

Abstract
This phase I trial sought to determine a biologically safe and effective dose of AR-42, a novel histone deacetylase inhibitor, which would lead to a doubling of miR-29b prior to decitabine administration. Thirteen patients with previously untreated or relapsed/refractory AML were treated at 3 dose levels (DL): AR-42 20 mg qd on d1,3,5 in DL1, 40 mg qd on d1,3,5 in DL2 and 40 mg qd on d1,3,4,5 in DL3. Patients received decitabine 20 mg/m2 on d6–15 of each induction cycle and 20 mg/m2 on d6–10 of each maintenance cycle. One DLT of polymicrobial sepsis and multi-organ failure occurred at DL3. Two patients achieved a CRi and one patient achieved a CR for an ORR of 23.1%. The higher risk features of this patient population and the dosing schedule of AR-42 may have led to the observed clinical response and failure to meet the biologic endpoint.
Keywords: Acute myeloid leukemia, HDAC inhibitor, AR-42, REC-2282, decitabine, miR-29b
Introduction
Acute myeloid leukemia (AML) is a biologically heterogeneous hematologic malignancy characterized by aberrant blast proliferation, peripheral blood cytopenias, increased risk for infection and bleeding complications. Despite an improved understanding of disease pathogenesis, patients continue to experience inferior disease free (DFS) and overall survival (OS) rates [1]. With the approval of agents that target recurrent molecular mutations, there are now alternatives to cytotoxic chemotherapy for both de novo and relapsed AML. Unfortunately, not all patients possess a targetable mutation and it is for this subset of patients that additional therapies are needed.
Pharmacologically reversible epigenetic alterations due to DNA methylation and/or histone acetylation are believed to play a role in leukemogenesis and have led to the use of both DNA methyltransferase (DNMT) inhibitors and histone deacetylase inhibitors (HDACis), alone or in combination [2]. The hypomethylating agents (HMAs), decitabine and azacitidine, are now considered standard lower intensity therapies for patients with newly diagnosed AML older than 60 years [3]. Given the modest activity of HDACis as single agents in AML, recent trials have combined these agents with cytotoxic chemotherapy or HMAs, concurrently or sequentially, with variable response [4,5].
HDACis currently available for clinical use or in development vary with regard to potency, isoenzyme specificity, and effects on acetylation of non-histone substrates [6-8]. One possible explanation for the lesser potency observed in some HDACis is the inability to access the active pocket and chelate the zinc cation. Based on this hypothesis, a new class of HDACis was designed and synthesized that links hydroxamate to short chain acids [7]. One of these new compounds, AR-42, exhibits high potency with an IC50 at the submicromolar level in cancer cells [7]. In addition to inducing cell cycle arrest at both S and G2/M phases and dose dependent apoptosis in AML cell lines, sequential administration of AR-42 followed by decitabine resulted in stronger anti-leukemic activity compared to either drug alone [9]. Furthermore, AR-42 was found to increase miR-29b transcription in vitro, likely due to inhibition of the Sp1/NFkB transactivation complex that plays a role in DNMT and tyrosine kinase expression [10]. Indeed, the Sp1/NFkB transactivation complex physically interacts with HDAC1 and HDAC3 to repress miR-29b expression.
We hypothesized that HDAC inhibition with AR-42 would induce upregulation of expression of miR-29b and increase response to decitabine given the observation that pretreatment levels of miR-29b may be associated with clinical response [9,11]. Herein we tested the feasibility of increasing endogenous expression of miR-29b via treatment with AR-42 followed by decitabine. Specifically, we sought to determine a biologically effective and tolerable dose (BED) of AR-42 in combination with a 10-day schedule of decitabine in patients with newly diagnosed or relapsed/refractory AML that would lead to a doubling of miR-29b levels prior to the administration of decitabine.
Materials and methods
Study design and patient enrollment
Adult patients age ≥ 18 years with relapsed or refractory AML or age ≥ 60 years with previously untreated AML who were not candidates for or refused standard/conventional induction chemotherapy were eligible. Patients who received decitabine or 5-azacitidine as prior treatment for myelodysplastic syndrome (MDS) or AML were eligible unless treatment occurred within 3 months of study entry. Patients with secondary or therapy related AML were also eligible. The studies described herein were performed in accordance with the World Medical Association Declaration of Helsinki. Informed written consent approved by The Ohio State University Human Studies Committee was obtained prior to study entry. This trial was registered with the NCI clinical trials network (NCT01798901).
Patients were required to have a total bilirubin <2.0 mg/dL, creatinine <2.0 mg/dL, ALT/AST <2.5×upper limit of normal, NYHA CHF Class II or better, Eastern Cooperative Oncology Group performance status ≥2. Patients with a prior diagnosis of a prolonged QT syndrome or mean QTc > 450 msec in males and > 470 ms in females were excluded.
