

Abstract
Cancer-associated bodyweight loss (cachexia) is a hallmark of many cancers and is associated with decreased quality of life and increased mortality. Hepatic function can dramatically influence whole-body energy expenditure and may therefore significantly influence whole-body health during cancer progression. The purpose of this study was to examine alterations in markers of hepatic metabolism and physiology during cachexia progression. Male C57BL/6J mice were injected with 1 × 106 Lewis Lung Carcinoma cells dissolved in 100 μL PBS and cancer was allowed to develop for 1, 2, 3, or 4 weeks. Control animals were injected with an equal volume of phosphate-buffered saline. Livers were analyzed for measures of metabolism, collagen deposition, protein turnover, and mitochondrial quality. Animals at 4 weeks had ~30% larger livers compared with all other groups. Cancer progression was associated with altered regulators of fat metabolism. Additionally, longer duration of cancer development was associated with ~3-fold increased regulators of collagen deposition as well as phenotypic collagen content, suggesting increased liver fibrosis. Mitochondrial quality control regulators appeared to be altered before any phenotypic alterations to collagen deposition. While induction of Akt was noted, downstream markers of protein synthesis were not altered. In conclusions, cancer cachexia progression is associated with hepatic pathologies, specifically liver fibrosis. Alterations to mitochondrial quality control mechanisms appear to precede this fibrotic phenotype, potentially suggesting mitochondrial mechanisms for the development of hepatic pathologies during the development and progression of cancer cachexia.
Novelty
Cachexia progression results in liver collagen deposition and fibrosis.
Alterations in mitochondrial quality control may precede liver pathologies during cachexia.
Keywords: mitochondrial dynamics, mitophagy, protein synthesis, fibrosis, cachexia, fat metabolism
Résumé :
La perte de poids corporel associée au cancer (cachexie) caractérise de nombreux cancers et est associée à une baisse de la qualité de vie et à une mortalité accrue. La fonction hépatique peut influer considérablement sur la dépense énergétique de tout le corps et, par conséquent, sur la santé globale pendant la progression du cancer. Examiner les altérations des marqueurs du métabolisme et de la physiologie hépatiques au cours de la progression de la cachexie. Des souris mâles C57BL/6J reçoivent une injection de 1 × 106 cellules de carcinome pulmonaire de Lewis dissoutes dans 100 μL de tampon phosphate salin; le cancer se développe pendant 1, 2, 3 ou 4 semaines. Les animaux témoins reçoivent une injection d’un volume égal de PBS. Les foies sont analysés pour mesurer le métabolisme, les dépôts de collagène, le renouvellement des protéines et la qualité des mitochondries. Les animaux de 4 semaines ont un foie environ 30 % plus gros comparativement aux autres groupes. La progression du cancer est associée à une altération des régulateurs du métabolisme des graisses. En outre, une plus longue durée de développement du cancer est associée à une augmentation d’environ 3 fois des régulateurs du dépôt de collagène ainsi que de la teneur en phénotype du collagène, ce qui suggère une fibrose hépatique accrue. Les régulateurs de contrôle de qualité mitochondriale semblent modifiés avant toute altération du dépôt en phénotype du collagène. Bien que l’induction de l’Akt soit notée, les marqueurs en aval de la synthèse des protéines ne sont pas altérés. En conclusion, la progression de la cachexie du cancer est associée à des pathologies hépatiques, notamment la fibrose du foie. Des modifications des mécanismes de contrôle de la qualité mitochondriale semblent précéder ce phénotype de la fibrose, suggérant potentiellement des mécanismes mitochondriaux pour le développement de pathologies hépatiques lors du développement et de la progression de la cachexie cancéreuse. [Traduit par la Rédaction]
Les nouveautés
La progression de la cachexie entraîne un dépôt de collagène hépatique et une fibrose.
Des altérations du contrôle de qualité mitochondriale peuvent précéder les pathologies du foie lors de la cachexie.
Mots-clés: dynamique mitochondriale, mitophagie, synthèse protéique, fibrose, cachexie, métabolisme des graisses
Introduction
Cancer remains a significant cause of death in the United States (Centers for Disease Control and Prevention (CDC)/National Center for Health Statistics 2015). A major proportion of these deaths are thought to be mediated by excessive weight loss (cancer cachexia), which often limits chemotherapeutic treatment capacity (Prado et al. 2007; van Vledder et al. 2012). This excessive weight loss is thought to be mediated by multi-organ aberrations contributing to reduced health outcomes and increased mortality (Bennani-Baiti and Walsh 2009; Hauser et al. 2006; Wallengren et al. 2013). Due to its multi-factorial pathology, thorough understanding of mechanisms contributing to cachexia have been challenging, making the advent of clinical interventions for this pathology difficult. Cachexia is partially mediated by a disparity between energy requirements and energy availability (Petruzzelli and Wagner 2016; Ryan et al. 2016). As the liver is the predominant location of energy production and storage, alterations in hepatic physiology may be a mediating factor for cancer-associated body weight loss (Argilés et al. 2015). To our knowledge, few studies have investigated the influence of cancer on liver metabolism. Narsale et al. (2015, 2016) found cachexia in the ApcMin/+ mouse resulted in decreased liver glycogen content and increased hepatic inflammation, as well as a more glycolytic phenotype, demonstrating that cancer has a substantial impact on hepatic metabolism and these alterations may affect the progression of cachexia (Narsale et al. 2015). Additionally, works using the C26 model have also found alterations in hepatic fat and cholesterol metabolism, which appear to mediated by tumor-secreted factors activating hepatic TSC22D4, resulting in decreased serum triglyceride and lipids compared with healthy control mice (Jones et al. 2013).
Overall, taken together these studies appear to strongly suggest hepatic manifestations occur with cancer associated muscle loss. However, to our knowledge, no prior study has investigated the initial development of cancer-cachexia in relation to measures of hepatic metabolism and health, leaving a crucial aspect in development of cancer-related pathologies largely uninvestigated. Understanding hepatic alterations during the early stages of cancer may be imperative for halting hepatic aberrations before they lead to irreversible pathologies. Therefore, the purpose of this study was to examine changes in physiologic and metabolic processes in the liver during the development and progression of cancer cachexia.
Materials and methods
Animals and interventions
We have published the animal interventions and muscle weights from these animals and this particular study includes a subset of animals from our prior works (Brown et al. 2017a). All methods were approved by the University of Arkansas Institutional Animal Care and Use Committee. Male mice were housed at the University of Arkansas animal care facility in a secure, temperature, and humidity-controlled environment on a 12-h light/ 12-h dark cycle. All animals had free access to chow and water for during the course of the study. Briefly, at 8 weeks of age C57BL/6J mice were anesthetized with isoflurane and injected with 1 × 106 Lewis Lung Carcinoma cells (ATCC, cat. no. CRL-1642) in the left hind flank. This is a commonly used method for inducing cancer cachexia in mice and has previously been demonstrated to induce a cachectic phenotype in mice (Choi et al. 2013; Iwata et al. 2016; Wang et al. 2011). Control animals were injected with phosphate buffered saline (PBS) as a sham control. Cancer was allowed to progress, and cohorts of animals were humanely euthanized and tissues collected for analysis. Animals were euthanized by cervical dislocation while under isoflurane anesthesia. Cohorts included PBS and 1, 2, 3, and 4 weeks of cancer progression. In our prior reports using these same animals we have noted lowered muscle size at 4 weeks of cancer progression, depending on the tissue of interest (Brown et al. 2017a). PBS animals were age-matched and harvested with 4-week animals.
Histology
Histological staining was performed as previously described (Brown et al. 2017a; Rosa-Caldwell et al. 2017, 2016) with minor modifications. Sections of liver were cut at 10–12 μm using a Leica CM1859 clinical cryostat (Leica Biosystems, Buffalo Grove, Ill., USA). Sections were stained for succinate dehydrogenase (SDH), Oil Red O, Sirius Red, and periodic acid-Schiff (PAS). Images were averaged within each sample for analysis. All images were taken at 20× magnification.
Oil Red O stain
Oil Red O staining was completed to measure lipid content within the liver. Slides were first fixed in 3.7% formaldehyde for 10 min at room temperature and then rinsed in 3 dips of dH2O water. After which, slides were placed in a 60% isopropyl alcohol: dH2O for 5 min. Slides were then incubated in Oil Red O working solution for 10 min (working solution 2:3 dilution of dH2O:Oil Red O stock solution, stock solution: 5 mg/mL Oil Red O powder (Amresco, Solon, Ohio; cat. no. 0684-250G) in isopropyl alcohol). Slides were incubated for 30 min at room temperature in Oil Red O solution, then rinsed 3×1 min in dH2O. Slides were mounted in 10% glycerol:PBS solution. Slides were quantified by percent area stained with Oil Red O. Images were averaged within each animal for analysis.
