

Abstract
Bronchial asthma is a heterogeneous disease whose definition and treatment are based on evidence of variable airway obstruction and airway inflammation. Despite the enormous increase in the amount of information on the pathogenesis of this disease, diagnosis is still an unresolved problem, as we still lack sensitive and specific biomarkers. On the other hand, at the turn of the 20th and 21st century, there was a rapid development of therapeutic modalities based on the principle of biological therapy. The first authorized drug matching these characteristics was omalizumab – a monoclonal antibody directed against immunoglobulin E (IgE). It has been used for the treatment of severe forms of bronchial asthma for more than 15 years, which is a sufficient time to acquire ways of its effective use and to assess whether the treatment with omalizumab has met our expectations. However, we continue to discover new and surprising facts about the effects of omalizumab treatment which leads to widening of therapeutic indications. In this work, a basic overview of the very complex role of the IgE molecule in the organism (with a special emphasis on allergic asthma) is discussed, and the most important practical and clinical consequences resulting from its modulation by targeted therapy with omalizumab are summarized.
Keywords: biological therapy, IgE receptors, immunoglobulin E, omalizumab
Introduction
Bronchial asthma is a heterogeneous disease characterized by chronic inflammation and bronchial remodelling, associated with their hyperresponsiveness and variable, often reversible airflow obstruction. It manifests itself as recurrent episodes of wheezing, cough, breathlessness and chest tightness.1 It is assumed that bronchial asthma currently affects more than 300 million people worldwide, of which approximately 180,000 die from the consequences of bronchial asthma annually (the probability of death increases with age). Also, the prevalence of asthma increases by up to 50% every 10 years; therefore, asthma became one of the most common chronic diseases in general.2
Both the definition and diagnostics of bronchial asthma are based on two cornerstones: the evidence of reversible airway obstruction and airway inflammation. Although we currently have powerful tools to detect these both two phenomenons (e.g. pulmonary function tests, laboratory markers of eosinophilic inflammation or fraction of NO in exhaled breath (FeNO)), the diagnosis may be problematic in real clinical practice due to the variability of clinical phenotypes and other possible comorbidities masquerading asthma features. Thus, according to the general definition and diagnostic obstacles, it is not possible to describe asthma as a uniform disease; it should be rather specified as a syndrome defined by clinical, functional and partially also laboratory findings.
Despite the mentioned difficulties, the therapeutic procedures have made tremendous progress. From the time when asthma therapy focussed exclusively on bronchodilation, we moved forward to first inhaled (topical) anti-inflammatory steroids in the 1970s which became nothing less than a therapeutic revolution. However, very rapidly it became clear that not even this therapy was effective in all patients. Apparently, there was still a group of patients (according to some studies about 17% of all patients) whose asthma was not fully controlled despite treatment, even with high-dose glucocorticosteroids use, and who remained symptomatic and tended to experience severe exacerbations. For their asthma, we started to use the term ‘difficult-to-treat asthma’. In some patients, the disease cannot be controlled by therapy due to the presence of unrecognized, inappropriately treated or untreated comorbidities or because of poor therapeutic compliance and adherence, or due to incorrect inhalation technique. The rest (about 3.6% of all patients with asthma)3 suffers from bronchial asthma with primary resistance to treatment (severe asthma), defined as a subset of difficult-to-treat asthma (i.e. asthma that is uncontrolled despite adherence with maximal optimized therapy and treatment of contributory factors, or that worsens when high-dose treatment is decreased)4,5. Despite the proportion of severe asthma patients is seemingly low, this group consume a high proportion of healthcare resources in terms of treatment costs and hospital admissions, as well as the wider societal costs of time of work and school.6 Moreover, this burden is present not only in adults but also in children.7
In the second half of the 20th century, there were introduced not only anti-inflammatory therapies with inhaled steroids but also the very first theoretical and experimental concepts for the use of monoclonal antibodies targeting pathogenetically significant mediators, intended mainly for the group of patients defined above. From the 1990s, trials with biotechnologically prepared drugs were carried out, and the first of them (omalizumab, a monoclonal antibody targeting IgE) was approved for this indication by the US Food and Drug Administration (FDA) in 2003 and 2 years later also by the European Medicines Agency (EMA) for the treatment of the most severe forms of asthma.
Remarkably, it was exactly the research of biologic drugs that triggered a new era of personalized medicine (a process of seeking clinical phenotypes and molecular endotypes of asthma). Inclusion of biological therapy (targeting either the IgE molecule – omalizumab, more recently also IL-5/IL-5Rα – mepolizumab, reslizumab, benralizumab, or the most recently IL-4Rα – dupilumab) into the therapeutic algorithms for patients with refractory forms of bronchial asthma became a turning point in their therapy. The use of biologics is since 2015, according to international recommendations by Global Initiative for Asthma (GINA), also indicated even before the initiation of permanent systemic therapy with corticosteroids that have been so far considered as the “ultimum refugium”.
