP D664A also demonstrated neuroprotection from this Aβ-induced synaptic toxicity paradigm as compared to control cultures from WT mice.
Figure 4. The D664A Point Mutation Also Rescues C99ΔC-Induced Dendritic Spine Loss.
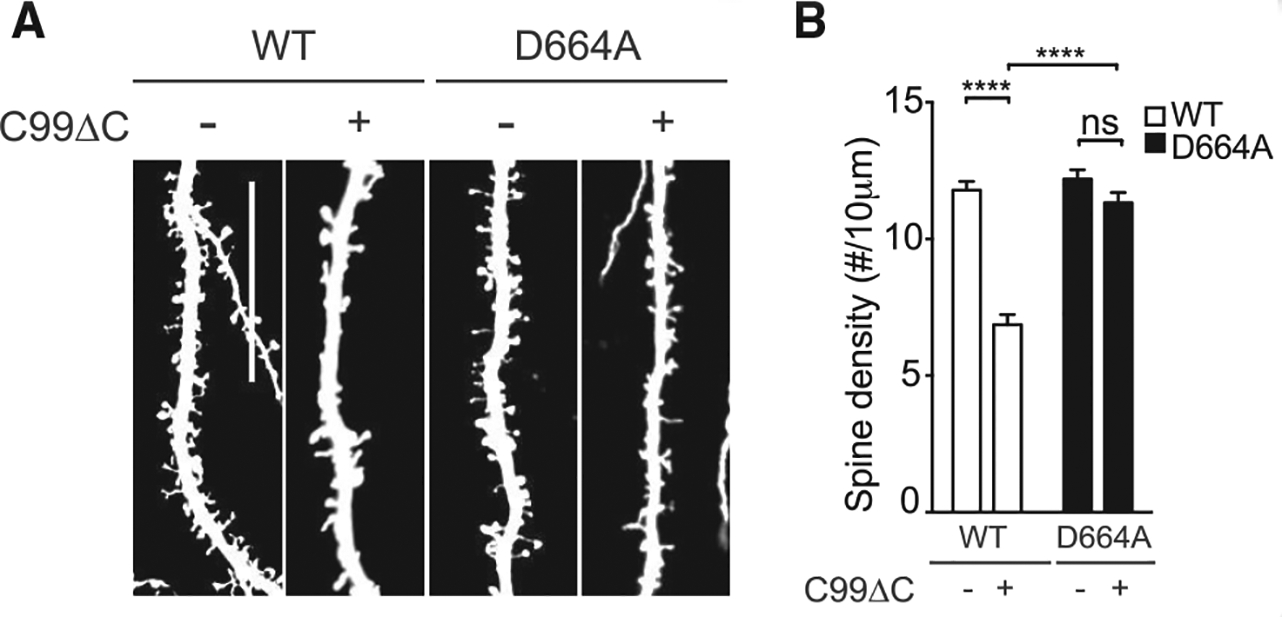
(A) Representative dendritic segments of hippocampal neurons in OTSCs derived from WT or APP D664A KI mice with C99ΔC as compared to control tdTomato virus. C99ΔC induced spine loss in WT cultures but not in OTSCs from APP D664A KI mice. Scale bar, 20 μm. (B) Quantification of dendritic spine densities from (A). Expression of C99ΔC induced about 50% spine loss after 24 h but was not detected in cultures from APP D664A mice and comparable to untreated control tdTomato virus-infected OTSCs. n = 15 neurons (control tdTomato) and n = 15 neurons (C99ΔC) from 10 APP D664AKI mice; n = 16 neurons (control tdTomato) and n = 18 neurons (C99ΔC) neurons from 7 WT littermates. NS, not significant; ****p ≤ 0.0001 by one-way ANOVA followed by Tukey’s multiple comparisons test.
In the above studies, spine counts were performed in tdTomato-labeled dendrites that identified individual neurons where C99ΔC was expressed. In this setting, it is unclear whether the Aβ-induced toxicity was intracellular in origin or derived from Aβ that was released from these C99Δ- expressing neurons and in turn induced synapse/spine injury in an autocrine manner. To test whether secreted Aβ can induce toxicity in an exocrine manner (i.e., in postsynaptic neurons that are connected to presynaptic cells expressing C99ΔC), we took advantage of the fact that CA3 neurons project their axons via the Schaffer collaterals to synapse onto dendrites of CA1 neurons (Collingridge et al., 1983). Thus, expressing C99ΔC in CA3 neurons but analyzing dendrites of CA1 neurons in the vicinity of fluorescently labeled CA3 axons should identify neurons that may be exposed to Aβ secreted from CA3 axons as reported previously (Wei et al., 2010). In this experiment, OTSCs were dually infected with Sindbis virus expressing C99ΔC linked to tdTomato constructs as before or tdTomato alone in CA3 neurons and concomitantly with GFP encoding virus in CA1 neurons, the latter to label dendrites of postsynaptic neurons (Figures 5A and 5B). In this setting, there was a ~60% reduction in apical spines of GFP-labeled CA1 dendrites that were in the vicinity of tdTomato-positive axons originating from infected CA3 neurons after 48 h, but not in control cultures infected with only control tdTomato virus. This indicated that in this setting, the spine loss was likely due to Aβ secreted from axons of CA3 neurons whose axonal projections were either in the vicinity of or actually contacting the postsynaptic dendrites of CA1 neurons, as suggested previously (Wei et al., 2010). In GFP-positive CA1 dendrites in microscopic fields where only one tdTomato-labeled axon was visible, a gradient in spine density was evident where maximal reduction in spine loss was in the region closest to the tdTomato-labeled axon, returning to near-normal levels by 50–60 μm away from the crossing axon (Figures 5C–5F). Importantly, in OTSCs derived from APP D664A mice and infected with dual viruses as above, there was no appreciable loss of spines in GFP-labeled CA1 dendrites in microscopic fields with tdTomato-innervated axons (Figures 5C and 5D). Lastly, CA1 dendrites that did not receive labeled CA1 axonal projections did not demonstrate synapse loss (Figure 5D). These findings strongly support the concept that Aβ-induced synaptic toxicity can be derived from Aβ secreted from projections of afferent (incoming) axons, and this toxicity was attenuated in OTSCs derived from APP-D664A-expressing mice.
Figure 5. The D664A Point Mutation Attenuated C99ΔC-Induced Dendritic Spine Loss in Postsynaptic CA1 Neurons.
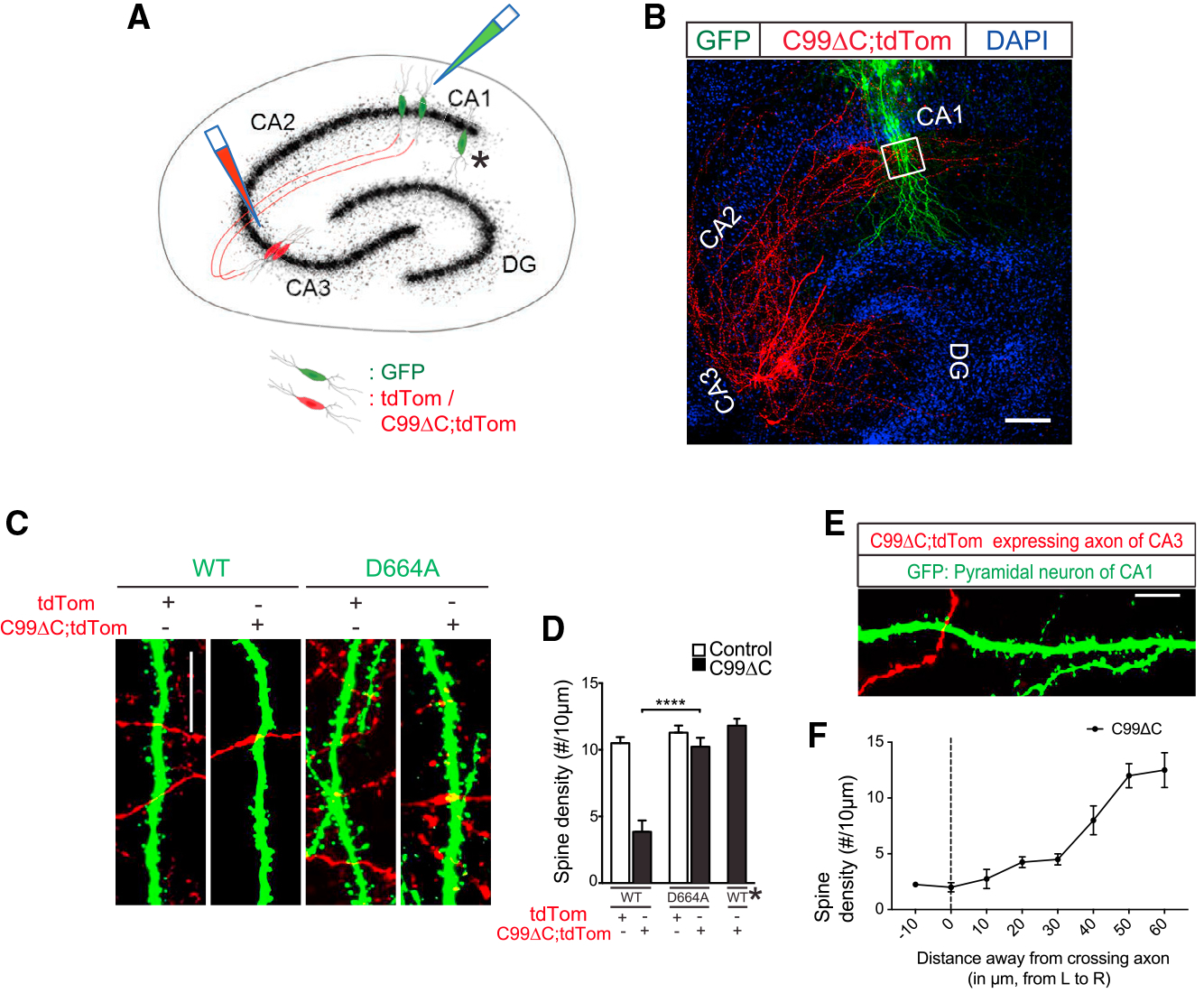
(A) Schematic of OTSCs infected with dual Sindbis viruses expressing C99ΔC and tdTomato or tdTomato alone in CA3 and GFP in CA1 neurons, respectively. Asterisk indicates region where CA1 pyramidal neurons were not innervated by tdTomato-labeled axonal projections from CA3 neurons. (B) Representative low-magnification image of OTSCs expressing two different fluorescent proteins in CA1 and CA3 neurons diagrammed in (A). Cell bodies and projects of axons and dendrites can be readily seen (red). The axons of CA3 pyramidal neurons labeled by tdTomato project to dendritic fields and cell bodies of GFP-labeled CA1 neurons (green) in this low-magnification image. DAPI was used to counterstain nuclei of hippocampal neurons. White box indicates region where spine densities were quantified. Scale bar, 100 μm. (C) Representative labeled dendritic segments of CA1 dendrites in OTSCs of WT and APP D664A KI mice showing tdTomato-labeled axons (red) from CA3 neurons within the same microscopic fields of GFP-labeled CA1 dendrites (green). Scale bar, 20 μm. (D) Quantification of dendritic spines from (C). The C99ΔC expression induced about 60% spine loss in CA1 dendrites but was absent in slices from APP D664A KI mice. CA1 dendrites not receiving afferent inputs from tdTomato-positive axons (asterisk here and in A) did not show spine loss. n = 12 neurons (tdTomato) and n = 14 neurons (C99ΔC) from 12 APP D664AKI mice; n = 9 neurons (tdTomato), n = 15 neurons (C99ΔC), and n = 11 neurons (non-innervated region designated by asterisk) from 13 WT littermate control mice. NS, not significant; ****p ≤ 0.0001 by two-way ANOVA followed by Tukey’s multiple comparisons test. (E) Representative image of dendritic fields of CA1 pyramidal neuron expressing GFP but with few CA3 axons expressing tdTomato. Scale bar, 20 μm. (F) Distance-dependent spine density loss. Quantification of image shown in (E) showed that proximity of dendrites closer to crossing axons (μm from left [L] to right [R]) infected with C99ΔC was negatively correlated with spine density (n = 15 neurons from 13 WT mice).