PAS stain
For PAS staining of glycogen, slides were first fixed in a solution containing 60 mL 100% ethanol, 30 mL chloroform, and 10 mL clacial acetic acid for 10 min. After rinsing 3×1 min in distilled water, slides were then incubated in 10% periodic acid solution (1 g periodic acid powder (MilliporeSigma, St. Louis, Mo., USA; cat. no. 7878-25G) dissolved in 10 mL dH2O for 10 min. Slides were then rinsed with distilled water and incubated in Schiff Reagent (MilliporeSigma, Burlington, Mass., USA; cat. no. 6073-71) for 10 min before rinsing with tap water. Slides were then dehydrated in ascending ethanol: distilled water solutions (50%, 70%, 80%, 95%, and 100%) and rinsed with xylenes. Slides were mounted in 10% glycerol:PBS solution. Slides were quantified by percent area stained with PAS stain. Images were averaged within each animal for analysis.
Sirius Red stain
For Sirius Red collagen staining, slides were incubated in picro-Sirius Red solution (0.5 g Sirius Red (Spectrum, Gardena, Calif., USA; cat. no. S1066) into 500 mL picric acid (EM Science, Hatfield, Pa.; cat. no. px1001-1) for 60 min. After which, slides were washed in acid–alcohol solution (2.5 mL of glacial acetic acid diluted to 500 mL using dH2O). Slides were then dehydrated and cleaned in 100% ethanol and xylenes, respectively, and then mounted in with toluene-based mounting media. Slides were quantified for percent area stained with Sirius Red stain per image and images were averaged within each animal for analysis.
SDH stain
SDH staining was competed to determine oxidative phenotype of the liver as we have previously described (Brown et al. 2017a; Rosa-Caldwell et al. 2017). Slides were quantified by intensity of the stain. SDH stain was quantified by the total stain intensity for each image taken per sample (~4–5 images/sample).
All images were collected with Nikon Sight DS-Vi1 camera mounted on an Olympus CKX41 inverted microscope and analyzed with Nikon Basic Research Imaging Software (Melville, N.Y., USA). All images were mounted with a glycerol-based mounting media and a coverslip. Total area or intensity of the stain of interest was quantified with Nikon Basic Research Imaging Software and is expressed as average percent stained or intensity stained, respectively.
Isolation of protein and immunoblotting
Isolation of protein and immunoblotting were performed as previously described (Greene et al. 2015). Briefly, ~40 mg of liver was homogenized in glass Dounce-type homogenizers in 0.30 mL of complete protein loading buffer (50 mmol/L Tris-HCl, pH 6.8, 1% sodium dodecyl sulfate (SDS), 10% glycerol, 20 mmol/L dithiothreitol, 127 mmol/L 2-mercaptoethanol, and 0.01% bromophenol blue supplemented with protease inhibitors (Roche) and phosphatase inhibitors (MilliporeSigma, St. Louis, Mo., USA) according to Alliance for Cellular Signaling protocols and as described elsewhere (Greene et al. 2015). After homogenization, samples were transferred to sterile 1.5-mL microcentrifuge tubes, heated for 5 min at 95 °C to denature protein and then centrifuged for 5 min at 13 000 rpm (10 000g). Protein concentrations were determined using a commercially available RC/DC assay kit following manufacturer instructions (Bio-Rad, Hercules, Calif., USA). Forty micrograms of liver protein were loaded into 6%–15% SDS-polyacrylamide gels, depending on size of protein of interest, and transferred onto polyvinylidene fluoride membranes (Thermo Scientific, Rockford, Ill., USA). Membranes were blocked for an hour in 5% milk solution in 0.5% Tris-buffered saline - Tween 20 (TBS-T), and then incubated in primary antibody blocking solution at 4 °C. After 12–48 h of primary incubation, membranes were washed in 0.5% TBS-T and incubated with appropriate secondary antibody solutions for 1 h (Li-Cor Biosciences, Lincoln, Nebr., USA). Membranes were imaged using a FlourChem M (Protein Simple, San Jose, Calif., USA) and protein content normalized to Ponceau S. Additionally, prior to analysis, we measured Ponceau stain for the entire lane and ensured that variability between groups was within an acceptable range (<10%–14%). Primary antibodies included COX-IV (Cell Signaling, Danvers, Mass., USA; cat. no. 4850), BCL2 interacting protein 3 (BNIP3) (Cell Signaling; cat. no. 44060), Akt (Cell Signaling; cat. no. 4685), phosphorylated (p)-AktSer473 (Cell Signaling; cat. no. 4060), p70 (Cell Signaling; cat. no. 2708), p-p70Thr389 (Cell Signaling, cat. no. 9234), 4EBP-1 (Cell Signaling; cat. no. 9644), p-4EBP-1Thr37/46 (Cell Signaling; cat. no. 9459), light-chain 3 (LC3) (Cell Signaling, cat. no. 3868), p62 (MilliporeSigma, St. Louis, Mo., USA; cat. no. P0067).
RNA isolation, complementary DNA (cDNA) synthesis, and qualitative real-time polymerase chain reaction
Tissues were homogenized, RNA isolated, and reverse transcribed into cDNA as we have previously described (Rosa-Caldwell et al. 2016). cDNA was analyzed by the −ΔΔCT method as we have previously described (Brown et al. 2017b; Greene et al. 2015; Rosa-Caldwell et al. 2016). Taqman probes or SYBR primers for stearoyl-coenzyme A desaturase-1 (Scd1), Srebp1, 3-hydroxy-3-methylglutaril coenzyme A (HMG-CoA) reductase, peroxisome proliferator activated receptor alpha (Pparα), cytochrome c oxidase 4 (Cox4), proliferatoractivated receptor-γ coactivator-1α (Pgc-1α), mitochondrial transcription factor A (Tfam), Bnip3, mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), optic atrophy protein 1 (Opa1), dynamin-1-like protein (Drp1), mitochondrial fission factor (Mf), mitochondrial fission 1 protein (Fis1), Beclin, autophagy related 7 (Atg7), Lc3, p62, Deptor, Collagen 1, Collagen 3, matrix metalloproteinase 9 (Mmp9), matrix metalloproteinase 2 (Mmp2), TIMP metallopeptidase inhibitor 1 (Timp1), and transforming growth factor beta (Tgf-β) were analyzed. SYBR primers have previously been reported (Greene et al. 2015; Rosa-Caldwell et al. 2016).
Statistical analysis
Independent variables included time groups (PBS vs. 1 week vs. 2 weeks vs. 3 weeks vs. 4 weeks). Results were analyzed by 1-way ANOVA, with α set at 0.05. Additionally, due to the time course nature of the data an additional trend analysis was performed as described (Maxwell and Delany 2004). This specific analysis differs from traditional regression analysis through key overall goals of the analysis and mathematical computations used for significance testing. Simple regression analysis seeks predict the dependent variable based on an independent variable (typically both independent and dependent variables are continuous). Trend analysis is a specific extension of the general linear model, using orthogonal contrast statements to test for polynomial relationships between a quantitative but not necessarily continuous independent variable (in this case weeks of cancer progression) and the dependent variable. Mathematically, simple regression has a model degrees of freedom of 1 for calculating the F statistic. For this particular design, a model degrees of freedom of 1 is not appropriate because we have 5 distinct groups, therefore our model degrees of freedom is 4. Trend analysis first utilizes a general linear model ANOVA and F test to determine if the variance between groups exceeds the variance within groups, using the appropriate degrees of freedom. If the global F test is significant, then trend analysis attempts to explain how the dependent variable changes between different factors of the independent variable (in this experiment how dependent variables change with increased duration of cancer progression). Specifically, higher order polynomial functions (quadratic, cubic, quartic etc.) are only added into the model when they increase the explanatory power of the overall model more than a simple linear trend. This analysis was utilized as an additional measure to delineate fluctuations in the data that appeared dependent on development of cancer that did not reach statistical significance for pairwise comparisons. When significant F tests were noted, F-tables were used to determine deviation from linearity to determine if higher order polynomials explained more variance than simple linear models (Maxwell and Delany, 2004). All data were analyzed using the Statistical Analysis System (SAS; version 9.3, Cary, N.C., USA) and expressed as means ± SEM. When significant trends were noted (p < 0.05), the trend line was overlaid on bar graphs of means with 95% confidence intervals for that specific trend analysis. All graphs have the ratio of sum of squares of the model to the total sum of squares (R2) noted to distinguish the strength of association between dependent variables and the duration of cancer progression.