To determine an optimal biologic and tolerable dose, dose escalation was driven by the incidence of dose limiting toxicities (DLTs) and at least a 100% increase in mature miR-29b or pri-miR-29b expression in bone marrow or peripheral blood on day 5 of treatment during cycle 1 over baseline. We utilized a standard rule-based phase I trial design that simultaneously evaluates tolerability and evidence of biologic activity at each dose level. Specifically, this design combined the standard cohorts of 3 assessment of a maximum tolerated dose (MTD) based on DLT with the optimal biological dose-finding design proposed by Hunsberger [12] using a similar rule-based design with 3–6 patients per dose level. If <2 of 6 patients enrolled to a dose level have DLTs, and if 5 or more out of 6 patients achieve biologic activity, that dose would be declared the recommended dose for the Phase II trial.
Patients enrolled to dose level 1 (DL1) received AR-42 20 mg daily on days 1, 3, 5, DL2 received AR-42 40 mg daily on days 1,3,5, and DL3 received AR-42 40 mg daily on days 1, 3, 4, 5. Decitabine was administered 20 mg/m2 IV daily on days 6–15 of each induction cycle (Table 1). Patients could receive up to 4 cycles of induction. Once there were < 5% blasts in the marrow by morphology only, AR-42 was administered at the same dose and schedule, followed by decitabine 20 mg/m2/d on days 6–10 only of each 28-day cycle. Patients could continue to receive treatment until the development of intolerable toxicity, relapse or death.
Table 1.
Adult cohort induction regimen.
| Dose level (DL) |
AR-42 | Decitabine |
|---|---|---|
| 1 | 20 mg po daily days 1,3,5 | |
| 2 | 40 mg po daily days 1,3,5 | 20 mg/m2/d IV days 6–15 |
| 3 | 40 mg po daily days 1,3,4,5 |
Dosing regimens for AR-42 and decitabine by dose level. Three patients enrolled to DL1, 3 to DL2 and 6 to DL 3.
Disease response was determined using International Working Group criteria [13]. Patients at any dose level who achieved a complete remission (CR) or a CR with incomplete count recovery (CRi) could proceed to allogeneic stem cell transplantation (alloSCT). Patients who did not complete the first cycle of therapy for reasons other than unacceptable toxicity were replaced. Hydroxyurea was permitted during the first cycle to maintain a white blood cell (WBC) count less than 40,000/μL, but no other antileukemic therapies were permitted. Patients requiring treatment with hydroxyurea after completion of cycle 1 were considered to have refractory disease and removed from the trial.
Definition of dose limiting toxicity (DLT)
Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. DLT was defined during cycle 1. Any non-hematologic toxicity ≥ CTCAE grade 3 at least possibly related to AR-42 during cycle 1 with the exception of grade 3 alopecia, nausea and vomiting controllable with anti-emetic therapy, infection (grade 3 or 4), and fatigue were considered DLT. An amendment to the protocol clarified that toxicities to be recorded but not considered DLTs included life-threatening (even fatal) infections during neutropenia including pneumonia with respiratory failure and sepsis with hypotension and subsequent renal failure. Hematologic toxicity for induction was defined as: failure to recover neutrophil and/or platelet counts by day 42 in patients with < 5% blasts in the bone marrow, absence of myelodysplastic changes, and/or absence of evidence of disease by flow cytometry in the bone marrow.
Pharmacokinetic analysis
Plasma samples were collected from each patient in pre-chilled heparinized tubes and stored on ice prior to centrifugation. Plasma was removed and placed on dry ice prior to storage at −80 °C until analysis. The following time points were assayed using a previously validated liquid chromatography-mass spectrometry assay [14]: pre-dose, 30 min, 1 h, 1.5 h, 2 h, 4 h, 8 h, 24 h on days 1 and 5 of cycle 1. Using the resulting concentration vs. time data, pharmacokinetic (PK) parameters were estimated by WinNonlin (V 6.0, Pharsight, Mountain View, CA) computer software, using noncompartmental analysis (NCA) and plasma concentration-time data as described previously [14]. Uniform weighting and the best fit method were used for terminal phase regression. For 5 patients on day 1 and 6 patients on day 5, time range was selected for terminal phase regression with a start time of 4 h and end time of 24 h.
Anthropometric measures
Patients were weighed to the nearest kg and height was measured to the nearest 0.1 cm. Lean body mass (LBM) was estimated using: LBM (kg) = (9.27 × 103 × weight (kg))/(6.68 × 103 + 216 × BMI (kg/m2)) for males and LBM (kg) = (9.27 × 103 × weight (kg))/(8.78 × 103 + 244 × BMI (kg/m2)) for females [15].
qRT-PCR of miR-29b
RNA extraction and real-time PCR were performed as we have previously described [9].
