

Abstract
We report the synthesis and physicochemical analysis of mixed-substituent dipeptide-based polyphosphazene polymers, poly[(glycineethylglycinato)x(phenylphenoxy)yphosphazene] (PNGEGxPhPhy) and poly[(ethylphenylalanato)x(glycineethylglycinato)yphosphazene] (PNEPAxGEGy), using glycylglycine ethyl ester (GEG) as the primary substituent side group and cosubstituting with phenylphenol (PhPh) and phenylalanine ethyl ester (EPA), respectively. The suitability of the cosubstituted polyphosphazenes to regenerative engineering was evaluated. The physicochemical evaluation revealed that the molecular weights, glass transition temperatures, hydrophilicity, and mechanical properties could be modulated by varying the compositions of the side groups to obtain a variety of properties. The PNEPA25GEG75 and PNGEG75PhPh25 polymers exhibited the most promising physicochemical properties. These two polymers were further subjected to in vitro hydrolysis and cell proliferation studies using poly(lactic-co-glycolic acid) (PLAGA) as a control. The hydrolysis experiments revealed that the two polymers hydrolyzed to near-neutral pH media (~5.3 to 7.0) in a relatively slow fashion, whereas a pH value as low as 2.2 was obtained for the PLAGA media over 12 weeks of degradation study. Furthermore, the two polymers showed continuous MC3T3 cell proliferation and growth in comparison to PLAGA over a 21-day culture period. These findings establish that cosubstitution of different side groups of polyphosphazenes and exploitation of the hydrogen-bonding capacity of peptide bonds in GEG offer a flexible tool that can be employed to make new and fascinating polymeric biomaterials with different and tailored properties that can suit different regenerative needs.
Keywords: regenerative biomaterials, biodegradable polymers, polyphosphazenes, structure–properties relationships, polymer synthesis, physicochemical evaluation
Graphical Abstract
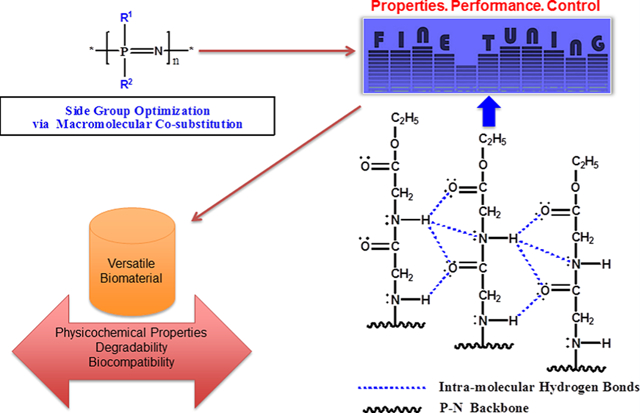
INTRODUCTION
Synthetic biodegradable polymers have found promising applications in tissue regeneration.1–5 This class of polymers can be employed in developing three-dimensional porous structures as scaffolds for regenerative engineering.2,3,6 Because biological tissues are complex structures with unique cell composition, chemistry, and mechanical properties,7 the requirements for tissue regeneration applications become unpredictable, and each of these applications demands materials with specific physical, chemical, biomechanical, and degradation properties to provide a positive outcome.7–11 Thus, it becomes crucial to customize polymers during synthesis to meet specific requirements for regenerative engineering.8 Poly(organophosphazenes) (PPHOS) have received a great deal of attention in this regard due to their outstanding synthetic flexibility and ability to exhibit a wide range of properties through the optimization of side group chemistry.6,12–17 PPHOS are inorganic–organic hybrid polymers with a unique inorganic backbone of alternating phosphorus and nitrogen atoms with two organic units attached to the phosphorus.6,16,18 Various functionalities can be linked to the backbone to modify and tailor the physicochemical properties to specific applications.6,13,19–23 The nature and compositions of the side groups can have tremendous effects on the overall properties of the polymeric products.13 For tissue regeneration, hydrolytically active side groups are utilized because of their ability to sensitize the polymers to hydrolysis, which allow them to break down into nontoxic small molecules.6,14,24 A few numbers of biodegradable polyphosphazene, particularly those with amino acid esters, glucosyl, glyceryl, glycolate, lactate, and imidazole side groups, have been extensively investigated as matrices for bone tissue regeneration.6,14,24 These side groups have been shown to sensitize the PPHOS backbone to hydrolysis. The labile nature of phosphorus–chlorine bonds in poly(dichlorophosphazene) (PDCP) has provided a versatile platform for fine-tuning and controlling the physicochemical properties to yield a rich library of polymeric biomaterials useful in biomedical applications such as regenerative engineering, tissue engineering, drug delivery systems, and so on.6,14,19,24–26 In our previous study, the first-generation biodegradable polyphosphazenes for tissue regeneration were designed with amino acid esters as side groups.27,28 The buffering ability and osteocompatibility of various amino acid ester PPHOS were demonstrated.27–29 When blended with PLAGA, they showed the feasibility of neutralizing the acidic degradation products of PLAGA. However, the blends of amino acid ester-substituted PPHOS and PLAGA were mostly partially miscible which adversely affected the mechanical properties. In the second generation, dipeptide side groups were employed to overcome the miscibility issues.30,31 The dipeptide-substituted PPHOS was highly osteocompatible with strong hydrogen-bonding ability.15,30,31 It formed completely miscible blends with PLAGA and exhibited unique degradation behavior by which the polymer blend changed from a coherent film to an assemblage of microspheres with interconnected porous structures upon degradation.31 However, the mechanical properties were not appropriate for tissue regeneration. The goal of this study was to develop a new generation of polyphosphazenes with high strength by optimizing the properties and evaluating their suitability as potential biomaterials for regenerative engineering. Our design strategy narrowly focused on designing dipeptide-based mixed-substituent polyphosphazenes and cosubstituting them with specific side groups such as phenylalanine ethyl ester and phenylphenoxy moieties at different ratios. Phenylalanine ethyl ester was chosen for its biocompatibility and its hydrophobic characteristics due to the presence of the aromatic ring in its chemical structure. On the other hand, the phenylphenol selection was based on the results of our previous studies that exploited its hydrophobic characteristics in tuning the degradation mechanism of the polymer.31 We hypothesized that the cosubstitution of polyphosphazenes using glycylglycine ethyl ester as the main substituent and phenylalanine ethyl ester or phenylphenol as cosubstituent at different compositions of the side groups would yield PPHOS polymers with a wide range of physicochemical and biological properties that can meet different regenerative requirements. Equimolar quantities of glycylglycine ethyl ester and phenylphenol (50:50) were employed in our previous study.30 In this study, we aimed to vary the molar composition of glycylglycine ethyl ester and phenylphenol (100:0, 25:75, 50:50, 75:25, and 0:100) for the optimization of their properties. Moreover, we also synthesized glycylglycine ethyl ester and phenylalanine-cosubstituted PPHOS at different side group ratios (100:0, 25:75, 50:50, 75:25, and 0:100) and assessed their physicochemical properties as regards to their regenerative engineering potentials.
EXPERIMENTAL SECTION
Materials.
All syntheses were performed using standard Schlenk-line techniques under a dry argon atmosphere. Glassware was first treated in acid and base baths, then dried for 24 h in an oven at 111 °C, and flame-dried before use. Hexachlorocyclotriphosphazene (HCCTP) was purchased from Sigma-Aldrich and purified before use. Tetrahydrofuran (THF) and triethylamine (TEA) were dried using solvent purification columns. Hexane (Sigma-Aldrich), heptane (Sigma-Aldrich), sodium hydride (60% w/w in material oil, Sigma-Aldrich), glycylglycine ethyl ester (Mw = 196.63 g/mol, CHEM-IMPEX), l-phenylalanine ethyl ester hydrochloride (Mw = 229.70 g/mol, Sigma-Aldrich), 4-phenylphenol (170.21 g/mol, Sigma-Aldrich), and PLAGA (MW = 60800 g/mol) were used as received.
Purification of Hexachlorocyclotriphosphazene.
The commercial HCCTP contains trimer, tetramer, and traces of physical impurities. Recrystallization and sublimation were used to obtain a pure trimer. First, recrystallization was performed by dissolving the commercial HCCTP in hot hexane at 60 °C or hot heptane at 80 °C and was subsequently filtered to remove traces of metals and other physical impurities. The obtained filtrate (free of physical contaminants) was allowed to cool to room temperature for the sample to recrystallize out of the solvent. The recrystallized HCCTP was dried in a vacuum oven for 24 h. In the second step, slow sublimation under vacuum at ~55 °C was performed to separate the tetramer from the trimer. The sublimate (trimer) was collected for the next step of action, and the leftover (tetramer) was discarded. Phosphorus-31 nuclear magnetic resonance (31P NMR) analysis was used to confirm that the obtained sample was a pure trimer (Figure S1).
Thermal Ring-Opening Polymerization.
Poly(organophosphazenes) were synthesized in a two-step reaction process: In the first step, PDCP was made from pure HCCTP using the thermal ring-opening polymerization method. The procedures were as follows: The sublimed and recrystallized HCCTP was weighed out and discharged in a glass tube. The tube containing the sample was evacuated with a vacuum pump and sealed with a flame. The sealed tube with the sample was then placed in an oven (equipped with a mechanical shaker) under 250 °C. The reaction was monitored periodically to check the extent of polymerization, and the polymerization was done when the chemical was no longer free-flowing. Stopping the process at this point is very important to prevent cross-linking. Figure S2 shows the reaction mechanism of the ring-opening polymerization of HCCTP to air-sensitive PDCP.
Small Molecule Model Reaction.
The model reactions between the trimer and each of the selected three organic nucleophilic groups (phenylalanine ethyl ester, glycylglycine ethyl ester, and phenylphenol) were performed to ascertain the feasibility of having a smooth synthetic substitution of the chlorine atoms during the macromolecular substitution of PDCP. An ideal nucleophilic group used as side group will smoothly react with cyclic trimeric hexachlorocyclotriphosphazene, substituting all its chlorine atoms, and would exhibit a single 31P NMR peak.18 The reactions of the selected side groups with cyclic trimeric hexachlorocyclotriphosphazene progressed with ease, and 31P NMR analysis identified the individual peak for each of the groups. The lack of complication in the model reactions demonstrated that the side groups could be successfully linked to the polyphosphazene backbone.
Nucleophilic Macromolecular Substitution.