Results
Cancer-cachexia development and progression induced marked alterations in hepatic size and overall phenotype
We have previously published phenotypic data on these animals, demonstrating larger tumor masses (0.8–3.5 g) in our 3-week and 4-week animals as well as 10%–15% lower muscle masses of the tibialis anterior, gastrocnemius, plantaris, and soleus in 4-week animals compared with all other groups, validating a cachectic phenotype in our 4-week animals (Brown et al. 2017a). Here, we report that liver weights were ~35% greater in 4-week animals compared with all other groups, no further differences were noted and liver weights across time demonstrated a quadratic trend with cancer development (R2 = 0.38, p < 0.001, Fig. 1A). PAS staining demonstrated a cubic trend in glycogen content with approximately 50% stained at 1 and 2 weeks of cancer development compared with approximately 40% stained in control animals. PAS area was then dramatically lower in 3- and 4-week animals, (~15%–20% stained, R2 = 0.29, p = 0.027, Fig. 1B and 1F). Oil Red O staining was not altered across any of the groups with approximately 1.5%–2% stained with Oil Red O. (R2 = 0.06, p = 0.750, Fig. 1C and 1F). Slight aberrations were noted in SDH intensity, with a quadratic trend noted across groups; however, no pairwise comparisons reached statistical significance (R2 = 0.18, p = 0.043, Fig. 1D and 1F). Finally, Sirius Red stain, a marker of collagen deposition, became progressively greater across cancer groups, with duration of cancer development having an exponential relationship with collagen content. Collagen deposition a was ~500% larger in 4-week animals compared with all other groups (~1.5% compared with 7.5% collagen deposition) with no further statistical differences noted (R2 = 0.78, p < 0.001, Fig. 1E and 1F).
Fig. 1.

Phenotypic data from the present study. (A) Liver mass. (B) Glycogen content of the liver measured by periodic acid Schiff (PAS) stain. (C) Lipid content of the liver measured by Oil Red O. (D) Oxidative capacity of the liver measured by intensity of succinate dehydrogenase (SDH) stain. (E) Collagen deposition measured by Sirius Red staining. (F) Representative images of PAS, Oil Red O, SDH, and Sirius Red. All images were taken at 20× magnification. All graphs are means ± SEM, n = 7–10/group for each analysis. Significant trends are plotted against means with 95% confidence intervals. 1wk, 2wk, 3wk, 4wk, 1, 2, 3, and 4 weeks of cancer progression, respectively; PBS, phosphate-buffered saline.
Cancer-cachexia development and progression resulted in alterations to messenger RNA (mRNA) content of lipid and cholesterol metabolism
The liver serves as one of the primary energy mediators in the body, serving as both a storage and synthesis site for many energy substrates. Therefore, alterations in these metabolic processes may contribute to cancer-induced body weight loss. More so, recent works have characterized serum metabolites associated with lipid and cholesterol altered in both cachectic and pre-cachectic patients (Der-Torossian et al. 2013; Yang et al. 2018). Therefore, we first measured markers of fatty acid and cholesterol metabolism to investigate possible phenotypic outcomes of the development and progression of cancer cachexia. Scd1, a catalyst for the synthesis of phospholipids and triglycerides, mRNA content demonstrated a linear trend downward with 4-week animals having ~60% less Scd1 mRNA content (R2 = 0.24, p = 0.001, Fig. 2A). Similarly, Srebp1, an important transcription factor for lipid and cholesterol synthesis, mRNA content also demonstrated a linear decrease with cancer progression, with statistical differences only detected between PBS and 1-week and 3-week animals (R2 = 0.33, p = 00.004, Fig. 2B). HMG-CoA reductase, a significant mediator for cholesterol synthesis, mRNA content demonstrated a quadratic relationship with cancer progression, with a trough at 1 wk of cancer development, that gradually became greater in 2, 3, and 4-week animals (R2 = 0.16, p = 0.032, Fig. 2C). Pparα, a transcription factor important for lipid catabolism and usage, mRNA content also demonstrated a quadratic trend during cancer progression; however, there was a peak in Pparα mRNA content in 2-week animals that then became lower in 3- and 4-week animals (R2 = 0.27, p = 0.002, Fig. 2D).
Fig. 2.

RNA content of markers for fatty acid and cholesterol metabolism. (A) Scdl content. (B) Srebp1 content. (C) HMG CoA reductase content. (D) Pparα content. All graphs are means ± SEM, n = 7–10/group for each analysis. Significant trends are plotted against means with 95% confidence intervals. 1wk, 2wk, 3wk, 4wk, 1, 2, 3, and 4 weeks of cancer progression, respectively; PBS, phosphate-buffered saline.
Cancer progression resulted in large increases in mediators of collagen deposition and degradation
Collagen deposition can lead to hepatic fibrosis and eventually liver failure (Bataller and Brenner 2005). Due to the strong relationship noted between collagen deposition and duration of cancer progression (Fig. 1E and 1F), we measured potential mediators of collagen deposition. Collagen 1, associated as the more flexible structural component of collagen, mRNA content demonstrated a cubic relationship with cancer progression, with 4-week animals having 3.5-fold more Collagen 1 mRNA content compared with all other groups (R2 = 0.48, p = 0.001, Fig. 3A). Similarly, Collagen 3, typically regarded as the more stiff structural component of collagen, mRNA content also demonstrated a cubic relationship with cancer progression, and 4-week animals having ~4-fold more Collagen 3 mRNA content compared with all other groups (R2 = 0.57, p = 0.002, Fig. 3B). The ratio of Collagen 1/3 can provide insight of the fibrotic phenotype of a tissue. In the current study, we find that Collagen 1/3 ratio did not have any trends or differences with cancer progression in the liver (R2 = 0.07, p = 0.590, Fig. 3C). Next, to determine regulation or Collagen turnover we investigated extracellular matrix (ECM) degradative regulators matrix metalloproteinases (Mmp2 and Mmp9) and associated inhibitor of Mmp9, Timp1. A quadratic relationship was noted between Mmp9 mRNA content and cancer progression, with 4-week animals having ~20-fold more mRNA content of Mmp9 compared with PBS and 1-week and 2-week animals and ~10-fold content compared with 3-week animals (R2 = 0.54, p = 0.003, Fig. 3D). Mmp2 content did not differ between any cancer groups (R2 = 0.16, p = 0.095, Fig. 3E). Timp1 mRNA content demonstrated a quadratic relationship with cancer progression with 4-week animals having ~4-fold more Timp1 mRNA content compared with all other groups (R2 = 0.50, p < 0.001, Fig. 3F). Finally, as collagen deposition is generally a response to inflammatory stimuli, we investigated markers of inflammation, including Tgf-β and tumor necrosis factor alpha (Tnf-α). Tgf-β mRNA content progressively became greater with cancer progression (R2 = 0.19, p = 0.028, Fig. 3G). Additionally, Tnf-α mRNA content demonstrated a similar trend with Tnfα content linearly increasing with cachexia progression (R2 = 0.39, p = 0.002, Fig. 3H). Due to our findings of exponential increases in measures of mediators of collagen content as well as phenotypic alterations to collagen deposition, we next sought to investigate potential mechanisms contributing to the increases in these measures of collagen deposition and fibrosis.
Fig. 3.
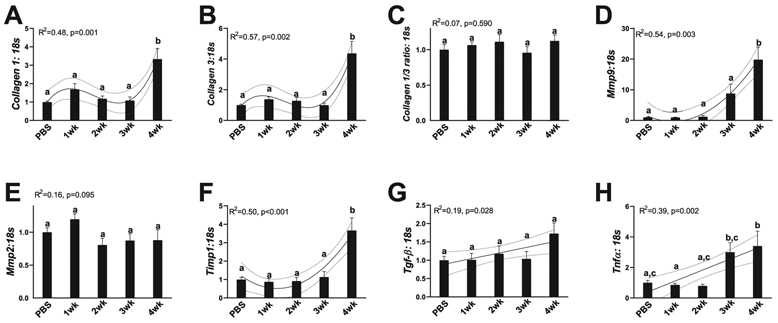
RNA content of markers of collagen deposition and breakdown. (A) Collagen 1 content. (B) Collagen 3 content. (C) Collagen 1/3 ratio. (D) Mmp9 content. (E) Mmp 2 content. (F) Timp1 content. (G) Tgf-β content; (H) Tnf-α content. All graphs are means ± SEM, n = 7–10/group for each analysis. Significant trends are plotted against means with 95% confidence intervals. 1wk, 2wk, 3wk, 4wk, 1, 2, 3, and 4 weeks of cancer progression, respectively; PBS, phosphate-buffered saline.