Statistical methods
Patient characteristics were summarized using medians and ranges or frequencies based on the type of data. Toxicities were summarized as the number of patients experiencing a certain toxicity at its maximum grade. The response rate with a 95% CI was calculated, assuming a binomial distribution. To assess dose proportionality, PK parameters, AUC0- and Cmax were log transformed and analyzed by the power model, ln(PK) = β0 + β1ln(dose) where β0 = the intercept and β1 = the slope. The power model assumes a linear relationship between natural log-transformed PK exposure parameters and natural log-transformed dose [16]. The proportionality constant, β1, and its 90% confidence interval were calculated across the 20–40mg dose range using nonlinear regression in GraphPad software (Prism 7, La Jolla, CA). The null hypothesis, β1=1, was tested and the PK parameter was determined as dose proportional if the null hypothesis was true (p > 0.05).
Results
Patients
Patient characteristics are shown in Table 2. Four patients with previously untreated AML, 3 patients with de novo and one patient with secondary AML after MDS, were enrolled. Of the remaining 9 patients, 6 had relapsed disease with 4 having a CR1 duration of less than 12 months. Two patients had primary refractory AML and one patient had relapsed refractory disease. Prior therapies included anthracycline and cytarabine induction (‘7 + 3’), high dose cytarabine containing salvage regimens (mitoxantrone, etoposide, cytarabine (MEC) or fludarabine, cytarabine (FLAG)), hypomethylating agents, allogeneic stem cell transplantation and phase I clinical trials that included lenalidomide and bortezomib. The median number of treatments received prior to enrollment was 3 (range 1–4) and 60% of patients had relatively chemotherapy resistant (i.e. primary refractory or CR1 duration < 12 months) disease.
Table 2.
Summary of patient characteristics.
| Patient characteristics (N = 13 patients) | |
|---|---|
| Age, years | |
| Median | 71 |
| Range | 42–83 |
| Sex | |
| Male | 5 |
| Female | 8 |
| Disease Type | |
| De Novo | 3 |
| Secondary AML | 1 |
| Relapsed | 6 |
| Primary Refractory | 2 |
| Relapsed Refractory | 1 |
| ELN category | |
| Favorable | 1 |
| Intermediate-I | 5 |
| Intermediate-II | 1 |
| Adverse | 6 |
| Number of prior therapies | |
| < 2 | 4 |
| ≥ 2 | 6 |
| Prior allogeneic SCT | 4 |
13 patients were treated at 3 dose levels. ELN refers to European LeukemiaNet; AML refers to acute myeloid leukemia; SCT refers to stem cell transplant.
Treatment and toxicity
Three patients enrolled to DL1. Of these, 2 patients completed 4 cycles of induction chemotherapy without response and one patient had less than 5% blasts following induction. There were no DLTs and an increase of at least 100% in miR-29b expression was not observed. Dose escalation to DL2 occurred and 3 patients enrolled. Two patients received only 2 cycles of induction chemotherapy, one withdrew to proceed with hospice and one developed sepsis and multi-organ failure and died. The final patient completed one cycle and withdrew. All 3 had persistent disease at the end of the treatment. There were no DLTs and an increase in miR-29b expression of at least 100% was not observed. Dose escalation to DL3 occurred and 3 patients enrolled. One patient, a 79 year old male with primary refractory complex karyotype AML with an inv(3)(q21q26.2) abnormality developed polymicrobial sepsis and multi-organ failure prior to the end of cycle 1. This was determined to be a DLT by the data safety monitoring committee and the dose level was expanded to treat an additional 3 patients. There were no additional DLTs and an increase in miR-29b expression was not observed.
As is expected in this patient population treated with HMAs, the most commonly reported adverse events of all grades were cytopenias including thrombocytopenia, neutropenia and leukopenia. The most common grade 3 or higher non-hematologic toxicities were febrile neutropenia, infection, electrolyte and LFT abnormalities. Although none of the patients developed QTc prolongation, 4 patients had worsening hypertension and 6 patients developed grade 3 or higher hypoxia in the setting of pulmonary infections. Cycle 1 and 2 grade 3 or higher non hematologic toxicities are summarized in Table 3.
Table 3.
Non-hematologic toxicities, greade ≥ 3 during the first 2 cycles of treatment.
