

Abstract
Alzheimer’s Disease (AD) is the leading cause of age-related neurodegeneration and is characterized neuropathologically by the accumulation of insoluble beta-amyloid (Aß) peptides. In AD brains, plaque-associated myeloid (PAM) cells cluster around Aß plaques but fail to effectively clear Aß by phagocytosis. PAM cells were originally thought to be brain-resident microglia. However, several studies have also suggested that Aß-induced inflammation causes peripheral monocytes to enter the otherwise immune-privileged brain. The relationship between AD progression and inflammation in the brain remains ambiguous because microglia and monocyte-derived macrophages are extremely difficult to distinguish from one another in an inflamed brain. Whether PAM cells are microglia, peripheral macrophages, or a mixture of both remains unclear. CD11a is a component of the ß2 integrin LFA1. We have determined that CD11a is highly expressed on peripheral immune cells, including macrophages, but is not expressed by mouse microglia. These expression patterns remain consistent in LPS-treated inflamed mice, as well as in two mouse models of AD. Thus, CD11a can be used as a marker to distinguish murine microglia from infiltrating peripheral immune cells. Using CD11a, we show that PAM cells in AD transgenic brains are comprised entirely of microglia. We also demonstrate a novel Fluorescence-Assisted Quantification Technique (FAQT), which reveals a significant increase in T lymphocytes, especially in the brains of female AD mice. Our findings support the notion that microglia are the lead myeloid players in AD and that rejuvenating their phagocytic potential may be an important therapeutic strategy.
Keywords: CD11a, Microglia, Peripheral Immune Cells, Alzheimer’s Disease, Plaque-Associated Myeloid cells
Graphical Abstract
CD11a expression can distinguish microglia (CD11a-) from peripheral immune cells (CD11a+) in homeostasis, neuroinflammation, and Alzheimer’s Disease mice.
Plaque Associated Myeloid (PAM) cells do not express CD11a and are microglia.
Fluorescence-Assisted Quantification Technique (FAQT) reveals a significant increase in microglia numbers and in infiltrating T cells in the brains of AD female mice.
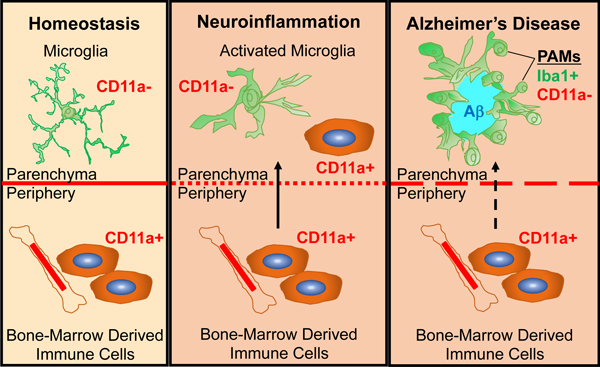
INTRODUCTION
Microglia are the resident immune cells of the central nervous system (CNS). They play critical roles in synapse formation (Parkhurst et al., 2013), maintaining the neuronal network by synaptic pruning (Stevens et al., 2007; Paolicelli et al., 2011; Schafer et al., 2012), as well as immune surveillance (Nimmerjahn et al., 2005). Microglia also clear apoptotic cells, myelin or synaptic debris through phagocytosis (Sierra et al., 2010). As the resident immune cells of the CNS, understanding microglial involvement in neuroinflammatory contexts, such as Alzheimer’s Disease (AD), is critical for understanding pathology and for targeting these cells for therapy. During many neuroinflammatory conditions, microglia become activated and the brain parenchyma can be infiltrated by peripheral immune cells. Indeed, single-cell studies have recently confirmed the presence of distinct bone marrow-derived immune cell types within compartments of the CNS in mouse models of AD (Mrdjen et al., 2018). The presence of these intruding cells further mystifies the exact role of infiltrating versus brain-resident immune cells in inflammation and neurodegeneration observed in AD.
The biggest hallmark of AD neuropathology is the deposition of insoluble beta amyloid (Aß) plaques throughout the brain. Aß plaques are frequently surrounded by myeloid immune cells, defined primarily by their expression of classical myeloid markers, including Iba1, CD11b, and CD45. However, these markers are not unique to microglia and any infiltrating peripheral monocytes and their macrophage progeny also express these markers (Bennett et al., 2018), complicating the ability to determine the origin(s) of plaque-associated cells. The significance of these plaques-associated myeloid (PAM) cells in plaque pathology is unclear, including whether this clustering is a cause or an effect of plaque formation. Furthermore, it is currently unknown whether PAM cells are brain-resident microglia or peripheral monocyte-derived macrophages that have infiltrated the brain parenchyma. The difficulty in distinguishing between these two populations has limited our understanding of each of their roles in neuroinflammation, thus underscoring a need for new strategies to discern them.
In a healthy brain, microglia and macrophages are easily distinguished through microscopy by their ramified and amoeboid morphology, respectively. Alternatively, these cells can also be differentiated by flow cytometry using relative expression of cell surface markers, such as CD45. Microglia were classically defined as CD11b+, CD45lo and infiltrating monocytes and macrophages as CD11b+, CD45hi (Sedgwick et al., 1991). Other proposed ‘microglia-specific’ markers include Tmem119, Sall1, P2ry12 and Siglec-H, were also recently identified (Bennett et al., 2016; Bennett et al., 2018; Buttgereit et al., 2016; Haynes et al., 2006; Konishi et al., 2017). While these markers are useful in healthy brains, during neuroinflammation, such as in AD, activated microglia and peripheral macrophages alter their respective morphology and marker expression patterns, confounding their distinction. Upon activation, microglia retract their processes and adopt a more macrophage-like, rounded morphology. Activated microglia also upregulate CD45 levels, making the conventional CD45lo/CD45hi strategy inadequate in distinguishing microglia through flow cytometry. Additionally, homeostatic microglial genes like Tmem119 and P2ry12 are downregulated and other markers such as Trem2 and Apoe are upregulated by microglia in an inflammatory milieu (Butovsky et al., 2014; Keren-Shaul et al., 2017).
To overcome the limitations of these markers, lineage tracing mouse models have been generated using the microglial expression of the fractalkine receptor, Cx3cr1 (Jung etal., 2000). One study crossbred the Cx3cr1GFP/GFP mice with the 5xFAD mouse model of AD and found that dying GFP positive cells contributed to Aß plaque growth by releasing previously phagocytosed Aß into the extracellular space (Baik et al., 2016). However, macrophages can also express Cx3cr1, thus the contribution of peripheral monocyte-derived macrophages cannot be ruled out. These questions can potentially be addressed using the inducible reporter Cx3cr1-CreERT2 (Yona et al., 2013; Parkhurst et al., 2013). While long-lived microglia will retain induced labeling after tamoxifen pulse, the short-live infiltrating macrophages will eventually be replaced by unmarked macrophages derived from unlabeled bone marrow monocytes. Thus, this labeling system can allow the distinction between these two populations in the brain. However, these genetic reporters may prove impractical or expensive in certain experimental setups, particularly when disease models are also employed, which require complex breeding.
Another strategy to genetically distinguish brain-resident microglia from peripheral bone marrow derived cells is to use bone marrow chimeras. Mice transplanted with hematopoietic stem cells (HSCs) post-irradiation can replenish all peripheral immune cell types, but not microglia (Mildner et al., 2007). Thus, transplanting HSCs from genetically distinct (e.g. GFP+) donor bone marrow can readily distinguish the peripheral immune cells from host microglia. This was performed in an AD model and revealed that the PAM cells contained peripheral macrophages (Simard et al., 2006). However, a major caveat of this approach is that robust donor HSC engraftment requires the elimination of the host blood system by high doses of irradiation, upwards of 10 Gy (or 1000 Rads). This can have long term effects on the brain environment, by activating microglia, modifying pathology and disrupting the blood brain barrier (BBB; Menzel et al., 2018). Moreover, a follow-up study from the same group and others showed that shielding the head during irradiation to reduce BBB disruption, or use of parabiosis resulted in little to no contribution of bone marrow-derived cells to the PAM population, despite robust bone marrow engraftment and donor peripheral immune cell reconstitution (Mildner et al., 2007; Lampron et al., 2012; Wang et al., 2016). The limitations of these strategies highlight the need for antibodies against surface markers that reliably distinguish microglia from peripheral macrophages in healthy and inflamed brains, circumventing the need for bone marrow chimeras.
Here we discuss and characterize a candidate peripheral immune cell-specific marker, CD11a (Itgal). CD11a, along with CD18, forms the leukocyte function-associated antigen 1 (LFA1) integrin. LFA1, and thus CD11a, is expressed on all peripheral immune cells, except red blood cells and platelets (Fathman et al., 2014). LFA1 is important for immunological synapse formation as well as extravasation into tissues during infection or injury (Monks et al., 1998; Lum et al., 2002). As microglia are brain-resident and not in circulation, we hypothesized that LFA1 would not be expressed on microglia. In support of this, a recent single-cell mapping study identified CD11a as a marker that is expressed only on immune cells in the periphery and not in microglia (Mrdjen et al., 2018). Another study also utilized CD11a, along with CD49d, to distinguish tumor-associated microglia from bone marrow-derived macrophages using flow cytometry (Bowman et al., 2016). While both of these studies suggest that CD11a is only expressed in peripheral immune cells through RNA-sequencing, we demonstrate its utility as a surface marker to distinguish microglia from peripheral immune cells through flow cytometry as well as immunofluorescence.