Historical view on bronchial asthma pathogenesis and formation of current classification
At the end of a 17th-century British physician, John Floyer published his compact monography of bronchial asthma which was already unique for the time. He became also the first associating this disease with airway narrowing. Later, in 1892, William Osler morphologically described asthma as a disease based on inflammation. These two basic associations formed two pillars for both the definition and treatment of asthma and their relevance remained undoubted until today. However, later we experienced dramatic development of our knowledge of the causative immunopathological mechanisms of bronchial asthma at the cellular and molecular levels, as well as we better understand their relationship to structural and functional airway defects.8
In spite of the signs of turning definition of asthma slightly away from the causality of in-flammation, which was suggested in the revised document by GINA in 2014 (where asthma is described, for the first time, as a heterogeneous disease usually characterized by chronic inflammation), it is certainly not possible to challenge its role in the rise and development of asthma in any way. This is true also despite accumulating uncertainties regarding the role of inflammation, for example, in the processes of pulmonary remodelling.9 Therapy targeting the destructive processes of airway inflammation therefore in the case of long-term maintenance therapy of bronchial asthma definitely remains superior to procedures affecting ‘only’ bronchial obstruction. We should also add that therapy based on combinations of drugs with anti-inflammatory and bronchodilator effects can lead to full control of the disease in most (approx. 95%–98%) patients with asthma. As already mentioned above, for the remaining circa 2%–5% of patients with asthma resistant to conventional therapy should be treated by targeted therapies based on the principles of biological therapy. Such treatment is characterized by highly phenotypically and endotypically specific effects.10
The idea of inflammatory phenotypes (sets of all observable characteristics of a specific type of asthma) or endotypes of asthma (the molecular biological mechanism for the formation of the corresponding asthma subtype; the concepts of phenotype and endotype do not have to coincide, one phenotype may contain more endotypes and vice versa) is by no means a matter of the last decades only. However, the most extensive expansion of these diagnostic methods (as mentioned above) was associated with the introduction of biological therapy at the turn of the 20th and 21st century. Crucial preconditions were exponential growth of knowledge about the immunopathogenesis of inflammation in asthma, including, among others, the discovery of subsets of T-helper cells (especially Th1, Th2 and Th17 subsets) in the 1980s, later the improved classification of innate lymphoid cells (ILC, particularly the so-called ILC2 subtype) and last but not least the full appreciation of the regulatory role of other natural immunity cells (e.g. eosinophils and mastocytes) which affects the onset, development and progression of the disease. By the end of the 20th century, the classification stabilized with the concepts of eosinophilic and non-eosinophilic asthma according to the representation of these elements in the airway mucosa or induced sputum.11
For the practical purpose of implementation of pheno- or endotyping of asthma as part of treatment decisions and procedures, it was of course necessary to create some generally acceptable and feasible nomenclature system. That is currently stabilized on the principle of differentiation of patients suffering from eosinophilic inflammatory phenotype to those in whom this inflammation is dominantly regulated with Th2 lymphocytes (usually allergic, eosinophilic asthma, referred to as Th2-high) and those where the dominant role is played by ILC2 cells (usually non-allergic, eosinophilic asthma, referred to as ILC2-high).12 Nevertheless, it is necessary to note that the activity of Th2 and ILC2 cells in allergic or non-allergic inflammation is not mutually exclusive (we can detect the activity of both cell lines in allergic as well as non-allergic asthma; however, the difference consists only of functional disbalance favouring either Th2 in allergic or ILC2 cells in non-allergic asthma), and this differentiation is rather academic (a practically most important feature of allergic asthma is the presence of clinically pertinent atopic reactivity).
The Th2-high and ILC2-high phenotypes may be further combined to a larger group of so-called eosinophilic type-2 high asthma. In contrast, non-eosinophilic phenotypes are analogically referred to as type-2 low, and it is assumed that the regulatory roles in these forms of asthma are taken over by Th1 or Th17 subsets (possibly ILC1 or ILC3 cells),12–15 but their real importance and pathogenesis are rather unclear to the present day. This approach has been anchored in the Czech inflammatory phenotype–based diagnostic algorithm.16
So far there have been only vague ideas regarding the incidence of individual inflammatory phenotypes or endotypes. In the general population of patients with asthma, the incidence of eosinophilic airway inflammation (defined by the increased representation of eosinophils in the induced sputum, that is, type-2 high endotype according to the new classification) is about 41%.17 According to the Belgic registry of patients suffering from severe asthma, eosinophilic asthma occurs in up to 57% patients.18 Increasing severity of eosinophilic asthma is additionally associated with a decreasing proportion of typical allergic eosinophilic asthma (Th2-high) in favour of eosinophilic and non-allergic asthma (ILC2-high).19 It is also highly probable that individual inflammatory patterns have a substantial level of plasticity, either due to therapy, external factors, or the natural development of the disease, and can thus mutually pass one into the other. Repeated examinations in the study by McGrath and Fahy described a stationary elevation of eosinophils in the sputum in only 22% patients with asthma even in patients not treated with inhaled steroids.20
It is also necessary to point out the fact that the borders between anticipated immunopathological mechanisms resulting in specific clinical manifestations are entirely arbitrary.
The role of IgE in the pathogenesis of bronchial asthma
History of the discovery and measurement of IgE molecule
The history of the discovery of immunoglobulins of class E begun in the 1960s. At that time, Japanese couple of Ishizaka (then based in Denver, Colorado) and the Swedish researchers Johansson a Bennich in Uppsala simultaneously worked on the discovery of a protein, until that time called reagin. They all hoped for explanation of both the basis of allergic ‘Prausnitz and Küstner reaction’21 (already known for more than 40 years) and also the immunological principles of type I hypersensitive reaction and anaphylactic reactions that were traditionally associated with asthma at that time. The whole process was, however, very difficult, mainly because of the exceptionally low levels of IgE in human serum (in tens to hundreds ng/ml, the IgE antibodies representing only about 0.002% of all immunoglobulins, because most of them is very likely bound on cell surfaces, with a biological half-life of approximately 2 days). At the end, it was possible to overcome this difficulty, probably by chance, thanks to the serum of a patient suffering from a very rare type of IgE myeloma.22 In 1968, this molecule was finally officially named immunoglobulin E in the WHO Bulletin.23
The structure of IgE antibodies does not differ significantly from other immunoglobulins; it forms monomers composed of two heavy and two light chains, the heavy chain contains one variable (Vε) and four constant domains (Cε1–Cε4), that is, it contains, similarly to IgM, one domain more in comparison with immunoglobulin classes G, A and D. The interaction between the IgE molecule and its receptors occurs via the Cε3 domain (see Figure 1). Similarly, to the IgD and non-aggregated IgA, IgE antibodies also do not fix complement.
Figure 1.
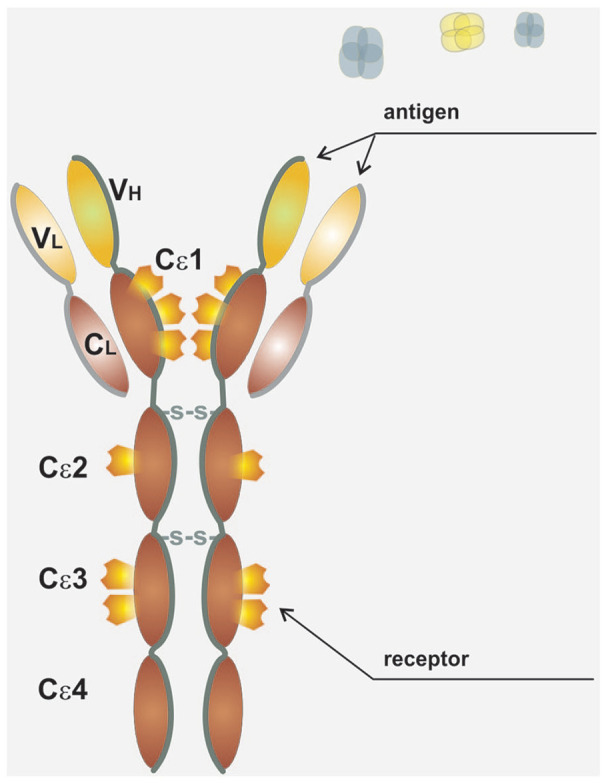
Structure of IgE. IgE antibody structure forms monomers composed of two heavy and two light chains, the heavy chain contains one variable (Vε) and four constant domains (Cε1–Cε4), that is, it contains, similarly to IgM, one domain more in comparison to immunoglobulin classes G, A, and D. The interaction between the IgE molecule and its receptors occurs via the Cε3 domain.