| Toxicity | Number of Patients with grade ≥ 3 in cycles 1 and 2 |
|---|---|
| Infection | |
| Catheter-related infection | 2 |
| Lung infection | 5 |
| Febrile neutropenia | 9 |
| Sepsis | 3 |
| Cardiac | |
| Hypertension | 4 |
| Hypotension | 2 |
| Heart failure | 1 |
| Cardiac troponin increased | 1 |
| Sinus tachycardia | 1 |
| Liver | |
| Alanine aminotransferase increased | 1 |
| Aspartate aminotransferase increased | 2 |
| Blood bilirubin increased | 1 |
| Metabolism and nutrition disorders | |
| Alkalosis | 1 |
| Hypokalemia | 3 |
| Hyponatremia | 5 |
| Hypophosphatemia | 2 |
| Hyperglycemia | 3 |
| Hyperuricemia | 1 |
| Hypoalbuminemia | 1 |
| Others | |
| Syncope | 1 |
| Hypoxia | 6 |
| Respiratory failure | 1 |
| Fall | 1 |
| Rash maculo-papular | 1 |
Clinical responses
Of the 13 patients enrolled, 2 patients achieved a CRi and one patient achieved a CR for an overall response rate (ORR) of 23.1% (95% CI: 5.0–53.8%). Reponses occurred in 2 patients with de novo AML and one patient with relapsed AML, all in DL3. Two patients had less than 5% blasts, 1 after 3 induction cycles (DL1) and one after 2 cycles (DL3). Both moved on to maintenance but did not meet criteria for CRi. In total, 5 patients received maintenance chemotherapy for a median of one cycle (range: 1–11). One patient was able to proceed to allogeneic stem cell transplant (Table 4).
Table 4.
Case series of patient outcomes.
| Case | Age/Sex | Prior MDS/MPN |
Relapsed/ Refractory |
CR1 duration | Cytogenetics | Dose level |
Response | Maintenance cycles |
Total Cycles |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 72/F | No | Relapsed | 20 mo | 46,XX | 1 | SD | 0 | 4 |
| 2 | 58/F | No | Relapsed | 4 mo | 45,XX,add(2)(q31), −3,−4,−5,add(7)(q21), der(16)del(16) (p13.1)del(16)(q21),−1 7,del(20)(q13.1),+21,+mar1, +mar2[14]/nonclonal w/clonal abnormalities[1]//46,XY[5] | 1 | <5% blasts | 1 | 3 |
| 3 | 74/F | MDS | Secondary | NA | 47,XX,+11[8]/48, sl,+8[5]/46,XX[7] | 1 | PD | 0 | 4 |
| 4 | 57/F | No | Relapsed/Refractory | 3 mo | 46,XX | 2 | PD | 0 | 2 |
| 5 | 66/M | No | De Novo | NA | 44,XY,del(5)(q13), −10,idic(11)(p11.2),−17,−18,der(18) t(10;18)(q21;q12)[6]/4 4,sl,del(2)(q31)[3]/44, sl,add(18)(p11.2) [3]/44-45,sl,-idic(11),+tric(?;11;11) (?::11q23.1 > 11p11. 2::11p11.2 > 11qter[cp2]/nonclonal w/clonal abnormalities[1]/46, XY[5].ishidic(11)(MLL++), tric(?;11;11)(MLL+) | 2 | PD | 0 | 2 |
| 6 | 18/M | De Novo | Relapsed | 8mo | Complex | 2 | SD, 50% blasts post cycle 1 | 0 | 1 |
| 7 | 79/M | No | Refractory | NA | 46,XY,inv(3)(q21q26.2), der(7)del(7)(p11)del(7) (q11.1)[20].nucish(D7Z1x2, D7S486x1)[131/209] | 3 | Unknown | 0 | 1 |
| 8 | 73/F | No | Refractory | NA | 46, XX | 3 | <5% blasts | 1 | 4 |
| 9 | 73/M | No | De Novo | NA | 46,XY,add(2) (p24),t(5;8)(q33;q13), del(12)(q22q24.1) [25].ishadd(2)(ALK+), t(5;8)(CMYC+;CMYC−) | 3 | CR | 6+ | 8+ |
| 10 | 70/F | No | De Novo | NA | 46,XX,t(1;15) (p36.3;q15),del(5)(q22q31), +8,psudic(17;3)(p13;p12) [16]/nonclonal w/clonal abnormalities[2]/46,XX[2] | 3 | CRi | 2 | 4 |
| 11 | 71/F | No | Relapsed | 10 mo | 46, XX | 3 | SD | 0 | 1 |
| 12 | 58/F | No | Relapsed from CR2 | CR1 = 31mo CR2 = 13mo | 46,XX | 3 | SD | 0 | 2 |
| 13 | 70/M | No | Relapsed | CR1 = 19mo | 46,XY | 3 | CRi | 1 | 5+ |
Case series table describing cytogenetics and outcomes of each patient enrolled. SD, stable disease; PD, progressive disease, CR, complete remission.
Correlative studies
Patient plasma pharmacokinetics were assessed from thirteen patients to determine time or dose-dependent effects on AR-42 disposition. Mean plasma concentration and time profiles of AR-42 in patients on days 1 and 5 are shown in Figure 1. The rate (tmax) and extent of AR-42 absoprtion (Cmax and AUC) were similar between days 1 and 5 (Table 5). We observed a significantly (approximately 10-fold) lower exposure in patient 1 compared to all other patients treated at the same dose level on cycle 1 day 1. Given that the PK data on day 5 was similar to that of other patients, it is unlikely that this difference was due to abnormal clearance. Furthermore, dose records confirmed that this patient received the intended 20 mg dose on day 1, and therefore cause of the 10-fold lower exposure on day 1 could not be explained. This patient′s day 1 PK was excluded from analysis.