As many seemingly reliable markers alter their expression during inflammation (e.g. CD45, P2ry12, Tmem119), it is critical to carefully evaluate the expression patterns of any new markers in a variety of neuroinflammatory contexts. Here we show the utility of CD11a in flow cytometry to mark infiltrating peripheral cells and distinguish them from microglia in steady state, in LPS-induced neuroinflammation, and in two different mouse models of Alzheimer’s Disease. In these cases, CD11a remains highly expressed on peripheral, infiltrating immune cells and remains unexpressed in microglia. As a result, CD11a allows reliable bifurcation of microglia, which are CD11a negative, and infiltrating monocytes, which are CD11a positive, by flow cytometry and fluorescence immunohistochemistry. Additionally, our immunohistochemistry data shows that PAM cells are CD11a negative and that infiltrating myeloid cells are rare in AD brains and do not cluster around Aß plaques, implicating the PAM cells as microglia. Lastly, we also present a novel flow cytometry-based spiking method to accurately quantify changes in infiltrating immune cells in aged WT and AD mice. We provide evidence of a statistically-significant increase in microglia numbers and infiltrating T cells in female AD mice compared to their WT littermates. Taken together, our data provides new insights into the infiltration of peripheral immune cells into the brains of AD mice using CD11a.
MATERIALS AND METHODS
Mice.
C57BL/6J (B6; stock no. 00664, JAX) strain was used for healthy, wild-type mouse experiments. 5xFAD (MMRRC stock no. 034848, JAX) heterozygous males were crossed with C57BL/6J females to obtain AD and WT littermates. CFP mice (Rosa26ECFP/ECFP, aka TM5; Ueno et al., 2006) were donated by Dr. Irving Weissman (Stanford University). RFP mice (Rosa26mTmG/mTmG, aka mT/mG, stock no., 007576, JAX, Muzumdar et al., 2007) were generated by Dr. Liqun Luo (Stanford University) and donated by Dr. Weissman. GFP mice (Rosa26mG/mG) mice were created at UC Irvine by crossing Rosa26mTmG/mTmG with a Cre strain that is expressed in the male germline (Lyve1-Cre; Pham et al., 2010). The mT/mG reporter will delete the mTomato expression cassette and express mGFP upon Cre-mediated excision. The progeny was further bred to remove Lyve1-Cre and create Rosa26mG/mG homozygous mice that were used. Cx3cr1+/GFP;Ccr2+/RFP double reporter mice (Saederup et al., 2010) were provided by Dr. Melissa Lodoen (UC Irvine). Arc48+/− (AD) and Arc48−/− (WT) littermates were a donation from Dr. Andrea Tenner (UC Irvine). All animal procedures were approved by the International Animal Care and Use Committee and University Laboratory Animal Resources of University of California, Irvine.
Microarray analysis of Itgal expression in immune populations.
Microarrays (Mouse 430 2.0) of mouse hematopoietic populations were downloaded and processed from the Gene Expression Commons (GEXC, gexc.riken.jp) database (Seita et al., 2012). The microglia microarrays were downloaded from the Gene Expression Omnibus database (Experiment: GSE29949, Arrays: GSM741192, GSM741193, and GSM741194 (Nayak et al., 2012) and processed by the GEXC. Itgal expression (probeset 1435560_at) was compared using the Gene Expression Activity tool, which compares the Normalized Signal Intensity of each microarray and reports it as a percentile of expression relative to a reference panel of 11,939 microarrays. For each probeset, the StepMiner algorithm analyzes the expression pattern across all microarrays of the reference panel and defines the cutoff between positive (high) and negative (low) expression, which is set as 0%.
Isolation of microglia and monocytes from brains.
Mice were euthanized with Euthasol® prior to intracardial perfusion of the circulating blood with ice-cold PBS (Hyclone™, GE Healthcare Life Sciences). Brains and spleens were immediately extracted and placed in 10% FBS in HBSS on ice. Microglial cells were isolated as described (Ford et al., 1995, and Carson et al., 1998) without enzymatic digestion. Briefly, one lobe of the brains was mechanically dissociated with manual chopping, passed through a 23G needle and sequentially forced through a 70μm filter mesh. Myelin was removed using a discontinuous layer 1.03/1.088 g/ml Percoll gradient in HBSS. Mononuclear cells were isolated from the interface, as well as the 1.03g/ml Percoll fraction after the floating myelin disk was aspirated.
Flow cytometry
Dissociated brains or spleens were analyzed on a BD LSR II Flow Cytometer (BD Biosciences), BD FACS Aria II Cell Sorter (BD Biosciences), or a BD Fortessa X20 cytometer. Data were analyzed with FlowJo software (TreeStar). Single-cell imaging flow cytometry was performed on Amnis ImageStreamX® Mark II Imaging Flow Cytometer (EMD Millipore) and data analysis was performed on the IDEAS® software.
Immunofluorescence staining and microscopy.
The second lobe of each mouse brain and spleen was drop-fixed in fresh 4% paraformaldehyde immediately after extraction and incubated at 4°C for 12–24 hours. Tissues were stored in 0.05% Sodium Azide in PBS (PBSN) or immersed in 30% sucrose for 24 hours prior to cutting. Floating 40μm sections of frozen brain or spleen tissue were cut on a sliding, freezing microtome (Leica SM 2010R) and stored in 24-well plates in PBSN till further use. For immunofluorescence staining, sections were washed with PBS and treated with AmyloGlo® (Biosensis) to stain β-Amyloid plaques. Sections were blocked with 10% goat serum in PBS (blocking buffer) prior to staining with primary antibodies. Primary antibodies were added to sections in blocking buffer and incubated at 4°C overnight. Secondary antibodies were diluted in PBS only and added to sections and incubated for one hour at room temperature. Tyramide Signal Amplification (TSA): CD11a antibody (Clone 2D7, BD Biosciences Cat# 553118) signal amplification was performed using the TSA kit (Invitrogen, #B40933) and manufacturer’s protocol. Tissue sections were mounted on charged glass slides with mounting media with or without DAPI. Microscopic analysis was performed on an Olympus FV3000 confocal microscope. It should be noted that brain tissues from each mouse and its controls were mounted on the same slide, imaged with identical microscope settings, treated with identical post-processing in Adobe Photoshop CS4 to ensure reliable comparisons.
Antibodies.
See Supplementary Table 1 for a full list of antibodies used for flow cytometry (FACS) and Immunofluorescence (IF) imaging. Technical note: While all three CD11a antibody clones appeared equally robust by flow cytometry, the 2D7 clone (BD Biosciences, Catalog# 553118) worked best for immunostaining when combined with TSA (described above), though the M17/4 clone was almost as effective.
Intracranial LPS injections.
10μg Lipopolysaccharide (LPS) from E. coli 011: B4 strain (Sigma-Aldrich, L4391) was diluted in 30μl sterile PBS and administered intracranially. The injection procedure for is as follows: Isoflurane (4%/min) mixed with oxygen (1L/min) were delivered to animals via an induction chamber. LPS or vehicle (PBS only) was injected into the right hemisphere of the brain using a 26-gauge needle inserted approximately 1 mm directly through the skull i.e. a hole is not drilled through the skull. Vehicle or LPS are generally delivered into ventricles by this method. Animals were analyzed 24 hours post-injection. Unpaired transplanted 5xFAD−/− (WT) littermates with robust chimerism (as described above) were used for these experiments at 6 to 8 weeks of age.
Toxoplasma gondii infection.
Type II GFP-expressing Prugniaud tachyzoites (Kim et al., 2007) were maintained by serial passage in human foreskin fibroblasts. For infection, parasites were lysed out of host cells by syringe passage and diluted in sterile PBS. C57BL/6 mice or Cx3cr1+/GFP;Ccr2+/RFP double reporter mice were infected by intraperitoneal injection of 200 tachyzoites. Mice were perfused with PBS intracardially and brains removed and fixed in 4% paraformaldehyde at 7 days post-infection for acute infection timepoints. To induce reactivation of latent infection, mice were injected I.P. with 2mg anti-interferon gamma or IgG control antibody (Bioxcell) at 28- and 32-days post infection (dpi). At 38 dpi, mice were perfused and brains were analyzed as described above.
Bone Marrow transplantation in neonatal mice.
To generate 5xFAD mice with labeled peripheral immune cells, fluorescently-labeled BM (GFP, RFP or CFP) was transplanted into neonatal (postnatal day 1, P1) 5xFAD+/− (5X) and 5xFAD−/− (WT) littermates. P1 5X and WT littermates were given ≤3.5 Gy (350 Rads) dose of X-ray irradiation (XRAD 320, Precision X-ray). Donor bone marrow was first ckit-enriched by staining with microbead-conjugated ckit antibodies (Miltenyi) and enriched using the positive selection (“possel”) setting on an autoMACS (Miltenyi). Each pup was given up to 200,000 ckit+ BM cells from RFP mice via facial vein injection. Pups were placed back with the mother, who was fed an antibiotic chow of Trimethoprim Sulfa (Uniprim, Envigo). After weaning, the recipients were switched to regular chow and genotyped with ear or tail snips. Chimerism was measured through tail vein bleeds at 5 weeks of age and immediately before harvest and confirmed by BM HSC analysis upon tissue harvesting. 5X and WT recipients with similar levels of donor chimerism were paired for comparison.