Then the three selected organic substituents, phenylalanine ethyl ester, glycylglycine ethyl ester, and phenylphenol groups, were used to replace the reactive chlorine atoms of PDCP. Substitutions of two different groups on the reactive PDCP were achieved by sequential substitution, allowing the bulkier group to react first followed by the smaller group in solution phase during chlorine substitution. A rigorous synthetic protocol was developed, and the synthesis procedures for 10 PPHOS polymers were described briefly as follows.
Synthesis of Poly[(ethylphenylalanato)x(glycineethylglycinato)y]phosphazene (PNEPAxGEGy).
PNEPAxGEGy with three different EPA:GEG side group compositions (75:25, 50:50, and 25:75) were synthesized under nitrogen via a Schlenk line (Figure S3 and Table 1). PDCP was dissolved in dry THF (for complete dissolution, it was allowed to stir for 24 h at room temperature). In a separate glass vessel, phenylalanine ethyl ester was suspended in THF and triethylamine was added. The mixture was refluxed for 24 h, then filtered, and added to the PDCP solution to replace part of the chlorine atoms. In a different vessel, glycylglycine ethyl ester hydrochloride was suspended in THF and triethylamine was added. This mixture was refluxed for 24 h, then filtered, and added to the polymer mixture to replace the remaining chlorine atoms. The reaction mixture was refluxed for 72 h, and complete substitution of the chlorine atoms was confirmed by using 31P NMR. The polymer solution was centrifuged, filtered, concentrated using a rotary evaporator, and then precipitated in hexanes. Subsequent precipitations into hexanes were used to purify the polymer further. The product was dried under vacuum to remove all solvents and then stored under argon for further use.
Table 1.
Ten Different Polyphosphazene Copolymers Synthesized with Various Side Group Compositions
| phenylalanine ethyl ester:glycylglycine ethyl ester | glycylglycine ethyl ester:phenylphenol |
|---|---|
| 100%:0% | 0%:100% |
| 75%:25% | 25%:75% |
| 50%:50% | 50%:50% |
| 25%:75% | 75%:25% |
| 0%:100% | 100%:0% |
Synthesis of Poly[(glycineethylglycinato)100phosphazene] (PNGEG100).
For the synthesis of PNGEG100 shown in Figure S4, PDCP was dissolved in THF in a separate vessel. An equimolar quantity of glycylglycine ethyl ester was suspended in THF and trimethylamine. The glycylglycine ethyl ester suspension was refluxed for 24 h, filtered, and then added dropwise to the polymer solution via an addition funnel. The resultant polymer solution was stirred for 72 h under reflux, placed in a centrifuge, filtered, and precipitated into hexanes. 31P NMR was used to characterize the polymer during and after substitution.
Synthesis of Poly[(ethylphenylalanato)100phosphazene] (PNEPA100).
For the synthesis of PNEPA100, PDCP was dissolved in THF. An equimolar quantity of phenylalanine ethyl ester was suspended in THF and triethylamine. The phenylalanine ethyl ester suspension was refluxed for 24 h, filtered, and then added dropwise to the polymer solution. The resultant polymer solution was stirred for 72 h under reflux, placed in a centrifuge, filtered, and precipitated into hexanes (Figure S5). 31P NMR was used to characterize the polymer during and after substitution.
Synthesis of Poly[(glycineethylglycinato)x(phenylphenoxy)yphosphazene] (PNGEGxPhPhy).
PDCP was dissolved in dry THF. In a separate reaction vessel, phenylphenol was added to a suspension of sodium hydride (60% dispersion in mineral oil) in THF over a period of 30 min and refluxed for 24 h. The sodium salt of phenylphenol was allowed to react with PDCP in THF to replace part of the chlorine atoms. The partially substituted polymers were then allowed to react with a large excess of glycylglycine ethyl ester and triethylamine to replace the remaining chlorine atoms (Figure S6). The polymerization resulted in fully substituted polyphosphazenes confirmed by 31P NMR (Figure S3). Then centrifugation was carried (3800 rpm for 10 min) to remove the residual salts (triethylammonium chloride). The polymers were concentrated by rotary evaporator and purified by repeated precipitations in hexanes (five times). The pure polymers were dried under vacuum and stored under argon before characterization.
Synthesis of Poly[(phenylphenoxy)100phosphazene] (PNPhPh100).
PDCP was dissolved in dry THF. An equimolar quantity of 4-phenylphenol was added to a suspension of sodium hydride (60% dispersion in mineral oil) in THF over a period of 30 min and refluxed for 24 h. The sodium salt of phenylphenol was allowed to react with PDCP in THF for 72 h to replace all of the chlorine atoms. The resulting polymer was centrifuged and filtered to remove the NaCl byproduct. The polymer filtrate was then concentrated using a rotary evaporator and purified via several precipitations in hexane (Figure S7).
Characterization.
Chemical Structural Analysis.
The NMR analysis was performed on a Bruker Avance ARX-300 spectrometer using deuterated THF as the solvent. 31P and 1H NMR were specifically performed on the obtained products to confirm the complete backbone substitutions with the intended side groups.
A Fourier-transform infrared (FTIR) spectrometer was used to identify the formation of new chemical bonds and the participation of the organic side groups in the chlorine substitutions. The analysis was operated in the wavelength range of 500–4000 cm−1 with a resolution of five wavenumbers and average of 16 scans.
Molecular weights and molecular weight distributions of the polymers were measured using gel permeation chromatography (GPC). Samples for GPC analysis were prepared in concentrations of ~1% (w/v) in THF and eluted at a rate of 1.5 mL/min through a size exclusion column calibrated with polystyrene standards with low polydispersities. Two columns (with diameter of 7.8 mm and 30 mm long) were used in series, and the relative (standardized) calibration method was employed against the narrowly distributed polystyrene.
Thermal Analysis.
The glass transition temperatures (Tg) of the polymer samples were determined by differential scanning calorimetry (DSC) using a TA Instruments DSC Q250 unit with thermal universal analysis software. Briefly, polymer samples (~10 mg) were heated from −50 to 150 °C at a heating rate of 10 °C/min under 50 mL/min nitrogen, and the results were analyzed with TA universal analysis. The Tg was determined from the half-height point of the heat capacity change in the thermograms, and the thermograms of the third heating cycle were used for the determination of Tg. Thermal stability and decomposition temperatures of the polymers were evaluated using thermal gravimetry analysis (TGA). The temperatures at which the polymers experience significant weight loss steps were determined. The peak maximum and char yield were also measured.
Film Casting.
The weight of ~1 g of each polymer was dissolved in 20 mL of THF and then poured into Teflon-coated Petri dishes. The solvent was allowed to evaporate slowly for 48 h. The films were further dried under high vacuum for 48 h. Films with thickness around 0.2 mm were obtained.
Surface Analysis.
The contact angles of droplets of distilled deionized (DI) water on the surface of the polymer films were measured using a digital contact angle measurement system equipped with a CCD camera (CAM 100 series, KSV Instruments). Temporal images of the water droplet on different polymer films were recorded. From these images, the contact angle values were obtained.
Mechanical Properties Analysis.
Tensile tests were performed on the “dog-bone shape” of 13.2 mm length, 6.6 mm width, and 0.1 mm thickness polymer strips cut from polymer films using the universal tensile testing machine (UTM) (3365, Instron) with a crosshead speed of 2 mm/min at ambient temperature and humidity. The samples were elongated to failure. The tensile testing was performed using at least five specimens for each sample and in accordance with ASTM D882–12. The ultimate tensile strength and Young’s modulus were calculated from the stress–strain curves.
Dynamic Mechanical Thermal Analysis.
The dynamic mechanical properties of the polymer samples were analyzed by using dynamic mechanical analysis (DMA, Q800). The dimensions of the specimens were L = 12.81 mm, W = 6.27 mm, and T = 0.13 mm. Two different methods were used to carry out this analysis: temperature and force ramps. For the temperature ramp, the polymer films were heated from 36 to 125 °C (below the decomposition temperature) with a heating increment of 3 °C/min. The strain amplitude was fixed to 0.1%, whose value was within the linear viscoelastic deformation, and the storage modulus, the loss modulus, and tan delta were determined. For the force ramp, stress–strain curves were generated at a constant temperature of 36 °C, and the ultimate tensile strength and Young’s modulus were calculated.
In vitro Hydrolysis.
The polymer disks with dimensions of 0.5 mm × 10 mm (T × D) and weighing ~100 mg each were incubated in 10 mL of PBS at pH 7.4. The vials were maintained at 37 °C in a water bath shaker for 12 weeks at 250 rpm. At specific time points (2, 4, 7, 10, and 12 weeks), the polymers were removed from PBS and were dried under vacuum for 2 weeks. The results were reported as percentage mass remaining versus time as calculated from the equation
where wt is the dry weight of the matrix at each predetermined time points and w0 is the initial matrix dry weight. Three samples were weighed from each group. The percentage molecular weight remaining was also determined. For each time point, the media were also collected, and the pH values were recorded by a pH meter. Attenuated total reflection infrared (ATR-IR) spectra of degrading polymers were recorded using an FT-IR spectrometer coupled with an ATR accessory at a resolution of 4 cm−1 and with an average of 16 scans.
In vitro Biocompatibility.
Cell seeding: Polymer films with dimensions of 0.1 mm × 10 mm (T × D) were sterilized by immersing in 70% ethanol for 30 min, washed twice with distilled DI water for 15 min, and exposed to UV light for 15 min on each side. Osteoblast-like MC3T3-E1 cells (obtained from the Calvaria of neonatal mouse) are well-known for their use in in vitro osteocompatibility study.32–34 The cell line was purchased from ATCC. The cells were seeded onto the polymers and the PLAGA control at a density of 6 × 104 cells per film. The seeded materials were cultured in α-MEM supplemented with 10% FBS and 1% penicillin–streptomycin and maintained in an incubator at 37 °C with 5% CO2 and 95% humidified air for 1, 3, 7, 14, and 21 days. Cell proliferation: Cell proliferation on the films was quantitatively analyzed with the use of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl-2-(4-sulfophenyl)-2H-tetrazolium) (MTS, Promega) mitochondrial reduction. This assay is based on the ability of metabolically active cells to bioreduce a tetrazolium-based compound, MTS, to a purple formazan product.35 The intensity of the resulting colored solution as measured by the absorbance at 490 nm is directly proportional to the number of viable cells.28,35,36 Cells seeded onto tissue-culture polystyrene (TCPS) were used as a control. Briefly, the films with cultured cells at predetermined time points were washed with phosphate-buffered saline (PBS), transferred into a new 48-well tissue culture plate with a mixture of the culture medium and MTS substrate (5:1), and incubated for 2 h in a humidified atmosphere with 5% CO2 at 37 °C. At the end of incubation, the absorbance of the resulting solution was read at 490 nm using a Tecan spectraFluo Plus plate reader. Live/dead cell viability: The viability of MC3T3 cells on the polymer films was investigated using a live/dead cell viability kit. Briefly, a bright green fluorescence is produced by the interaction of the intracellular esterase of the live cells with calcein AM, whereas a bright red fluorescence is produced upon the binding of ethidium homodimer-1 to nucleic acids of the dead cells with damaged membranes. The polymer films were imaged at 3 and 7 days by using a laser scanning confocal microscope (Zeiss LSCM 880).