Soon after the discovery of IgE, the first evidence was obtained about its direct role in the pathogenesis of bronchial asthma and its connection with skin reactivity to allergen exposition.24 It became apparent that the production of IgE is dominantly regulated by IL-425 and IL-13,26 and depends, in addition to cytokine microenvironment (signal 1), also on direct interaction between T- and B-lymphocytes through the mediation of bonds between the cell-surface molecules of T-lymphocytes (especially CD40L (CD154) and CD28) with receptors situated on the external membrane of B-lymphocytes (namely CD40 and B7-1 (CD80) or B7-2 (CD86)) (signal 2). It is only the combination of both these stimuli (sometimes also called as ‘immunological synapse’)27 that initiates both the process of maturation of B-lymphocyte to an active plasmatic cell and isotype switching and formation of this specific immunoglobulin class (see Figure 2). Interestingly, both signals can be provided to B-lymphocytes not only by T-lymphocytes but for example also by basophils. Furthermore, though this interaction occurs dominantly in the lymphatic tissue germinal centres, it has also been described in the peripheral non-lymphatic tissues, for example, in airway mucosa in patients with bronchial asthma or in the gastrointestinal tract mucosa in patients with food allergy (so-called local IgE production).28
Figure 2.
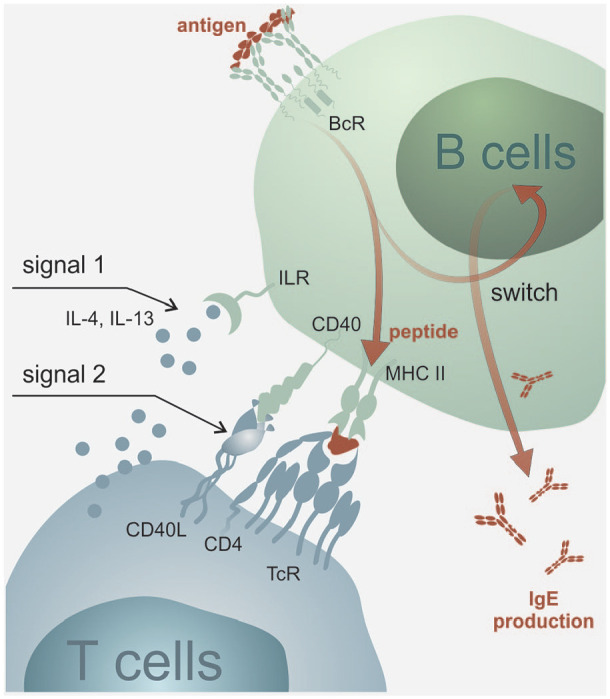
IgE production. Production of IgE antibodies depends on cytokine microenvironment (signal 1) and also on direct interaction between T- and B-lymphocytes through the mediation of bonds between the superficial molecules of T-lymphocytes CD40 L (CD154), resp. CD28 and CD40 receptors, resp. B7-1 (CD80) or B7-2 (CD86) situated on the external membrane of B-lymphocytes (signal 2). The combination of both stimuli initiates the process of maturation of B-lymphocyte into an active plasmatic cell and isotype switching and formation of this specific immunoglobulin class.
Today it is possible to routinely examine the concentration of total IgE and its specific components targeting particular antigens (mixed or molecular) in the form of specific IgE. The importance of this examination consists especially of the demonstration of atopic reactivity or specific sensitization as part of allergy diagnostics. However, the clinical consequences of known IgE levels are doubtful. Overall IgE levels are also used as a key parameter to calculate the dose of biologic therapy targeting this specific molecule (omalizumab), but in relation to other biologics it probably appears to be ineffective both for the estimate of efficacy and monitoring of treatment effect, which applies both in the case of therapy targeting IL-5 (mepolizumab)29 and IL-13 together with s IL-4 (dupilumab).30 Notably, decline of IgE levels has been described in connection with anti-IL-13 therapy (lebrikizumab),31 but the significance of this dependence is unclear. In general, examination of serum concentration of IgE is therefore currently employed especially as a supportive parameter for Th2-high subtype of type-2 high asthma (in particular associated with IL-4 and IL-13 activity).
A significant increase of total IgE levels related to allergic reactivity occurs especially in patients with atopic eczema, and further (in descending order) in patients with allergic asthma, persistent rhinitis bound to perennial allergens and intermittent rhinitis – its levels culminate approximately 4–6 weeks after peak of the pollen season. High levels are also measured in patients suffering from allergic bronchopulmonary aspergillosis (ABPA) (serum levels can exceed 1000 kIU/L), and further in most eosinophilic syndromes, especially secondary because of the joint increase of IL-4, IL-13 and IL-5 activities.28
Current view on the general role of the IgE molecule in the pathogenesis of bronchial asthma
The physiological role of IgE in the organism has been traditionally associated with anthelmintic immunity, and activity of mastocytes, eosinophils and basophils (to which they are functionally related, especially in term of the early allergic reaction (see Figure 3), but is still rather unclear today and also surprising through its comprehensiveness.32 There are also data available supporting the role of IgE antibodies in antitumor surveillance.33
Figure 3.