Fluorescence-Assisted Quantification Technique (FAQT):
To quantify relative numbers of immune cell populations in wildtype (WT) and 5xFAD (5X) brains, fluorescently labeled brains were added to each brain sample immediately after harvest and prior to processing. Specifically, each WT sample contained one lobe of a WT brain and one lobe of a fluorescent reporter brain (GFP, RFP, or CFP). The 5X sample was prepared similarly with the remaining lobe of the same fluorescent reporter brain. Thus, each WT and 5X sample pair received equal amounts (one lobe each) of fluorescently labeled brain tissue added to it. The samples were then processed for single cell suspension and myelin removal as described above. During FACS analysis, each cell population was sub-divided into fluorescent or non-fluorescent gates (for example, CFP+ and CFP-). Assuming that each brain sample received an equal number of fluorescent brain cells, the frequency of WT versus 5X populations could then be normalized to one another by comparing to the frequency of that population in the fluorescent cells. To do this, the individual ratios of 5X and WT cell populations in the final sample were obtained by first dividing percentages of CFP- cells (5X or WT) by CFP+ cells, producing the ratios of 5X: CFP and WT: CFP cells, respectively. Next, to obtain the relative changes in diseased mice, the 5X:CFP ratio of each cell population was divided by the WT:CFP ratio. No difference in immune cell populations between WT or 5X mice would result in a relative change of one. To gauge the baseline sensitivity of this method, right and left lobes of WT mice (WT lobe-to-lobe) were processed and analyzed as described above and used as the control group for statistical comparison.
Statistical Analysis.
Statistical analysis was performed with GraphPad Prism 5 software (La Jolla, CA).
RESULTS
CD11a is expressed on peripheral immune cells in the brain, but not on microglia
A microarray database screen of markers that are differentially expressed in peripheral immune cells and brain-resident microglia cells revealed that integrin alpha L chain, Itgal or CD11a, was not expressed in microglia, but highly expressed in all circulating immune cell types (Figure 1A). Therefore, we sought to confirm cell surface expression of CD11a in these cells via flow cytometry (Figure 1B). Cells from adult B6 mouse brains were harvested and stained for surface marker expression. In the CD11b+, CD45lo microglia population, we did not detect CD11a surface expression when compared to the Fluorescence Minus One (FMO) negative control. Conversely, CD11a was highly and uniformly expressed in the CD11b+, CD45hi population which contains primarily monocyte-derived macrophages. The CD11b- CD45hi population, which contains mainly lymphocytes, was also uniformly CD11a+ (data not shown). We found identical expression patterns using three distinct clones of antibodies that recognize CD11a. These include the M17/4 and 2D7 clones of the CD11a antibody, along with clone H155–78 that recognizes LFA-1, the complex of CD11a with CD18 (Figure 1B, i - iii). To test the expression of CD11a on peripheral immune cells, CD45+ splenocytes were also stained with the same antibody cocktail as the brain cells. Splenocytes of myeloid and lymphoid lineages express all three clones of CD11a at high levels (Figure S1A).
Figure 1: CD11a is not expressed in steady-state microglia.
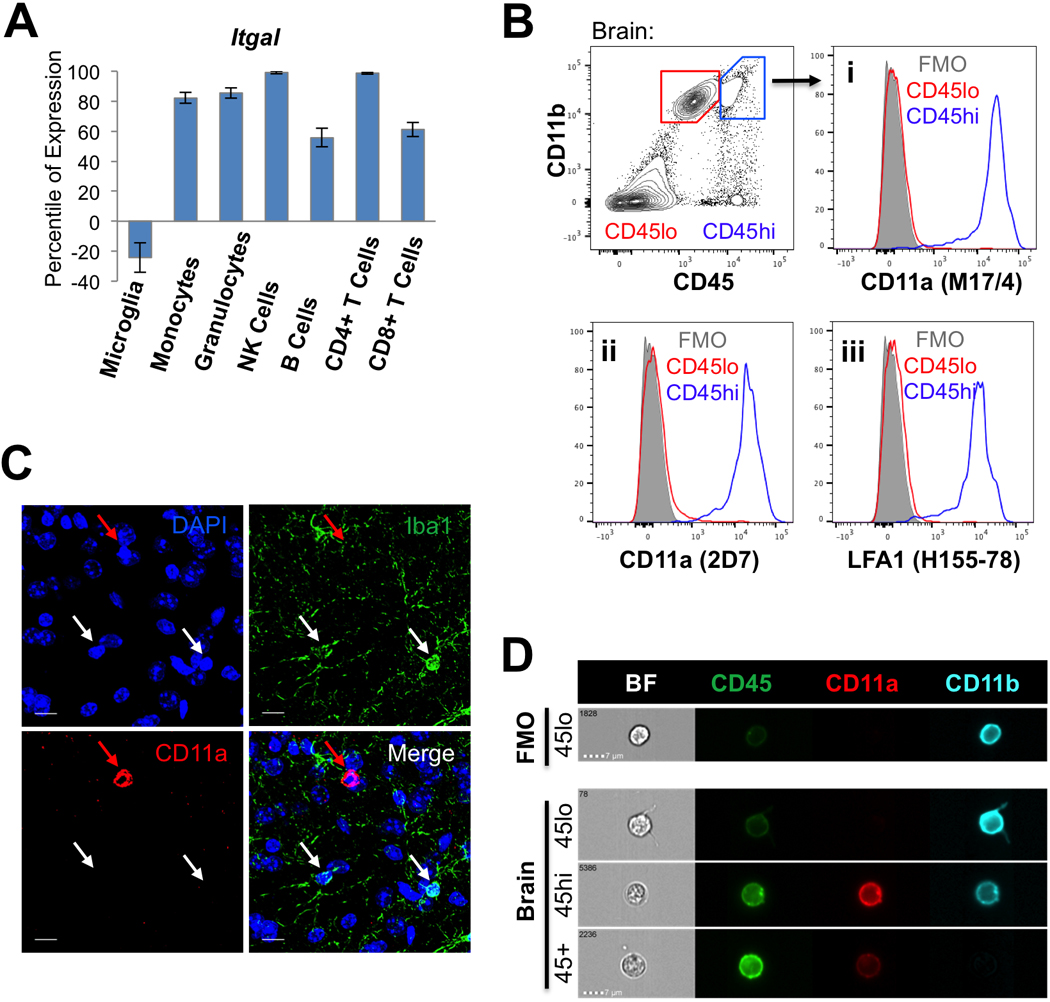
(A)Gene expression profile of Itgal (CD11a) in microglia and peripheral hematopoietic cells. Percentile of expression is based on comparison to a reference panel of nearly 12,000 microarrays (www.gexc.riken.jp). Values below zero are considered unexpressed. (B) Flow cytometry plots of adult mouse brain cells. CD11b+ CD45lo microglia (“CD45lo”, red gate) and CD11b+ CD45hi macrophages (“CD45hi”, blue gate) were analyzed for cell surface expression of CD11a using antibody clones M17/4 (i), 2D7 (ii) against CD11a, and H155–78 (iii) against LFA-1 (CD11a/CD18). Histograms show CD45lo microglia (red), CD45hi macrophages (blue), and fluorescence minus one control (“FMO”, grey). (C) Immunofluorescence image of C57/B6 brain. White arrows denote representative microglia which are Iba1+ but CD11a-. The red arrow denotes an example of a CD11a+ immune cell, which is Iba1-. Images were taken at 120X, scale bars = 10μm. (D) Imaging flow cytometry of brain cells. Representative images of cells within CD11b+ CD45lo microglia (“45lo”), CD11b+ CD45hi macrophages (“45hi”) and CD11b- CD45+ lymphocytes (45+) populations in each channel (see Figure S1C for splenocyte controls). Exact same voltage correction settings were applied to all samples.
Immunofluorescence analysis of CD11a expression in fixed brain tissue was difficult to assess using all clones and required tyramide signal amplification in order to detect CD11a+ cells. After amplification, Iba1+ microglia cells in adult B6 brain did not express CD11a, but we observed rare round CD11a+ cells that are likely infiltrating lymphocytes, based on a lack of Iba1 staining (Figure 1C). To visualize the differential expression of CD45 and CD11a in microglia (CD11b+ CD45lo), macrophages (CD11b+ CD45hi) and lymphoid cells (CD11b- CD45+), brain and spleen samples were also stained and analyzed on an imaging flow cytometer to obtain high resolution images of single cells. Consistent with our observations by FACS, CD11a was not detected on the surface of microglia, but readily expressed on monocytes and lymphocytes in the brain and spleen (Figure 1D, S1B-C).
The fractalkine receptor Cx3cr1, is strongly expressed by microglia in the CNS and has been crucial in studying microglia development and functions in homeostatic and neuroinflammatory conditions (Mizutani et al., 2012; Jung et al., 2000; Cardona et al., 2006). Thus, we also compared CD11a expression in the commonly-used Cx3cr1-GFP reporter mice (Saederup et al., 2010). As expected, GFP-labeled cells in the brain, which consist mostly of microglia, did not express CD11a (Figure S1D-E). Together, these data show that in healthy brains, CD11a remains highly expressed in all peripheral hematopoietic cells, but remains unexpressed in brain-resident microglia. Thus, CD11a meets the requirement for an on-off marker that can clearly distinguish between microglia (off) and peripheral immune cells (on) in a homeostatic mouse brain.