Role of Caspase Activation in Aβ-Induced Spine Loss
The rationale for mutating the APP D664 residue was to inhibit caspase-mediated cleavage of APP, thus abrogating the release of C31, an intracellular APP fragment that we hypothesize is synaptotoxic. That caspases may participate in synaptic injury in AD has been proposed previously (D’Amelio et al., 2011; Jellinger, 2006; Louneva et al., 2008; Pozueta et al., 2013; Stadelmann et al., 1999). In particular, a recent study reported that within dendritic spines in the Tg2576 line of APP transgenic mice, calcineurin was activated by caspase-3 activity, which in turn led to the dephosphorylation and removal of GluR1 from postsynaptic sites and ultimately led to spine degeneration and behavioral impairment (D’Amelio et al., 2011). To determine whether caspases and caspase activation play a role in synaptic injury that is APP dependent, we first confirmed that GluR1 phosphorylation was indeed reduced in our model of acute Aβ-mediated synaptic injury, which, at first blush, may be distinct from chronic Aβ-induced toxicity seen in APP transgenic mice (Figure S5). However, this finding of a similar reduction in phosphorylated GluR1 subunit of the AMPA receptor reported by D’Amelio et al. (2011) is consistent with the notion that caspase activation may initiate these downstream alterations after C99ΔC expression as well. Furthermore, this decrease in GluR1 was attenuated in cultures from APP D664A mice.
We next asked whether caspase activation contributed to C99ΔC-induced toxicity by treating OTSCs infected with C99ΔC with a pan-caspase inhibitor, zVAD-FMK, or inhibitors to caspase-3, caspase-4, caspase-6, or caspase-8, where previous studies have implicated these caspases in the cleavage of APP (Gervais et al., 1999; Lu et al., 2003b; Pellegrini et al., 1999; Tesco et al., 2003). The zVAD-FMK treatment preserved dendritic spines in neurons expressing C99ΔC, similar to the protective effects seen in OTSCs from APP D664A KI mice or in WT mice treated with GSI (Figures 3A and 6). However, caspase-3 and caspase-8 inhibitor treatment showed partial attenuation of spine toxicity, while caspase-4 and caspase-6 inhibitors had no effect on the Aβ-induced spine loss (Figures 6A and 6B). Similar to the results seen in the GSI experiment described above, treatment with zVAD-FMK at 6 h after Sindbis infection completely restored spine loss, but the ability of zVAD-FMK to limit synaptic loss was reduced with shorter treatment times (Figures 6C and 6D). These results indicate that caspase activation plays a role in this pathway, which could be attenuated by either GSI or caspase inhibitors when given adequate time to block the effects of Aβ-induced synaptic injury.
Figure 6. Caspase Inhibitors Reduced C99ΔC-Induced Dendritic Spine Loss.
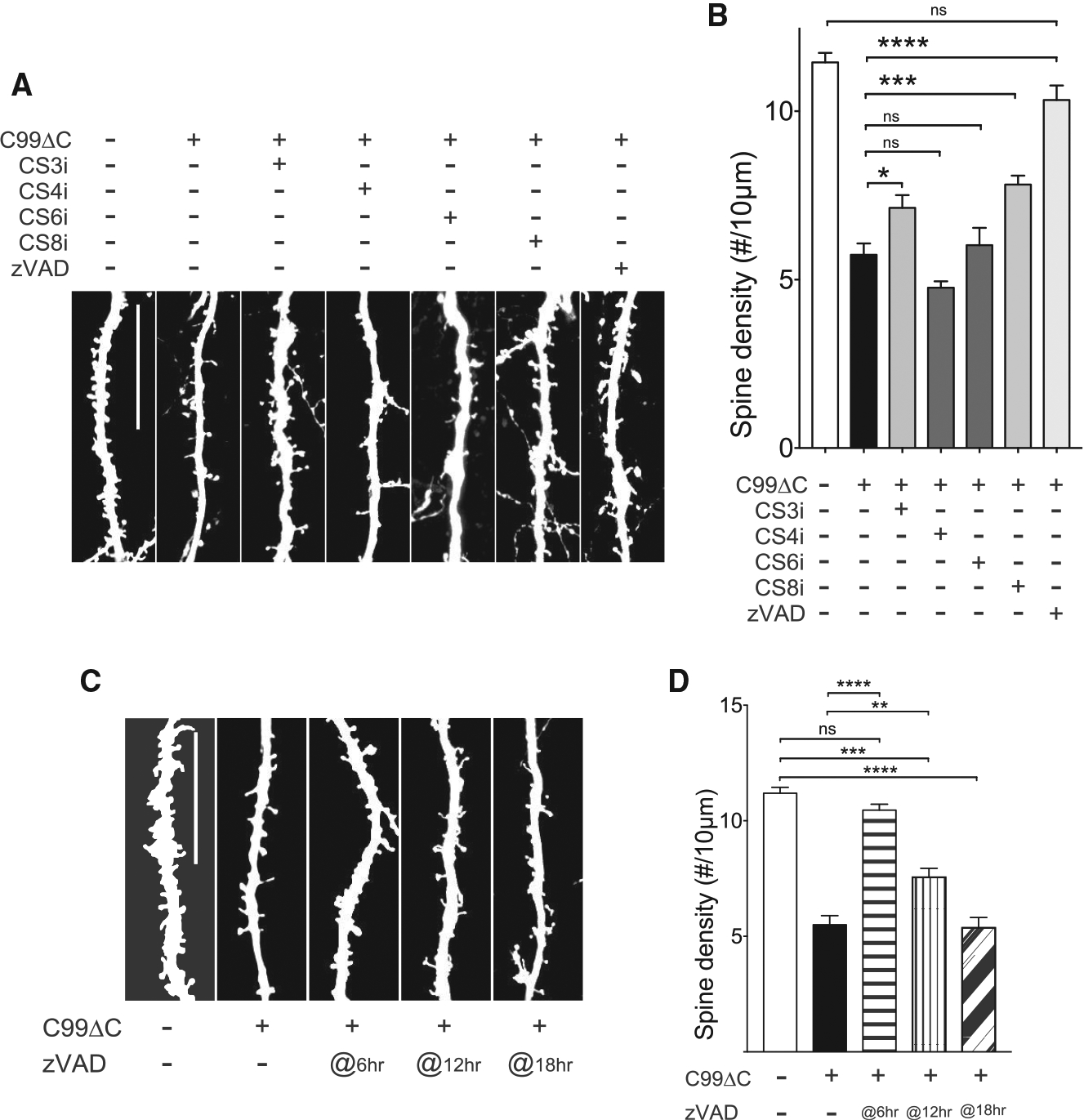
(A) Representative images of dendritic segments of CA1 hippocampal neurons in OTSCs infected with C99ΔC Sindbis virus and treated with caspase inhibitors for 24 h. Scale bar, 20 μm. (B) Quantification of dendritic numbers from (A). The C99ΔC-induced spine loss was attenuated by caspase-3 (CS3i) and caspase-8 inhibitors (CS8i) and unchanged with zVAD-FMK as compared to control GFP-infected cultures. Caspase-4 (CS4i) and caspase-6 (CS6i) inhibitors had no detectable effects on attenuating spine loss. n = 20 neurons (non-treated tdTomato), n = 25 neurons (C99ΔC control without drug), n = 15 neurons (CS3i-treated C99ΔC), n = 20 neurons (CS4i-treated C99ΔC), n = 21 neurons (CS6i-treated C99ΔC), n = 17 neurons (CS8i-treated C99ΔC), n = 18 neurons (zVAD-treated C99ΔC) from a total of 21 WT mice. NS, not significant; *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001 by one-way ANOVA followed by Tukey’s multiple comparisons test. (C) Representative images of dendritic segments of CA1 hippocampal neurons in OTSCs treated with zVAD at different time points after Sindbis infection. Scale bar, 20 μm. (d) The addition of zVAD showed a time-dependent protective effect. n = 13 neurons (non-treated tdTomato), n = 7 neurons (non-treated C99ΔC), n = 12 neurons (zVAD at 6 h), n = 18 neurons (zVAD at 12 h), n = 15 neurons (zVAD at 18 h) from a total of 18 WT mice. NS, not significant; **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 by one-way ANOVA followed by Tukey’s multiple comparisons test.