The early and late phase of allergic reaction. The key elements for the course of the early phase of an allergic reaction are basophils and mastocytes. These cells express large amounts of FcεRI on their surface that bind IgE molecules through the mediation of their Cε3 domains. In the case interaction of the variable part of IgE with a soluble allergen occurs, as well as the so-called bridging of two adjacent IgE molecules, it is triggered the activation cascade inside the cells via intracellular ITAM domains and tyrosine kinases. Activated basophils and mastocytes then rapidly release large amounts of preformed, biologically highly active mediators, such as histamine, heparin and prostaglandins. This reaction usually lasts about 2–3 h. In addition to above-mentioned early phase of an allergic reaction, mastocytes, basophils and other cells (such as eosinophils) can infiltrate the inflamed tissue and produce a number of other substances (the synthesis of which occurs only after their activation, with their levels increasing within minutes to hours after the insult). These other substances include lipid mediators such as cysteine leukotrienes (CysLT) or prostaglandin D2 (PgD2), or large amounts of cytokines (e.g. IL-4, IL-5, IL-13 and IL-9), all contributing to the overall modulating of the immune system towards Th2 reactivity and subsequently to remodelling. The classical late phase of allergic reaction begins within 4–6 hours and lasts 18–24 hours.
To understand the complex effects of IgE both under physiological conditions and, for example, in bronchial asthma it was necessary to discover also the relevant receptor structures responsible for its broad field of action.
Two basic receptors for the IgE molecule are currently known. These are (1) a membrane high-affinity receptor FcεRI (expressed especially on the surface of mastocytes, basophils, smooth muscle cells, and dendritic cells in the form of an αβγ2 tetramer and further and in lesser amounts, on the surface of antigen-presenting cells /dendritic cells, Langerhans cells, monocytes, or eosinophils/ in the form of an αγ2 trimer)34 and (2) a cellular or soluble low-affinity receptor, FcεRII (CD23, Ca-dependent lectin, first described on the surface of B-lymphocytes and some haematopoietic cells, later also on T-lymphocytes, dendritic cells, alveolar macrophages, eosinophils, platelets, smooth muscle cells or epithelial cells) (see Figure 4).35 The relationship between IgE and its receptors is bidirectional; the receptors mediate the biological effects of IgE and are, at the same time, mutually positively regulated.36 It has turned out that the expression of both receptors (the high-affinity FcεRI receptor and the low-affinity FcεRII) in basophils (but also in dendritic cells and monocytes) correlates directly with serum levels of total IgE (that is stabilized by FcεRI),37 the production of FcεRII is additionally increased under the influence of IL-4. The soluble form of the lectin FcεRII results from cleaving the extracellular part away from the membrane-bound form by endogenous or exogenous proteases (that include e.g. the protease of Der p1 allergen, the main allergen produced by the mite Dermatophagoides pteronyssinus), its binding on free IgE antibodies inhibits their biological activity,38 but can also paradoxically stimulate, under certain circumstances, the IgE production.28
Figure 4.

IgE receptors structure. There are two basic receptors for the IgE molecule: (1) a membrane high-affinity receptor FcεRI (expressed especially on the surface of mastocytes, basophils, smooth muscle cells and dendritic cells in the form of an αβγ2 tetramer and further and in lesser amounts, on the surface of antigen-presenting cells /dendritic cells, Langerhans cells, monocytes or eosinophils/ in the form of an αγ2 trimer) and (2) a cellular or soluble low-affinity receptor, FcεRII (CD23, Ca-dependent lectin, first described on the surface of B-lymphocytes and some haematopoietic cells, later also on T-lymphocytes, dendritic cells, eosinophils, platelets, smooth muscle cells or epithelial cells). Receptor transmembrane signalling and subsequent cell activation are mediated by intracellular domains – ITAM (Immunoreceptor Tyrosine-based Activation Motif) that are contained in the intracellular parts of β and γ chains.
Role of IgE in the immunopathogenesis of asthma as related to cellular structures
We have already mentioned above that the relationship between IgE and its receptors does not consist only in their mutually positive regulation of synthesis but especially in the transfer of corresponding information (resulting from the bond of an allergen to immunoglobulin E) inside the cells carrying the receptor. In this regard, an important role is played by the high-affinity receptor for IgE (FcεRI) that is expressed either in the form of an αβγ2 tetramer (on basophils, mastocytes, smooth muscle cells and dendritic cells) or in smaller amounts in the form of an αγ2 trimer (on the surface of antigen-presenting cells /dendritic cells, monocytes or eosinophils/), with the β chain significantly enhancing receptor transmembrane signalling and subsequent cell activation thanks to intracellular domains of ITAM – Immunoreceptor Tyrosine-based Activation Motif that are contained in the intracellular parts of β and γ chains.34 In comparison, we know far less about the processes of signalling of the low-affinity receptor FcεRII; it probably acts as an adhesive molecule and its free form inactivates IgE. However, it becomes apparent that this receptor structure can influence a far broader range of biological processes in the organism, for example, antigen presentation, regulation of growth and differentiation of B- and T-lymphocytes, cellular apoptosis, the release of cytotoxic and anti-inflammatory mediators, and last but not least also the transcytosis of IgE-allergen complexes.39
The IgE molecule is a key element already in the very introduction of the interaction between the airway mucosa and an allergen, thanks to the possibility of transcytosis across the epithelial cells all along from the submucosal connective tissue into the airway lumen. This process is mediated by the low-affinity FcεRII receptor expressed on the epithelial cells in the proximity of the basement membrane. It is very likely that the backward transition of IgE molecules can occur in a very similar manner from the airway lumen into the submucosal connective tissue carrying a bound allergen (see Figure 5, part 1).39,40
Figure 5.

Overview of potential roles of IgE in asthma pathology (adapted and modified from).36
Part 1: The IgE molecule is able to transcytosis across the epithelial cells. This process is mediated by the low-affinity FcεRII receptor expressed on the epithelial cells in the proximity of the basement membrane.
Part 2: The complexes of IgE molecule with allergen bind to the FcεRI receptor on the surface of the projections of dendritic cells that penetrate from subepithelial spaces up to the airway lumen.
Part 3: The presence of FcεRI on the surface of dendritic cells is strengthening the processes of antigen presentation to naïve Th-lymphocytes (with their subsequent maturation towards the Th2 subset), up to 1000 times.
Part 4: The key elements for the further course of allergic reaction mediated by IgE immunoglobulins are basophils and mastocytes that express large amounts of FcεRI on their surface. In the case interaction of the variable part of IgE with a soluble allergen occurs, the activation cascade inside the cells appears and leads subsequently to the early phase of allergic reaction initiation.
Part 5: Recently, the expression of both receptor types for IgE has been discovered on the surface of smooth muscle cells in the airways. The interaction with IgE molecules leads to a proliferation of the smooth muscle cells and type I, III, VII collagen and fibronectin production during remodelling processes.