Microglia and monocytes/macrophages do not alter CD11a expression in activation states.
CD45 expression levels are commonly used to distinguish microglia (CD45lo) from infiltrating peripheral monocytes (CD45hi) by flow cytometry. However, CD45 expression increases in microglia cells with activation in neuroinflammatory contexts (Sedgwick et al., 1991; Greter et al., 2015). To determine whether activated microglia similarly upregulate CD11a expression, we injected the inflammatory molecule lipopolysaccharide (LPS) or vehicle control intracranially into 6–8-week-old B6 mice to activate microglia. LPS is known to cause a massive inflammatory response in the brain and induces a rapid influx of infiltrating immune cells into the brain (Cazareth et al., 2014). Brains of vehicle- and LPS-treated mice were harvested 24 hours post-injection. As expected, CD45 expression slightly increased in all CD11b+ cells with LPS treatment, partially obscuring the normal separation between CD45lo microglia and CD45hi monocytes (Figure 2A, left panels). However, CD11a expression remained absent on microglia, while infiltrating monocytes highly expressed this marker (Figure 2A, middle panels). Furthermore, an influx of monocytes into LPS-injected brains further blurred the distinction between microglia and monocytes using CD45, which becomes apparent when CD11a- and CD11a+ populations are overlaid using the conventional CD11b vs. CD45 gating strategy (Figure 2A, right panels).
Figure 2: CD11a expression remains consistent in activated microglia and infiltrating macrophages.
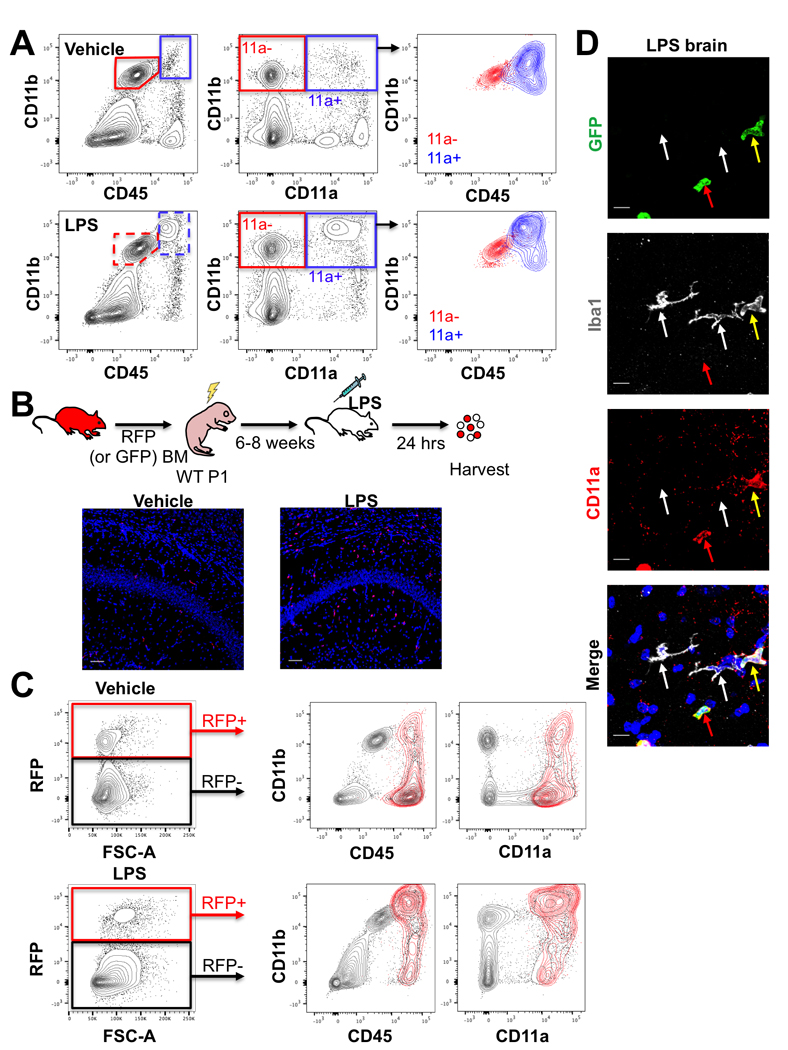
(A) FACS plots of the brains of C57/B6 adult mice 24 hours after intracranial injection of vehicle (top row) or 10 μg of LPS (bottom row). Leftmost plots show traditional CD11b vs CD45 gating scheme with microglia (CD11b+ CD45lo, red gates) and macrophages (CD11b+ CD45hi, blue gates). Middle plots show CD11b versus CD11a and revised gating for microglia (CD11b+ CD11a-, “11a-”, red gate) and macrophages (CD11b+ CD11a+, “11a+”, blue gate). Rightmost plots show overlays of CD11a- (red) and CD11a+ (blue) populations (from the middle plots) on CD11b vs. CD45 plots. (B) Schematic of transplantation of adult RFP+ bone marrow into P1 C57/B6 (WT) pups, followed by intracranial LPS (or vehicle) injection 6–8 weeks after transplant, and analysis 24 hours post-injection. 20X images of one brain lobe following vehicle (left) or LPS (right) injection showing RFP+ (red) BM-derived cells in the brain versus DAPI-stained nuclei (blue). Scale bars = 50μm. (C) Flow cytometry analysis of the other brain lobe 24 hours after vehicle (top row) or LPS (bottom row) injection. The leftmost plots show gating on RFP+ BM-derived donor cells versus RFP- host cells. The middle and right plots show CD11b versus CD45 (middle plots) or versus CD11a (right plots). RFP+ cells are shown in red and host cells in grey. (D) Representative immunofluorescence images of brain sections from LPS-treated mice after transplantation of adult GFP+ bone marrow cells into neonatal recipients (120X, scale bars = 10μm). All GFP+ infiltrating cells (green) co-stain with CD11a (red). White arrows denote Iba1+ microglia, which do not express CD11a or GFP. Red arrows indicate GFP+ cells that are CD11a+ Iba1-. Yellow arrows indicate GFP+ cells that express Iba1 as well as CD11a.
It is formally possible that some infiltrating monocytes rapidly downregulate CD45 and CD11a upon infiltration and would be included in the “microglia” gate. To ensure that infiltrating monocytes and their descendent macrophages did not downregulate CD11a expression, we repeated the LPS and vehicle injection in 6–8 weeks old B6 mice that had been irradiated and transplanted as neonates with bone marrow cells from adult GFP+ or RFP+ donors (Figure 2B). This neonatal transplantation strategy allowed for effective HSC engraftment with low doses of irradiation (≤3.5 Gy) compared to the higher required dose for adult recipients (8–10 Gy). It also allowed for the blood-brain barrier to recover and the bone marrow compartment to stabilize by the time of LPS injection. 6–8 weeks post-transplant, vehicle or LPS was injected intracranially and the brains were harvested 24 hours post-injection. Fluorescent imaging of one lobe of each brain showed infiltrating RFP+ cells throughout the brain only in the LPS-treated mice (Figure 2B). Flow cytometric analysis was performed on the other lobe of the brain and showed that all donor bone-marrow derived RFP+ cells lied within the CD11a positive gate (Figure 2C). This confirms that infiltrating RFP+ cells did not downregulate CD11a. Microglia, which are not replenished by the bone marrow, remained RFP- and CD11a-. Immunostaining of brains transplanted with GFP bone marrow cells (instead of RFP), showed Iba1+ microglia remained CD11a- even after LPS-treatment, while all GFP+ cells were CD11a+, including both Iba1+ (myeloid) or Iba1- (likely lymphoid) populations (Figure 2D, S2A-B). Together, these data indicate that activated microglia remain CD11a negative and infiltrating immune cells remain CD11a+ in an inflammatory milieu. Thus, CD11a is a stable marker that can distinguish peripheral immune cells from brain-resident microglia.
Whereas microglia in LPS- injected mice did not express CD11a, they did upregulate its expression in chronic Toxoplasma gondii infection, a parasite known to infect the brain and induce a strong immune reaction. Through flow cytometry analysis, microglia showed slight upregulation of CD11a in the acute stage of T. gondii infection (7 dpi) compared to mock-infected mice (Figure S3A). However, this shift in microglia was not detectable via immunofluorescence staining, despite tyramide amplification (Figure S3B). In contrast, during chronic infection, when the parasites are encysted in the brain, CD11a was detectable on microglia by both flow cytometry and immunofluorescence microscopy and highly upregulated in peripheral immune cells (Figure S3C-E). This finding aligns with previous reports that activated microglia and infiltrating cells express CD11a during chronic infection (Biswas et al., 2015). In this context, CD11a is comparable to CD45 as a marker. Thus, CD11a upregulation is context-dependent and must be tested in each model of neuroinflammation.
Microglia in AD model mice do not express CD11a.