Statistical Analysis.
All analyses were run in triplicate or more per sample, and quantitative data were presented as mean ± standard deviation (n ≥ 3). Statistical analysis was performed using Minitab software. The comparisons of the means were performed using a two-sample t test with a significant difference of p < 0.05.
RESULTS AND DISCUSSION
Synthesis.
Yields around 83% were obtained for all polymers under study. As shown in Figures S2, 31P NMR spectra were similar to the one reported for amino acid ester-substituted polyphosphazenes.14 The chemical shift for P–Cl in PDCP is sharp and at about −17 ppm, and that of the P-amino units is very broad and commonly found around 0 ppm. The disappearance of P–Cl in the PDCP peak at δ = −17 ppm and the appearance of a new peak around δ = 0 ppm confirmed the total replacement of the chlorine units by the substituent groups in the polymers. Figures S8a–c show the spectra of partially substituted PDCP during polymerization. These 31P measurements were taken after the macromolecular substitution of the first cosubstituent, and the spectra showed two or more peaks indicating the presence of some P–Cl units in the backbone. The spectra for the polymers were modified into a single broad peak at 0 ppm, after the macromolecular substitutions of the second side groups. The case is opposite for the completely substituted PNGEGxPhPhy groups as the 31P spectra show three peaks corresponding to three different segments of the polymers. The broad peak slightly above 0 ppm corresponds to phosphorus in the backbone bonded with only glycylglycine ethyl ester; the second broad peak slightly below 0 ppm corresponds to phosphorus with both glycylglycine ethyl ester and phenylphenoxy moieties. The third peak at 15 ppm indicates the presence of phosphorus attached with two phenylphenoxy units through an oxygen linkage (Figure S9). As shown in Figures S10a–d, 1H NMR spectra revealed the formation of secondary amine and the establishment of the linkage between the nitrogen of the amino acid ester or peptide ester and phosphorus of the PPHOS backbone.
For the FTIR spectra in Figure S11, peaks of the starting side groups and PDCP prepolymer were modified entirely with the formation of new absorption bands for the polymers. The disappearance of the peaks for primary amines (3268 cm−1) and formation of the weak absorption band for secondary amines (3350 cm−1) confirmed the participation of the terminal amines of the dipeptide esters and amino acid esters in the chlorine substitutions. For the PNGEGxPhPhy spectra, the disappearance of O–H stretch (3398 cm−1) of phenylphenoxy group indicates the formation of oxygen and phosphorus linkage (Figures S11 and S12).
Molecular weight and molecular weight distribution of the polymers were determined with GPC. The polymers were found to have moderate molecular weights (Table 2), and their molecular weight distributions were reasonably narrow (low polydispersity indices). Most of the polymers exhibited monomodal peaks with polydispersity indices varying from 1.6 to 2.9, indicating uniform compositions of the polymer molecules and could have significant effects on degradation. However, the PNEPA100GEG0, PNGEG0PhPh100, and PNGEG25PhPh75 exhibited bimodal peaks due to the aggregation of polymer molecules (Figure 1). The aggregation is presumably as a result of the higher content of bulky aromatic side groups in the PNEPA100GEG0, PNGEG0PhPh100, and PNGEG25PhPh75 and the presence of π–π stacking.37–41
Table 2.
Measurements of Molecular Weight and Molecular Distributions
| ratio (x:y) | Mw (kDa) | Mn (kDa) | Ð |
|---|---|---|---|
| phenylalanine ethyl ester:glycylglycine ethyl ester (PNEPAxGEGy) | |||
| 100%:0% | 127 | 34 | 3.7 |
| 75%:25% | 151 | 80.41 | 1.9 |
| 50%:50% | 112 | 58 | 1.9 |
| 25%:75% | 125 | 78 | 1.6 |
| 0%:100% | 176 | 60 | 2.9 |
| glycylglycine ethyl ester:phenylphenol (PNGEGxPhPhy) | |||
| 0%:100% | 136 | 51 | 2.7 |
| 25%:75% | 125 | 78 | 2.8 |
| 50%:50% | 118 | 68 | 1.7 |
| 75%:25% | 162 | 92 | 1.8 |
| 100%:0% | 176 | 60 | 2.9 |
Figure 1.
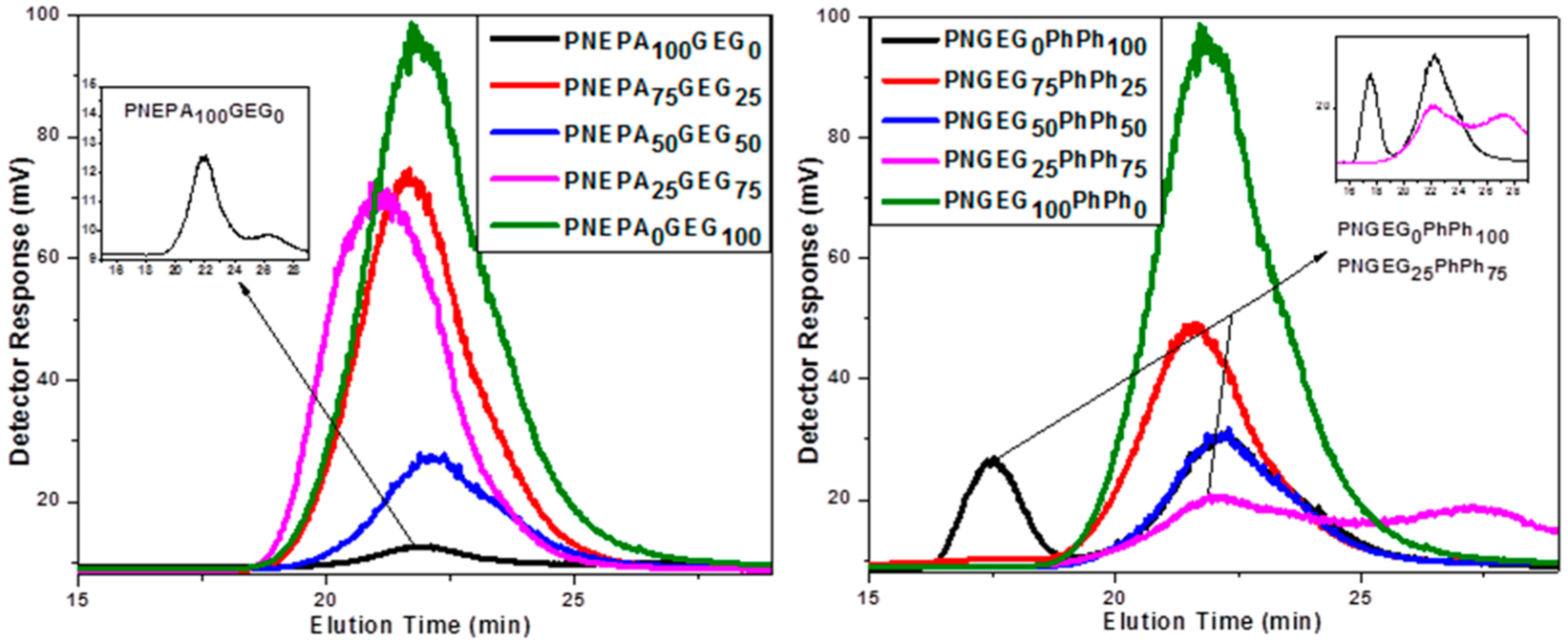
GPC chromatograms of PNEPA-GEG and PNGEG-PhPh polymers with different compositions.
Thermal Analysis.
The inherent backbone flexibility of polyphosphazenes can hinder the development of polymers with high Tg and excellent mechanical properties. However, the DSC results showed that optimizing the side group chemistry (which entails the nature and molar compositions of side groups) will yield polymers with high glass transition temperatures and ultimately desirable mechanical properties. The glass transition temperatures of the polymers are shown in Figure 2. The PNEPAxGEGy and PNGEGxPhPhy polymers have Tgs that are higher than the physiological temperature of 36 °C, suggesting that the average body temperature will not present any material deformation when the polymers are under load. The Tg increased as the molar concentration of glycylglycine ethyl ester was increased. These trends were found to be the same for both PNEPAxGEGy and PNGEGxPhPhy polymers. This is presumably due to the barrier to torsion of polyphosphazene backbone by glycylglycine ethyl ester being more dominant than the ones caused by phenylalanine ethyl ester and phenylphenol. Peptide bonds in the glycylglycine ethyl ester may have allowed more intramolecular hydrogen bond formations in the polymers.
Figure 2.