Part 6: Increased expression of FcεRI associated with elevated IgE levels has been shown also on the eosinophils of atopic patients. IgE molecules thus support their survival and inhibit their apoptosis. The complex mutual relationship between IgE molecules and eosinophils is mediated also indirectly, probably through increased production of prostaglandin D2 (PgD2) by activated mastocytes.
Alternatively, the complexes of IgE molecule with allergen bind to the FcεRI receptor on the surface of the projections of dendritic cells (that penetrate from subepithelial spaces up to the airway lumen) (see Figure 5, part 2). The presence of FcεRI on the surface of dendritic cells is very likely strengthening the processes of antigen presentation to naïve Th-lymphocytes (with their subsequent maturation towards the Th2 subset),41,42 up to 1000 times, resulting in significant reduction of the triggering threshold for the allergic reaction, already in this early phases43 (see Figure 5, part 3). Surprisingly, the IgE molecule is (alone or in combination with activated mast cells) a potent promotor of intestinal ILC2 cells activity in murine model of food allergy.44
It was already mentioned that these Th2-lymphocytes (and at least in part also ILC2 cells) are responsible for the formation of a cytokine microenvironment (IL-4 and IL-13) which is necessary for the maturation of B-lymphocytes, their transformation to antibody-producing plasma cells and isotype switching leading to the production of IgE class antibodies (see Figure 2).
The key elements for the further course of allergic reaction mediated by IgE immunoglobulins are of course basophils and mastocytes, that also express large amounts of FcεRI on their surface that bind IgE molecules through the mediation of their Cε3 domains. In the case interaction of the variable part of IgE with a soluble allergen occurs, as well as so-called bridging of two adjacent IgE molecules, it is triggered the activation cascade inside the cells via intracellular ITAM domains and tyrosine kinases Lyn, Fyk and Syk45 (see Figure 3 and Figure 5, part 4). Activated basophils and mastocytes then rapidly release large amounts of preformed, biologically highly active mediators, such as histamine, heparin, and mastocytes further more exclusively release neutral proteases (chymase and serine protease – tryptase).46 In addition to the mediators mentioned above, mastocytes and basophils can produce a number of other substances (the synthesis of which occurs only after their activation, with their levels increasing within minutes to hours after the insult). These other substances include lipid mediators such as cysteine leukotrienes (CysLT) or prostaglandin D2 (PgD2), or large amounts of cytokines (e.g. IL-4, IL-5, IL-13 and IL-9), all contributing to the overall modulating of the immune system towards Th2 reactivity.28 The classic rapid phase of allergic reaction mediated through IgE in the airways is thus characterized by increased vascular permeability and oedema, increased expression of adhesive molecules and cellular tissue infiltration, increased production of mucus and increased contractility of smooth muscle cells47 (see Figure 3).
However, recently it has become clear that these acute processes developing in the pulmonary tissue are by far not the last effects of IgE molecules. We have already known for some time that activated mastocytes can also affect the long-term remodelling processes through their interaction with smooth muscle cells.48 As a new, relatively recent development, expression of both receptor types for IgE has been discovered on the surface of smooth muscle cells in the airways49,50 and a direct correlation was found between IgE levels and proliferation of the smooth muscle cells and type I, III, VII collagen and fibronectin production51 (see Figure 5, part 5). Experimental evidence has also been provided that this effect does not depend on the interaction IgE with an allergen, and furthermore, it can be affected by therapy consisting of the administration of a blocking antibody – omalizumab.52 Recently it has been demonstrated in vitro that IgE antibodies are able to directly (regardless of antigen presence) affect the activity of smooth muscle cells through increased production of the so-called miRNA (microRNA-21-5p – single chains of non-coding RNA comprising 21–23 nucleotides that participate in the regulation of gene expression).53
Increased expression of FcεRI associated with IgE levels has been shown also on the eosinophils of atopic patients54 (see Figure 5, part 6). It has been even documented that increased levels of total IgE support their survival and inhibit their apoptosis.55 It is believed that the complex mutual relationship between IgE molecules and eosinophils is mediated also indirectly, probably through increased production of prostaglandin D2 (PgD2) by activated mastocytes. It has been known for some time that exactly this prostaglandin is capable of attracting and activating eosinophils, basophils, but also Th2-lymphocytes and ILC2 cells inside the allergic (as well as non-allergic) inflammation through the mediation of a CRTh2 (DP2) receptor.56,57 Affecting of signalling by this receptor thus appears to be a promising way how to effectively step in the pathogenesis of eosinophilic inflammation and its later phases in particular.58–60
Possibilities of targeted therapeutic affecting of IgE action
The theoretic concept of the use of blocking antibodies against the IgE molecule appeared in the middle of the 1980s (almost 20 years after its discovery).61 Less than 10 years later, the effects of this blockade were successfully clinically confirmed in the first clinical trial (antibody CGP51901, omalizumab).62 As a result of an unprecedented success in registration studies,63–65 this molecule was approved by the FDA in 2003 for the treatment of severe forms of bronchial asthma, and later earned approval in the European Union as well by EMA (European Medicine Agency) and became the first drug, matching the characteristics of a biologic drug, used for the treatment of bronchial asthma.
Omalizumab is a humanized antibody derived from recombinant DNA (IgG1, κ), targeting the Fc fragment of IgE molecules (domain Cε3). Generally, it is possible to say that bond of omalizumab on the IgE molecule prevents mainly its subsequent bonding on the corresponding high-affinity FcεRI receptor found particularly on the surface of mast cells and basophils (resulting in inhibition of their degranulation within the early phase of allergic response).
As was demonstrated above, the biological effects of the IgE molecule are very broad and that is why the impact of its blockade is far more complex. From the view of immunopathogenesis of asthma, omalizumab interferes with the processes of mutual interaction of IgE antibodies with both receptors, and in addition to affecting early phase of allergic reaction. However, very probably omalizumab influences also late phases of allergic reaction and the complex processes of airway remodelling.52,66 Omalizumab furthermore (1) reduces the levels of the free fraction of IgE67 (while it paradoxically increases the levels of total IgE (up to 5 times) as a result of increased stability and extended biological half-life of the bound molecules)68 and, at the same time, (2) inhibits the expression of FcεRI on the surface of mast cells, Langerhans cells, eosinophils and basophils, indirectly reducing their reactivity to allergenic stimuli.69 In addition to that (3) omalizumab also decreases the surface expression of IL-4R and germline Cε mRNA levels of B-lymphocytes which together reduces their responsiveness to antigenic stimulation and further the production of IgE.70
Interestingly, there was not registered a direct effect of omalizumab on circulating ILC2 cells, basophils and myeloid dendritic cells (mDC) in long-term treated asthma patients.71 Although the effect of omalizumab is manifested also as the strengthening the production of interferon α (IFNα) which leads to increased antiviral immunity and reduced tendency to exacerbations induced by viral infections,72 plasmacytoid dendritic cells (pDC, a potent source of type I interferons) seem to be lowered. In this view, the low number of these cells may be considered as a cellular biomarker of low risk of exacerbations and of good disease control.71
Last but not least, it should be mentioned that treatment with omalizumab has great treatment potential in conditions with evidently non-allergic aetiology, such as chronic idiopathic urticaria resistant to therapy with antihistamines73 and to chronic rhinosinusitis with nasal polyps (CRSwNP).74 At present, two phase III studies have been registered at ClinicalTrials.gov, NCT03280550 (POLYP 1) and NCT03280537 (POLYP 2) with extension NCT03478930. Results are still awaited.