If inhibiting caspase activity is beneficial, then there should be evidence of caspase activation in the OTSCs. Therefore, we next sought to determine whether localized caspase activation is present after C99ΔC expression. Using a caspase-3 FLICA (fluoro-chrome inhibitor of caspases) reagent (Smolewski et al., 2002; Wensveen et al., 2012), a fluorogenic indicator of caspase-3 activation, fluorescent signal was not detectable at 12 h after infection by C99ΔC virus (i.e., at a time was synapse loss was not yet detectable; Figures 7 and S6). However, by 18 h, scattered positive signals were present within ~10% of dendritic spine heads but not yet evident in main dendritic shafts (Figures 7A, 7B, S6A, and S6B). This correlated to a ~20% decrease in the number of dendritic spines as compared to control cultures. By 24 h, at the conclusion of the experiment when spine loss was maximal, not only was the percent of spines positive for caspase activity marker increased (~30%), but there was also signal present within dendritic shafts (Figures 7A, 7B, and S6C), suggesting a spread of caspase activation from spine heads to shafts. Rotation of the images confirmed that the fluorescent signals, especially when present as linear profiles, were within dendritic shafts and did not emanate from overlying spines (Figures S6A and S6B). These findings indicated that caspase activation might first begin in dendritic spines before “spreading” to the interior of the dendritic shafts. Concurrent treatment of OTSCs with zVAD-FMK for 24 h markedly reduced the FLICA reporter signals, almost to the level of control cultures (Figures S6D and S6E). The results derived from the FLICA caspase-3 reagent were confirmed by immunostaining with an antibody specific to activated caspase-3 species (Figure S6F). Lastly, as expected, the FLICA signal was essentially negative in OTSCs derived from APP D664A KI mice infected with C99ΔC virus (Figures 7C and 7D), consistent with the neuroprotective effects of the D664A mutation. Taken together, these findings strongly support the concept that APP itself contributes to Aβ-induced synaptic injury and that this pathway involves local caspase activation within dendrites and requires an intact APP caspase cleavage sequence in the cytosolic domain.
Figure 7. C99ΔC-Induced Spine Loss Was Accompanied by Local Caspase Activation.
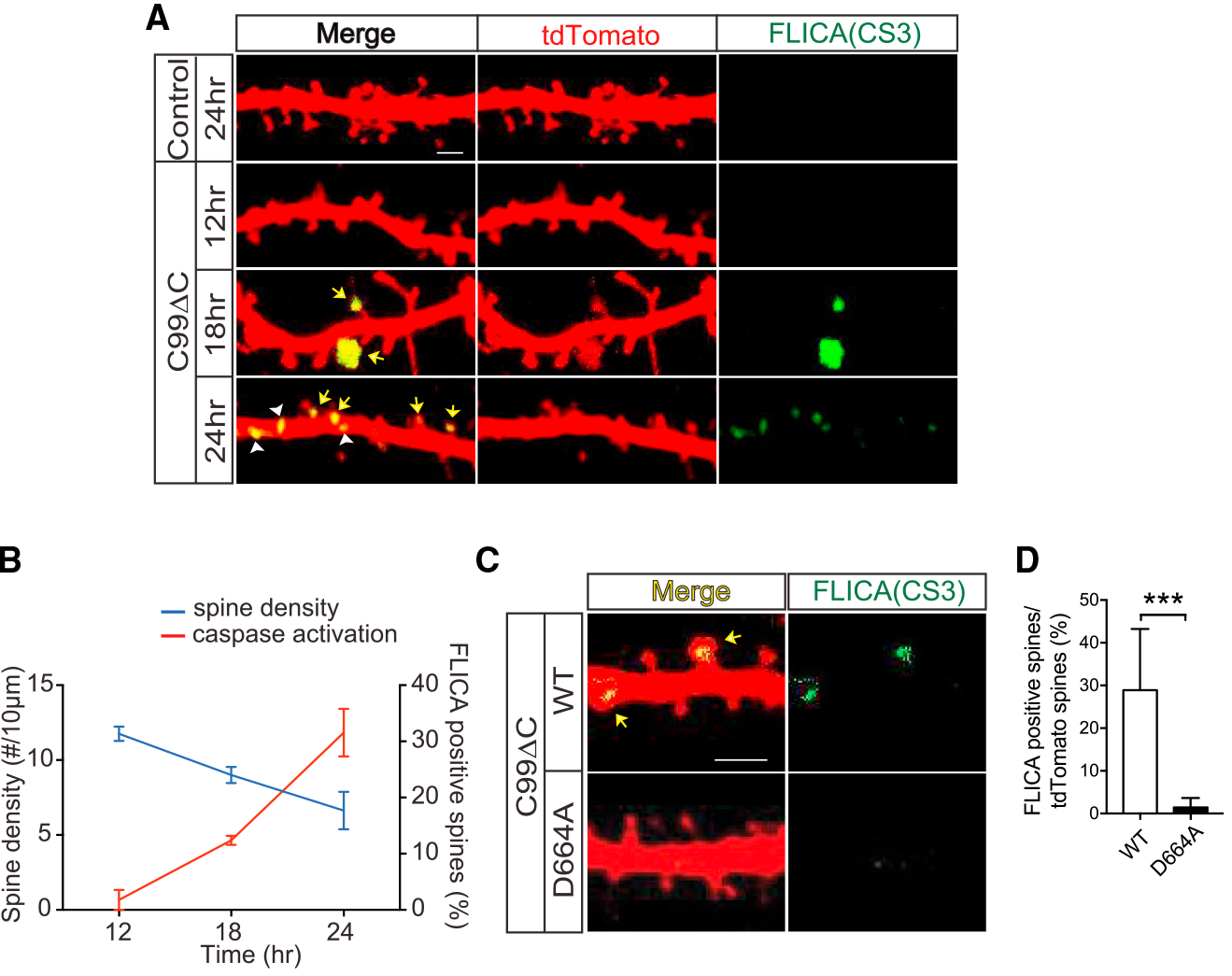
(A) Representative images of dendritic segments of CA1 hippocampal neurons from OTSCs infected with Sindbis virus expressing C99ΔC and incubated with caspase-3 FLICA reagent. The FLICA signal can be seen in dendritic spines and dendritic shafts. Yellow arrows indicate positive FLICA signal overlapping with tdTomato-positive dendrites in the merged images, which can be seen separately in the FLICA-only channel (green). Arrowheads show the positive signals in a dendritic shaft. Scale bar, 2 μm. (B) Quantification of tdTomato-labeled and FLICA-positive spines from (A) as a function of time after C99ΔC virus infection of the OTSCs. The number of caspase-positive spines by FLICA activity was increased in a time-dependent manner and negatively correlated with the reduction in spine density. n = 8 neurons (12 h C99ΔC), n = 19 neurons (18 h C99ΔC), n = 21 neurons (24 h C99ΔC) from a total of 14 WT mice. (C and D) Representative images (C) and quantification (D) of local caspase activation in dendritic spines of OTSCs of WT and APP D664A KI mice infected by Sindbis virus expressing C99ΔC for 24 h, as detected by FLICA reagent. The C99ΔC-induced caspase activation (yellow arrow) was virtually absent in APP D664A KI mice. n = 7 neurons from 4 APP D664AKI mice and n = 6 neurons from 3 WT littermate control mice. ***p ≤ 0.001 by two-tailed Student’s t test. Scale bar, 20 μm.
DISCUSSION
In this study, we examined the role of caspase-mediated cleavage of APP and caspase activation in Aβ-induced synaptic injury and asked whether these two events are related. This potential interaction was tested in a newly generated KI transgenic mouse line with a D664A substitution in the endogenous APP gene designed to abrogate cleavage of APP at that position. Our results showed that neurons expressing the D664A mutation were protected from Aβ-induced impairment in synaptic plasticity as well as dendritic spine loss. Pharmacologic inhibition of caspase activity attenuated Aβ-induced synaptic toxicity in OTSCs from WT mice, similar to the neuroprotection seen in OTSCs derived from APP D664A mice. In cultures from WT mice, the time course of dendritic spine loss after Sindbis virus infection of C99ΔC to locally express Aβ was inversely correlated with the activation of caspase-3 within dendritic spines. In OTSCs from APP D664A mice, evidence of caspase-3 activation was noticeably absent, concomitant with the preservation of dendritic spines. While APP is recognized as one of many substrates cleaved by caspases, our results showed that Aβ-induced synaptic damage appears to require both the localized activation of caspase-3 and an intact caspase cleavage domain in APP.