Figure 1.
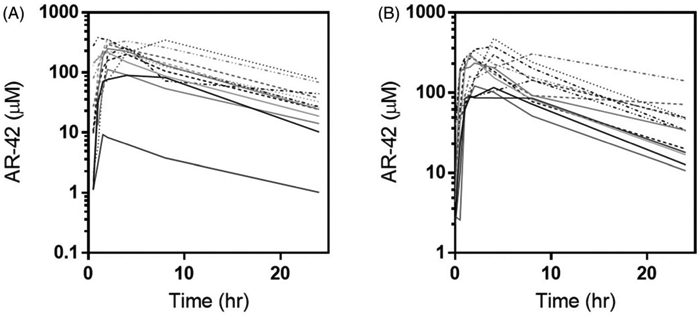
Individual patient plasma-time concentration profiles by dose level (DL1 – solid lines; DL2 – dashed lines; DL3 – dotted lines) on day 1 (A) and day 5 (B).
Table 5.
Summary of pharmacokinetic parameters on day 1 and day 5.
| Dose (mg) | 20 (day 1, 3, 5) | 40 (day 1, 3, 5) | 40 (day 1, 3, 4, 5) | |||
|---|---|---|---|---|---|---|
| Parameter by day | Day 1 | Day 5 | Day 1 | Day 5 | Day 1 | Day 5 |
| Tmax (h) | 2.00 (1.32) | 2.00 (1.32) | 2.00 (1.15) | 1.50 (0.29) | 2.00 (2.36) | 4.00 (2.60) |
| Cmax (μM) | 0.15 (0.17) | 0.35 (0.05) | 0.71 (0.08) | 0.86 (0.12) | 1.01 (0.18) | 0.96 (0.32) |
| AUC0–∞ (μM h) | 1.77 (2.34) | 4.63 (0.71) | 7.90 (1.10) | 7.71 (1.10) | 12.22 (4.24) | 14.42 (7.91) |
| T1/2 (h) | 7.36 (0.42) | 6.91 (1.11) | 6.63 (0.29) | 6.93 (1.26) | 7.63 (1.14) | 9.98 (6.79) |
| AUC0–4 (μM h) | 1.59 (2.13) | 4.19 (0.54) | 7.20 (1.06) | 6.94 (0.64) | 11.33 (3.54) | 11.13 (3.36) |
| Cl/F (L/h) | 36.11 (114.88) | 13.84 (2.18) | 16.22 (2.15) | 16.61 (2.22) | 10.48 (2.98) | 8.88 (3.23) |
| Vz/F (L) | 381.33 (1238.57) | 137.94 (14.39) | 155.67 (27.03) | 166.11 (9.73) | 120.53 (35.87) | 127.81 (54.36) |
Tmax: time to reach maximum concentration; Cmax: maximum concentration; AUC0–∞: area under the curve from time 0 to extrapolated to infinite time; T1/2: half-life; AUC0–24: area under the curve during 24 h; Cl/F: apparent oral clearance; Vz/F: apparent volume of distribution. All values are represented as geometric means (SD) except for Tmax which are represented as media.
Similar to the first-in-human (FIH) trial, we observed a 2.4- and 1.7-fold increase in Cmax and AUC values, respectively, on day 5 between the 20 and 40 mg dose levels [14]. In addition, AUC and Cmax values increased with increasing dose of AR-42 (Table 5) and were proportional to dose. The estimate of the proportionality constant (90% CI) for AUCall and Cmax were 1.313 (0.669, 1.956) and 1.492 (1.032, 1.951), respectively, for day 1 and 1.369 (0.658, 2.081) and 1.413 (0.917, 1.909), respectively, for day 5 (Figure 2). As each 90% CI contained one (except for Cmax on day 1), we conclude proportionality was maintained between dose levels. Similarly, the relevant 90% CIs for Cmax between day one and day 5 were 0.0482 (−0.155, 0.251), 0.116 (−0.017, 0.249) and 0.097 (−0.202, 0.144) for dose levels 1, 2 and 3 respectively; AUCall between day 1 and day 5 were −8.73e-17 (−0.167, 0.167), −0.015 (−0.163, 0.133) and 0.103 (−0.111, 0.317) for dose levels 1, 2 and 3 respectively, suggesting AR-42′s pharmacokinetics are time independent over 5 days (Supplemental Figure 1).
Figure 2.