Cancer progression caused increases in Akt content and phosphorylation without dramatically influencing downstream measures of protein synthesis
The liver serves as one of the primary sites for whole-body protein metabolism, allowing for the synthesis of new amino acids from various substrates (Sunny et al. 2015). Patients with chronic liver disease exhibit significant alterations to protein and amino acid turnover, which appears to result in chronic protein deficiencies (Charlton 1996; Miwa and Moriwaki 2004) and changes in serum branched-chain amino acids have been associated with poorer prognosis of liver patients (Tajiri and Shimizu 2013). Therefore, alterations in hepatic protein turnover could have significant implications for maintenance of muscle mass in patients with cancer. We find Deptor mRNA content progressively decreased with cancer progression without any pairwise comparisons reaching statistical significance (R2 = 0.14, p = 0.023, Fig. 4A). Akt protein content and p-Aktser473 appeared to be linearly related to weeks of cancer progression, with 4-week animals having 2–7-fold more Akt or p-Akt, respectively (Akt: R2 = 0.43, p < 0.001, Fig. 4B and 4K; p-Aktser473: R2 = 0.28, p = 0.008, Fig. 4C and 4K). p-Aktser473/Akt ratio was altered quadratically in relation to cancer progression, with 1, 2, 3, and 4-week animals having 50%–60% less p-Aktser473/Akt compared with PBS animals. Downstream marker of protein synthesis p70, p-p70Thr389, and p-p70Thr389/p70 ratio did not change with cancer progression (p70: R2 = 0.12, p = 0.193, Fig. 4E and 4K; p-p70Thr389: R2 = 0.09, p = 0.129, Fig. 5F and 5K; p-p70Thr389/p70 ratio: R2 = 0.129, p = 0.647, Fig. 4G and 4K). 4E-BP1 protein content did not demonstrate alterations with cancer progression (R2 = 0.10, p = 0.211, Fig. 4H and 4K). p-4E-BP1Thr37/38 content did demonstrate a linear relationship with cancer progression, with 4-week animals having ~2.5-fold more p-4E-BP1Thr37/38 content compared with PBS animals (R2 = 0.21, p = 0.013, Fig. 4I and 4K). However, p-4E-BP1Thr37/38/4E-BP1 content did not demonstrate any differences or trends with cancer progression (R2 = 0.18, p = 0.210, Fig. 4J and 4K).
Fig. 4.

Markers of protein synthesis and inhibitor of protein synthesis Deptor. (A) RNA content of Deptor. (B) Protein content of Akt. (C) Protein content of p-AktSer473. (D) p-AktSer473/Akt ratio. (E) p70 protein content. (F) p-p70Thr389 protein content. (G) p70/p-p70Thr389 protein content. (H) 4EBP-1 protein content. (I) p-4EBP-1Thr37/46 protein content. (J) 4EBP-1/ p-4EBP-1Thr37/46 protein content. (K) Representative images for measured Western blot targets. All graphs are means ± SEM, n = 7–10/group for each analysis. Significant trends are plotted against means with 95% confidence intervals. 1wk, 2wk, 3wk, 4wk, 1, 2, 3, and 4 weeks of cancer progression, respectively; PBS, phosphate-buffered saline.
Fig. 5.

Markers of autophagy initiation, autophagsomal formation and resolution. (A) RNA content of Beclin. (B) RNA content of Atg7. (C) RNA content of Lc3. (D) Total LC3 protein. (E) LC3II/I ratio. (F) p62 RNA content. (G) p62 protein content. (H) Representative images for measured Western blot targets. All graphs are means ± SEM, n = 7–10/group for each analysis. Significant trends are plotted against means with 95% confidence intervals. 1wk, 2wk, 3wk, 4wk, 1, 2, 3, and 4 weeks of cancer progression, respectively; PBS, phosphate-buffered saline.
Cancer progression corresponded to decreases in autophagy machinery as well as decreased autophagy initiation and resolution
Prior works have strongly suggested the importance of autophagy for hepatic functionality, whereby decreased autophagy is typically associated with poorer outcomes (Baena et al. 2015; González-Rodríguez et al. 2014; Jiang et al. 2013). Beclin mRNA content did not demonstrate any relationship to cancer progression (R2 = 0.07, p = 0.212, Fig. 5A). Atg7 mRNA content had a linear relationship with cancer progression with 3- and 4-week animals having 30%–40% less Atg7 content compared with PBS animals (R2 = 0.31, p < 0.001, Fig. 5B). Lc3 mRNA content also demonstrated a linear decrease with cancer progression, with 4-week animals having ~40% less Lc3 content compared with PBS animals (R2 = 0.27, p = 0.008, Fig. 5C). However, total LC3 protein content did not appear affected by cancer progression (R2 = 0.02, p = 0.78, Fig. 5D and 5H). LC3II/I ratio, a surrogate measure of autophagosome formation (Klionsky et al. 2016), demonstrated a quadratic relationship with cancer progression with 1-, 2-, and 3-week animals having 40%–50% less LC3II/I (reaching statistical significance at 3 weeks), which then began to become greater in 4-week animals (R2 = 0.26, p = 0.044, Fig. 5E and 5H). p62 mRNA content did not differ across any cancer group (R2 = 0.10, p = 0.871, Fig. 5F); however, p62 protein content progressively became greater with longer durations of cancer progression (R2 = 0.24, p = 0.036; Fig. 5G and 5H).
Cancer progression altered mRNA content of mitochondrial biogenesis and mitophagy without altering total mitochondrial content
It is generally well accepted that mitochondrial health is a strong mediator of cellular and overall hepatic health (Grattagliano et al. 2011; Gusdon et al. 2014). Broadly, mitochondrial function, measured by efficiency of adenosine triphosphate production, can be influenced by mitochondrial quality control mechanisms, such as mitochondrial biogenesis and total content, the fusion of healthy mitochondria for more efficient energy production, and the fission and removal of unhealthy mitochondrial components for removal by autophagy (mitophagy) (Yan et al. 2012). Aberrations in any of these processes can influence mitochondrial function and lead to excessive reactive oxygen species (ROS) production and subsequent reductions in overall cellular health (Auger et al. 2015; Meyer et al. 2017). Prior work has shown that disruptions to mitochondrial health are hallmark features of many chronic hepatic diseases, such as nonalcoholic fatty liver disease (NAFLD) (García-Ruiz et al. 2013) and liver cirrhosis (Lane et al. 2016), and moderate aberrations to hepatic mitochondrial quality control markers can be seen in a pre-NAFLD state (Rosa-Caldwell et al. 2016). Therefore, disruptions to measures of mitochondrial quality control measures and associated metabolism may underlie liver pathologies during the development of cancer cachexia. Indeed, we find marked alterations in many measures of mitochondrial quality control mechanisms. A quadratic relationship between Cox4 mRNA content and cancer progression was noted, with a slight peak in Cox4 content in 2-week animals that then became progressively lower in 3- and 4-week animals (R2 = 0.34, p = 0.004, Fig. 6A). However, these differences in mRNA content did not translate to differences in COX-IV protein content (a surrogate marker of mitochondrial content), as no differences or trends were detected in COX-IV protein content (R2 = 0.09, p = 0.700, Fig. 6B and 6G). Pgc1α mRNA content demonstrated a gradual linear trend downward with cancer progression (R2 = 0.19, p = 0.018, Fig. 6C). Tfam mRNA content had a quadratic relationship with cancer progression, with 2-week animals having 50% greater Tfam mRNA content compared with PBS animals, which then progressively decreased in 3- and 4-week animals (R2 = 0.20, p = 0.013, Fig. 6D). A quadratic trend was also found between Bnip3 mRNA content and cancer progression, peaking in 2-week animals, having ~75% more Bnip3 content compared with PBS, then gradually becoming lower in 3- and 4-week animals back to PBS levels (R2 = 0.30, p = 0.003, Fig. 6E). In BNIP3 protein, there was a cubic relationship noted, with 3-week animals having ~2-fold more BNIP3 content compared with any other group (R2 = 0.39, p = 0.002, Fig. 6F and 6G).
Fig. 6.

Markers for mitochondrial turnover. (A) Cox4 RNA content. (B) COX-IV protein content. (C) Pgc-1α RNA content. (D) Tfam RNA content. (E) Bnip3 RNA content. (F) BNIP3 protein content. (G) Representative images for measured Western blot targets. All graphs are means ± SEM, n = 7–10/group for each analysis. Significant trends are plotted against means with 95% confidence intervals. 1wk, 2wk, 3wk, 4wk, 1, 2, 3, and 4 weeks of cancer progression, respectively; PBS, phosphate-buffered saline.