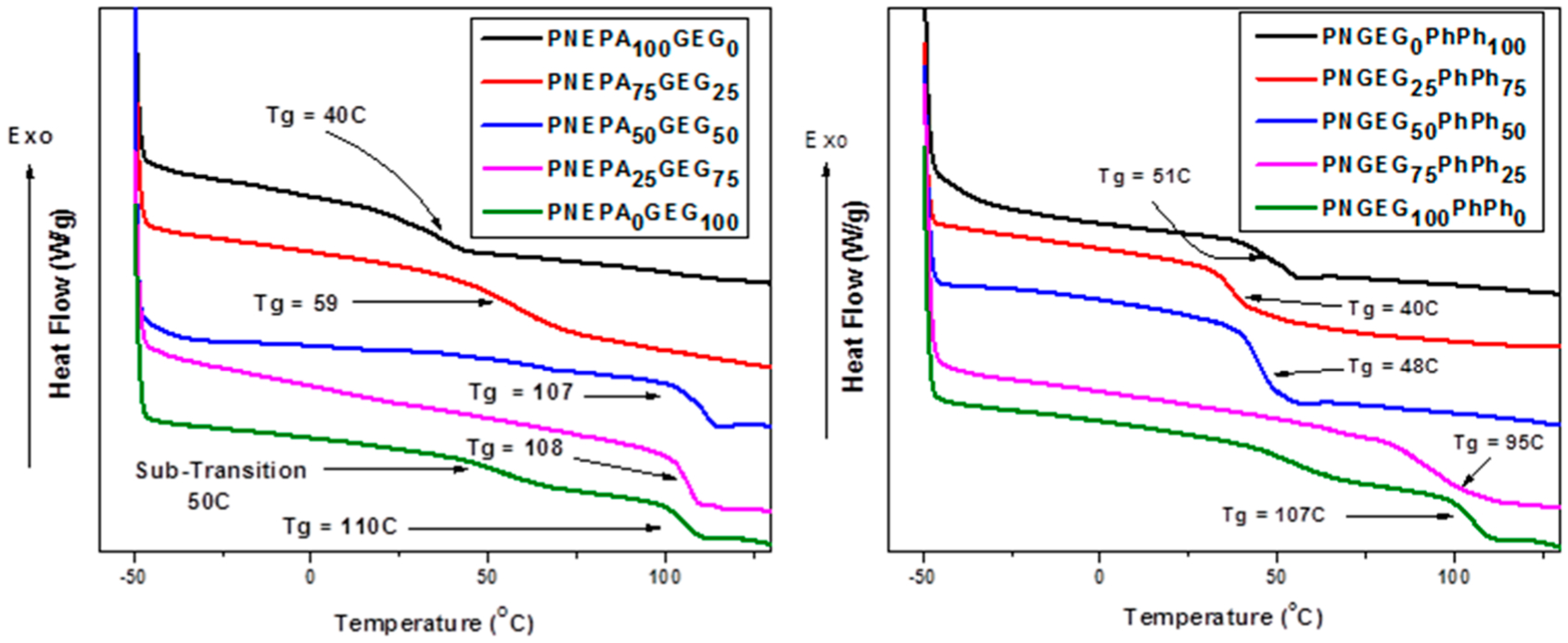
Thermal transitions temperature of the synthesized polyphosphazene-based polymers from DSC thermograms. The results indicate that the polymers have relatively high glass transition temperatures, which are above the physiologically temperature of 36 °C.
The thermal characteristics of the polyphosphazene polymers were further assessed by TGA analyses as shown in Figures S13 and S14. Two significant weight loss steps were observed in the thermal stability examination, and both corresponded to the side groups and the polymer backbone. For instance, the first weight loss step in PNEPA75GEG25 and PNGEG50PhPh50 was due to the decomposition of the side groups at 264 and 240 °C, respectively. The second stage that appeared at higher temperature was due to the decomposition of the polyphosphazene backbone (P=N) at 409 and 390 °C, respectively. After heating to 700 °C, char yields of the PNEPAxGEGy polymers were found to vary from 2.9 to 30%, and those for the PNGEGxPhPhy ranged from 3 to 72%. The presence of aromatic and bulky groups (phenylphenoxy group) caused the higher char yields in PNGEGxPhPhy polymers.
Surface Analysis.
Contact angles are directly related to the hydrophilicity and hydrophobicity of a material surface. The hydrophilicity and hydrophobicity of polymer components are typically utilized to fine-tune degradation rates, and the degradation is triggered or controlled by the hydrolytic sensitivity of the side groups. Material surface is considered hydrophilic if the contact angle is smaller than 90°, and if it is larger than 90°, the surface is deemed to be hydrophobic.
As shown in Table 3, all the polyphosphazene polymers synthesized showed contact angles smaller than 90° except PNGEG0PhPh100 with an average contact angle of 90.5°. This is probably due to the presence of a hydrophobic phenylphenoxy group that was used in 100% side group composition.
Table 3.
Measurements of Contact Angles of PNEPAxGEGy and PNGEGxPhPhy Polymers with Different Compositions
| PNEPAxGEGy | PNGEGxPhPhy | ||
|---|---|---|---|
| composition | av contact angle (deg) | composition | av contact angle (deg) |
| 100%:0% | 88.9 | 0%:100% | 90.5 |
| 75%:25% | 82.8 | 25%:75% | 86.2 |
| 50%:50% | 79.1 | 50%:50% | 82 |
| 25%:75% | 65 | 75%:25% | 75 |
| 0%:100% | 58 | 100%:0% | 58 |
The decrease in contact angles indicated that the hydrophilicity of the materials was increased by increasing the composition of glycylglycine ethyl ester (Figure 3). The increase in the composition of phenylalanine ethyl ester and phenylphenol led to the rise in contact angle, which in turn decreased the hydrophilicity of the polyphosphazenes.
Figure 3.

Photographs of the water droplet taken immediately after water contact showing the effect of change in side group composition on hydrophilicity of PNEPAxGEGy and PNGEGxPhPhy polymers.
Hydrolytically active groups such as glycylglycine ethyl ester would induce hydrolysis within the polyphosphazene backbone, whereas the presence of hydrophobic groups such as phenylphenoxy side groups would inhibit hydrolysis. Amino acid ester with a bulky group just like the phenylalanine ethyl ester used in this study would hydrolyze slower than the one with only hydrogen at its α-position. Therefore, macromolecular cosubstitution of PDCP with a hydrolytically active group and a hydrophobic group will allow the modulation of the degradation pattern to meet specific tissue types and disease states.
Mechanical Analysis.
The testing of tensile properties of biomaterials gives information about the mechanical performance of the new bone scaffolding materials. Mechanical performance is an essential factor that directly affects the success of bone tissue regeneration. For bone regenerative engineering applications, the Young’s modulus and tensile strength of materials should be in the range of the native bone. The average tensile Young’s modulus and ultimate tensile strength of the polymer matrices determined by the stress–strain curves from tensile tests on thin strips of PNEPAxGEGy and PNGEGxPhPhy polymers are shown in Figure 4. The ultimate tensile strengths were found to vary from 5.4 ± 0.3 to12.8 ± 1.9 and from 3.8 ± 0.1 to 11.7 ± 0.6 for PNEPAxGEGy and PNGEGxPhPhy, respectively. The elastic modulus values were found to vary from 325 ± 29 to 1786 ± 37 and from 380 ± 19 to 620 ± 22 for PNEPAxGEGy and PNGEGxPhPhy respectively, indicating that a wide range of mechanical properties can be obtained by optimizing the side group chemistry of cosubstituted polyphosphazene polymers. The tensile testing results revealed that an increase in the content of glycylglycine ethyl ester could cause a rise in the ultimate tensile strengths of the PNEPAxGEGy and PNGEGxPhPhy polymers. A similar trend was found for the tensile modulus. The increase in the ultimate tensile strength and tensile modulus of the polymers is presumably due to the intramolecular hydrogen bond formations of glycylglycine ethyl ester. The tensile modulus values for the PNEPAxGEGy and PNGEGxPhPhy polymers were higher than the reported value of 75 MPa for trabecular bone.42 Similarly, the tensile strength values for the same polymers were higher than the reported value of 1.2 MPa for trabecular bone.42 Interestingly, PNEPA25GEG75 and PNGEG75PhPh25 stood out among other polymers synthesized as they were able to exhibit tensile strength value that is higher than 10 and highest in their various groups. Polyphosphazene polymers with 100% glycylglycine ethyl ester could not be tested because PNEPA0GEG100 and PNGEG100PhPh0 films were so brittle and kept breaking.
Figure 4.

Effect of different compositions of side groups on mechanical properties of PNEPAxGEGy and PNGEGxPhPhy polymers. (A) Ultimate tensile strength decreased as the EPA and PhPh content increased. (B) Tensile modulus decreased with increased content of EPA and PhPh.
DMTA.
Dynamic mechanical, thermal analysis (DMTA) is a direct technique used in characterizing storage modulus, loss modulus, and tan delta. The storage modulus and loss modulus correspond to stored and dissipated energy, respectively. The ratio of dissipated energy to stored energy gives the tan delta. One necessary result of the dynamic mechanical analysis was glass transition temperature that could be determined from the peak or onset of the tan delta curve, the onset of the storage modulus drop, or the onset or peak of the loss modulus curve.43
DMTA was employed to draw some comparison between the glass transition temperatures obtained using DSC with that of the DMTA, and the results are shown in Table 4. It was found that the PNEPA25GEG75 and PNGEG75PhPh25 have the highest Tg for each group (Figure S15). This is in good agreement with the DSC results. It was revealed that the glass transition temperatures increased with increasing content of glycylglycine ethyl ester in the polymers. Polyphosphazene polymers with 100% glycylglycine ethyl ester could not be tested with DMA because PNEPA0 GEG100 and PNGEG100PhPh0 could not yield self-standing films. The stress–strain responses of the polymers were also investigated (Figure S16). The tensile strengths and moduli obtained in this analysis were in good agreement with the ones obtained by using UTM as the nature, and the molar composition of the side groups had an influence on the mechanical properties of the PNEPAxGEGy and PNGEGxPhPhy polymers. The tensile strength and modulus were found to increase as the composition of glycylglycine ethyl ester side group increased. This is as a result of extensive intramolecular hydrogen bonding in glycylglycine ethyl ester. Similar to DSC and UTM, it was also revealed that the PNEPA25GEG75 and PNGEG75PhPh25 have the highest value of glass transition temperatures and good tensile properties and seemed like the most promising set among the polymers synthesized.
Table 4.
Glass Transition Temperatures, Ultimate Tensile Strength, and Tensile Young’s Modulus Obtained from DMA, Demonstrating the Effects of Varying Compositions of Side Groups on Physicochemical Analysis
| ratio | Tg (°C) | ultimate tensile strength (MPa) | tensile Young’s modulus (MPa) |
|---|---|---|---|
| phenylalanine ethyl ester:glycylglycine ethyl ester (PNEPAxGEGy) | |||
| 100%:0% | 48 | 5.5 ± 0.5 | 105.0 ± 5.8 |
| 75%:25% | 53 | 6.4 ± 0.3 | 466 ± 12 |
| 50%:50% | 57 | 8.8 ± 0.2 | 920 ± 26 |
| 25%:75% | 87 | 11.1 ± 0.2 | 1620 ± 43 |
| 0%:100% | |||
| glycylglycine ethyl ester:phenylphenoxy (PNGEGxPhPhy) | |||
| 0%:100% | 56.8 | 6.5 ± 0.7 | 117.0 ± 8 |
| 25%:75% | 51 | 5.8 ± 0.4 | 228.4 ± 6.3 |
| 50%:50% | 57 | 6.2 ± 0.2 | 350.0 ± 14 |
| 75%:25% | 76 | 6.2 ± 0.1 | 428.6 ± 9.3 |
| 100%:0% | |||
In Vitro Hydrolysis.