In a prior study, the APP transgenic mouse lines overexpressing full-length APP with the D664A mutation did appear to demonstrate the anticipated reduction in synaptic pathology, despite abundant amyloid deposits in brain (Galvan et al., 2006; Saganich et al., 2006). However, subsequent re-analyses of these mouse lines did not confirm the initial observations, and whether there is indeed neuroprotection in the D664A mice previously seen in cultured cells remains unresolved (Harris et al., 2010). One difficulty in studying these APP-D664A-overex-pressing transgenic mice was the imbalance in transgene expression levels across the different mouse lines, especially in comparing the APP D664A mice with APP-expressing mice (Bredesen et al., 2010). In this study, the KI technology of the endogenous APP gene obviated the need to compare different transgenic mouse lines with their variable transgene expression levels and different insertional sites. In this setting, our results nevertheless strongly supported the hypothesis—first noted in cultured systems and subsequently in the aforementioned APP D664A transgenic mice—that the APP D664A substitution is neuroprotective against Aβ-induced synaptic toxicity.
In our experimental system, although it is clear that Aβ released from the APPΔC-expressing virus contributes significantly to synaptic toxicity, it is unclear where Aβ exerts its toxicity. The loss of spines in CA1 neurons following injection of APPΔC virus in CA3 neurons strongly argues that the synaptic toxicity is a consequence of secreted Aβ, hence originating from extracellular Aβ. This finding is strikingly similar to the results from Wei et al. (2010), where a comparable CA3-to-CA1-Aβ-induced postsynaptic toxicity to dendritic spines of CA1 neurons was first reported. On the other hand, it is difficult to explain the mechanism of Aβ-induced toxicity when spine loss occurred in neurons that were infected with the APPΔC virus, as many of our results demonstrated. In many ways, our results are reminiscent of the reduction in AMPA and NMDA currents in neurons that were infected with an APPC99 virus when compared to adjacent control neurons that were not infected by virus (Kamenetz. et. al., 2003). But in this instance, where Aβ initiates this diminution of AMPA and NMDA currents is unknown. In the report from Kamenetz et al. (2003) and in this study, we can envision that the electrophysiological and anatomic injury is the consequence of either secreted Aβ molecules binding to the same neuron where the Aβ was synthesized or, alternatively, originating from intracellular Aβ. To date, while there is substantial literature favoring a role of intracellular Aβ in neuronal toxicity (Giménez-Llort et al., 2007; Gouras et al., 2010; Takahashi et al., 2017), it is unclear whether these Aβ molecules are derived from internalization of extracellular Aβ or if these non-fibrillar Aβ species detected by immunochemical means represent molecules that are awaiting secretion from the neurons. Thus, the precise site of Aβ toxicity from this and previous studies remains unresolved.
As described, one pathway of Aβ-induced cell death in cultured cells was attributed to the cleavage of APP at the VEVD sequence, which is similar to the consensus caspase-3 “DEVD” motif, leading to the release of the putatively cytotoxic C31 fragment (Banwait et al., 2008; Lu et al., 2000). Thus, in APP-deficient cells, Aβ toxicity would be predicted to be significantly reduced given the absence of APP, a situation that is somewhat analogous to the APP D664A mutation. Indeed, this precise finding has been reported by multiple laboratories (Fogel et al., 2014; Kirouac et al., 2017; Lorenzo et al., 2000; Lu et al., 2003a; Wang et al., 2017). The observations reported here further validate this APP-dependent pathway of Aβ-induced synaptic injury. In addition, one interpretation of this APP-dependent mechanism is that both the presence and cleavage of APP are required to transmit this synaptic injury pathway. Although our results demonstrated that an intact VEVD motif is critical to induce synaptic injury, presumably to preserve the caspase cleavage site, the neuroprotective property of the APP D664A substitution cannot be attributed definitively to the loss of C31, which, in turn, attenuated synaptic and dendritic spine injury. This is because while C31 peptide indeed cannot be generated in cells expressing the D664A mutation (Galvan et al., 2006; Lu et al., 2000, 2003a; Figure S3C), concomitant with the attenuation of caspase-3 activation shown in the present study, it is conceptually possible that this D to A base substitution altered other cellular interactions that could also contribute to the synaptic injury/cell toxicity pathway (Barbagallo et al., 2011; Bórquez and González-Billault, 2012; Radzimanowski et al., 2008).
The finding that caspase-3 was activated in dendritic spines in a time course that correlated with dendritic spine loss indicated that these events could be causally related. Indeed, increasing evidence supports a role of caspases in AD and neurodegeneration—not only in neuronal cell death or apoptosis, as originally proposed, but also, more intriguingly, in non-apoptotic cell injury pathways (D’Amelio et al., 2011; Li and Sheng, 2012). While degeneration of characteristic groups of neurons is commonly seen in most neurodegenerative diseases, the mechanisms responsible for neuronal death remain unresolved. Many pathways have been proposed, but none are widely accepted to explain the loss of neurons in brains of AD individuals. The discovery of the caspase ced-3 pathway in programmed cell death has led many investigators to speculate whether a similar apoptotic mechanism underlies neuronal degeneration (Jellinger, 2006). However, the evidence has been wanting, and while caspase activation has been detected in AD brains, neuronal apoptosis as seen during brain development is likely a rare event (Stadelmann et al., 1999). On the other hand, there is considerably more evidence in support of caspases contributing in critical ways to multiple non-apoptotic roles in the nervous system (e.g., in neurogenesis [dendritic pruning, maturation of olfactory sensory neurons, and neurite outgrowth] and synaptic plasticity; D’Amelio et al., 2010, 2012; Hyman and Yuan, 2012; Li and Sheng, 2012; Mukherjee and Williams, 2017; Sheng et al., 2012; Unsain and Barker, 2015; Walsh and Selkoe, 2007). In this regard, increasing evidence favors a role of caspase activation in neuronal and synaptic dysfunction, such as the finding of activated caspase-3 in synaptic fractions and in synaptic terminals from AD brains (Louneva et al., 2008). More compelling is the finding in the Tg2576 line of APP-overexpressing transgenic mouse line where, in the absence of neuronal loss, there was an increase in caspase-3 activity in the hippocampus at the onset of memory impairment, together with a reduction in dendritic spines in these young animals prior to the deposition of extracellular amyloid (D’Amelio et al., 2011). Accordingly, pharmacologic inhibition of caspase-3 reduced the synaptic and behavioral deficits, as we confirmed here in OTSCs with respect to synaptic loss. Further, caspase-3 activity was localized to dendritic spines, highly reminiscent of what was observed in this study, where it was thought to elevate calcineurin levels. In turn, the dephosphorylation of GluR1, triggered by calcineurin and detected in the OTSC system described in this study, was thought to result in postsynaptic dysfunction. Thus, treatment with calcineurin inhibitors has been reported to be beneficial in multiple studies in cultured cells and in APP transgenic mice (Rozkalne et al., 2011; Shankar et al., 2007; Wu et al., 2010).
Our results are thus noteworthy because we have incorporated the cleavage of APP as an additional and necessary step within this pathway, where the proteolysis of APP presumably takes place downstream of caspase-3 activation. But interestingly, the cleavage of APP must result in positive feedback signals to amplify the caspase activation cascade. This is because abrogating this cleavage event by the D664A substitution not only rescued dendritic spine loss, but also significantly diminished caspase-3 activation. However, it is unclear how Aβ triggers this localized caspase activation within spines or dendrites. One can envision several scenarios of how this might occur. For example, Aβ may bind to surface proteins, including APP itself, that may be concentrated on the surface of dendritic spines. We, along with others, have shown that there is an APP-dependent component in Aβ toxicity (Fogel et al., 2014; Kirouac et al., 2017; Lorenzo et al., 2000; Lu et al., 2003a; Wang et al., 2017), and we have previously proposed that the interaction of Aβ with cell-surface APP dimerizes APP, which results in the recruitment and activation of caspase-8 (Lu et al., 2003b; Saganich et al., 2006) and subsequent cleavage of APP at position 664. Second, there is likely a concentration gradient where Aβ levels in the microenvironment are maximal near its site of release from neurons, and it is conceptually possible that this is near synapses, especially the presynaptic axonal terminal, which has been proposed to be a source of activity-enhanced Aβ secretion (Cirrito et al., 2008). This mechanism could also explain the findings from the CA3-to-CA1 dendritic toxicity reported by Wei et al. (2010), where Aβ must be released from presynaptic axonal terminals (see Figure 5), and from the postulated diffusion of oligomeric Aβ from the periphery of Aβ plaque deposits in APP transgenic mice (Koffie et al., 2009). Finally, results from the Sheng laboratory also showed that localized activation of caspases by photosimulation of mitochondrial toxicity resulted in local pruning of dendritic spines and dendritic retraction via a caspase-3-dependent mechanism without neuronal death (Ertürk et al., 2014). In short, this localized activation of caspases within dendrites is an intriguing observation and may represent an underlying susceptibility of dendrites to Aβ toxicity that is likely mediated by multiple mechanisms and will require further study. Aβ has consistently been shown to inhibit LTP in vitro and in vivo and cause synapse loss (Rowan et al., 2005; Sheng et al., 2012; Walsh et al., 2002; Walsh and Selkoe, 2007). While Aβ toxicity can be prevented by a number of different approaches (Cavallucci et al., 2012; Sheng et al., 2012), one effective strategy pertinent to this is by pharmacologic inhibition or genetic knockout of caspase-3 (D’Amelio et al., 2011). The restoration of LTP and dendritic spines in slice cultures from D664A mice would be consistent with these observations from Jo et al. (2011). While LTP is associated with growth of dendritic spines and strengthening of synapses, long-term depression (LTD) is associated with the shrinkage and loss of spines (Sheng et al., 2012; Zhou et al., 2004). In this regard, it has been reported that mitochondrial caspase-3 is required for NMDA-dependent LTD but not for LTP, and this caspase activation does not lead to neuronal cell death (Ertürk et al., 2014; Li et al., 2010). Interestingly, LTD is either unaffected or enhanced by Aβ. Thus, Aβ-induced synaptic injury plays dual roles by impairing LTP while augmenting LTD through caspase-3-dependent mechanisms (Li et al., 2011; Li and Sheng, 2012). Interwoven into this mechanism is the contribution of the APP cytosolic domain at position D664, where it serves as a substrate for proteolysis by caspase-3. Thus, in the absence of APP or in the presence of the D664A mutation, Aβ-induced inhibition of LTP is attenuated.