In the 5xFAD mouse model of Alzheimer’s disease, a tandem of two transgenes contain a total of five familial mutations in human Amyloid Precursor Protein (APP) and Presenilin-1 (PSEN1) genes. Together they cause a rapid accumulation of Aβ in the brain as early as three months, with AD plaque-like pathology being observable at 4 months, and end-stage equivalent at 6 months of age (Oakley et al., 2006). Similar to activated microglia, CD45 expression in aged 5xFAD mice is upregulated in all CD11b+ cells, making it difficult to distinguish monocytes from microglia using the conventional CD11b vs CD45 gating scheme. With disease progression, it becomes increasingly unclear whether the abundant population of CD45hi cells are microglia or infiltrating monocytes (Figure 3A). However, these populations could still be distinguished using a CD11b vs. CD11a gating strategy (Figure 3B). Using CD11a also highlighted the abundant CD45hi population in aged 5xFAD brains, which also upregulates CD11b but is almost entirely CD11a- (Figure 3C). Therefore, these cells likely represent microglia that have upregulated CD45, and not a massive influx of infiltrating monocytes/macrophages. We also observed a similar trend in a second independent AD transgenic model, Arc48 mice, which express a different combination of familial mutations than the 5xFAD mouse model (Cheng et al., 2004; Figure S4). While there were slight increases in CD11a expression on microglia with age and disease progression in AD mice, a similar trend was also noticed in their WT littermates, as well as in the monocyte/macrophage populations of WT and AD mice, and the separation between CD11a- and CD11a+ populations was still distinct (Figure 3B).
Figure 3: Microglia in 5xFAD mice do not express CD11a.
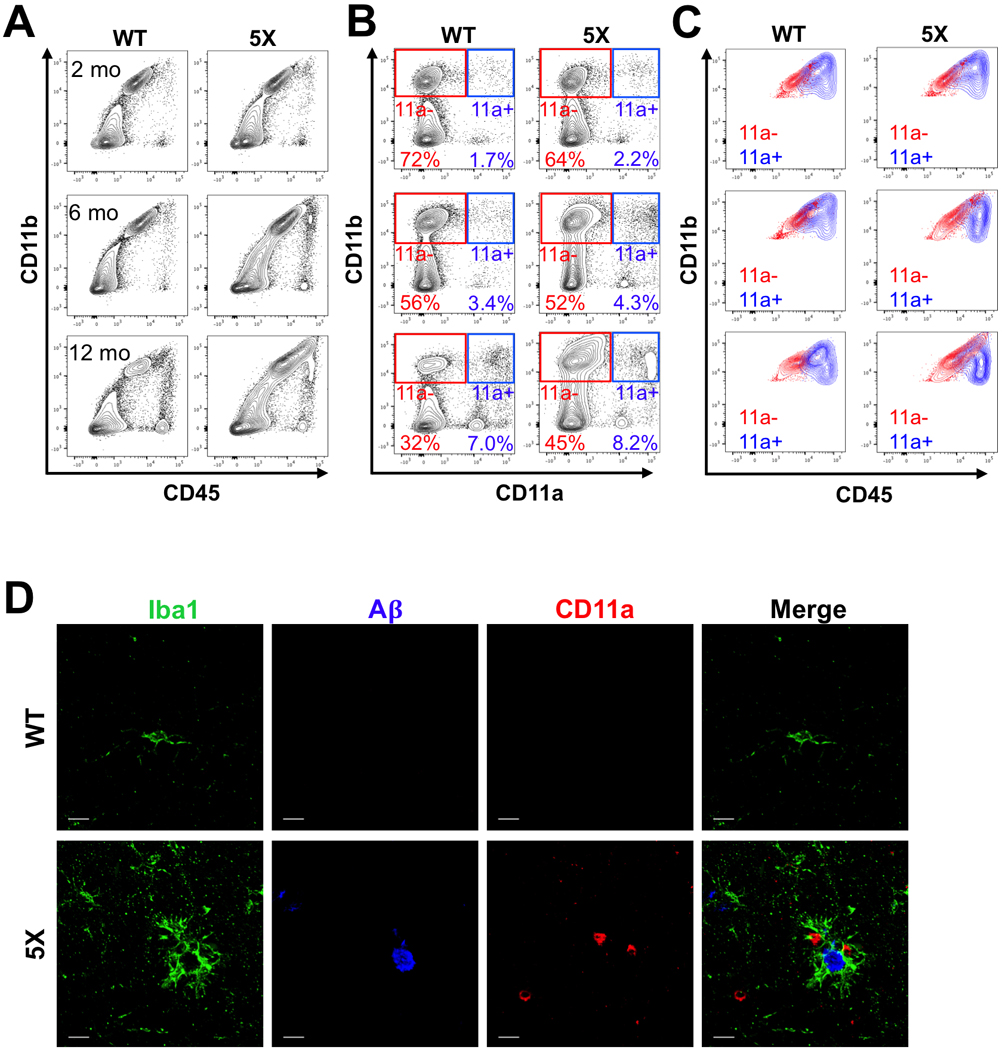
(A-C) Representative FACS plots of 5xFAD−/− (WT, left columns) and 5xFAD+/− (5X, right columns) littermates at 2 months (top row), 6 months (middle row), and 12 months (bottom row) of age. (A) Traditional CD11b versus CD45 plots show increasing overlap between microglia and macrophages in 5X mice with age. (B) CD11b versus CD11a plots show a clear distinction between microglia (CD11b+ CD11a-, “11a-”, red gate) and macrophages (CD11b+ CD11a+, “11a+”, blue gate). (C) Traditional CD11b versus CD45 plot showing an overlay of microglia (“11a-”, red) and macrophage (“11a+”, blue) populations using the CD11a gating strategy from B. (D) Representative immunofluorescent images (120X, scale bar = 10μm) of 12-month-old WT (top) and 5X (bottom) brains. Iba1+ cells (green) surrounding beta amyloid plaques (A𝛃, blue) are CD11a-. Extremely rare instances of CD11a+ cells (red) found near plaques do not express Iba1.
Plaque-associated myeloid cells are microglia, not peripheral monocytes/macrophages.
Immunofluorescence imaging of microglia in WT and AD mice consistently showed that microglia (Iba1+) in the brain parenchyma were not labeled with CD11a. Additionally, plaque-associated Iba1+ cells did not co-label with CD11a even at 12 months of age (Figure 3D). On rare occasions, CD11a+ cells were found near plaques, but they did not co-stain with Iba1, meaning they were not of myeloid lineage. Together, these data show that microglia in two AD mouse models, 5xFAD and Arc48, remain CD11a negative despite the increase in plaque burden with age. Moreover, peripheral myeloid cells have nominal contribution to the overall CD45hi population, indicating there is not a massive influx of peripheral monocytes in the AD brain.
Peripheral immune cells do not downregulate CD11a upon infiltration in AD brains.
Given the low amounts of CD45hi and CD11a+ cells observed in 12-month-old 5xFAD mice, we wanted to confirm that infiltrating monocytes/macrophages did not downregulate CD11a. To this end, we transplanted neonatal 5X and WT littermates with RFP+ bone marrow cells (Figure 4A). After 12 months of age (and 12 months post-transplantation), we harvested one lobe for flow cytometry and the other for immunofluorescence imaging. By flow cytometry, almost all RFP cells remained in the CD11a+ fraction whereas the CD11a- cells were all negative for RFP (Figure 4B). There was little evidence of robust infiltration of peripheral cells, as the percentage of RFP+ cells in the brain was low in both WT and 5X mice. Donor chimerism was high in all recipient mice, as measured by HSC chimerism in the BM (Figure 4B). Thus, the absence of RFP+ cells in the brain was not due to low engraftment levels of RFP+ cells. As seen in LPS-treated chimeric mice, infiltrating RFP+ cells in 5X and WT mice did not downregulate CD11a (Figure 4B). Importantly, RFP cells maintained CD11a expression one year after transplant. Therefore, CD11a- cells in the 5X brains are unlikely to be from bone marrow-derived cells infiltrating from the periphery. This transplantation approach also allowed us to quantify the relative abundances of different immune populations (CD11b+ CD11a- microglia, CD11b+ CD11a+ myeloid cells, and CD11b- CD11a+ lymphoid cells) in 5X and WT brains (Figure 4C). No significant difference was observed in the distribution of bone marrow-derived cells in 5X mice compared to their WT littermates. To assess whether the RFP+ cells clustered around plaques, we also imaged sections of the other lobe of these transplanted 5x mice. Observation of RFP cells was extremely rare in 5X brains as well as WT mice (Figure 4D). Thus, it is unlikely that any RFP+ cells contributed to the PAM population. Taken together, these data provide strong evidence that infiltrating cells do not downregulate CD11a and that infiltration of CD11a+ peripheral immune cells is not dramatically increased in AD mice compared to their wildtype littermates. This also provides further evidence that the plaque-associated myeloid cells are CD11a-, brain-resident microglia.
Figure 4: Plaque-associated myeloid cells are not derived from bone marrow.