PNEPA25 GEG75 and PNGEG75PhPh25 polymers were further subjected to in vitro hydrolysis study to ascertain their degradation behaviors.
The main advantage of degradable polyphosphazene over the commonly used polyester biomaterials is its self-neutralizing ability and tunability of its degradation rates. Polyphosphazene with hydrolytically active side groups degrade into the ammonia, phosphate, and corresponding side groups.4,6,31,44–47 These degradation products constitute a natural buffer.48 The pH profile of the degradation media (PBS) in which the polymers were incubated was used to determine the neutralizing effects of the systems. The initial pH of the PBS medium was maintained at 7.4 to mimic the physiological environment. The resulting media for PNEPA25GEG75 and PNGEG75PhPh25 polymers exhibited near neutral pH values (~5.3 to 7.0) throughout the degradation period, whereas the pH values of the resulting media for PLAGA were as low as 2.2 after 12 weeks (Figure 5). A significant decrease in the pH of the degradation media for PLAGA was observed after 2 weeks as compared to the PNEPA25GEG75 and PNGEG75PhPh25 polymers, indicating the neutral bioactivity of the polyphosphazene-based polymers. This is crucial in regenerative engineering applications as neutral degradation products and bioactivity are desired for optimal tissue growth and development. Meanwhile, the significant drop of pH for the PLAGA media was due to the accumulation of lactic and glycolic acids from its rapid degradation.
Figure 5.

In vitro hydrolytic degradation profile of polymers in comparison with PLAGA in PBS medium at physiological conditions (pH 7.4, 37 °C) over 12 weeks. The medium for the PNEPA25GEG75 and PNGEG75PhPh25 polymers showed a near neutral pH as compared to PLAGA.
As shown in Figures 5 and 6, PNEPA25GEG75 and PNGEG75PhPh25 polymers showed a slower degradation rate than the PLAGA control. The slower degradation rates of the polyphosphazene-based polymers are due to the intramolecular hydrogen bonding within the polymers, near-neutral pH of the media, and the presence of hydrophobic aromatic ring in the side groups (phenylalanine and phenylphenol). The intramolecular hydrogen bonding may require more time to be broken down, the near-neutral pH of the media will retard the degradation, and the hydrophobic groups will shield the backbone from hydrolytic attack, thereby preventing the permeation of water.
Figure 6.
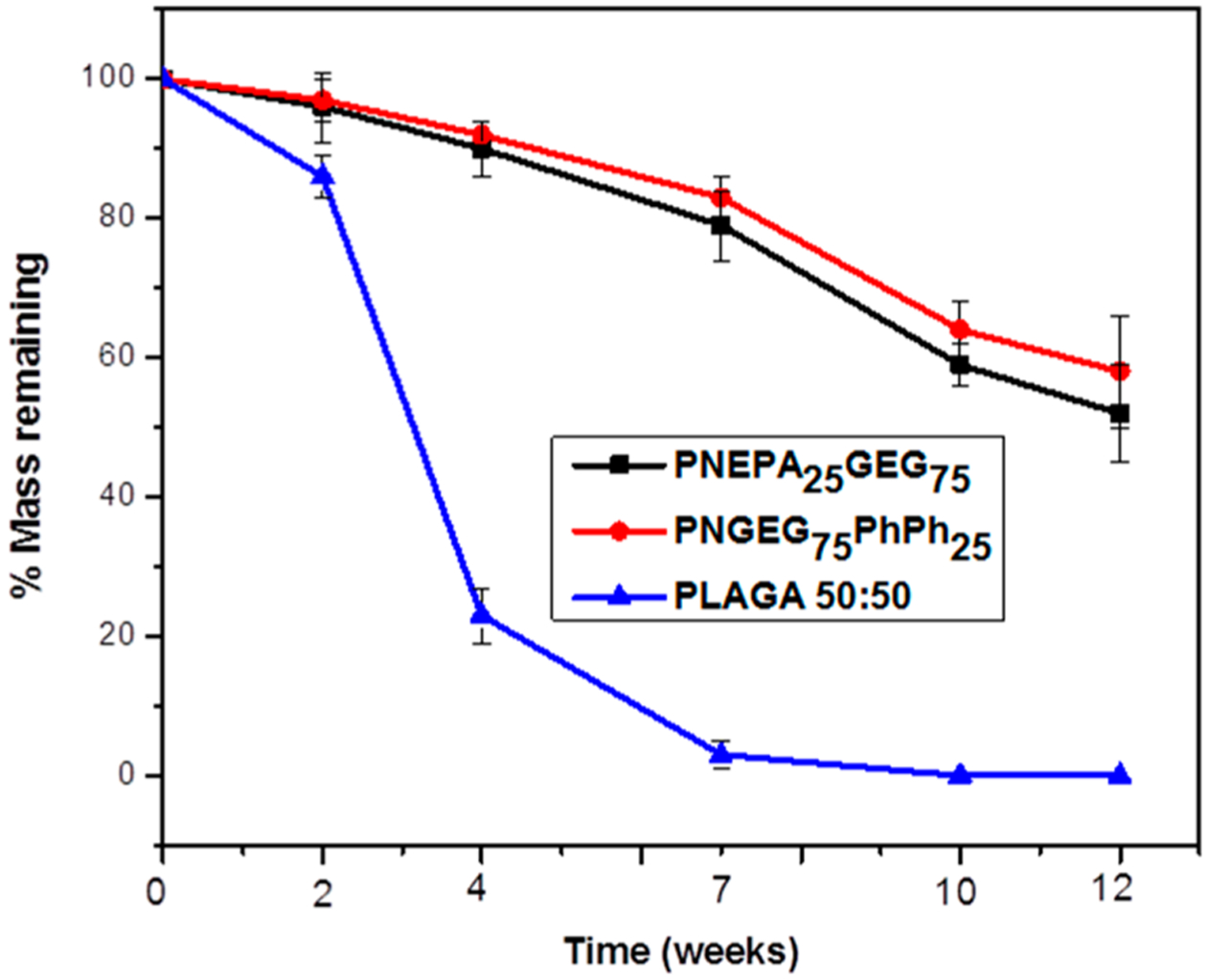
Percent film mass remaining of PNEPA25GEG75, PNGEG75PhPh25, and PLAGA polymers. Only minimal mass loss was observed for both PNEPA25GEG75 and PNGEG75PhPh25 polymers over 12-week degradation. Significantly more mass loss of the PLAGA than the two polyphosphazene-based polymers was observed for all time points. p < 0.05.
However, PNGEG75PhPh25 degraded slightly slower than the PNEPA25GEG75 polymers, presumably due to the higher hydrophobicity of phenylphenoxy units as compared to phenylalanine. The difference in the percent mass remaining between the polyphosphazene-based polymers and PLAGA increased with time and reached a maximum at 7 weeks, at which PLAGA hydrolyzed completely. After the 7 weeks, the percentage mass remaining for PNEPA25GEG75 and PNGEG75PhPh25 polymers were 79 ± 5% and 83 ± 3%, respectively, and the remaining masses for the polyphosphazene polymers were above 50% at end of the degradation period (Figure 6).
The molecular weight decline of the polymers with respect to degradation time had a similar trend as the mass loss with respect to time. Figure 7 shows that the PNEPA25GEG75 and PNGEG75PhPh25 polymers retained about 25% of its original molecular weight after 12 weeks of degradation, whereas PLAGA appeared to have hydrolyzed completely within 7 weeks of hydrolysis. This suggests that the acidic degradation products from PLAGA accelerated its hydrolysis and near-neutral pH degradation products of the polyphosphazene polymers might have contributed to its slower degradation.
Figure 7.
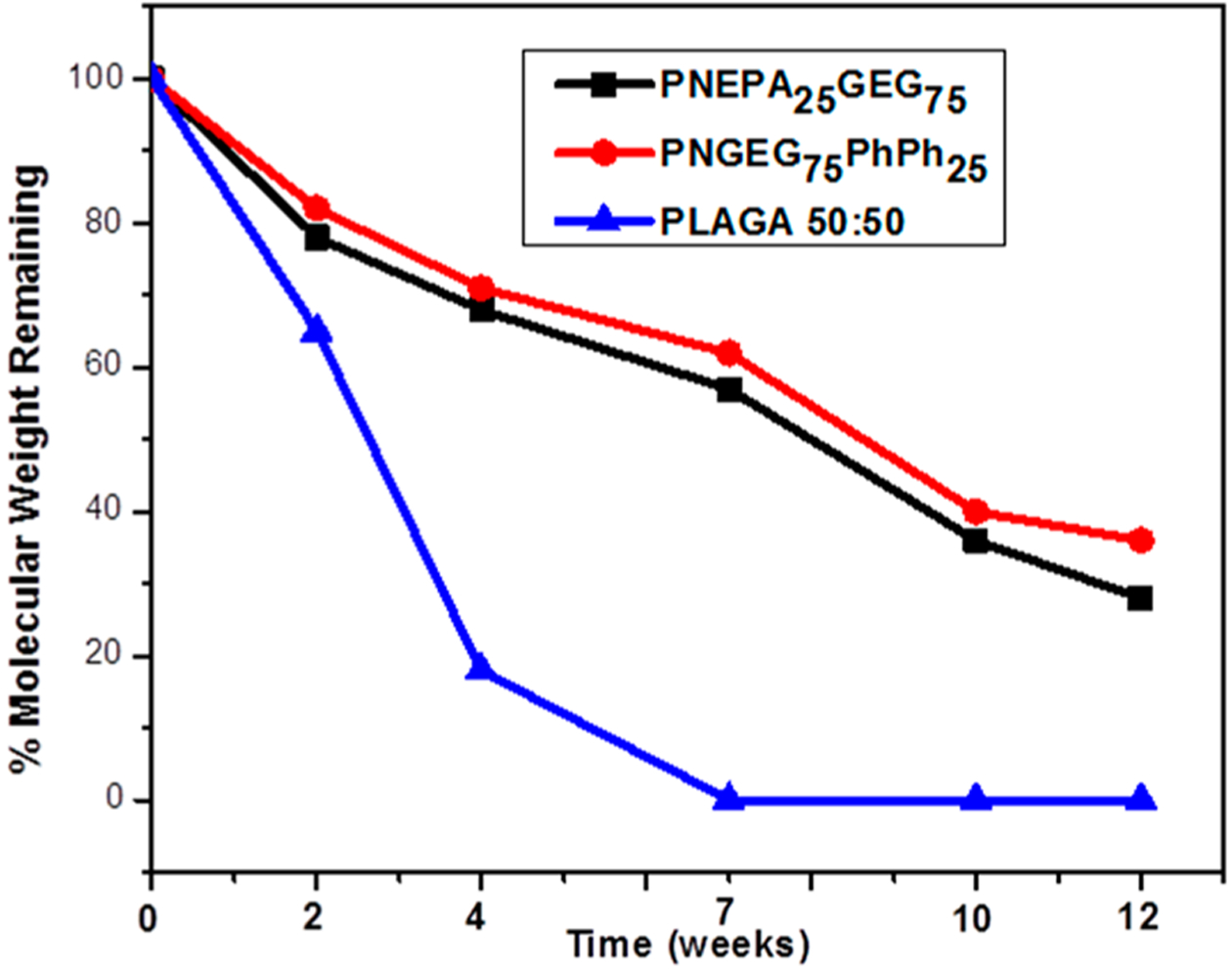
Molecular weight degradation profile of PNEPA25GEG75, PNGEG75PhPh25, and PLAGA polymers with respect to degradation time. A faster degradation rate was observed in the PLAGA control than in the two polyphosphazene-based polymers.