In summary, we have highlighted the role of the APP cytosolic domain, specifically at position D664, as a contributor to the molecular mechanisms underlying Aβ-induced synaptic injury. Interestingly, our results have pointed to an intricate and complicated interplay among APP (which is a substrate for caspase cleavage), caspase activation, and dendritic spine loss. How Aβ elicits these cellular changes—in particular, whether Aβ directly or indirectly causes the caspase activation or APP cleavage—is unclear. Nonetheless, the findings reported in this study, together with previously published reports, demonstrate that both caspases and APP contribute significantly to Aβ-induced synaptic damage and offer the potential that synaptic injury may be amenable to pharmacologic approaches targeting this pathway.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources or reagents should be directly to Lead Contact, Edward Koo (edkoo@health.ucsd.edu) at UCSD.
Materials Availability
Plasmid DNAs, virus, and mice generated during the project are available upon request by contacting Dr. Koo’s lab.
Data and Code Availability
This study did not generate new datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
were maintained in cages caring for 24hr light/dark cycle with the normal chow diet and water. The experimental procedure for mouse hippocampal slice culture were performed in compliance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC) of University of California, San Diego. The wild-type and mutant mice were confirmed by genotyping the littermate for experiments. Both male and female mice at 8 to 12 week-old were maintained to mate for further experiments. Wild-type C57BL/6 mice and GFP-M mice were purchased from Jackson Laboratory. The APPD664 KI mice were developed by UCSD in collaboration with Ingenious Targeting Laboratory (Ronkonkoma, NY).
METHOD DETAILS
Generation of APP D644A KI and GFP-M mice
A 9.75kb genomic DNA was used to construct the targeting vector from a positively identified B6 BAC clone (RP23:126H12). The region was designed such that the long homology arm (LA) extends about 6.35kb 5′ to the point mutation in 18th exon. The LoxP/FRT-Neo cassette was inserted 187 bp downstream of exon 18. The sequencing result confirmed that no the other mutations were introduced into the PCR-modified region. The point mutation GAC > GCT within exon 18 was generated by a 3-step PCR muta-genesis. The total size of the targeting construct (including vector backbone) was 13.9kb. 10 μgs of the targeting vector was linearized by NotI and then transfected by electroporation into C57BL/6 (B6) embryonic stem cells. After selection with G418 antibiotic, surviving clones were expanded for PCR analysis to identify recombinant ES clones. Secondary confirmation of positive clones identified by PCR was performed by Southern Blotting analysis where the expected sizes of the HpaI digested fragments are ~18kb for a negative clone and ~16kb for a positive clone. Positive clones were further confirmed by Southern Blotting analysis using an internal probe against the 3′ end of the short homology arm. Targeted iTL IC1 (C57BL/6N) embryonic stem cells were microinjected into BALB/c blastocysts. Resulting chimeras with a high percentage black coat color were mated to wild-type C57BL/6N mice to generate F1 heterozygous offspring. Tail DNA was analyzed from pups with black coat color with confirmation of the presence of the point mutations in the PCR product. Four heterozygous mice were identified for targeted integration and confirmed with the introduced point mutations were bred with the B6.129S4-Gt(ROSA)26Sortm2(FLP*)Sor/J line (Jackson Laboratory, stock #012930) for the excision of the Neo cassette and subsequently maintained in either homozygous or heterozygous state of the recombined gene. For adult spine morphology and density experiments, APP D664A KI mice were crossed with GFP-M mice (Jackson Lab, stock #007788) to produce mice heterozygous for the DA mutation and GFP marker. These doubly heterozygous mice were crossed with each other to produce APP D664A KI homozygotes or control WT littermates that were also GFP heterozygotes for the study. Genotyping for the GFP transgene was performed by PCR using a forward primer (AAG TTC ATC TGC ACC ACC G) and reverse primer (TCC TTG AAG ATG GTG CG) to produce a band of 173 bp as recommended by the Jackson Lab. For genotyping of the mice with D664A KI mutation, the forward primer AMBE4 (ATGAACACCGATGGGTAGTGAAGC) and two reverse primers (WT16 and KI16) were used for PCR-based identification. WT16 (TGCTTTCTAGGTCGAC) and KI16 (TGCTTTCTAGGTCGCT) are primers that specifically bind to wild-type or D664A KI sequences, respectively, to generate a fragment of 191 bp.
Immunohistochemical staining and spine analysis in GFP mice
GFP-M and APP D664A crossed to GFP-M mice were perfused with saline and fixed in 4% PFA/ 30% sucrose. After sectioning at 50 μm, the tissues were immunostained with chicken anti-GFP primary antibody (#GFP-1020, Aves Lab) in free-floating sections. The sections were subsequently immunostained with donkey anti-chicken secondary antibody conjugated with Alexa Flour 488 followed by counterstaining with DAPI. After dehydration in graded alcohol, the tissues were mounted with Harleco’s Krystalon mounting medium (64969, Millipore). The sections were imaged with an Olympus DSU IX-81 microscope equipped with a spinning disk attachment using 60x/1.2 N.A. water immersion objective. The GFP positive neurons, which were not overlapping, were selected randomly from the CA1 region for imaging. Dendritic segments selected for analysis had to fulfill the following criteria: 1) the selected segment must be at least 50-μm away from the soma; 2) the selected segment must be between second and third branch point of the dendrite. All imaging and analyses of dendritic spine density was carried out in a blinded fashion. Dendritic spine density was manually counted after processing with Slidebook and ImagePro programs.
Preparation of hippocampal organotypic slice culture (OTSC)
As previously described (Opitz-Araya and Barria, 2011), hippocampi were collected from p5 – p7 postnatal day mouse pups and sectioned at 400 μm thickness (7~8 slices/brain) using a McIIwan Chopper for the ex vivo slice cultures. Following sectioning, the slices were placed onto cell culture inserts (Millicell® Cell Culture Inserts: PICM0RG50) with OTSC culture media {50% MEM-HEPES (GIBCO:12360-038), 25% Hanks’ Balanced Salt Solution (GIBCO: 24020-117), 25% heat inactivated horse serum (GIBCO: 16050-122), 27 mM Glucose 27mM, 0.5% PG-SM (GIBCO: 15140-148), 0.5% and 2 mM GlutaMAX (Sigma: G2150) at 5% CO2, 35°C, humidified incubator for up to 10 days. Slices obtained from one mouse brain were distributed among the different experimental conditions to minimize intra-animal variability.
Preparation of Sindbis virus
Preparation of Sindbis virus expressing GFP, tdTomato, or C99ΔC with tdTomato was described previously (Kamenetz et al., 2003). In brief, RNA was prepared from cDNA viral constructs (AM1340, mMessage mMachine SP6, Ambion, Life Technologies, Carlsbad, CA) and electroporated into BHK cells along with helper virus and incubated for 36 hours. The resultant media were centrifuged at 150,000 g for 90 min at 4°C. The supernatant was collected and frozen until use without dilution for dense infection and diluted at 1:50 for sparse infection.
Analysis of dendritic spine numbers after 7PA2-conditioned medium treatment
Conditioned media preparation: Chinese hamster ovary (CHO) cell stably expressing human APP751 with the Val717Phe mutation (7PA2 cell) (Podlisny et al., 1995; Walsh et al., 2002) were cultured in 10% fetal bovine serum (FBS) (FB-02, Omega) in 1x DMEM (MT10013CV, Corning Cellgro). Cells were grown to near confluence and washed briefly with PBS, and then incubated in DMEM for ~16 h. The 7PA2 conditioned media (7PA2-CM) were concentrated 10-fold using YM-3 (3kDa cutoff) Centriprep filters (4303, Amicon, Beverly) (Calabrese et al., 2007). The concentration of Aβ was determined by Aβ ELISA assay, aliquoted and stored at −80°C. The mAb of Aβ42 (Levites et al., 2006) (kindly provided by Todd Golde, UFL) and HRP-conjugated 6E10 (SIG-39320, Covance) were used for the Aβ ELISA assay as previously described (Eggert et al., 2009).