During cancer progression, mRNA content of mitochondrial fusion progressively became lower, while mRNA content of markers of mitochondrial fission demonstrated quadratic relationship with cancer progression
Mitochondrial dynamics refers to the fusion of the mitochondrial network for more efficient energy production and fission of unhealthy mitochondria from the mitochondrial network (Yan et al. 2012). Mitochondrial fusion is mediated by the protein Mfn1, Mfn2, and Opa1, whereas fission is mediated by Drp1, Mff, and Fis1 (Yan et al. 2012). These specific processes have been reviewed elsewhere, but broadly, increased mitochondrial fusion is related to a healthier cell phenotype and increased fission is related to an unhealthy cell phenotype (Yan et al. 2012). mRNA content of Mfn1 progressively became lower during cancer development, with 4-week animals having ~40% less Mfn1 mRNA content (R2 = 0.21, p = 0.031, Fig. 7A). Mfn2 mRNA content similarly became reduced with cancer progression, with 4-week animals also having ~40% less Mfn2 mRNA content (R2 = 0.31, p < 0.001, Fig. 7B). Opa1 mRNA content also had a linear decrease with cancer progression, with 3- and 4-week animals having ~50% and 55% less Opa1 mRNA content compared with PBS animals (R2 = 0.29, p < 0.001, Fig. 7C). Regarding mitochondrial fission, Drp1 mRNA content did not differ across any cancer timepoints (R2 = 0.13, p = 0.14, Fig. 7D). However, Mff and Fis1 both demonstrated quadratic relationships with cancer progression. With 2-week animals having ~25%–40% greater Mff and Fis1 mRNA content compared with PBS animals, which then progressively became lower in 3- and 4-week animals (Mff: R2 = 0.30, p = 0.004, Fig. 7E; Fis1: R2 = 0.39, p = 0.004, Fig. 7F).
Fig. 7.

Makers for mitochondrial dynamics. (A) RNA content of Mfn1. (B) RNA content of Mfn2. (C) RNA content of Opa1. (D) RNA content of Drp1. (E) RNA content of Mff.(F) RNA content of Fis1. All graphs are means ± SEM, n = 7–10/group for each analysis. Significant trends are plotted against means with 95% confidence intervals. 1wk, 2wk, 3wk, 4wk, 1, 2, 3, and 4 weeks of cancer progression, respectively; PBS, phosphate-buffered saline.
Discussion
To our knowledge, this is one of the early studies to time-course hepatic pathophysiological alterations during the development of cancer cachexia. We find hepatic hypertrophy with longer durations of cancer development that corresponds to increased hepatic fibrosis. This hepatic fibrosis also corresponds to alterations in markers of hepatic metabolism. Interestingly, we also note alterations in mitochondrial quality control mechanisms that preceded the development of hepatic hypertrophy and associated maladies. Taken together, these results suggest potential mitochondrial maladies preceding the development of hepatic fibrosis and growth and warrant further investigation on hepatic alterations during the development of cancer cachexia.
Cancer cachexia development and progression corresponds to alterations in markers of fatty acid and glucose metabolism
The tissue weights from these animals has been previously reported with animals showing reductions in muscle size and cross-sectional area after 3–4 weeks of cancer development (Brown et al. 2018; Brown et al. 2017a). In the current study, we find 4 weeks of cancer development results in hepatic hypertrophy, which is similar to prior works using a colorectal cancer model (Narsale et al. 2015). As the liver is one of the primary regulators of whole-body metabolism, we began by first investigating markers of fatty acid and glucose metabolism. Oil Red O did not detect alterations in overall fat storage; however, because of the catabolic stimuli associated with the experimental design, it is likely that our method was not sensitive to detect fat losses in an atrophic model. However, we find marked alterations in various regulators of fatty acid metabolism and cholesterol synthesis. With a progressive decrease in Scd1 and Srebp1 and an increase in HMG CoA reductase mRNA content with cancer progression. These results suggest progressive aberrations to hepatic fatty acid mobilization and utilization and complements prior works in the C26 model finding lowered levels of these fatty acid regulators in cachectic mice (Jones et al. 2013). As the liver is one of the key organs for fatty acid metabolism and the primary site of fatty acid synthesis, these results imply hepatic alterations could contribute to energy deficits associated with cancer cachexia. More so, due to reduced fatty acid mobilization, and subsequent reduced circulating lipids (Jones et al. 2013), it is plausible the liver may then shift to more readily available energy sources, such as glucose. Indeed, we find marked depletion in hepatic glycogen content, suggesting a shift towards a more glycolytic phenotype. This aligns with prior work finding shifts in hepatic metabolism towards a more glycolytic phenotype using an ApcMin/+ model of colorectal cancer (Narsale et al. 2015) and potentially suggests a conserved mechanism across different cancer types for hepatic alterations. Additionally, increased cholesterol production has been linked to excessive cholesterol accumulation in the mitochondrial membrane and decrease in mitochondrial function (Garcia-Ruiz et al. 2017; Martin et al. 2016). As such, our results, may also suggest excessive cholesterol synthesis that may contribute to hepatic mitochondrial deficits. However, more direct measures of cholesterol accumulation and mitochondrial function in relation to cancer progression are necessary to fully understand these relationships.
Cancer progression corresponds to increases in hepatic fibrosis, despite greater content of collagen degradative genes
We noted large increases in mRNA content of collagen 1 and collagen 3 as well as subsequent increase in deposition of collagen in the liver as measured by Sirius Red staining. These data demonstrate a clear fibrotic phenotype with the progression of cancer-cachexia, which is similar to works found in human models (Judge et al. 2018; Pinter et al. 2016). More so, this collagen deposition appears to occur concurrently with increased mRNA content of genes responsible for remodeling ECM, Mmp9 and Timp1, suggestive of degradation of ECM regulatory system. Collagen deposition is predominantly facilitated by hepatic stellate cells (HSCs), which account for 5%–8% of liver cells (Yin et al. 2013). During liver injury, HSCs activate to accumulate hepatic ECM as a protective mechanism from further damage (Puche et al. 2013). However, with continuous trauma, increased HSC activation causes pathological fibrosis and sinusoidal contraction with a concurrent disruption in hepatic endothelial function and blood flow (Mallat 1998; Zhang et al. 1994, 1995). These alterations in vasculature integrity can lead to decreased oxygen transport within the liver and have direct impacts on nutrient exchange and metabolism (Marra and Pinzani 2002). For example, advanced liver fibrosis is known to increase energy demand of the tissue (Ganapathy-Kanniappan et al. 2014), which could intensify the energy deficiency characteristic of cancer cachexia. Additionally, recent works have tied the alterations in muscle metabolism to collagen deposition in muscle (Graae et al. 2019). As such metabolic alterations may have largely contributed to the hepatic fibrosis noted in our works; therefore, based on our findings regarding hepatic fibrosis, we further investigated hepatic cellular processes that plausibly could contribute to the development of this fibrotic phenotype.
Cancer cachexia progression had limited impacts on markers of protein synthesis and degradation
Overall, the progression of cancer cachexia did not appear to influence hepatic protein synthesis but may have influenced autophagic degradation of proteins. While we noted a small decrease in Deptor mRNA content and a decrease in p-Aktser473/Akt ratios, these relationships did not appear to hold to further downstream markers of protein synthesis such p-p70Thr389/p70 or p4EBP-1Thr37/46/EBP1 ratios, which overall suggest negligible alterations to the cellular signaling mechanisms of protein synthesis. While increased p-Aktser473 and Akt can be suggestive of increased protein synthetic signaling, Akt serves as a major signaler for many metabolic processes including glycogen synthesis and storage (Morales-Ruiz et al. 2017). Taken in aggregate with our histological data, the linear increase in Akt and p-Aktser473 content may suggest an attempted protective mechanism against glycogen depletion, whereby Akt content and activity increases to meet energetic demands of tumor growth. However, the increase in tumor development and energy expenditure likely outpaces the increases in Akt activation, as despite increased Akt and p-Aktser473 at 4 weeks of tumor development, we still see lowered hepatic glycogen content. However, we did not directly measure energy expenditure of these mice in relation to tumor development, as such this hypothesis requires further evaluation.