FTIR spectroscopy of the degrading PNEPA25GEG75 and PNGEG75PhPh25 polymers at each time point is shown in Figure 8. A gradual formation of absorption bands at 3200–3700 cm−1 over 12 weeks was observed for both polyphosphazene-based polymers. Moreover, the absorption was strongest after 12 weeks of degradation. These strong absorptions at 3200–3700 cm−1 were due to the stretching vibrations of P–NH, P–NH2, and P–OH functional groups in [N=P(OH)(NHR)1]. The [N=P(OH)(NHR)1] precursor compound hydrolytically breaks down to phosphate and ammonia after the migration of its proton from the hydroxyl unit to the skeletal nitrogen.13–15,49,50 Ammonium phosphate is an amphoteric compound that behaves like an acid in the presence of a base and behaves as a base in the presence of an acid.47 For the PNEPAxGEGy polymers, the formation of the absorption bands at this range was subtle before 12 weeks. This could be attributed to the absence of aromatic OH units which constitute a strong stretch.
Figure 8.
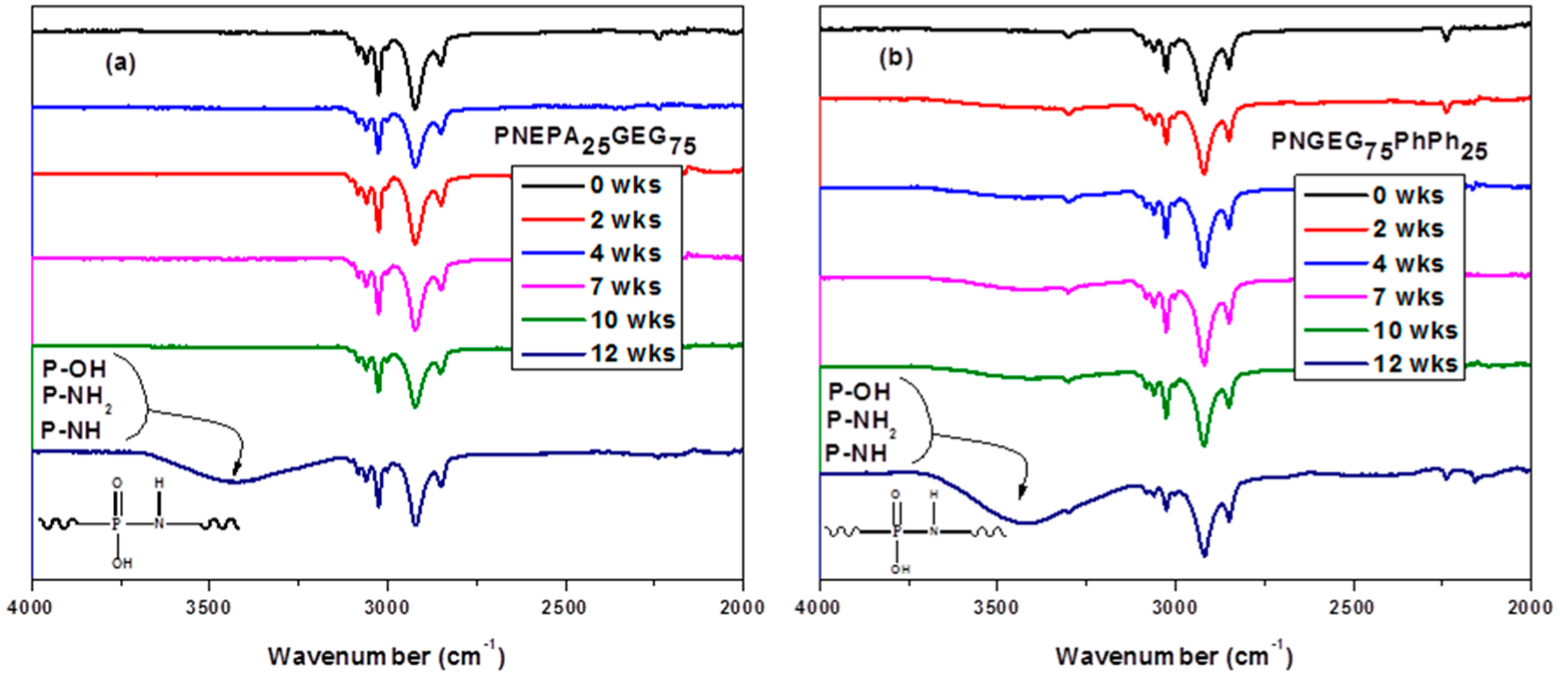
FTIR spectroscopy of (a) PNEPA25GEG75 and (b) PNGEG75PhPh25. The NH, NH2, and OH stretches at 3200–3700 cm−1 indicated the formation of [O=P(OH)(NHR)1], an intermediate structure known to be prone to proton migration from the hydroxyl unit to the skeletal nitrogen and subsequent chain cleavage by water into phosphate and ammonia.
Cell Proliferation and Viability.
The MTS results revealed continuous cell growth on the two polyphosphazene-based polymers as compared to the PLAGA. PNGEG75PhPh25 showed higher cell growth rates than the PLAGA after 14 days (Figure 9). The cell growth for PNGEG75PhPh25 was also higher than that for PNEPA25GEG75. This is supported by our in vitro hydrolysis data as PNGEG75PhPh25 maintained higher pH values than that of PNEPA25GEG75 and PLAGA. The higher pH value that resulted from the slow degradation rate of PNGEG75PhPh25 presented a huge increment after 14 days.
Figure 9.
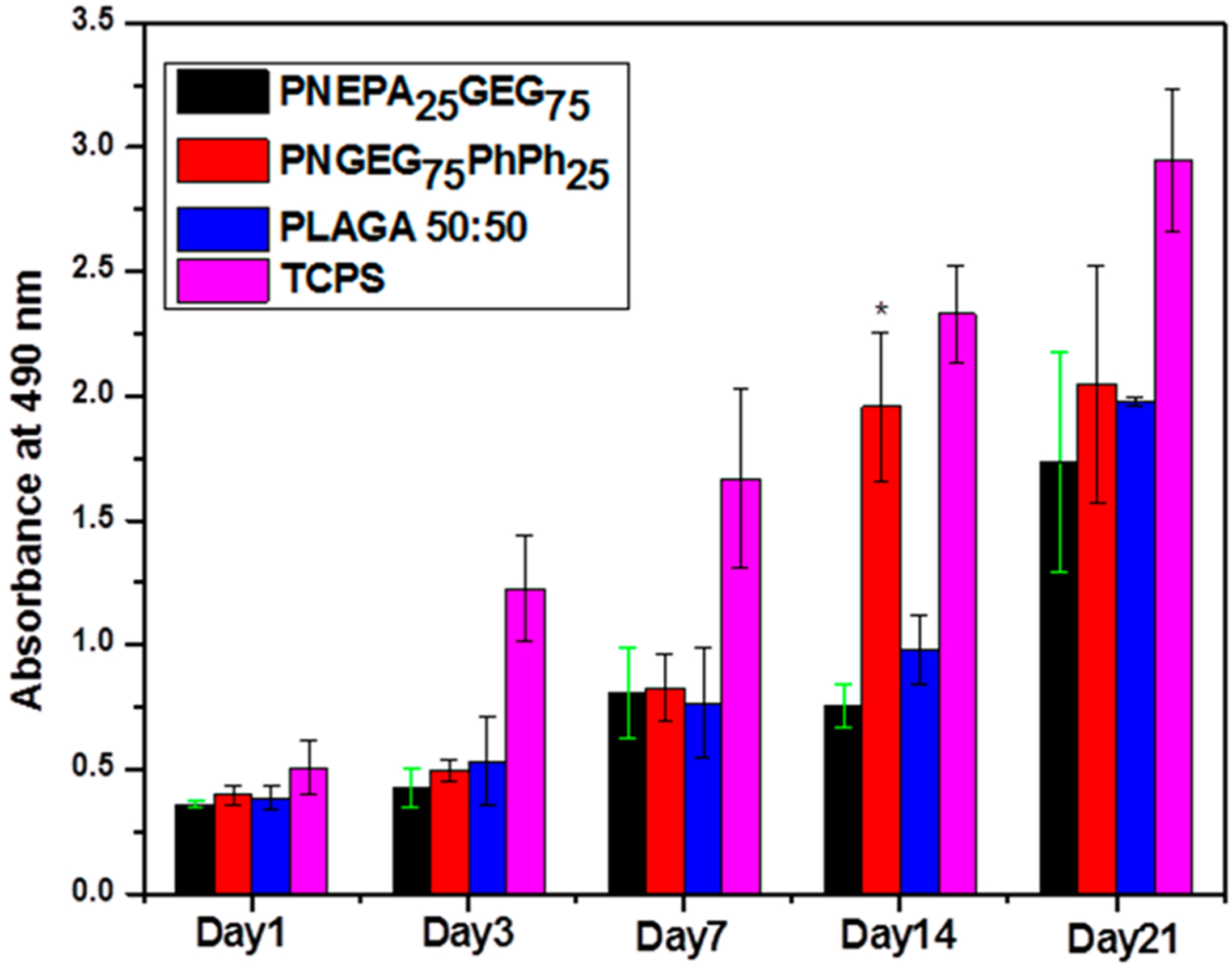
Cell proliferation measured by MTS assay. The cell numbers on the PNEPA25GEG75 and PNGEG75PhPh25 polymers at all time points were comparable to PLAGA. At day 14, PNGEG75PhPh25 showed significantly higher cell numbers than the PNEPA25GEG75 and PLAGA polymers. (*) denotes significant difference (p < 0.05) as compared to PLAGA. Throughout the 21-day culture, cell numbers on TCPS were significantly higher than polymer groups, indicating a healthy population of MC3T3 cells.