Treatment of Aβ and injection of Sindbis virus: After 3–4 days in vitro (DIV), conditioned media from 7PA2 cells were added to the OTSCs at two different concentrations: 150 and 250 pM of Aβ42 previously determined by ELISA diluted in OTSC culture media. Treatment durations were for 5 or 10 days. GFP expressing Sindbis virus was injected 24 hours prior to the endpoint of experiment, using micro injector (Wei et al., 2010) containing glass micropipette (Warner Instruments, Hamden, CT), Picospritzer II (General Valve Corporation, Fairfield, NJ), and the SD9 Stimulator (Grass Technologies, Natus Neurology, Middleton, WI) under the Zeiss Stemi 1000 dissecting microscope system (Zeiss, Germany). After the incubation, slices were fixed in 4% paraformaldehyde and 3% sucrose. Slices were then cut out from the inserts and mounted in ProLong Gold Antifade reagent (Life Technologies, Carlsbad, CA) and then imaged by confocal microscope.
Confocal imaging and analyses of dendritic spines: Dendritic branches were sampled following criteria described previously (Midthune et al., 2012), and imaged using the Leica SP5 system (Leica Microsystems, Germany), with a 63X / 1.3 N.A. Plan-Apochromat glycerol immersion objective, digital zoom 6X. Confocal stacks were then deconvolved using an iterative blind deconvolution algorithm (AutoQuantX / AutoDeblur Gold CF version x2.2.2; MediaCybernetics, Bethesda, MD, USA). In the analyses, a total of 33 D664A and 33 wild-type littermate mice were used to generate the OTSCs and then subsequently treated with the respective experimental conditions. As many neurons that met the sampling criteria were imaged from the OTSCs, analyzed, with each dendrite manually inspected and appropriate contrast corrected using NeuronStudio interface for ease of visualization and counting. Spines were manually counted from these images in a blinded fashion. For 5-day old cultures, results were obtained from 30–50 neurons imaged from sections derived from a total of 9 APP D664A and 10 wild-type mice. For the 10 day old cultures, results were obtained from 10 neurons for each condition derived from slices cultured from 2 APP D664A KI and 2 wild-type mice.
Electrophysiology of acute hippocampal slices: Electrophysiology of extracellular field potential recordings from acute hippocampal slices was performed as reported previously (Tyan et al., 2012). The only difference is the addition of conditioned medium from 7PA2 cells at an Aβ42 concentration of 150 pM in artificial CSF (ACSF: 125mM NaCl, 2.4mM KCl, 1.2mM NaH2PO4, 1mM CaCl2, 2mM MgCl2, 25mM NaHCO3, and 25mM glucose) to the hippocampal slices for 20 minutes during the basal recording period followed immediately by the tetanic stimulation. Media from untransfected CHO cells diluted in aCSF were used as control. LTP measurements were carried out in pairs of APP D664A KI and wild-type control mice as well as experimental (7PA2) and control CHO media. The raw data was analyzed by using pClamp 10 software (Molecular Devices, Sunnyvale, CA, USA).
Analysis of dendritic morphology after C99ΔC expression
At 12 to 24 hours following infection with C99ΔC or control tdTomato Sindbis virus, OTSCs were fixed in 4% paraformaldehyde and 3% sucrose. The tissue slices were gently dissected from the membranes of the tissue culture inserts and mounted (H-1000, vector laboratories) on glass slides for imaging by confocal microscopy using an Olympus FV1000 microscope, with a 100X / 1.40 N.A. oil immersion objective, digital zoom 1X. Dendritic morphology was assessed in fluorescently labeled neurons using Imaris software based on the following criteria in a blinded fashion generally as described previously (Wei et al., 2010): 1) main apical dendritic trunk from the second or third dendritic branch of CA1 pyramidal neurons that were clearly isolated from any adjacent labeled neurons or dendrites originating from nearby neurons were randomly chosen (see Figures S4C and S4D); 2) The dendritic segments were free from overlapping dendrites with well-defined dendritic and spine morphology, and > 1 μm dendritic thickness. Typically, a segment of dendrite approximately 70 – 100 μm in the secondary and tertiary branches were imaged and assessed. 3) Spine numbers were manually counted by two observers blinded to genotype. The values from the two observers were averaged and expressed as number of spines/10 μm of dendritic segment. Average of coefficient of variation (CV) between observers was ~5%, indicating high concordance between the two observers. 4) For all experiments, a minimum 6 neurons were assessed, but in general, an average of ~14 neurons (range of from 6 to 25 neurons) were quantified from multiple slices obtained from 3 to 21 mice within each condition. To characterize the individual spine type, 3D images were reconstructed and analyzed with the Automated Filament Tracer module in Imaris software (Bitplane). This module enabled the automatic detection of dendritic spines after subtracting the dendritic shafts, the identification of which were based on an algorithm based Spines were classified as stubby (length ≤ 1 μm; neck width/head width < 1 μm), mushroom (neck width/head width ≥ 1 μm; length ≤ 5 μm), thin (1 μm < length ≤ 2 μm; neck width/head width ≤ 1 μm) and filopodia (5 μm ≤ length; neck width/head width ≤ 1 μm).
Protein sample extraction and immunoassays
Briefly, adult 3–5 month old mice were perfused with saline after which hemi-brains were homogenized with Dounce homogenizer in ice cold 1% CHAPSO in PBS together with protease inhibitor (S8820, Sigma-Aldrich). Following centrifugation (100,000 g) at 4°C for 1hr, the soluble fraction was analyzed for protein concentration and frozen. 20 μg of total protein was fractionated on an 8% Bis/Tris gel and transferred to nitrocellulose for western blotting. Antibodies used were: CT15 (APP680-695) for full length APP (Park et al., 2009); 6E10 which recognizes the N-terminal region of Aβ as well as APP (SIG-39320, Signet Laboratories), 82E1 which is specific to the cleaved N terminus of Aβ and C99 CTF (JP10323, IBL international), β-Tubulin (CP07, Calbiochem Novabiochem), p-GluR1 (Ser845, 36-8300, Thermo Fisher), NMDA R-1 (NBP1-48559, Novus), activated caspase-3 (9664, Cell Signaling) and β-actin (A5316, Sigma-Aldrich) .
Treatment of g-secretase and caspase inhibitors
After the Sindbis virus injection expressing control tdTomato or C99ΔC with tdTomato as bicistronic constructs, the slices were incubated for 24 hr without changing media. At 12 or 18 hours after viral infection, GSI or caspase inhibitors were added to the OTSCs: 10 μM DAPT (565770, EMD Biosciences), 10 μM zVAD (FMK001, pan-caspase inhibitor, R&D Systems), 10 μM zDEVD (FMK004, caspase-3 inhibitor, R&D Systems), 10 μM zYVAD (FMK005, caspase-4 inhibitor, R&D Systems), 10 μM zVEID (FMK006, caspase-6 inhibitor, R&D Systems), and 10 μM zIETD (FMK007, caspase-8 inhibitor, R&D Systems). A drug concentration of 10uM for caspase inhibitors was selected based on the prior determination that was no observable toxicity in dendritic spine density when given to control cultures and this concentration is at or above the Ki of all the inhibitors (data not shown).
In vitro caspase-3 reaction
HEK293T cells were transiently transfected with C99 WT, C99 D664A, or C99ΔC plasmids using TransIT LT-1 Transfection reagent (MIR2304, Mirus) following manufacturer’s instructions and incubated for 2 days. The cells were lysed in RIPA buffer, centrifuged at 14,000 g for 10 min at 4°C and the supernatants incubated with 1X caspase buffer (20 mM PIPES, 0.1% (w/v) CHAPS, 100 mM NaCl, 10 mM dithiothreitol, 10% (w/v) sucrose, 1 mM EDTA, pH 7.2) and 1 μL purified active caspase-3 (offered from Salvesen’s lab) for 1hour at 37°C as previously described (Scott et al., 2008). The reaction was stopped with addition of sample loading buffer and subsequently fractionated by SDS-PAGE and immunoblotted with 82E1 antibody to detect the cleaved APP fragments.
Immunohistological analysis of caspase activation
To detect caspase activation in dendrites, a FLICA reagent, DEVD-ultrabright green dye (13101, Vergent Bioscience,) and an activated caspase-3 specific antibody (9664, Cell Signaling) were used. Briefly, for FLICA analysis, the FLICA reagent (0.1 μM) following manufacturer’s instructions was added to OTSC cultures, incubated for the final hour prior to termination of the experiment, washed, fixed with 4% PFA solution containing 3% sucrose, and mounted on glass slides and imaged by confocal microscopy as before. For caspase 3 immunohistochemistry, the fixed OTSCs were permeabilized with 0.5% Triton X-100 and stained following published protocol (Gogolla et al., 2006). The number of caspase-positive spines visualized by FLICA and active caspase 3 positive signal and total number of spines (tdTomato positive) within ~50 μm length of dendrites (in 2nd or 3rd dendritic branches) were manually counted in a blinded manner. Further, positive caspase 3 signals within dendritic shafts were measured and expressed as a percent of the length of the dendritic segments. 3-dimensional reconstructions were carried out with Imaris software to ascertain whether the positive FLICA signal emanated from within dendritic shafts or from overlying dendritic spines.