A meta-analysis of seven controlled trials with omalizumab has confirmed the clinically significant effect of therapy of severe asthma – results demonstrated reduced rate of exacerbations by 38% in comparison with placebo, in spite of reduced overall corticoid dose. Furthermore, it was indisputably demonstrated that patients treated with omalizumab experience reduced rates of physician and emergency room visits and hospital admissions because of exacerbations.75 Increased availability of omalizumab after its approval made it possible to observe its effect in real-world clinical practice in adults (e.g. real-world data trials PERSIST,76 eXpeRience77,78 or for example in local Czech CAR (Czech Anti-IgE Registry) registry)79 as well as in children,80–82 – these observations confirmed the real impact of this treatment on patient quality of life.
Last but not least, it is certainly worth mentioning that omalizumab has shown its efficacy, in the sense of the reduction of exacerbations and improved physician’s overall assessment of clinical response, GETE (Global Evaluation of Treatment Effect) – these results were demonstrated regardless of the baseline counts of eosinophils as was shown in a 12-month retrospective, observational real-world trial STELLAIR.83 Favourable effect of omalizumab (in the sense of reduced rates of exacerbations and hospital admissions, and improvement in the asthma control test) was further described in the 48-week prospective observational study PROSPERO,84 regardless of the baseline eosinophils counts and fractional nitric oxide (NO) concentration in exhaled breath (FeNO). This characteristics distinguishes omalizumab from other biologic products, since it seems that except the demonstration of atopic reactivity with bond to a perennial allergen, there is no other limiting pheno- or endotypic classification of the patients to predict therapeutic effect.85
Finally, it seems plausible that therapy targeting IgE may influence the natural history of the disease and potentially the remodelling process as well.66,86–88 All these facts open up the question of conceivable sustained effect of omalizumab after its cessation.89 One long-term study has highlighted the possible role of anti-IgE therapy in improving the course of asthma, with clinical improvements that were still seen 3 years after long-lasting (6 years) treatment withdrawal.90,91 However, other clinical observations suggest an increased risk of severe asthma exacerbation already during the first year after omalizumab discontinuation or even upon dose reduction.92–94 Busse et al. performed a Phase IV multinational randomized, placebo controlled clinical trial (XPORT) primarily aimed to evaluate the persistency of response to omalizumab in moderate-to-severe asthma patients (52 weeks after omalizumab therapy discontinuation). Preliminary results showed that nearly 40% of the patients remained free of severe asthma exacerbation after 1 year of discontinuing long-term omalizumab treatment.95 Definite results confirmed the existence of a substantial group of responders who tend to benefit from therapy, despite the adjusted odds ratio (age, sex, ICS use) of continuing omalizumab treatment for risk of acute exacerbation was 0.44 (95% CI 0.23–0.82).96
Conclusion
The IgE molecule is the latest defined immunoglobulin class, discovered more than 50 years ago. However, we are still discovering new and surprising facts about its very complex role in the organism, both under physiological and pathological circumstances. The same applies in the case of therapy targeting this antibody, which is currently indicated for allergic forms of severe bronchial asthma or chronic idiopathic urticaria.
However, as it was demonstrated above, it is very likely that anti-IgE therapy affects also processes that do not have to be causally associated with allergy. It is supported, for example, by clinical data demonstrating its very effective use in the non-allergic diseases, like in the above-mentioned chronic idiopathic urticaria and promisingly in the future in chronic rhinosinusitis with nasal polyps (CRSwNP). Moreover, there are many still not-resolved questions regarding its plausible disease-modifying and long-lasting effect sustaining despite treatment cessation. We are also lacking reliable biomarkers depicting patients with best treatment response. Nonetheless, it seems that exactly the broad range of IgE roles currently guarantees very well-established position of anti-IgE therapy in the cases of refractory forms of bronchial asthma.
Discussion, review of study limitation
The aim of this review was to analyse our theoretical and practical knowledge through the prism of more than 10 years of experience with using omalizumab for severe allergic asthma treatment. The scope of our work was limited by our expertise in the treatment of severe asthma patients in National Centre for Severe Asthma and therefore we did not closely discuss the clinical application of omalizumab in chronic idiopathic urticaria or in other still not approved indications, like in chronic rhinosinusitis. We focused especially at the clinical (and less biological) impact of omalizumab treatment in the light of our practice. We hope, this point of view may be helpful for other specialists dealing with a similar topic.
Footnotes
Authors contributions: Both Jakub Novosad and Irena Krčmová wrote the manuscript, prepared figures (with generous help of Hana Kotlandová regarding graphical processing) and were responsible for the literature review and interpretation.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grant PROGRESS Q40 dedicated by Charles University, Czech Republic.
ORCID iD: Jakub Novosad  https://orcid.org/0000-0003-3290-3821
https://orcid.org/0000-0003-3290-3821
References
- 1. Boulet L-P, Bateman ED, Brusselle G, et al. Global strategy for asthma management and prevention (2019 Update), www.ginasthma.org (2019, accessed 27 June 2019).