Additionally, we found a slight decrease in autophagic capacity as well as a decrease in autophagy resolution measured by Lc3 mRNA, LC3II/I ratio, and p62 protein content. As autophagy is well known to be dysregulated in many forms of liver disease (Lavallard and Gual 2014; Levine and Kroemer 2008; Werling 2011), this suggests a progressive decrease in autophagy activation and resolution with the progression of cancer cachexia, which likely contributes to decreased hepatic health. Importantly, we observed an increase in p62 protein content, a surrogate measure of autophagy resolution where increased p62 in the absence of lysosomal inhibition is indicative of impaired autophagy resolution (Klionsky et al. 2016), without any change in p62 mRNA content. This accumulation of p62 protein suggests an impediment in the autophagy resolution process. However, based on the current study it is difficult to directly delineate how these aberrations in autophagy directly influence hepatic function and warrant further investigation.
Aberrations of mitochondrial quality control occurred during the progression of cachexia, specifically alterations to mitochondrial dynamics preceded hepatic pathologies
Of note, we observed multiple alterations in makers of mitochondrial quality control. Pgc-1α, the master regulator of mitochondrial biogenesis, exhibited a small but steady decrease in mRNA content throughout the duration of cancer progression. However, this was not accompanied by a decrease of mitochondrial content, measured by COX-IV protein. Interestingly, COX-IV protein did not change despite a quadratic relationship between cancer progression and Cox4 mRNA content, with a peak at 2 weeks of cancer progression suggesting alterations to hepatic Cox4 transcript through the development of cancer. This may be partially accounted for by increases in Bnip3 mRNA and protein content at 2 weeks and 3 weeks of cancer development, respectively, which may indicate enhanced autophagic removal of mitochondria. Of note, multiple measures of mitochondrial quality control demonstrated this quadratic relationship, including Cox4 mRNA, Tfam mRNA, Bnip3 mRNA, Mff mRNA, and Fis1 mRNA. Phenotypically, we also noted this same relationship in SDH stain intensity. Taken together these aberrations at 2 weeks strongly suggest significant metabolic and mitochondrial shifts occurring before significant phenotypic changes in liver size or fibrotic phenotype. There are multiple possible interpretations for this finding. First, the increase in mitochondrial fission markers and mitophagy may be a compensatory response to inflammatory or pathological stimuli signaling coming from within the liver or from other tissues. This increase in mitophagic signaling may serve as a protective mechanism to remove dysfunctional mitochondrial components to preserve mitochondrial function. Prior work has demonstrated mitophagy may serve as a protective mechanism for mitochondrial function (Glick et al. 2012); therefore, this increase in mitophagy and fission markers may be a protective mechanism against cancer-associated pathologies. However, an alternative interpretation is that the increase in mitophagic signaling in conjunction with the progressive decrease in mitochondrial fusion and biogenesis may be indicative of an already degenerating mitochondrial network and a predecessor to hepatic pathologies such as fibrosis. This finding would be in agreement with our prior works finding mitochondrial degeneration preceding significant muscle pathologies in this model (Brown et al. 2017a). More so, mitochondrial function has been strongly related to liver injury and subsequent fibrosis (Sokol et al. 2001), whereby alterations to mitochondrial morphology cause a fibrotic phenotype (Kang et al. 2016). This aberration in the mitochondrial network then potentiates increased ROS formation, facilitating fibrosis (Vilaseca et al. 2017). Prior work has found increased hepatic mitochondrial fission elicits hepatocyte apoptosis while decreasing fission is protective against hepatocyte death (Frank et al. 2001; Lee et al. 2004; Yu et al. 2014). As such, it is plausible that increased fission at 2 weeks may be the first initiating signal in hepatic pathologies during the progression of cancer cachexia. Importantly, we note increases in mitochondrial fission and mitophagy precede the increase in collagen mRNA content and collagen deposition. Although not directly causative, this appears to suggest a temporal relationship between increased alterations in mitochondrial dynamics, specifically mitochondrial fission, mediating the development of a fibrotic phenotype during the progression of cancer-cachexia. We have previously demonstrated muscle mitochondrial function and quality can begin to deteriorate at 1–2 weeks of cancer development in the muscle (Brown et al. 2017a) and recent works have demonstrated strong interplay between muscle metabolism and muscle collagen accumulation (Graae et al. 2019). As such it is plausible that mitochondrial degeneration may be the first initiating step in the development of cancer-associated pathologies in other tissues. It should be noted that we did not directly measure mitochondrial function or directly measure the process of mitochondrial fission or mitophagy in these livers. Therefore, more work investigating early stages of cancer development are needed to more thoroughly validate the role of mitochondrial function and fission in the progression of hepatic pathologies during the progression of cancer.
Trend analysis as a novel tool for time-course experimental designs
To our knowledge, this is also one of the first studies in biomedical sciences to utilize trend analysis as a statistical modeling tool for the analysis of data. As the field of biomedical sciences advances, so to do statistical techniques necessary to appropriately model the experimental design. As noted, many markers of hepatic physiology are altered prior to the development of hepatic hypertrophy and fibrosis. Therefore, more studies will likely be needed utilizing a time-course based experimental design to more thoroughly elucidate these time-course aberrations. Trend modeling statistics allows for investigating overall patterns within the data that normal ANOVAs with pairwise comparisons are unable and not designed to detect. Therefore, moving forward, the use of these statistical techniques may allow new insight to data interpretation in future research utilizing similar time-course-based experimental designs.
In conclusion, to our knowledge, this is the first study to directly investigate time-course alterations in hepatic physiology during the development and progression of cancer-cachexia. Interestingly, we have observed the development of hepatic fibrosis across cancer development. We find that multiple aspects of mitochondrial quality control, specifically fission and mitophagy, are then altered before the development of the hepatic fibrotic phenotype. Overall, these data potentially suggest mitochondrial mechanisms may underlie liver pathologies during the development and progression of cancer cachexia and these aberrations in mitochondrial quality control may in fact occur before any noticeable hepatic damage. Taken together, this strongly suggests the need for more research into early interventions for the treatment of hepatic cancer-cachexia associated maladies prior to the onset of fibrosis.
Acknowledgements
The authors would like to thank Ms. Sarah Ramey and Mr. Kyle Turner for their assistance with microscopy analysis for this project. Additionally, the authors would also like to thank the fantastic students, faculty, and staff at the University of Arkansas Exercise Science Research Center for their continued administrative support. This study was funded by the National Institutes of Health, award number R15AR069913/AR/NIAMS, as well as the Arkansas Biosciences Institute.
Footnotes
Conflict of interest statement
The authors have no conflicts of interest to report.
Contributor Information
Megan E. Rosa-Caldwell, Integrative Muscle Metabolism Laboratory, Exercise Science Research Center, University of Arkansas, Fayetteville, Arkansas 72701, USA.
Jacob L. Brown, Integrative Muscle Metabolism Laboratory, Exercise Science Research Center, University of Arkansas, Fayetteville, Arkansas 72701, USA.
David E. Lee, Integrative Muscle Metabolism Laboratory, Exercise Science Research Center, University of Arkansas, Fayetteville, Arkansas 72701, USA.
Michael P. Wiggs, Department of Health and Kinesiology, University of Texas at Tyler, Tyler, TX 75799, USA.
Richard A. Perry, Jr., Exercise Muscle Biology Laboratory, Exercise Science Research Center, University of Arkansas, Fayetteville, Arkansas, USA.
Wesley S. Haynie, Exercise Muscle Biology Laboratory, Exercise Science Research Center, University of Arkansas, Fayetteville, Arkansas, USA.
Aaron R. Caldwell, Exercise Science Research Center, Department of Health, Human Performance, and Recreation, University of Arkansas, Fayetteville, Arkansas, USA.
Tyrone A. Washington, Exercise Muscle Biology Laboratory, Exercise Science Research Center, University of Arkansas, Fayetteville, Arkansas, USA.
Wen-Juo Lo, Department of Rehabilitation, Human Resources, and Communication Disorders, University of Arkansas, Fayetteville, Arkansas, USA..
Nicholas P. Greene, Integrative Muscle Metabolism Laboratory, Exercise Science Research Center, University of Arkansas, Fayetteville, Arkansas 72701, USA.