QUANTIFICATION AND STATISTICAL ANALYSIS
The results of immunoblot and immunohistology were quantified using NIH ImageJ software. Either Student’s t test, One-way or Twoway ANOVA followed by Tukey’s multiple comparisons test were used to determine statistical significance (GraphPad Prism, San Diego, CA). Imaging experiments using OTSCs were performed from 2 to 5 times and results are expressed as averages of all experiments ± standard error of mean (SEM). A probability of less than 0.05 was considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-GFP | Aves Lab | #GFP-1020; RRID:AB_10000240 |
| CT15 | UCSD | APP680-695 |
| 6E10 | Signet Laboratories | S G-39320; RRID:AB_662798 |
| 82E1 | IBL international | JP10323; RRID:AB_1630806 |
| β-Tubulin | Calbiochem | CP07; RRID:AB_565242 |
| p-GluR1 | Thermo Fisher | 36-8300; RRID:AB_2533280 |
| NMDA R-1 | Novus | NBP1-48559; RRID:AB_10011203 |
| activated caspase-3 Ab | Cell Signaling | 9664; RRID:AB_2070042 |
| β-actin | Sigma-Aldrich | A5316; RRID:AB_476743 |
| HRP-conjugated 6E10 | Covance | SIG-39320; RRID:AB_662798 |
| Bacterial and Virus Strains | ||
| Sindbis Virus-GFP | UCSD | N/A |
| Sindbis Virus-Tomato | UCSD | N/A |
| Sindbis Virus-C99ΔC | UCSD | N/A |
| Sindbis Virus-C99ΔC_Tomato | UCSD | N/A |
| mMessage mMachine SP6 | Ambion, Life Technologies | AM1340 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DEVD-ultrabright green dye (FLICA) | Vergent Bioscience | 13101 |
| Hanks’ Balanced Salt Solution | GIBCO | 24020-117 |
| heat inactivated horse serum | GIBCO | 16050-122 |
| MEM-HEPES | GIBCO | 12360-038 |
| PG-SM | GIBCO | 15140-148 |
| GlutaMAX | Sigma-Aldrich | G2150 |
| DAPT | EMD Biosciences | 565770 |
| zVAD | R&D Systems | FMK001 |
| zDEVD | R&D Systems | FMK004 |
| zYVAD | R&D Systems | FMK005 |
| zVEID | R&D Systems | FMK006 |
| zIETD | R&D Systems | FMK007 |
| active caspase 3 protein | Salvesen’s lab, SBP | N/A |
| Vector shield | vector laboratories | H-1000 |
| Harleco’s Krystalon mounting medium | Millipore | 6496964969 |
| protease inhibitor | Sigma-Aldrich | S8820 |
| Critical Commercial Assays | ||
| TranslT LT-1 Transfection reagent | Mirus | MIR2304 |
| Experimental Models: Organisms/Strains | ||
| B6.129S4-Gt(ROSA)26Sortm2(FLP*)Sor/J | Jackson Laboratory | #012930 |
| GFP-M | Jackson Laboratory | #007788 |
| APP D664A KI | UCSD | N/A |
| Hippocampal organotypic slice | 5–7 day old mouse hippocampi culture, UCSD | N/A |
| Oligonucleotides | ||
| GFP forward primer | (AAG TTC ATC TGC ACC ACC G) | N/A |
| GFP reverse primer | (TCC TTG AAG ATG GTG CG) | N/A |
| APP D664A AMBE4 | (ATG AAC ACC GAT GG G TAG TGA AGC) | N/A |
| APP D664A WT16 | (TGC TTT CTA GGT CGA C) | N/A |
| APP D664AKI16 | (TGC TTT CTA GGT CGC T) | N/A |
| Software and Algorithms | ||
| pClamp 10 | Molecular Devices | N/A |
| NeuronStudio | BISE | N/A |
| Imaris | Bitplane | N/A |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism | GraphPad | N/A |
| Other | ||
| FBS | Omega | FB-02 |
| DMEM | Corning Cellgro | MT10013CV |
| Cell Culture Inserts | Millicell® | PICM0RG50 |
| Centriprep filters | Amicon, Beverly | 4303 |
| glass micropipette | Warner Instruments | N/A |
| Picospritzer II | General Valve Corporation | N/A |
| SD9 Stimulator | Grass Technologies | N/A |
Highlights.
Aβ-dependent caspase activation cleaves the APP C terminus, leading to synaptic injury
Inhibition of APP C-terminal cleavage ameliorates Aβ-dependent synaptic injury
APP D664A mutation prevents APP C terminus cleavage and impairs caspase activation
Synaptic injury by Aβ is rescued in APP D664A knock-in mice
ACKNOWLEDGMENTS
These studies were supported in part by NIH grant NS84324 (E.H.K.), NMRC STaR award 009/2012 (E.H.K.), UCSD ADRC Pilot project award P50 5P50AG005131 (G.P.), and BrightFocus ADR award A2018212F (G.P.). We thank Dr. Roberto Malinow for providing technical guidance on Sindbis virus preparation and OTSC methodology; Dr. Guy Salvesen for his scientific input and for sharing reagents; Dr. Wei Guo, Dr. Karen Chiang, Dr. I-Fang Ling, Kyle Kim, Yifan He, Jennifer Santini, and Scott Snipas for helpful discussions and technical assistance; and Dr. Todd Golde for providing Aβ ELISA reagents. We are grateful for the assistance of Ingenious Targeting Laboratory (Ronkonkoma, NY) for generating the D664A knock-in mice. We also acknowledge the critical support of the UCSD Microscopy Core, supported by NINDS grant P30 NS047101.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107839.
REFERENCES
- Banwait S, Galvan V, Zhang J, Gorostiza OF, Ataie M, Huang W, Crippen D, Koo EH, and Bredesen DE (2008). C-terminal cleavage of the amyloid-beta protein precursor at Asp664: a switch associated with Alzheimer’s disease. J. Alzheimers Dis 13, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbagallo AP, Wang Z, Zheng H, and D’Adamio L (2011). The intracellular threonine of amyloid precursor protein that is essential for docking of Pin1 is dispensable for developmental function. PLoS ONE 6, e18006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, and Holtzman DM (2011). Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci 14, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bórquez DA, and González-Billault C (2012). The amyloid precursor protein intracellular domain-fe65 multiprotein complexes: a challenge to the amyloid hypothesis for Alzheimer’s disease? Int. J. Alzheimers Dis 2012, 353145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredesen DE, John V, and Galvan V (2010). Importance of the caspase cleavage site in amyloid-β protein precursor. J. Alzheimers Dis 22, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese B, Shaked GM, Tabarean IV, Braga J, Koo EH, and Halpain S (2007). Rapid, concurrent alterations in pre- and postsynaptic structure induced by naturally-secreted amyloid-beta protein. Mol. Cell. Neurosci 35, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallucci V, D’Amelio M, and Cecconi F (2012). Aβ toxicity in Alzheimer’s disease. Mol. Neurobiol 45, 366–378. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, and Holtzman DM (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, and Holtzman DM (2008). Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, and McLennan H (1983). Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J. Physiol 334, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amelio M, Cavallucci V, and Cecconi F (2010). Neuronal caspase-3 signaling: not only cell death. Cell Death Differ. 17, 1104–1114. [DOI] [PubMed] [Google Scholar]
- D’Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, et al. (2011). Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci 14, 69–76. [DOI] [PubMed] [Google Scholar]
- D’Amelio M, Sheng M, and Cecconi F (2012). Caspase-3 in the central nervous system: beyond apoptosis. Trends Neurosci. 35, 700–709. [DOI] [PubMed] [Google Scholar]
- Dyrks T, Weidemann A, Multhaup G, Salbaum JM, Lemaire HG, Kang J, Müller-Hill B, Masters CL, and Beyreuther K (1988). Identification, transmembrane orientation and biogenesis of the amyloid A4 precursor of Alzheimer’s disease. EMBO J. 7, 949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert S, Midthune B, Cottrell B, and Koo EH (2009). Induced dimerization of the amyloid precursor protein leads to decreased amyloid-beta protein production. J. Biol. Chem 284, 28943–28952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertürk A, Wang Y, and Sheng M (2014). Local pruning of dendrites and spines by caspase-3-dependent and proteasome-limited mechanisms. J. Neurosci 34, 1672–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, O’Malley T, Slomowitz E, Berdichevsky Y, Walsh DM, et al. (2014). APP homodimers transduce an amyloid-β-mediated increase in release probability at excitatory synapses. Cell Rep. 7, 1560–1576. [DOI] [PubMed] [Google Scholar]
- Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, et al. (2006). Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc. Natl. Acad. Sci. USA 103, 7130–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, et al. (1999). Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 97, 395–406. [DOI] [PubMed] [Google Scholar]
- Gillingwater TH, and Wishart TM (2013). Mechanisms underlying synaptic vulnerability and degeneration in neurodegenerative disease. Neuropathol. Appl. Neurobiol 39, 320–334. [DOI] [PubMed] [Google Scholar]
- Giménez-Llort L, Blázquez G, Cañete T, Johansson B, Oddo S, Tobeña A, LaFerla FM, and Fernández-Teruel A (2007). Modeling behavioral and neuronal symptoms of Alzheimer’s disease in mice: a role for intraneuronal amyloid. Neurosci. Biobehav. Rev 31, 125–147. [DOI] [PubMed] [Google Scholar]
- Gogolla N, Galimberti I, DePaola V, and Caroni P (2006). Staining protocol for organotypic hippocampal slice cultures. Nat. Protoc 1, 2452–2456. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tampellini D, Takahashi RH, and Capetillo-Zarate E (2010). Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 119, 523–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot-Sestier MV, Sunyach C, Ferreira ST, Marzolo MP, Bauer C, Thevenet A, and Checler F (2012). α-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid β (Aβ)-associated cell death. J. Biol. Chem 287, 5021–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada J, and Sugimoto M (1999). Activation of caspase-3 in beta-amyloid-induced apoptosis of cultured rat cortical neurons. Brain Res. 842, 311–323. [DOI] [PubMed] [Google Scholar]
- Harris JA, Devidze N, Halabisky B, Lo I, Thwin MT, Yu GQ, Bredesen DE, Masliah E, and Mucke L (2010). Many neuronal and behavioral impairments in transgenic mouse models of Alzheimer’s disease are independent of caspase cleavage of the amyloid precursor protein. J. Neurosci 30, 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, Maki M, Itohara S, and Saido TC (2005). Distinct mechanistic roles of cal-pain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J. Biol. Chem 280, 15229–15237. [DOI] [PubMed] [Google Scholar]