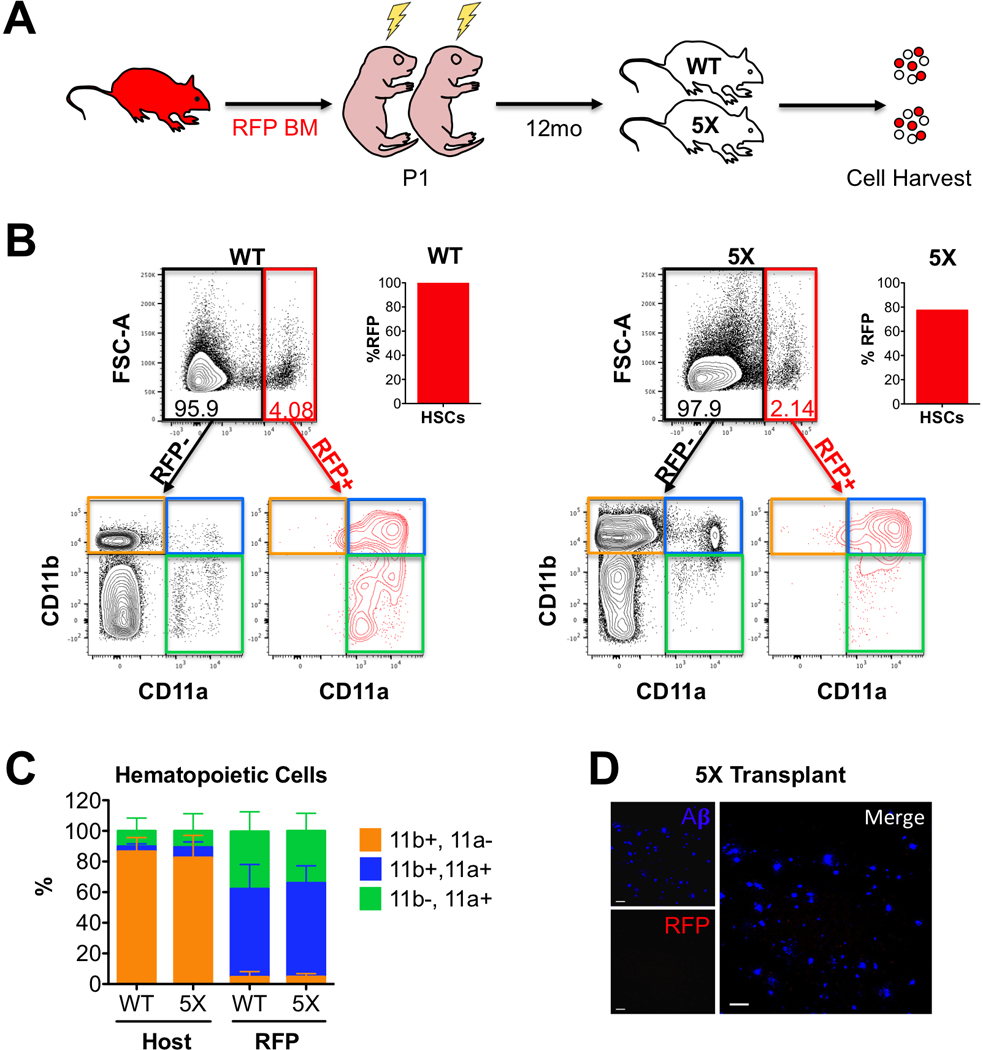
(A) 5xFAD−/− (WT, n=4) and 5xFAD+/− (5X, n=4) littermates were transplanted with RFP+ bone marrow as neonates (P1), then analyzed 12 months later. (B) Representative flow cytometry plots of WT (left) and 5X (right) brains at 12 months of age. Total RFP- (black gate) and RFP+ (red gate) cells were analyzed for CD11b vs. CD11a expression (lower FACS plots). The bar graph (right) indicates the donor HSC chimerism in the BM of the transplanted mice at 12 months. The bottom CD11b versus CD11a plots show the distribution of immune populations in the RFP- host cells (left, black) and RFP+ BM-derived cells (right, red). Quadrants Q1 (CD11b+ CD11a-, orange quadrant) represent microglia, Q2 (CD11b+ CD11a+, blue quadrant) macrophages, and Q3 (CD11b- CD11a+, green quadrant) lymphocytes. (C) Bar graph showing the distribution of immune cell types in the host (left bars) and RFP+ donor (right bars) in WT and 5X brains. The sum of the three immune cell quadrants was set to 100%. Error bars are S.D. (N=4 mice each). (D) Representative image of the brain of a 5X mouse transplanted with RFP+ BM 12 months post-transplant show no infiltrating RFP+ cells (red). Aß plaques are shown in blue. Images taken at 20X magnification, scale bars = 50μm.
Fluorescence-Assisted Quantification Technique (FAQT) reveals an increase in T cell infiltration in 5xFAD females
Our transplantation approach in 5X mice did not show any major influx of peripheral immune cells (Figure 4C and 4D), especially compared to LPS-injected brains (Figure 2B). Given the improved distinction between microglia and peripheral immune cells that the CD11b vs. CD11a gating strategy provides, we aimed to quantify the relative number of infiltrating cells in unmanipulated, aged 5X mice compared to their wildtype littermates. However, 5X brains, which suffer from chronic inflammation, have differences in cell viability and display different properties on flow cytometry analysis than WT brains, making it difficult to quantitatively compare microglia, monocytes or lymphocytes between WT and 5X brains as a percentage of total cells in the brain. Furthermore, the final cell yield, which requires mechanical digestion and centrifugation through a Percoll gradient, is inconsistent and can lead to huge variability in the absolute numbers of cells harvested from prep to prep. Lastly, counting beads, which can be added to sample tubes to estimate absolute numbers, are depleted via Percoll gradient, and therefore cannot be added during the prep stage to accurately control for differences in prep yield.
As a solution to these issues, we developed a method called “Fluorescence-Assisted Quantification Technique” (FAQT) to spike in fluorescently labeled brain tissue during the initial harvesting step and before the Percoll gradient steps. As such, any discrepancies in yield due to preparation would equally affect the fluorescently labeled cells. As long as an equal amount of fluorescently-labeled brain tissue (i.e. one lobe each from the same brain) was added to each prep of WT and 5X brain, the yields of brain populations could be normalized to the fluorescent control and the relative abundances of each population calculated. As illustrated in Figure 5A, we harvested a pair WT and 5X littermates, and added one lobe of each brain to separate tubes. We then harvested a CFP+ brain and separated the two lobes, then added one lobe to the WT tube and one lobe to the 5X tube, so each tube received an identical amount of fluorescent brain tissue. We then proceeded with the normal preparation and Percoll gradient. We then stained and analyzed each brain prep by flow cytometry, gated on each immune population (e.g. microglia, monocytes, T cells, B cells, granulocytes), then separated them into CFP+ and CFP- fractions (Figure S5). Thus, any variability in cell yield will be reflected in both the CFP+ and CFP- cells within the sample. We could then compare the percentage of fluorescently-labeled cells between the WT and 5X population to estimate the ratios of each population to the fluorescent control (Figure 5B). If we divide the ratio of WT:CFP microglia by the ratio of 5X:CFP microglia, then we can estimate the abundance of the 5X microglia relative to its WT littermate (which is set to 1). For example, if 50% of microglia in the WT brain are CFP+ (a 1:1 WT:CFP ratio), but only 25% of microglia in the 5X brain are CFP+ (a 3:1 5X:CFP ratio), then this would suggest that the microglia in the 5X brain are three times as abundant as the WT microglia, because we added the same number of CFP+ microglia to each tube. As a control, we first performed this using only WT brains, comparing the abundance of immune populations between the left lobe and right lobe (“WT L/R”), which we assume to be equal (Figure 5C-H). The differences we observe between the left and right lobes represent the variation inherent in this system and offer a control group to the 5X:WT ratios for statistical comparisons.
Figure 5: Fluorescence-Assisted Quantification Technique (FAQT).
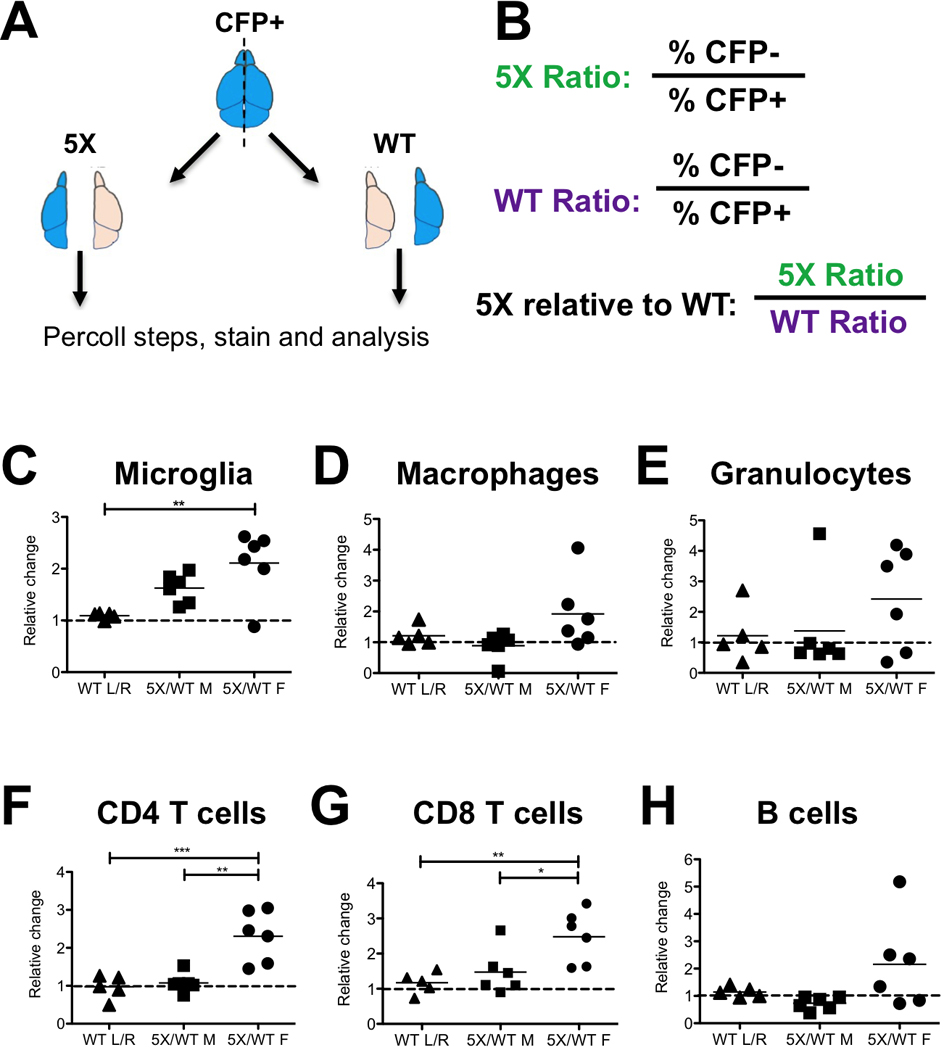
(A) Schematic of FAQT. Fluorescent brains (e.g. CFP) are divided into lobes and mixed with one lobe from a 5X mouse and one lobe of a WT littermate, then processed and analyzed by flow cytometry. After FACS analysis, immune cell populations were each gated and then separated into CFP+ and CFP- fractions. See gating strategy for each immune cell type in Figure S5.(B) Quantification strategy. The ratio of CFP- to CFP+ cells was calculated for both the 5X and WT pair, then was used to calculate the ratio of 5X to WT cells (5X:WT), canceling out the CFP+ spiking cells. (C-H) For each immune cell population, the leftmost column shows the relative ratio of immune cells between the left and right lobes of a WT brain (WT L/R), which represents the WT lobe-to-lobe control. The 5X:WT ratio of each pair of 5X/WT lobes is plotted, showing male pairs (middle) and female pairs (right). The value is the ratio of 5X immune cells relative to WT (which is set to one and indicated by the dashed line). Statistical analysis (one-way ANOVA, Tukey’s multiple comparison test) compares the three groups. Asterisks represent a p-value of <0.05.