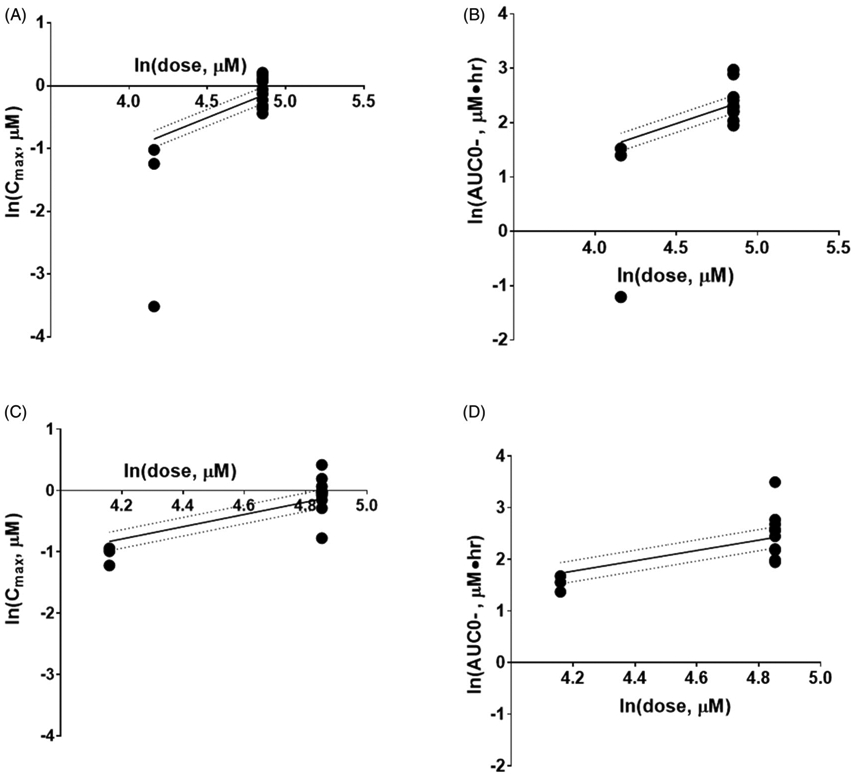
Relationship between extent of exposure and dose following AR-42 administration 20–40 mg. Day 1 Cmax (A) and AUC0- (B) and Day 5 Cmax (C) and AUC0- (D). The circles represent observed values, solid lines are the fitted values based on the power model, and the dotted lines are the 90% CI. AUC0-: area under the plasma concentration-time curve from time 0 to infinity; Cmax: measured peak plasma concentration.
MiR-29b expression in bone marrow was normalized to patient baseline samples and nanostring′s internal controls. Expression was evaluated against outcome measures (toxicities, total cycles, months survival and response) and PK parameters. Results showed a weak correlation between miR-29b ΔΔCT values versus Cmax values that was statistically significant (p < 0.05), suggesting that increased levels of AR-42 resulted in bone marrow miR-29b induction (Figure 3). However, neither Cmax nor miR-29b ΔΔCT values correlated with outcomes.
Figure 3.
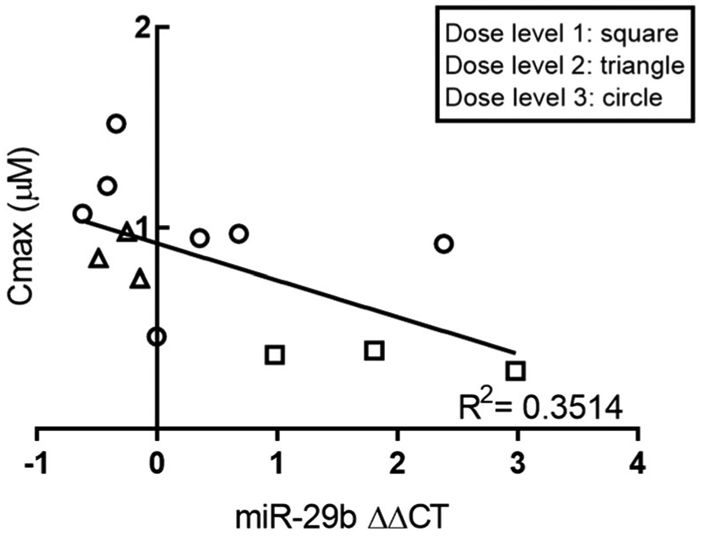
Relationship between miR-29b expression in bone marrow and patient Cmax. Results showed a weak correlation between miR-29b ΔΔCT values and Cmax (p < 0.05; linear regression), suggesting that increased levels of circulating AR-42 resulted in bone marrow miR-29b induction. ΔΔCT: the comparative CT method; Cmax: measured peak plasma concentration.
In an effort to better understand apparent interpatient differences in AR-42 pharmacokinetics, relationships between pharmacokinetic parameters and other variables including patient demographics (age, sex, bodyweight, BMI) and clinical outcomes (toxicities, total cycles, months survival and response) were explored. No differences were observed in CL/F, Vz/F, t1/2, Cmax or AUC values when evaluated against outcome measures and the aforementioned demographic parameters.