References
- Argilés JM, Stemmler B, López-Soriano FJ, and Busquets S 2015. Nonmuscle tissues contribution to cancer cachexia. Mediators Inflamm. 2015: 182872. doi: 10.1155/2015/182872. PMID:26523094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger C, Alhasawi A, Contavadoo M, and Appanna VD 2015. Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front. Cell Dev. Biol 3: 40. doi: 10.3389/fcell.2015.00040. PMID:26161384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baena M, Sanguesa G, Hutter N, Sánchez RM, Roglans N, Laguna JC, and Alegret M 2015. Fructose supplementation impairs rat liver autophagy through mTORC activation without inducing endoplasmic reticulum stress. Biochim. Biophys. Acta, 1851(2): 107–116. doi: 10.1016/j.bbalip.2014.11.003. PMID:25463011. [DOI] [PubMed] [Google Scholar]
- Bataller R, and Brenner DA. 2005. Liver fibrosis. J. Clin. Invest 115(2): 209–218. doi: 10.1172/JCI24282. PMID:15690074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennani-Baiti N, and Walsh D 2009. What is cancer anorexia-cachexia syndrome? A historical perspective. J. R. Coll. Physicians Edinb 39(3): 257–262. PMID:20608345. [PubMed] [Google Scholar]
- Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA., et al. 2017a. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle, 8(6): 926–938. doi: 10.1002/jcsm.12232. PMID:28845591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Rosa-Caldwell ME, Lee DE, Brown LA, Perry RA, Shimkus KL, et al. 2017b. PGC-1alpha4 gene expression is suppressed by the IL-6-MEK-ERK 1/2 MAPK signalling axis and altered by resistance exercise, obesity and muscle injury. Acta Physiol. (Oxf) 220(2): 275–288. doi: 10.1111/apha.12826. PMID: 27809412. [DOI] [PubMed] [Google Scholar]
- Brown JL, Lee DE, Rosa-Caldwell ME, Brown LA, Perry RA, Haynie WS, et al. 2018. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J. Cachexia Sarcopenia Muscle, 9(5): 987–1002. doi: 10.1002/jcsm.12354. PMID:30328290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC/National Center for Health Statistics. 2015. Leading Causes of Death. Available from https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm. U.S. Department of Health & Human Services. [Google Scholar]
- Charlton MR 1996. Protein metabolism and liver disease. Baillieres Clin. Endocrinol. Metab 10(4): 617–635. doi: 10.1016/S0950-351X(96)80771-3. PMID: 9022955. [DOI] [PubMed] [Google Scholar]
- Choi E, Carruthers K, Zhang L, Thomas N, Battaglino RA, Morse LR, and Widrick JJ 2013. Concurrent muscle and bone deterioration in a murine model of cancer cachexia. Physiol. Rep 1(6): e00144. doi: 10.1002/phy2.144. PMID:24400146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der-Torossian H, Wysong A, Shadfar S, Willis MS, McDunn J, and Couch ME 2013. Metabolic derangements in the gastrocnemius and the effect of Compound A therapy in a murine model of cancer cachexia. J. Cachexia Sarcopenia Muscle, 4(2): 145–155. doi: 10.1007/s13539-012-0101-7. PMID:23344889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. 2001. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell, 1(4): 515–525. doi: 10.1016/S1534-5807(01)00055-7. PMID:11703942. [DOI] [PubMed] [Google Scholar]
- Ganapathy-Kanniappan S, Karthikeyan S, Geschwind JF, and Mezey E 2014. Is the pathway of energy metabolism modified in advanced cirrhosis? J. Hepatol 61(2): 452. doi: 10.1016/j.jhep.2014.04.017. PMID:24810232. [DOI] [PubMed] [Google Scholar]
- García-Ruiz C, Baulies A, Mari M, García-Rovés PM, and Fernandez-Checa JC 2013. Mitochondrial dysfunction in non-alcoholic fatty liver disease and insulin resistance: cause or consequence? Free Radic. Res 47(11): 854–868. doi: 10.3109/10715762.2013.830717. PMID:23915028. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz C, Ribas V, Baulies A, and Fernandez-Checa JC 2017. Mitochondrial cholesterol and the paradox in cell death. Handb. Exp. Pharmacol 240: 189–210. doi: 10.1007/164_2016_110. PMID:28035533. [DOI] [PubMed] [Google Scholar]
- Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, et al. 2012. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol. Cell. Biol 32(13): 2570–2584. doi: 10.1128/MCB.00167-12. PMID: 22547685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. 2014. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 5: e1179. doi: 10.1038/cddis.2014.162. PMID:24743734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graae AS, Grarup N, Ribel-Madsen R, Lystbaek SH, Boesgaard T, Staiger H, et al. 2019. ADAMTS9 regulates skeletal muscle insulin sensitivity through extracellular matrix alterations. Diabetes, 68(3): 502–514. doi: 10.2337/db18-0418. PMID:30626608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grattagliano I, Russmann S, Diogo C, Bonfrate L, Oliveira PJ, Wang DQ, and Portincasa P 2011. Mitochondria in chronic liver disease. Curr. Drug Targets, 12(6): 879–893. doi: 10.2174/138945011795528877. PMID:21269263. [DOI] [PubMed] [Google Scholar]
- Greene NP, Lee DE, Brown JL, Rosa ME, Brown LA, Perry RA., et al. 2015. Mitochondrial quality control, driven by PGC-1α, is dysregulated by Western Diet-induced obesity and partially restored by moderate physical activity in mice. Physiol. Rep 3: e12470. doi: 10.14814/phy2.12470. PMID:26177961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusdon AM, Song KX, and Qu S 2014. Nonalcoholic Fatty liver disease: pathogenesis and therapeutics from a mitochondria-centric perspective. Oxid. Med. Cell Longev 2014: 637027. doi: 10.1155/2014/637027. PMID: 25371775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser CA, Stockler MR, and Tattersall MH 2006. Prognostic factors in patients with recently diagnosed incurable cancer: a systematic review. Support. Care Cancer, 14(10): 999–1011. doi: 10.1007/s00520-006-0079-9. PMID: 16708213. [DOI] [PubMed] [Google Scholar]
- Iwata Y, Suzuki N, Ohtake H, Kamauchi S, Hashimoto N, Kiyono T, and Wakabayashi S 2016. Cancer cachexia causes skeletal muscle damage via transient receptor potential vanilloid 2-independent mechanisms, unlike muscular dystrophy. J. Cachexia Sarcopenia Muscle, 7(3): 366–376. doi: 10.1002/jcsm.12067. PMID:27239414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Huang Z, Zhao H, and Wei T 2013. Hydrogen peroxide impairs autophagic flux in a cell model of nonalcoholic fatty liver disease. Biochem. Biophys Res. Commun 433(4): 408–414. doi: 10.1016/j.bbrc.2013.02.118. PMID: 23537653. [DOI] [PubMed] [Google Scholar]
- Jones A, Friedrich K, Rohm M, Schafer M, Algire C, Kulozik P, et al. 2013. TSC22D4 is a molecular output of hepatic wasting metabolism. EMBO Mol. Med 5(2): 294–308. doi: 10.1002/emmm.201201869. PMID:23307490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge SM, Nosacka RL, Delitto D, Gerber MH, Cameron ME, Trevino JG, and Judge AR 2018. Skeletal muscle fibrosis in pancreatic cancer patients with respect to survival. JNCI Cancer Spectr. 2(3): pky043. doi: 10.1093/jncics/pky043. PMID:30637373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JW, Hong JM, and Lee SM 2016. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J. Pineal. Res 60(4): 383–393. doi: 10.1111/jpi.12319. PMID:26882442. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. 2016. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy, 12(1): 1–222. doi: 10.1080/15548627.2015.1100356. PMID:26799652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane M, Boczonadi V, Bachtari S, Gomez-Duran A, Langer T, Griffiths A, et al. 2016. Mitochondrial dysfunction in liver failure requiring transplantation. J. Inherit. Metab. Dis 39(3): 427–436. doi: 10.1007/s10545-016-9927-z. PMID:27053192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavallard VJ, and Gual P 2014. Autophagy and non-alcoholic fatty liver disease. Biomed. Res. Int 2014: 120179. doi: 10.1155/2014/120179. PMID:25295245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, and Youle RJ 2004. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell, 15(11): 5001–5011. doi: 10.1091/mbc.e04-04-0294. PMID:15356267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, and Kroemer G 2008. Autophagy in the pathogenesis of disease. Cell, 132(1): 27–42. doi: 10.1016/j.cell.2007.12.018. PMID:18191218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallat A 1998. Hepatic stellate cells and intrahepatic modulation of portal pressure. Digestion, 59(4): 416–419. doi: 10.1159/000007501. PMID:9693221. [DOI] [PubMed] [Google Scholar]