- 2. Braman SS. The global burden of asthma. Chest 130: 4S–12S. [DOI] [PubMed] [Google Scholar]
- 3. Hekking PPW, Wener RR, Amelink M, et al. (2015) The prevalence of severe refractory asthma. The Journal of Allergy and Clinical Immunology 135(4): 896–902. [DOI] [PubMed] [Google Scholar]
- 4. Global Initiative for Asthma (GINA). Difficult-to-treat severe asthma in adolescent and adult patients GINA pocket guide for health professionals diagnosis and management, 2019, www.ginasthma.org
- 5. Chung KF, Wenzel SE, Brozek JL, et al. (2014) International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma task force report ERS/ATS guidelines on severe asthma Executive Summary. European Respiratory Journal 43: 343–373. [DOI] [PubMed] [Google Scholar]
- 6. Fleming L, Heaney L. (2019) Severe asthma – Perspectives from adult and pediatric pulmonology. Frontiers in Pediatrics 7: 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Montella S, Baraldi E, Cazzato S, et al. (2016) Severe asthma features in children: A case-control online survey. Italian Journal of Pediatrics 42: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Akdis A, Agache I, Asher MI, et al. (eds) Global Atlas of Asthma. EAACI, 2014, http://www.eaaci.org/resources/global-atlas-of-allergy.html
- 9. Tagaya E, Tamaoki J. (2007) Mechanisms of airway remodeling in asthma. Allergology International 56: 331–340. [DOI] [PubMed] [Google Scholar]
- 10. Viswanathan RK, Busse WW. (2018) Biologic therapy and asthma. Seminars in Respiratory and Critical Care Medicine 39: 100–114. [DOI] [PubMed] [Google Scholar]
- 11. Wenzel SE, Schwartz LB, Langmack EL, et al. (1999) Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. American Journal of Respiratory and Critical Care Medicine 160(3): 1001–1008. [DOI] [PubMed] [Google Scholar]
- 12. Brusselle GG, Maes T, Bracke KR. (2013) Eosinophilic airway inflammation in nonallergic asthma. Nature Medicine 19(8): 977–979. [DOI] [PubMed] [Google Scholar]
- 13. Wenzel SE. (2012) Asthma phenotypes: The evolution from clinical to molecular approaches. Nature Medicine 18: 716–725. [DOI] [PubMed] [Google Scholar]
- 14. Robinson D, Humbert M, Buhl R, et al. (2017) Revisiting Type 2-high and Type 2-low airway inflammation in asthma: Current knowledge and therapeutic implications. Clinical & Experimental Allergy 47(2): 161–175. [DOI] [PubMed] [Google Scholar]
- 15. Godar M, Blanchetot C, de Haard H, et al. (2018) Personalized medicine with biologics for severe type 2 asthma: Current status and future prospects. MAbs 10(1): 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Terl M, Sedlák V, Cap P, et al. (2017) Asthma management: A new phenotype-based approach using presence of eosinophilia and allergy. Allergy 72: 1279–1287. [DOI] [PubMed] [Google Scholar]
- 17. Schleich FN, Manise M, Sele J, et al. (2013) Distribution of sputum cellular phenotype in a large asthma cohort: Predicting factors for eosinophilic vs neutrophilic inflammation. BMC Pulmonary Medicine 13: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schleich F, Brusselle G, Louis R, et al. (2014) Heterogeneity of phenotypes in severe asthmatics. Respiratory Medicine 108(12): 1723–1732. [DOI] [PubMed] [Google Scholar]
- 19. Katial RKK, Bensch GWW, Busse WWW, et al. (2017) Changing paradigms in the treatment of severe asthma: The role of biologic therapies. The Journal of Allergy and Clinical Immunology: In Practice 5(2S): S1–S14. [DOI] [PubMed] [Google Scholar]
- 20. McGrath KW, Icitovic N, Boushey HA, et al. (2012) A large subgroup of mild-to-moderate asthma is persistently noneosinophilic. American Journal of Respiratory and Critical Care Medicine 185: 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prausnitz C, Küstner H. (1921) Studien über Uberempfindlicht. Centralb Baketerial 1 Abt Orog 86: 160–169. [Google Scholar]
- 22. Johansson SGO, Stockholm S. (2016) The discovery of IgE. The Journal of Allergy and Clinical Immunology 137: 1671–1673. [DOI] [PubMed] [Google Scholar]
- 23. Bennich HH, Ishizaka K, Johansson SGO, et al. (1968) Immunoglobulin E, A new class of human immunoglobulin. Bulletin of the World Health Organization 38: 151–152. [PMC free article] [PubMed] [Google Scholar]
- 24. Burrows B, Martinez FD, Halonen M, et al. (1989) Association of asthma with serum IgE levels and skin-test reactivity to allergens. The New England Journal of Medicine 320: 271–277. [DOI] [PubMed] [Google Scholar]
- 25. Snapper CM, Finkelman FD, Paul WE. (1988) Regulation of IgG1 and IgE production by interleukin 4. Immunological Reviews 102: 51–75. [DOI] [PubMed] [Google Scholar]
- 26. Bacharier Leonard D, Geha Raif S. (2000) Molecular mechanisms of IgE regulation. The Journal of Allergy and Clinical Immunology 105: 547–558. [DOI] [PubMed] [Google Scholar]
- 27. Dustin ML. (2014) The immunological synapse. Cancer Immunology Research 2: 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stone KD, Prussin C, Metcalfe DD. (2010) IgE, mast cells, basophils, and eosinophils. The Journal of Allergy and Clinical Immunology 125: S73–S80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ortega H, Chupp G, Bardin P, et al. (2014) The role of mepolizumab in atopic and nonatopic severe asthma with persistent eosinophilia. European Respiratory Journal 44(1): 239–241. [DOI] [PubMed] [Google Scholar]
- 30. Rabe KF, Nair P, Brusselle G, et al. (2018) Efficacy and safety of dupilumab in glucocorticoid-dependent severe asthma. The New England Journal of Medicine 378: 2475–2485. [DOI] [PubMed] [Google Scholar]
- 31. Corren J, Lemanske RF, Hanania NA, et al. (2011) Lebrikizumab treatment in adults with asthma. The New England Journal of Medicine 365: 1088–1098. [DOI] [PubMed] [Google Scholar]
- 32. Gould HJ, Sutton BJ. (2008) IgE in allergy and asthma today. Nature Reviews Immunology 8(3): 205–217. [DOI] [PubMed] [Google Scholar]
- 33. Karagiannis SN, Wang Q, East N, et al. (2003) Activity of human monocytes in IgE antibody-dependent surveillance and killing of ovarian tumor cells. European Journal of Immunology 33(4): 1030–1040. [DOI] [PubMed] [Google Scholar]