Discussion
In this phase I trial of patients with newly diagnosed and relapsed/refractory AML, we sought to determine the feasibility of dose escalation based on a real time assessment of a pharmacodynamic endpoint in addition to toxicity. The previously established MTD from the FIH study of AR-42 was 40 mg administered 3 times weekly (Monday, Wednesday, and Friday) for 3 weeks of a 28 day cycle [14]. In the current trial, AR-42 was given for either 3 or 4 days and not continued during the decitabine chemotherapy or the remainder of the treatment cycle. Our pharmacodynamic assessment included both a measure of the mature miR-29b transcript as well as the primary transcript as this can be a more direct measure of transcription of the miR-29b gene. Specifically, the primary transcript may be more stable than the mature as the mature miR-29b may engage immediately with the target through the RNA induced silencing complex. With the current schedule we did not observe a doubling in either the mature or primary miR at day 5 prior to decitabine administration. However, Cmax values were significantly correlated with mature miR-29b ΔΔCT values at the higher dose levels, which suggests that a greater induction of miR-29b might occur with a higher dose of AR-42 and result in achievement of the biologic endpoint. From a clinical standpoint, of the 13 patients treated, 2 achieved a CRi and one achieved a CR for an ORR of 23.41%. Two patients had <5% blasts but did not meet criteria for CRi. The observed ORR is lower than what has previously been reported in patients with newly diagnosed AML and likely reflects the high risk features of the heavily pretreated patients who were enrolled to the trial [10].
The most commonly reported adverse events of all grades were cytopenias including thrombocytopenia, neutropenia and leukopenia. Of the six patients who experienced neutropenia in cycle 1, 4 patients also experienced a grade 3 or 4 infection. Of 27 patients treated with single-agent AR-42 in the FIH trial, the only other clinical dataset published to date for this agent, only one neutropenic infection was observed [14]. Other toxicities that were observed included diarrhea, nausea and fatigue which is a known side effect of HDACi therapy. In this trial, one patient experienced grade 3 fatigue in cycle 4, and three patients (23%) experienced grade 1/2 fatigue in cycle 1. This is lower than what has been observed with other HDACi, as 47.4% of patients treated with single agent panobinostat and 52% treated with single agent vorinostat experienced fatigue [17,18]. Given that AR-42 was only administered for 3 or 4 days per cycle, AR-42 associated fatigue and its contribution to other toxicities are likely modest.
Based on early clinical research with the HDACi romidepsin, QTc prolongation and cardiac toxicity were considered to be a class side effect of HDACi therapy [19]. These toxicities have not been confirmed in other studies and currently, romidepsin, vorinostat and panobinostat are FDA approved with safe and minimal cardiac toxicity. Similar to other FDA approved HDACi′s, minimal cardiac toxicities were reported in patients treated with AR-42. Recently, an HDAC-6 specific inhibitor, quisinostat, was evaluated in patients with relapsed multiple myeloma in which grade 3 and 4 cardiac events were reported [20]. Whether cardiac toxicities are characteristic of HDAC specific and/or pan inhibitors requires further investigation.
From in vitro evidence, AR-42 upregulated miR-29b expression in Kasumi-1 AML cells [9], and a weak correlation between ΔΔCT values and Cmax was observed in this study (Figure 3; p < 0.05). It is possible that adequate levels of unbound AR-42 that would elicit a clinically meaningful miR-29b induction at the doses administered were not achieved. Cmax values of AR-42 were 0.15–0.35 μM at DL1, 0.71–0.86 μM at DL2 and 0.96–1.01 μM at DL3, whereas 1 μM AR-42 was shown to result in miR-29b induction in AML cells after 24 h [9]. Given that DL3 was well tolerated with dosing on days 1, 3, 4 and 5, daily dosing of AR-42 at 40 mg daily or higher may be an ideal dosing regimen for miR-29b induction and increased clinical response.
Body composition, specifically adipose and lean tissue has been shown to predict survival and risk of toxicity in cancer patients [21]. BMI was not related to cycle 1 toxicities in this study (Supplemental Figure 2), however, limited data exists surrounding AR-42 and the impact of body composition in AML patients. Alternate measures of body composition, other than BMI, may better predict treatment toxicity and overall outcome [22]. As patients experience weight loss with disease progression, future trials may include monitoring of body composition changes with toxicity and efficacy outcomes. Ideally, these would include pharmacokinetic measures of the chemotherapeutic agent to determine if lower doses would be necessary for patients who experience weight loss.