We used this technique to measure the relative abundance of several immune populations between the brains of WT and 5X littermates at approximately 12 months of age. We separately analyzed male and female brains due to the known differences in AD pathology between the genders in this mouse model (Oakley et al., 2006; Sadleir et al., 2015). In the males, the differences between WT and 5X brains were relatively minor for most populations and were not statistically significant compared to the WT lobe-to-lobe control. In the female 5X brains, where pathology is greater, there was a significant increase in microglia, but not in macrophages or granulocytes, which were slightly elevated but not statistically significant (Figure 5C-5E). Furthermore, we saw a statistically significant increase in CD4+ and CD8+ T cell populations compared to either the WT only lobe-to-lobe ratios or the male 5X:WT ratios (Figure 5F-G). B cells appeared elevated in 5X females but were not statistically significant (Figure 5H). Using CD11a allowed us to determine that a majority of the CD45hi cells in AD brains were in fact microglia that also upregulated CD11b. Using CD11a and FAQT, we show evidence of microglial proliferation in AD brains, with no significant infiltration of peripheral myeloid cells.
DISCUSSION
In this study, we demonstrate the utility of using CD11a as a marker to distinguish healthy, activated, and Alzheimer’s disease-associated microglia from CD11a+ infiltrating peripheral immune cells. As CD11a is a negative marker for microglia, it can be combined with existing positive markers (e.g. Tmem119, P2ry12) to enhance microglia/monocyte distinction, or be used in lieu of markers such as CD45 to simplify microglia identification. When combined with just CD11b, microglia can be readily identified as CD11b+ and CD11a-. CD11a overcomes several limitations of the CD45 vs CD11b gating strategy. First, we observed no significant upregulation of CD11a by activated microglia in our LPS-induced inflammation model, which is not the case for CD45. Second, we determined that microglia in two different AD mouse models do not upregulate CD11a, unlike CD45, even at ages with peak plaque burden. Thus, using CD11a, along with CD11b, for flow cytometry will allow isolation of microglia in the CD11a- fraction and peripheral myeloid cells in the CD11a+ fraction. While CD11a is excellent in distinguishing microglia and infiltrating myeloid cells in AD models, we showed that its expression on microglia was upregulated in the chronic stages of Toxoplasma gondii infection. Thus, the utility of CD11a in distinguishing microglia from peripheral myeloid populations must be tested on a case-by-case basis. The signaling events that lead to LFA1 upregulation on microglia still remain to be investigated. As LFA1 is also important for antigen presentation, it is possible that LFA1 upregulation on microglia may play a role in facilitating a T cell response in the brain, as T cell-mediated immunity is well-established for T. gondii infection.
Using CD11a as a marker has allowed us to address outstanding questions regarding the roles of microglia versus infiltrating monocytes in AD pathology. One such question regards the identity of the myeloid cells that surround Aß plaques. We have determined that PAM cells are CD11a-, and therefore our findings support the notion that PAM cells are microglia, rather than infiltrating monocytes recruited to plaques. While it remains formally possible that monocytes, and subsequent macrophages, recruited to plaques eventually downregulate CD11a, we saw no evidence of this in our transplantation system, despite the fact that we analyzed 5X brains one year after transplantation. Although nearly 100% of the BM-derived cells were RFP+ or GFP+, we saw no fluorescent cells clustered around the plaques through immunofluorescent staining. Additionally, all fluorescent cells we observed in the brain were CD11a+ through flow cytometry. Together, our data strongly supports the notion that PAM cells are entirely composed of microglia, and not peripheral macrophages.
Our findings align with previous studies supporting microglia as the sole players in an AD CNS. Such studies have shown no significant infiltration based on lack of BBB disruption (Bien-Ly et al., 2015). However, previous studies replacing the peripheral immune system by transplanting reporter-expressing HSCs have shown evidence of bone marrow-derived monocytes infiltrating the CNS in AD models (Simard et al 2006; Mildner et al., 2007). However, HSC transplantation in adult mice requires high doses of irradiation, which can alter the brain environment by activating microglia and disrupting the BBB. We circumvented the artifacts of high dose irradiation by transplanting 5X and WT littermates as neonates with fluorescently labeled, ckit-enriched bone marrow cells. This approach allowed us to attain high levels of chimerism with much lower doses of irradiation (≤3.5 Gy). Additionally, it allowed substantial time for BBB recovery: 6–8 weeks for LPS experiments or one full year for AD experiments and analysis. These findings suggest that neonatal transplantation allows the BBB to remain intact during the onset and progression of the disease and that the lack of infiltration is physiologically relevant.
A second question we attempted to address using CD11a was to what extent do peripheral immune cells infiltrate the brain in AD? By analyzing CD11a expression in 5X mice, we saw little evidence of a robust infiltrating monocyte population in 5X brains compared to WT, even after one year. The abundant CD45hi population in 5X brains was CD11a-, suggesting they are microglia. Furthermore, in the transplantation system, these CD45hi cells were also RFP-, suggesting they are not BM-derived. Together, this strongly argues against a massive influx of monocytes into the brain but does not rule out mild increases or gender-specific differences in infiltrating populations.
To provide a sensitive method of quantifying the abundance of immune populations in the brain, we developed a technique called FAQT (Fluorescence-Assisted Quantification Technique). Pairs of WT and 5X brains are spiked with an equal quantity of fluorescently-labeled brain tissue, then after flow cytometry analysis, each population is normalized to the fluorescent control. As long as we can assume that the abundance of each population is consistent between the right lobe and left lobe of the fluorescent mice, the age, gender, or strain of the spiking brains is irrelevant after normalization. This makes it easy to add fluorescent brains to any brain prep to allow comparison between any two mice. This method is preferable to adding fluorescent counting beads, which would either be lost during Percoll processing if added before, or neglect sample-specific differences in cell yield if added before analysis. Using this strategy, we were able to quantify and isolate gender-specific differences in peripheral immune cell populations of all aged 5X mice relative to their WT littermates. Aged male 5X mice showed no significant increases in any cell populations. Whereas peripheral myeloid populations in the females were only mildly increased, T- lymphocyte infiltration was significantly increased in 5X female brains relative to WT as well as their male counterparts. A significant increase in T-lymphocytes supports a role for the adaptive immune system in AD, as we have previously shown using an AD mouse model that lacks an adaptive immune system (Marsh et al., 2016). Moreover, FAQT can be useful in a variety of neuroinflammatory contexts to accurately quantify infiltrating immune cell populations.
While there is strong evidence that PAM cells are CD11a negative microglia in mouse models of AD, whether this finding translates to human AD patients has yet to be determined. It is known that CD11a is expressed in peripheral human immune cells (Watanabe et al., 1998). A recent microglial gene-expression study found CD11a is not expressed in brain tissues resected during surgery (Gosselin et al., 2017). We are currently investigating whether human peripheral cells infiltrate the parenchyma and cluster around Aß plaques, or whether microglia are the main players in human AD, as seen in mouse models of AD.
In this study, we have established CD11a as a stable and reliable marker to distinguish microglia and peripheral myeloid populations in the brain. This commercially available marker is inexpensive and widely available in a variety of fluorochromes. For immunofluorescent staining of fixed brain sections, we highly recommend tyramide signal amplification to greatly improve signal intensity. While CD11a expression must be validated for each neuroinflammatory context prior to its use, we find that in many contexts (e.g. LPS, 5xFAD, Arc48), but not all (e.g. Toxoplasma infection), CD11a dramatically improves the ability to distinguish microglia from other infiltrating immune cell types. Therefore, we contend this marker should be included in the neuroimmunologist’s standard toolkit.