- Marra F, and Pinzani M 2002. Role of hepatic stellate cells in the pathogenesis of portal hypertension. Nefrologia, 22(Suppl 5): 34–40. PMID:12107915. [PubMed] [Google Scholar]
- Martin LA, Kennedy BE, and Karten B 2016. Mitochondrial cholesterol: mechanisms of import and effects on mitochondrial function. J. Bioenerg. Biomembr 48(2): 137–151. doi: 10.1007/s10863-014-9592-6. PMID:25425472. [DOI] [PubMed] [Google Scholar]
- Maxwell SE, and Delany HD 2004. Designing experiments and analyzing data: a model comparison perspective (2nd ed.). Psychology Press Taylor and Francis Group, New York, N.Y., USA. [Google Scholar]
- Meyer JN, Leuthner TC, and Luz AL 2017. Mitochondrial fusion, fission, and mitochondrial toxicity Submitted for consideration for the Special Issue of Toxicology on “Chemical Mitochondrial Toxicity”. Toxicology, 391: 42–53. doi: 10.1016/j.tox.2017.07.019. PMID:28789970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa Y, and Moriwaki H 2004. Nocturnal energy and BCAA supplementation in patients with liver cirrhosis. Hepatol. Res 30s: 63–66. doi: 10.1016/j.hepres.2004.08.012. PMID:15607141. [DOI] [PubMed] [Google Scholar]
- Morales-Ruiz M, Santel A, Ribera J, and Jimenez W 2017. The role of Akt in chronic liver disease and liver regeneration. Semin. Liver Dis 37(1): 11–16. doi: 10.1055/s-0036-1597819. PMID:28201844. [DOI] [PubMed] [Google Scholar]
- Narsale AA, Enos RT, Puppa MJ, Chatterjee S, Murphy EA, Fayad R, et al. 2015. Liver inflammation and metabolic signaling in ApcMin/+ mice: the role of cachexia progression. PLoS One, 10(3): e0119888. doi: 10.1371/journal.pone.0119888. PMID:25789991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narsale AA, Puppa MJ, Hardee JP, VanderVeen BN, Enos RT, Murphy EA, and Carson JA 2016. Short-term pyrrolidine dithiocarbamate administration attenuates cachexia-induced alterations to muscle and liver in ApcMin/+ mice. Oncotarget, 7(37): 59482–59502. doi: 10.18632/oncotarget.10699. PMID: 27449092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruzzelli M, and Wagner EF 2016. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 30(5): 489–501. doi: 10.1101/gad.276733.115. PMID:26944676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter M, Trauner M, Peck-Radosavljevic M, and Sieghart W 2016. Cancer and liver cirrhosis: implications on prognosis and management. ESMO. Open, 1(2): e000042. doi: 10.1136/esmoopen-2016-000042. PMID:27843598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado CM, Baracos VE, McCargar LJ, Mourtzakis M, Mulder KE, Reiman T, et al. 2007. Body composition as an independent determinant of 5-fluorouracil-based chemotherapy toxicity. Clin. Cancer Res 13(11): 3264–3268. doi: 10.1158/1078-0432.CCR-06-3067. PMID:17545532. [DOI] [PubMed] [Google Scholar]
- Puche JE, Saiman Y, and Friedman SL 2013. Hepatic stellate cells and liver fibrosis. Compr. Physiol 3(4): 1473–1492. doi: 10.1002/cphy.c120035. PMID: 24265236. [DOI] [PubMed] [Google Scholar]
- Rosa-Caldwell ME, Lee DE, Brown JL, Brown LA, Perry JRA, Greene ES, et al. 2016. Moderate physical activity promotes basal hepatic autophagy in diet-induced obese mice. Appl. Physiol. Nutr. Metab 42(2): 148–156. doi: 10.1139/apnm-2016-0280. PMID:28084795. [DOI] [PubMed] [Google Scholar]
- Rosa-Caldwell ME, Brown JL, Lee DE, Blackwell TA, Turner KW, Brown LA, et al. 2017. Autophagy activation, not peroxisome proliferator-activated receptor gamma coactivator 1alpha, may mediate exercise-induced improvements in glucose handling during diet-induced obesity. Exp. Physiol 102(9): 1194–1207. doi: 10.1113/EP086406. PMID:28639297. [DOI] [PubMed] [Google Scholar]
- Ryan AM, Power DG, Daly L, Cushen SJ, Ni Bhuachalla E, and Prado CM 2016. Cancer-associated malnutrition, cachexia and sarcopenia: the skeleton in the hospital closet 40 years later. Proc. Nutr. Soc 75(2): 199–211. doi: 10.1017/S002966511500419X. PMID:26786393. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Straka MS, Dahl R, Devereaux MW, Yerushalmi B, Gumpricht E, et al. 2001. Role of oxidant stress in the permeability transition induced in rat hepatic mitochondria by hydrophobic bile acids. Pediatr. Res 49(4): 519–531. doi: 10.1203/00006450-200104000-00014. PMID:11264436. [DOI] [PubMed] [Google Scholar]
- Sunny NE, Kalavalapalli S, Bril F, Garrett TJ, Nautiyal M, Mathew JT, et al. 2015. Cross-talk between branched-chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab 309(4): E311–E319. doi: 10.1152/ajpendo.00161.2015. PMID: 26058864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajiri K, and Shimizu Y 2013. Branched-chain amino acids in liver diseases. World J. Gastroenterol 19(43): 7620–7629. doi: 10.3748/wjg.v19.i43.7620. PMID:24282351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vledder MG, Levolger S, Ayez N, Verhoef C, Tran TC, and Ijzermans JN 2012. Body composition and outcome in patients undergoing resection of colorectal liver metastases. Br. J. Surg 99(4): 550–557. doi: 10.1002/bjs.7823. PMID:22246799. [DOI] [PubMed] [Google Scholar]
- Vilaseca M, Garcia-Caldero H, Lafoz E, Ruart M, Lopez-Sanjurjo CI, Murphy MP, et al. 2017. Mitochondria-targeted antioxidant mitoquinone deactivates human and rat hepatic stellate cells and reduces portal hypertension in cirrhotic rats. Liver Int. 37(7): 1002–1012. doi: 10.1111/liv.13436. PMID: 28371136. [DOI] [PubMed] [Google Scholar]
- Wallengren O, Lundholm K, and Bosaeus I 2013. Diagnostic criteria of cancer cachexia: relation to quality of life, exercise capacity and survival in unselected palliative care patients. Support Care Cancer, 21(6): 1569–1577. doi: 10.1007/s00520-012-1697-z. PMID:23314651. [DOI] [PubMed] [Google Scholar]
- Wang H, Lai YJ, Chan YL, Li TL, and Wu CJ 2011. Epigallocatechin-3-gallate effectively attenuates skeletal muscle atrophy caused by cancer cachexia. Cancer Lett. 305(1): 40–49. doi: 10.1016/j.canlet.2011.02.023. PMID: 21397390. [DOI] [PubMed] [Google Scholar]
- Werling K 2011. Role of autophagy in the pathogenesis of liver diseases. Orv. Hetil 152(49): 1955–1961. doi: 10.1556/OH.2011.29269. PMID:22106162. [DOI] [PubMed] [Google Scholar]
- Yan Z, Lira VA, and Greene NP 2012. Exercise training-induced regulation of mitochondrial quality. Exerc. Sport. Sci. Rev 40(3): 159–164. doi: 10.1097/JES.0b013e3182575599. PMID:22732425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Zhao J, Hao J, Li B, Huo Y, Han Y, et al. 2018. Serum and urine metabolomics study reveals a distinct diagnostic model for cancer cachexia. J. Cachexia Sarcopenia Muscle, 9(1): 71–85. doi: 10.1002/jcsm.12246. PMID: 29152916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C, Evason KJ, Asahina K, and Stainier DY 2013. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Invest 123(5): 1902–1910. doi: 10.1172/JCI66369. PMID:23635788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Wang L, Lee H, O’Brien DK, Bronk SF, Gores GJ, and Yoon Y 2014. Decreasing mitochondrial fission prevents cholestatic liver injury. J. Biol. Chem 289(49): 34074–34088. doi: 10.1074/jbc.M114.588616. PMID:25342755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JX, Pegoli W, and Clemens MG 1994. Endothelin-1 induces direct constriction of hepatic sinusoids. Am. J. Physiol 266(4 Pt. 1): G624–G632. doi: 10.1152/ajpgi.1994.266.4.G624. PMID:8179001. [DOI] [PubMed] [Google Scholar]
- Zhang JX, Bauer M, and Clemens MG 1995. Vessel- and target cell-specific actions of endothelin-1 and endothelin-3 in rat liver. Am. J. Physiol 269(2 Pt. 1): G269–G277. doi: 10.1152/ajpgi.1995.269.2.G269. PMID:7653568. [DOI] [PubMed] [Google Scholar]