In summary, AR-42 is a novel HDACi that has been shown to increase miR-29b expression in vitro, however in this study, the current schedule did not achieve the proposed biologic endpoint. Possible explanations for this, as well as the low response rate, include the small number of patients, high risk features of the patient population, as well as an inadequate dosing schedule. Given these limitations it is difficult to comment on the efficacy of the regimen, although an increase in the dose of AR-42 and continued administration during decitabine therapy may have resulted in an increase in miR-29b levels. While AR-42 will not be explored further in AML, investigation into combinations with other anti-cancer agents may provide support for use of HDACi therapy in this disease.
Supplementary Material
Acknowledgments
Funding
AR-42 was provided by Arno Therapeutics, Inc. This project was supported by The Ohio State University Comprehensive Cancer Center and Pharmacoanalytical Shared Resource; the Division of Cancer Prevention, National Cancer Institute grants [P30CA016058, K12CA133250] to CCC, [R01CA158350] to RG and GM and [R01CA201382] to MAP; an Eli Lilly Fellowship in Pharmaceutics to SGL; Pelotonia Award to ARW.
Footnotes
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. [DOI] [PubMed] [Google Scholar]
- [2].Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127(1):42–52. [DOI] [PubMed] [Google Scholar]
- [3].O′Donnell MR, et al. Acute myeloid leukemia, Version 3.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2017;15(7):926–957. [DOI] [PubMed] [Google Scholar]
- [4].Lubbert M, Kuendgen A. Combining DNA methyltransferase and histone deacetylase inhibition to treat acute myeloid leukemia/myelodysplastic syndrome: achievements and challenges. Cancer. 2015;121(4): 498–501. [DOI] [PubMed] [Google Scholar]
- [5].Quintas-Cardama A, Santos FP, Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia. 2011;25(2):226–235. [DOI] [PubMed] [Google Scholar]
- [6].Giles F, Fischer T, Cortes J, et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res. 2006;12(15):4628–4635. [DOI] [PubMed] [Google Scholar]
- [7].Lu Q, Yang Y-T, Chen C-S, et al. Zn2+-chelating motif-tethered short-chain fatty acids as a novel class of histone deacetylase inhibitors. J Med Chem. 2004; 47(2):467–474. [DOI] [PubMed] [Google Scholar]
- [8].Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. JCO. 2007;25(21):3109–3115. [DOI] [PubMed] [Google Scholar]
- [9].Mims A, Walker AR, Huang X, et al. Increased anti-leukemic activity of decitabine via AR-42-induced upregulation of miR-29b: a novel epigenetic-targeting approach in acute myeloid leukemia. Leukemia. 2013; 27(4):871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Garzon R, Liu S, Fabbri M, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113(25):6411–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Blum W, Garzon R, Klisovic RB, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci USA. 2010; 107(16) :7473–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hunsberger S, Rubinstein LV, Dancey J, et al. Dose escalation trial designs based on a molecularly targeted endpoint. Statist Med. 2005; 24(14) :2171–2181. [DOI] [PubMed] [Google Scholar]
- [13].Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. JCO. 2003;21(24):4642–4649. [DOI] [PubMed] [Google Scholar]
- [14].Sborov DW, Canella A, Hade EM, et al. A phase 1 trial of the HDAC inhibitor AR-42 in patients with multiple myeloma and T- and B-cell lymphomas. Leuk Lymphoma. 2017;58(10):2310–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moisey LL, Mourtzakis M, Kozar RA, et al. Existing equations to estimate lean body mass are not accurate in the critically ill: results of a multicenter observational study. Clin Nutr. 2017;36(6):1701–1706. [DOI] [PubMed] [Google Scholar]
- [16].Kim YH, Choi HY, Noh Y-H, et al. Dose proportionality and pharmacokinetics of carvedilol sustained-release formulation: a single dose-ascending 10-sequence incomplete block study. Drug Des Devel Ther. 2015;9: 2911–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007; 12(10):1247–1252. [DOI] [PubMed] [Google Scholar]
- [18].Wolf JL, et al. Phase II trial of the pan-deacetylase inhibitor panobinostat as a single agent in advanced relapsed/refractory multiple myeloma. Leukemia Lymphoma. 2012;53(9):1820–1823. [DOI] [PubMed] [Google Scholar]
- [19].Shah MH. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2006;12(13): 3997–4003. [DOI] [PubMed] [Google Scholar]
- [20].Moreau P, Facon T, Touzeau C, et al. Quisinostat, bortezomib, and dexamethasone combination therapy for relapsed multiple myeloma. Leuk Lymphoma. 2016;57(7):1546–1559. [DOI] [PubMed] [Google Scholar]
- [21].Mourtzakis M, Prado CMM, Lieffers JR, et al. A practical and precise approach to quantification of body composition in cancer patients using computed tomography images acquired during routine care. Appl Physiol Nutr Metab. 2008;33(5):997–1006. [DOI] [PubMed] [Google Scholar]
- [22].Hopkins JJ, Sawyer MB. A review of body composition and pharmacokinetics in oncology. Expert Rev Clin Pharmacol. 2017;10(9):947–956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.