- Hyman BT, and Yuan J (2012). Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat. Rev. Neurosci 13, 395–406. [DOI] [PubMed] [Google Scholar]
- Jellinger KA (2006). Challenges in neuronal apoptosis. Curr. Alzheimer Res 3, 377–391. [DOI] [PubMed] [Google Scholar]
- Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G, and Cho K (2011). Aβ(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci 14, 545–547. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, and Malinow R (2003). APP processing and synaptic function. Neuron 37, 925–937. [DOI] [PubMed] [Google Scholar]
- Kirouac L, Rajic AJ, Cribbs DH, and Padmanabhan J (2017). Activation of Ras-ERK Signaling and GSK-3 by Amyloid Precursor Protein and Amyloid Beta Facilitates Neurodegeneration in Alzheimer’s Disease. eNeuro 4, ENEURO.0149–16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, et al. (2009). Oligomeric Amyloid Beta Associates With Postsynaptic Densities and Correlates With Excitatory Synapse Loss Near Senile Plaques. Proc. Natl. Acad. Sci. USA 106, 4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Xu H, Li Z, Wang Z, O’Malley TT, Zhang D, Walsh DM, Xu P, Selkoe DJ, and Li S (2016). Soluble Aβ oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol. Dis 85, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Das P, Price RW, Rochette MJ, Kostura LA, McGowan EM, Murphy MP, and Golde TE (2006). Anti-Abeta42- and anti-Abeta40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J. Clin. Invest 116, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, and Sheng M (2012). Caspases in synaptic plasticity. Mol. Brain 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, and Selkoe D (2009). Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, and Sheng M (2010). Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141, 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, and Selkoe DJ (2011). Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci 31, 6627–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, and Yankner BA (2000). Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer’s disease. Nat. Neurosci 3, 460–464. [DOI] [PubMed] [Google Scholar]
- Louneva N, Cohen JW, Han LY, Talbot K, Wilson RS, Bennett DA, Trojanowski JQ, and Arnold SE (2008). Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am. J. Pathol 173, 1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, and Bredesen DE (2000). A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat. Med 6, 397–404. [DOI] [PubMed] [Google Scholar]
- Lu DC, Shaked GM, Masliah E, Bredesen DE, and Koo EH (2003a). Amyloid beta protein toxicity mediated by the formation of amyloid-beta protein precursor complexes. Ann. Neurol 54, 781–789. [DOI] [PubMed] [Google Scholar]
- Lu DC, Soriano S, Bredesen DE, and Koo EH (2003b). Caspase cleavage of the amyloid precursor protein modulates amyloid beta-protein toxicity. J. Neurochem 87, 733–741. [DOI] [PubMed] [Google Scholar]
- Midthune B, Tyan SH, Walsh JJ, Sarsoza F, Eggert S, Hof PR, Dickstein DL, and Koo EH (2012). Deletion of the amyloid precursor-like protein 2 (APLP2) does not affect hippocampal neuron morphology or function. Mol. Cell. Neurosci 49, 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, and Williams DW (2017). More alive than dead: non-apoptotic roles for caspases in neuronal development, plasticity and disease. Cell Death Differ. 24, 1411–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieweg K, Andreyeva A, van Stegen B, Tanriöver G, and Gottmann K (2015). Alzheimer’s disease-related amyloid-β induces synaptotoxicity in human iPS cell-derived neurons. Cell Death Dis. 6, e1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz-Araya X, and Barria A (2011). Organotypic hippocampal slice cultures. J. Vis. Exp, 2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SA, Shaked GM, Bredesen DE, and Koo EH (2009). Mechanism of cytotoxicity mediated by the C31 fragment of the amyloid precursor protein. Biochem. Biophys. Res. Commun 388, 450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L, Passer BJ, Tabaton M, Ganjei JK, and D’Adamio L (1999). Alternative, non-secretase processing of Alzheimer’s beta-amyloid precursor protein during apoptosis by caspase-6 and −8. J. Biol. Chem 274, 21011–21016. [DOI] [PubMed] [Google Scholar]
- Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Te-plow DB, and Selkoe DJ (1995). Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J. Biol. Chem 270, 9564–9570. [DOI] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, Ribe EM, Troy CM, Arancio O, and Shelanski M (2013). Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice. Nat. Commun 4, 1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzimanowski J, Simon B, Sattler M, Beyreuther K, Sinning I, and Wild K (2008). Structure of the intracellular domain of the amyloid precursor protein in complex with Fe65-PTB2. EMBO Rep. 9, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Vyas V, Hernandez-Estrada T, Nichol KE, Christie LA, and Head E (2008). Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J. Neurosci 28, 3051–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Wang Q, and Anwyl R (2005). Synaptic plasticity disruption by amyloid beta protein: modulation by potential Alzheimer’s disease modifying therapies. Biochem. Soc. Trans 33, 563–567. [DOI] [PubMed] [Google Scholar]
- Rozkalne A, Hyman BT, and Spires-Jones TL (2011). Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol. Dis 41, 650–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, and Heinemann SF (2006). Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J. Neurosci 26, 13428–13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott FL, Fuchs GJ, Boyd SE, Denault JB, Hawkins CJ, Dequiedt F, and Salvesen GS (2008). Caspase-8 cleaves histone deacetylase 7 and abolishes its transcription repressor function. J. Biol. Chem 283, 19499–19510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, and Sabatini BL (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci 27, 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Sabatini BL, and Südhof TC (2012). Synapses and Alzheimer’s disease. Cold Spring Harb. Perspect. Biol 4, a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolewski P, Grabarek J, Halicka HD, and Darzynkiewicz Z (2002). Assay of caspase activation in situ combined with probing plasma membrane integrity to detect three distinct stages of apoptosis. J. Immunol. Methods 265, 111–121. [DOI] [PubMed] [Google Scholar]
- Soriano S, Lu DC, Chandra S, Pietrzik CU, and Koo EH (2001). The amyloidogenic pathway of amyloid precursor protein (APP) is independent of its cleavage by caspases. J. Biol. Chem 276, 29045–29050. [DOI] [PubMed] [Google Scholar]
- Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Brück W, Jellinger K, and Lassmann H (1999). Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am. J. Pathol 155, 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Nagao T, and Gouras GK (2017). Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathol. Int 67, 185–193. [DOI] [PubMed] [Google Scholar]
- Tesco G, Koh YH, and Tanzi RE (2003). Caspase activation increases beta-amyloid generation independently of caspase cleavage of the beta-amyloid precursor protein (APP). J. Biol. Chem 278, 46074–46080. [DOI] [PubMed] [Google Scholar]
- Tyan SH, Shih AY, Walsh JJ, Maruyama H, Sarsoza F, Ku L, Eggert S, Hof PR, Koo EH, and Dickstein DL (2012). Amyloid precursor protein (APP) regulates synaptic structure and function. Mol. Cell. Neurosci 51, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsain N, and Barker PA (2015). New Views on the Misconstrued: Executioner Caspases and Their Diverse Non-apoptotic Roles. Neuron 88, 461–474. [DOI] [PubMed] [Google Scholar]
- Walsh DM, and Selkoe DJ (2007). A beta oligomers - a decade of discovery. J. Neurochem 101, 1172–1184. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, and Selkoe DJ (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- Wang Z, Jackson RJ, Hong W, Taylor WM, Corbett GT, Moreno A, Liu W, Li S, Frosch MP, Slutsky I, et al. (2017). Human Brain-Derived Aβ Oligomers Bind to Synapses and Disrupt Synaptic Activity in a Manner That Requires APP. J. Neurosci 37, 11947–11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, and Malinow R (2010). Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci 13, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann A, König G, Bunke D, Fischer P, Salbaum JM, Masters CL, and Beyreuther K (1989). Identification, biogenesis, and localization of precursors of Alzheimer’s disease A4 amyloid protein. Cell 57, 115–126. [DOI] [PubMed] [Google Scholar]
- Welzel AT, Maggio JE, Shankar GM, Walker DE, Ostaszewski BL, Li S, Klyubin I, Rowan MJ, Seubert P, Walsh DM, and Selkoe DJ (2014). Secreted amyloid β-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity. Biochemistry 53, 3908–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensveen FM, Derks IA, van Gisbergen KP, de Bruin AM, Meijers JC, Yigittop H, Nolte MA, Eldering E, and van Lier RA (2012). BH3-only protein Noxa regulates apoptosis in activated B cells and controls high-affinity antibody formation. Blood 119, 1440–1449. [DOI] [PubMed] [Google Scholar]
- Wertkin AM, Turner RS, Pleasure SJ, Golde TE, Younkin SG, Trojanowski JQ, and Lee VM (1993). Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular beta-amyloid or A4 peptides. Proc. Natl. Acad. Sci. USA 90, 9513–9517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, et al. (2010). Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci 30, 2636–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, and Poo MM (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new datasets or code.