Furthermore, the steady growth of the TCPS control indicated a healthy MC3T3 cell culture. Data revealed that TCPS had higher cell numbers than all the polymers at all time points. The results of the live/dead cell stain show that the MC3T3 cells exhibited a robust growth with a well-spread morphology on the PNEPA25GEG75 and PNGEG75PhPh25 polymers as compared to PLAGA (Figure 10). There was a normal morphological sequence of adhesion and proliferation on the polymer films as the cells showed a progressive growth with time. The more significant presence of green stains as compared to the red stains signifies that the polymer films were biocompatible, and this is in a good agreement with the MTS results.
Figure 10.
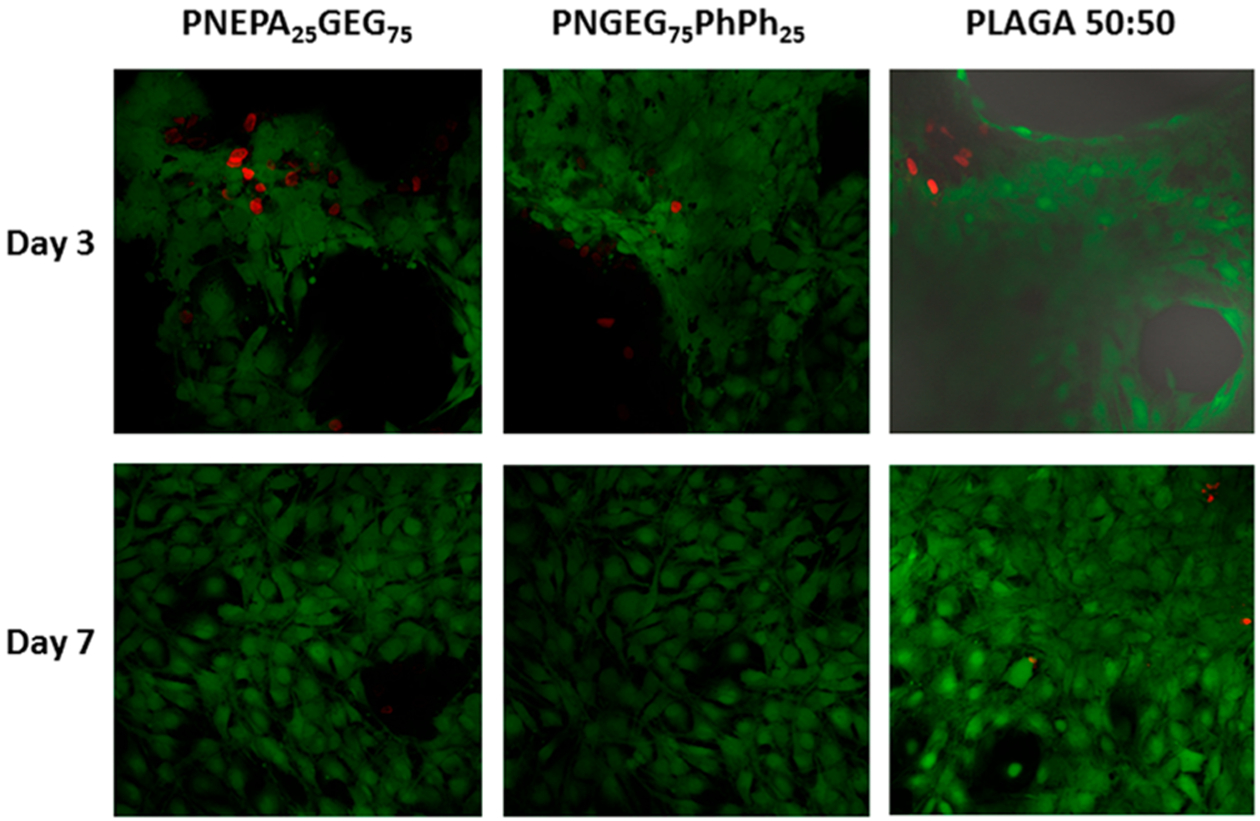
Fluorescence image of MC3T3 cells on polyphosphazene and PLAGA polymers with live/dead cell stain after 3 and 7 days of seeding. PNEPA25GEG75 and PNGEG75PhPh25 exhibited excellent cell growth with well-spread morphology as compared to PLAGA 50:50. Green stains = live cells, and red stains = dead cells.
CONCLUSION
This work demonstrates the design of novel peptide-based polyphosphazene biomaterials that can be customized to obtain polymers with a wide range of properties and neutral bioactivities capable of matching specific tissue types and disease states. Poly[(glycineethylglycinato)x(phenylphenoxy)yphosphazene] (PNGEGxPhPhy) and poly[(ethylphenylalanato)x(glycineethylglycinato)yphosphazene] (PNEPAxGEGy) with different side group compositions were synthesized and confirmed by 31P NMR and FTIR. The effects of side group optimization on physicochemical properties of the polymers were evaluated. Results from GPC, DSC, DMA, TGA, contact angle analysis, and UTM showed that the optimization of the side group chemistry of polyphosphazenes and exploitation of the hydrogen bonding capacity of glycylglycine dipeptide could yield polymers with a wide range of physicochemical properties such as glass transition temperatures, molecular weights, polydispersities, hydrophilicity/hydrophobicity, tensile modulus, and strength. Also, in vitro hydrolysis and cell proliferation studies were performed on the two polymers (namely, PNEPA25GEG75 and PNGEG75PhPh25) with most promising physicochemical properties and compared to PLAGA. It was shown that the degradation behaviors of the polymers could be modulated by employing different side groups, and the resulting hydrolysis media exhibited near-neutral pH values. The PNEPA25GEG75, PNGEG75PhPh25, and PLAGA displayed a similar progressive cell growth pattern as characterized by MTS assay and live/dead stain. Thus, the composition–structure–properties relationships have proven that these versatile polymers have high potentials for use in regenerative engineering due to their synthetic flexibility, property tunability, and neutral bioactivity (buffering degradation products). These characteristics are desirable in bone tissue regeneration.
Supplementary Material
ACKNOWLEDGMENTS
Support from NIH DP1 AR068147 and the Raymond and Beverly Sackler Center for Biomedical, Biological, Physical and Engineering Sciences is gratefully acknowledged. We thank Kennedy S. Ogueri of the Department of Chemistry, PennState, University Park, PA, for his help with the NMR analyses.
ABBREVIATIONS
- PPHOS
polyphosphazenes
- PNEPAxGEGy
poly[(ethylphenylalanato)x(glycineethylglycinato)yphosphazene
- PNGEGxPhPhy
poly[(glycineethylglycinato)x(phenylphenoxy)yphosphazene
- GEG
glycylglycine ethyl ester
- EPA
phenylalanine ethyl ester
- PhPh
phenylphenol
- PLAGA
poly(lactic-co-glycolic acid)
- FTIR
Fourier-transform infrared spectroscopy
- GPC
gel permeation chromatography
- DSC
differential scanning calorimetry
- DMA
dynamic mechanical analysis
- TGA
thermal gravimetry analysis
- UTM
universal tensile testing machine
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsapm.9b00333.