Supplementary Material
ACKNOWLEGMENTS
The authors would like to thank Dr. Andrea Tenner at UC Irvine for donating the Arctic48 mice and Dr. Kim Green for donating additional 5xFAD mice. We thank Yasamine Ghorbanian, Dr. Alborz Karimzadeh, Tannaz Faal, and Erika Varady of Dr. Inlay’s lab, as well as previous and current members of Dr. Blurton-Jones’ lab for continuing support and helpful discussions throughout the study, and Dr. Jun Seita for assistance with the Gene Expression Commons microarray analysis. The authors would also like to thank Dr. Jennifer Atwood at the Institute for Immunology for assistance with imaging flow cytometry, and Karin Grathwohl and Vanessa Scarfone from the Sue and Bill Gross Stem Cell Research Center (SCRC) flow cytometry core (supported by California Institute for Regenerative Medicine grant CL1–00520-1.2). This study was supported by National Institutes of Health grants R03 NS099969 and R56 HL133656 (to M.A.I.), R01 AG055524 and R01 AG048099 (to M.B.J.). L.LM. was supported by NIH T32 NS 082174 through the SCRC. A.K.S. and C.A.S.L were supported by NIH T32 AI 060573 through the Institute for Immunology at UC Irvine.
REFERENCES
- Baik SH, Kang S, Son SM, & Mook Jung I (2016). Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia, 64(12), 2274–2290. [DOI] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, … Barres BA (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences, 113(12), E1738–E1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, … Barres BA (2018). A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron, 98(6), 1170–1183.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien-Ly N, Boswell CA, Jeet S, Beach TG, Hoyte K, Luk W, … & DeVoss J (2015). Lack of widespread BBB disruption in Alzheimer’s disease models: focus on therapeutic antibodies. Neuron, 88(2), 289–297. [DOI] [PubMed] [Google Scholar]
- Biswas A, Bruder D, Wolf SA, Jeron A, Mack M, Heimesaat MM, & Dunay IR (2015). Ly6Chigh monocytes control cerebral toxoplasmosis. The Journal of Immunology, 194(7):3223–35. [DOI] [PubMed] [Google Scholar]
- Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, … Joyce JA (2016). Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Reports, 17(9), 2445–2459.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, … & Fanek Z (2014). Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nature neuroscience, 17(1), 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, … & Greter M (2016). Sall1 is a transcriptional regulator defining microglia identity and function. Nature immunology, 17(12), 1397–1406 [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, … & Lee JC (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nature neuroscience, 9(7), 917–924. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, & Lo D (1998). Mature microglia resemble immature antigen‐presenting cells. Glia, 22(1), 72–85. [DOI] [PubMed] [Google Scholar]
- Cazareth J, Guyon A, Heurteaux C, Chabry J, & Petit-Paitel A (2014). Molecular and cellular neuroinflammatory status of mouse brain after systemic lipopolysaccharide challenge: importance of CCR2/CCL2 signaling. Journal of neuroinflammation, 11(1), 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng IH, Palop JJ, Esposito LA, Bien-Ly N, Yan F, & Mucke L (2004). Aggressive amyloidosis in mice expressing human amyloid peptides with the Arctic mutation. Nature medicine, 10(11), 1190–1192. [DOI] [PubMed] [Google Scholar]
- Fathman JW, Fernhoff NB, Seita J, Chao C, Scarfone VM, Weissman IL, & Inlay MA (2014). Upregulation of CD11A on hematopoietic stem cells denotes the loss of long-term reconstitution potential. Stem cell reports, 3(5), 707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, & Sedgwick JD (1995). Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. The Journal of Immunology, 154(9), 4309–4321. [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JC, Sajti E, … & Pena M (2017). An environment-dependent transcriptional network specifies human microglia identity. Science, 356(6344), eaal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Lelios I, & Croxford AL (2015). Microglia versus myeloid cell nomenclature during brain inflammation. Frontiers in immunology, 6:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, & Julius D (2006). The P2Y 12 receptor regulates microglial activation by extracellular nucleotides. Nature neuroscience, 9(12), 1512. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, & Littman DR (2000). Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Molecular and cellular biology, 20(11), 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren Shaul H, Spinrad A, Weiner A, Matcovitch Natan O, Dvir Szternfeld R, Ulland TK, … Amit I (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell, 169(7), 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Kim SK, Karasov A, & Boothroyd JC (2007). Bradyzoite-specific surface antigen SRS9 plays a role in maintaining Toxoplasma gondii persistence in the brain and in host control of parasite replication in the intestine. Infection and immunity, 75(4), 1626–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H, Kobayashi M, Kunisawa T, Imai K, Sayo A, Malissen B, … & Kiyama H (2017). Siglec‐H is a microglia‐specific marker that discriminates microglia from CNS‐associated macrophages and CNS‐infiltrating monocytes. Glia, 65(12), 1927–1943. [DOI] [PubMed] [Google Scholar]
- Lampron A, Lessard M, & Rivest S (2012). Effects of myeloablation, peripheral chimerism, and whole-body irradiation on the entry of bone marrow-derived cells into the brain. Cell transplantation, 21(6), 1149–1159. [DOI] [PubMed] [Google Scholar]
- Lum AF, Green CE, Lee GR, Staunton DE, & Simon SI (2002). Dynamic regulation of LFA-1 activation and neutrophil arrest on intercellular adhesion molecule 1 (ICAM-1) in shear flow. Journal of Biological Chemistry, 277(23), 20660–20670. [DOI] [PubMed] [Google Scholar]
- Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H, … Blurton Jones, M. (2016). The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proceedings of the National Academy of Sciences, 113(9), E1316–E1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel F, Kaiser N, Haehnel S, Rapp F, Patties I, Schöneberg N, … & Bechmann I (2018). Impact of X‐irradiation on microglia. Glia, 66(1), 15–33. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, … & Prinz M (2007). Microglia in the adult brain arise from Ly-6C hi CCR2+ monocytes only under defined host conditions. Nature neuroscience, 10(12), 1544–1553. [DOI] [PubMed] [Google Scholar]
- Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, & Cardona AE (2012). The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. The Journal of Immunology, 188(1), 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks CR, Freiberg BA, Kupfer H, Sciaky N, & Kupfer A (1998). Three-dimensional segregation of supramolecular activation clusters in T cells. Nature, 395(6697), 82. [DOI] [PubMed] [Google Scholar]
- Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, … Becher B (2018). High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity, 48(2), 380–395.e6. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, & Luo L (2007). A global double‐fluorescent Cre reporter mouse. genesis, 45(9), 593–605. [DOI] [PubMed] [Google Scholar]
- Nayak D, Zinselmeyer BH, Corps KN, & McGavern DB (2012). In vivo dynamics of innate immune sentinels in the CNS. Intravital, 1(2), 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, & Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science, 308(5726), 1314–1318. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, … & Berry R (2006). Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. Journal of Neuroscience, 26(40), 10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, … & Ragozzino D (2011). Synaptic pruning by microglia is necessary for normal brain development. Science, 333(6048), 1456–1458. [DOI] [PubMed] [Google Scholar]
- Parkhurst C, Yang G, Ninan I, Savas J, Yates J, Lafaille J, … Gan W (2013). Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell, 155(7), 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham TH, Baluk P, Xu Y, Grigorova I, Bankovich AJ, Pappu R, … & Cyster JG (2010). Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. Journal of Experimental Medicine, 207(1), 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadleir KR, Eimer WA, Cole SL, & Vassar R (2015). Aβ reduction in BACE1 heterozygous null 5XFAD mice is associated with transgenic APP level. Molecular neurodegeneration, 10(1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, … & Charo IF (2010). Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PloS one, 5(10), e13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, … & Stevens B (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74(4), 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, & Ter Meulen V (1991). Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proceedings of the National Academy of Sciences, 88(16), 7438–7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seita J, Sahoo D, Rossi DJ, Bhattacharya D, Serwold T, Inlay MA, … & Weissman IL (2012). Gene Expression Commons: an open platform for absolute gene expression profiling. PloS one, 7(7), e40321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, … & Maletic Savatic, M. (2010). Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell stem cell, 7(4), 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, & Rivest S (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron, 49(4), 489–502. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, … & Sher A (2007). The classical complement cascade mediates CNS synapse elimination. Cell, 131(6), 1164–1178. [DOI] [PubMed] [Google Scholar]
- Ueno H, & Weissman IL (2006). Clonal analysis of mouse development reveals a polyclonal origin for yolk sac blood islands. Developmental cell, 11(4), 519–533. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, … & Cella M (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. Journal of Experimental Medicine, jem-20151948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, & Fan J (1998). Atherosclerosis and inflammation: mononuclear cell recruitment and adhesion molecules with reference to the implication of ICAM-1/LFA-1 pathway in atherogenesis. International Journal of Cardiology, 66, S45–S53. [DOI] [PubMed] [Google Scholar]
- Yoder MC, & Hiatt K (1997). Engraftment of embryonic hematopoietic cells in conditioned newborn recipients. Blood, 89(6), 2176–2183. [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, … & Hume DA (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity, 38(1), 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.