Mechanisms of ring-opening polymerization and macromolecular substitution reactions, 31P NMR spectra for the starting monomers, 31P NMR spectra of PNEPAxGEGy and PNGEGxPhPhy polymers during and after macromolecular substitutions, 1H NMR of spectra of PNEPAxGEGy and PNGEGxPhPhy polymers; FTIR, TGA, and DMTA results (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Chawla K Biomaterials for tissue engineering and regenerative medicine: Treatment of musculoskeletal injury and disease. Mater. Sci. Eng., A 2012, 557, 45–53. [Google Scholar]
- (2).Laurencin CT; Ambrosio A; Borden M; Cooper J Jr. Tissue engineering: orthopedic applications. Annu. Rev. Biomed. Eng 1999, 1 (1), 19–46. [DOI] [PubMed] [Google Scholar]
- (3).Laurencin CT; Khan Y Regenerative Engineering; American Association for the Advancement of Science: 2012. [Google Scholar]
- (4).Ogueri KS; Jafari T; Ivirico JLE; Laurencin CT Polymeric Biomaterials for Scaffold-Based Bone Regenerative Engineering. Regener. Eng. Transl. Med 2018, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Baillargeon AL; Mequanint K Biodegradable polyphosphazene biomaterials for tissue engineering and delivery of therapeutics. BioMed. Res. Int 2014, 2014, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ogueri KS; Ivirico JLE; Nair LS; Allcock HR; Laurencin CT Biodegradable polyphosphazene-based blends for regenerative engineering. Regener. Eng. Transl. Med 2017, 3 (1), 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Oliva N; Unterman S; Zhang Y; Conde J; Song HS; Artzi N Personalizing biomaterials for precision nanomedicine considering the local tissue microenvironment. Adv. Healthcare Mater 2015, 4(11), 1584–1599. [DOI] [PubMed] [Google Scholar]
- (8).Nair LS; Laurencin CT Biodegradable polymers as biomaterials. Prog. Polym. Sci 2007, 32 (8–9), 762–798. [Google Scholar]
- (9).Reed A; Gilding D Biodegradable polymers for use in surgery—poly (glycolic)/poly (Iactic acid) homo and copolymers: 2. In vitro degradation. Polymer 1981, 22 (4), 494–498. [Google Scholar]
- (10).Singh A; Krogman NR; Sethuraman S; Nair LS; Sturgeon JL; Brown PW; Laurencin CT; Allcock HR Effect of side group chemistry on the properties of biodegradable L-alanine cosubstituted polyphosphazenes. Biomacromolecules 2006, 7 (3), 914–918. [DOI] [PubMed] [Google Scholar]
- (11).Zhang Z; Ortiz O; Goyal R; Kohn J Biodegradable polymers In Handbook of Polymer Applications in Medicine and Medical Devices; Elsevier: 2014; pp 303–335. [Google Scholar]
- (12).Andrianov AK Water-soluble polyphosphazenes for biomedical applications. J. Inorg. Organomet. Polym. Mater 2007, 16(4), 397–406. [Google Scholar]
- (13).Allcock HR The expanding field of polyphosphazene high polymers. Dalton Transactions 2016, 45 (5), 1856–1862. [DOI] [PubMed] [Google Scholar]
- (14).Allcock HR; Morozowich NL Bioerodible polyphosphazenes and their medical potential. Polym. Chem 2012, 3 (3), 578–590. [Google Scholar]
- (15).Weikel AL; Krogman NR; Nguyen NQ; Nair LS; Laurencin CT; Allcock HR Polyphosphazenes that contain dipeptide side groups: synthesis, characterization, and sensitivity to hydrolysis. Macromolecules 2009, 42 (3), 636–639. [Google Scholar]
- (16).Zou W; Basharat M; Dar SU; Zhang S; Abbas Y; Liu W; Wu Z; Zhang T Preparation and performances of novel polyphosphazene-based thermally conductive composites. Composites, Part A 2019, 119, 145–153. [Google Scholar]
- (17).Huang Z; Gao C; Huang Y; Zhang X; Deng X; Cai Q; Yang X Injectable polyphosphazene/gelatin hybrid hydrogel for biomedical applications. Mater. Des 2018, 160, 1137–1147. [Google Scholar]
- (18).Allcock HR Chemistry and Applications of Polyphosphazenes; Wiley-Interscience: 2003. [Google Scholar]
- (19).Hackl CM; Schoenhacker-Alte B; Klose MH; Henke H; Legina MS; Jakupec MA; Berger W; Keppler BK; Brüggemann O; Teasdale I; Heffeter P; Kandioller W Synthesis and in vivo anticancer evaluation of poly (organo) phosphazene-based metallodrug conjugates. Dalton Trans. 2017, 46 (36), 12114–12124. [DOI] [PubMed] [Google Scholar]
- (20).Sundhoro M; Park J; Wu B; Yan M Synthesis of Polyphosphazenes by a Fast Perfluoroaryl Azide-Mediated Staudinger Reaction. Macromolecules 2018, 51 (12), 4532–4540. [Google Scholar]
- (21).Valenzuela ML; Jara-Ulloa P; Rodriguez L; Cisternas R Electrochemical Study of Poly (aryloxyphosphazenes) Functionalized with COOH and NO 2 Groups. In Search of New Applications. J. Inorg. Organomet. Polym. Mater 2019, 1–9. [Google Scholar]
- (22).Teasdale I Stimuli-Responsive Phosphorus-Based Polymers. Eur. J. Inorg. Chem 2019, 2019 (12), 1445–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ren Y; Yang K; Shan D; Tong C; Allcock HR Polyphosphazenes and Cyclotriphosphazenes with Propeller-like Tetraphenylethyleneoxy Side Groups: Tuning Mechanical and Optoelectronic Properties. Macromolecules 2018, 51 (23), 9974–9981. [Google Scholar]
- (24).Laurencin CT; Norman ME; Elgendy HM; El-Amin SF; Allcock HR; Pucher SR; Ambrosio AA Use of polyphosphazenes for skeletal tissue regeneration. J. Biomed. Mater. Res 1993, 27 (7), 963–973. [DOI] [PubMed] [Google Scholar]
- (25).Ullah RS; Wang L; Yu H; Haroon M; Elshaarani T; Fahad S; Khan A; Nazir A; Xia X; Teng L Synthesis of polyphosphazene and preparation of microspheres from polyphosphazene blends with PMMA for drug combination therapy. J. Mater. Sci 2019, 54 (1), 745–764. [Google Scholar]
- (26).Iturmendi A; Theis S; Maderegger D; Monkowius U; Teasdale I Coumarin-Caged Polyphosphazenes with a Visible-Light Driven On-Demand Degradation. Macromol. Rapid Commun 2018, 39 (18), 1800377. [DOI] [PubMed] [Google Scholar]
- (27).Andrianov AK Polyphosphazenes for Biomedical Applications; John Wiley & Sons: 2009. [Google Scholar]
- (28).Deng M; Nair LS; Nukavarapu SP; Kumbar SG; Brown JL; Krogman NR; Weikel AL; Allcock HR; Laurencin CT Biomimetic, bioactive etheric polyphosphazene-poly (lactide-coglycolide) blends for bone tissue engineering. J. Biomed. Mater. Res., Part A 2010, 92A (1), 114–125. [DOI] [PubMed] [Google Scholar]
- (29).Schacht E; Vandorpe J; Lemmouchi Y; Dejardin S; Seymour L Degradable polyphosphazenes for biomedical applications In Frontiers in Biomedical Polymer Applications of Polymers; Technomic: Lancaster, PA, 1998; pp 27–42. [Google Scholar]
- (30).Deng M; Nair LS; Nukavarapu SP; Jiang T; Kanner WA; Li X; Kumbar SG; Weikel AL; Krogman NR; Allcock HR; Laurencin CT Dipeptide-based polyphosphazene and polyester blends for bone tissue engineering. Biomaterials 2010, 31 (18), 4898–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Deng M; Nair LS; Nukavarapu SP; Kumbar SG; Jiang T; Weikel AL; Krogman NR; Allcock HR; Laurencin CT In situ porous structures: a unique polymer erosion mechanism in biodegradable dipeptide-based polyphosphazene and polyester blends producing matrices for regenerative engineering. Adv. Funct. Mater 2010, 20 (17), 2794–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lv F; Zhu L; Zhang J; Yu J; Cheng X; Peng B Evaluation of the in vitro biocompatibility of a new fast-setting ready-to-use root filling and repair material. International endodontic journal 2017, 50(6), 540–548. [DOI] [PubMed] [Google Scholar]
- (33).Elgendy HM; Norman ME; Keaton AR; Laurencin CT Osteoblast-like cell (MC3T3-E1) proliferation on bioerodible polymers: an approach towards the development of a bone-bioerodible polymer composite material. Biomaterials 1993, 14 (4), 263–269. [DOI] [PubMed] [Google Scholar]
- (34).Shea LD; Wang D; Franceschi RT; Mooney DJ Engineered bone development from a pre-osteoblast cell line on three-dimensional scaffolds. Tissue Eng. 2000, 6 (6), 605–617. [DOI] [PubMed] [Google Scholar]
- (35).Malich G; Markovic B; Winder C The sensitivity and specificity of the MTS tetrazolium assay for detecting the in vitro cytotoxicity of 20 chemicals using human cell lines. Toxicology 1997, 124 (3), 179–192. [DOI] [PubMed] [Google Scholar]
- (36).Deng M; Nair LS; Nukavarapu SP; Kumbar SG; Jiang T; Krogman NR; Singh A; Allcock HR; Laurencin CT Miscibility and in vitro osteocompatibility of biodegradable blends of poly [(ethyl alanato)(p-phenyl phenoxy) phosphazene] and poly (lactic acid-glycolic acid). Biomaterials 2008, 29 (3), 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Deng Y; Feng X; Yang D; Yi C; Qiu X Pi-pi stacking of the aromatic groups in lignosulfonates. BioResources 2012, 7 (1), 1145–1156. [Google Scholar]
- (38).Liao YH; Kwei TK; Levon K Investigation of the aggregation phenomenon of polyaniline in dilute solutions. Macromol. Chem. Phys 1995, 196 (10), 3107–3116. [Google Scholar]
- (39).Liu Y; Zhan G; Zhong X; Yu Y; Gan W Effect of pi–pi stacking on the self-assembly of azomethine-type rod–coil liquid crystals. Liq. Cryst 2011, 38 (8), 995–1006. [Google Scholar]
- (40).Zhong CJ; Kwan WSV; Miller LL Self-assembly of delocalized. pi.-stacks in solution. Assessment of structural effects. Chem. Mater 1992, 4 (6), 1423–1428. [Google Scholar]
- (41).Parent JS; Sengupta SS; Kaufman M; Chaudhary BI Coagent-induced transformations of polypropylene microstructure: Evolution of bimodal architectures and cross-linked nano-particles. Polymer 2008, 49 (18), 3884–3891. [Google Scholar]
- (42).Marra KG; Szem JW; Kumta PN; DiMilla PA; Weiss LE In vitro analysis of biodegradable polymer blend/hydroxyapatite composites for bone tissue engineering. J. Biomed. Mater. Res 1999, 47 (3), 324–335. [DOI] [PubMed] [Google Scholar]
- (43).Menard KP; Menard N Dynamic mechanical analysis In Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation; John Wiley & Sons, Ltd.: Chichester, UK, 2006; pp 1–25. [Google Scholar]
- (44).Allcock HR; Pucher SR; Scopelianos AG Poly [(amino acid ester) phosphazenes] as substrates for the controlled release of small molecules. Biomaterials 1994, 15 (8), 563–569. [DOI] [PubMed] [Google Scholar]
- (45).Allcock HR; Pucher SR; Scopelianos AG Poly [(amino acid ester) phosphazenes]: synthesis, crystallinity, and hydrolytic sensitivity in solution and the solid state. Macromolecules 1994, 27 (5), 1071–1075. [Google Scholar]
- (46).Andrianov AK; Marin A Degradation of polyaminophosphazenes: Effects of hydrolytic environment and polymer processing. Biomacromolecules 2006, 7 (5), 1581–1586. [DOI] [PubMed] [Google Scholar]
- (47).Ogueri KS; Laurencin CT Polyphosphazene-Based Biomaterials for Regenerative Engineering In Polyphosphazenes in Biomedicine, Engineering, and Pioneering Synthesis; American Chemical Society: Washington, DC, 2018; Vol. 1298, pp 53–75. [Google Scholar]
- (48).Krogman NR; Weikel AL; Kristhart KA; Nukavarapu SP; Deng M; Nair LS; Laurencin CT; Allcock HR The influence of side group modification in polyphosphazenes on hydrolysis and cell adhesion of blends with PLGA. Biomaterials 2009, 30 (17), 3035–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Allcock HR Synthesis, Structures, and Emerging Uses for Poly(organophosphazenes) In Polyphosphazenes in Biomedicine, Engineering, and Pioneering Synthesis; American Chemical Society: Washington, DC, 2018; Vol. 1298, pp 3–26. [Google Scholar]
- (50).Ulery BD; Nair LS; Laurencin CT Biomedical applications of biodegradable polymers. J. Polym. Sci., Part B: Polym. Phys 2011, 49 (12), 832–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.