- 34. Garman SC, Kinet JP, Jardetzky TS. (1998) Crystal structure of the human high-affinity IgE receptor. Cell 95: 951–961. [DOI] [PubMed] [Google Scholar]
- 35. Sutton BJ, Beavil RL, Beavil AJ. (2000) Inhibition of IgE-receptor interactions. British Medical Bulletin 56(4): 1004–1018. [DOI] [PubMed] [Google Scholar]
- 36. Pelaia G, Canonica GW, Matucci A, et al. (2017) Targeted therapy in severe asthma today: Focus on immunoglobulin E. Drug Design, Development and Therapy 11: 1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Foster B, Metcalfe DD, Prussin C. (2003) Human dendritic cell 1 and dendritic cell 2 subsets express FcεRI: Correlation with serum IgE and allergic asthma. The Journal of Allergy and Clinical Immunology 112: 1132–1138. [DOI] [PubMed] [Google Scholar]
- 38. Kikutani H, Yokota A, Uchibayashi N, et al. (1989) Structure and function of Fc epsilon receptor II (Fc epsilon RII/CD23): A point of contact between the effector phase of allergy and B cell differentiation. Ciba Foundation Symposium 147: 23–31, discussion; 31. [DOI] [PubMed] [Google Scholar]
- 39. Palaniyandi S, Liu X, Periasamy S, et al. (2015) Inhibition of CD23-mediated IgE transcytosis suppresses the initiation and development of allergic airway inflammation. Mucosal Immunology 8(6): 1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Palaniyandi S, Tomei E, Li Z, et al. (2011) CD23-dependent transcytosis of IgE and immune complex across the polarized human respiratory epithelial cells. Journal of Immunology 186: 3484–3496. [DOI] [PubMed] [Google Scholar]
- 41. Matucci A, Vultaggio A, Maggi E, et al. (2018) Is IgE or eosinophils the key player in allergic asthma pathogenesis? Are we asking the right question? Respiratory Research 19: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stingl G, Maurer D. (1997) IgE-mediated allergen presentation via fc epsilon rl on antigen-presenting cells. International Archives of Allergy and Immunology 113: 24–29. [DOI] [PubMed] [Google Scholar]
- 43. Maurer D, Ebner C, Reininger B, et al. (1995) The high affinity IgE receptor (Fc epsilon RI) mediates IgE-dependent allergen presentation. Journal of Immunology 154: 6285–6290. [PubMed] [Google Scholar]
- 44. Burton OT, Medina Tamayo J, Stranks AJ, et al. (2018) IgE promotes type 2 innate lymphoid cells in murine food allergy. Clinical & Experimental Allergy 48(3): 288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Colgan Isaiah L, Hankel Roy JJD, Carver LA. (2010) Rozhodující signální dráhy alergického zánětu v dýchacích cestách. Current Opinion in Allergy and Clinical Immunology 7: 42–47. [Google Scholar]
- 46. Vitte J. (2015) Human mast cell tryptase in biology and medicine. Molecular Immunology 63(1): 18–24. [DOI] [PubMed] [Google Scholar]
- 47. Kambayashi T, Koretzky GA. (2007) Proximal signaling events in FcεRI-mediated mast cell activation. The Journal of Allergy and Clinical Immunology 119: 544–552. [DOI] [PubMed] [Google Scholar]
- 48. Bradding P. (2007) Mast cell regulation of airway smooth muscle function in asthma. European Respiratory Journal 29(5): 827–830. [DOI] [PubMed] [Google Scholar]
- 49. Redhu NS, Gounni AS. (2013) The high affinity IgE receptor (FcεRI) expression and function in airway smooth muscle. Pulmonary Pharmacology & Therapeutics 26(1): 86–94. [DOI] [PubMed] [Google Scholar]
- 50. Belleau JT, Gandhi RK, McPherson HM, et al. (2005) Research upregulation of CD23 (FcεRII) expression in human airway smooth muscle cells (huASMC) in response to IL-4, GM-CSF, and IL-4/GM-CSF. Clinical and Molecular Allergy 3: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roth M, Zhong J, Zumkeller C, et al. (2013) The role of IgE-receptors in IgE-dependent airway smooth muscle cell remodelling. PLoS ONE 8(2): e56015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roth M, Zhao F, Zhong J, et al. (2015) Serum IgE induced airway smooth muscle cell remodeling is independent of allergens and is prevented by omalizumab. PLoS ONE 10(9): e0136549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fang L, Wang X, Sun Q, et al. (2019) IgE downregulates PTEN through microRNA-21-5p and stimulates airway smooth muscle cell remodeling. International Journal of Molecular Sciences 20: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sihra BS, Kon OM, Grant JA, et al. (1997) Expression of high-affinity IgE receptors (FcεRI) on peripheral blood basophils, monocytes, and eosinophils in atopic and nonatopic subjects: Relationship to total serum IgE concentrations. The Journal of Allergy and Clinical Immunology 99: 699–706. [DOI] [PubMed] [Google Scholar]
- 55. Kim IS, Kim MJ, Kim DH, et al. (2013) Different anti-apoptotic effects of normal and asthmatic serum on normal eosinophil apoptosis depending on house dust mitespecific IgE. Molecular Biology Reports 40(10): 5875–5881. [DOI] [PubMed] [Google Scholar]
- 56. Nagata K, Hirai H, Tanaka K, et al. (1999) CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s). FEBS Letters 459: 195–199. [DOI] [PubMed] [Google Scholar]
- 57. Hirai H, Tanaka K, Yoshie O, et al. (2001) Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. Journal of Experimental Medicine 193: 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pettipher R, Hansel TT, Armer R. (2007) Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nature Reviews Drug Discovery 6(4): 313–325. [DOI] [PubMed] [Google Scholar]
- 59. Kupczyk M, Kuna P. (2017) Targeting the PGD2/CRTH2/DP1 signaling pathway in asthma and allergic disease: Current status and future perspectives. Drugs 77: 1281–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Domingo C, Palomares O, Sandham DA, et al. (2018) The prostaglandin D2 receptor 2 pathway in asthma: A key player in airway inflammation. Respiratory Research 19: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Marshall JS, Bell EB. (1985) Induction of an auto-anti-IgE response in rats. I. Effects on serum IgE concentrations. European Journal of Immunology 15(3): 272–277. [DOI] [PubMed] [Google Scholar]
- 62. Corne J, Djukanovic R, Thomas L, et al. (1997) The effect of intravenous administration of a chimeric anti-IgE antibody on serum IgE levels in atopic subjects: Efficacy, safety, and pharmacokinetics. Journal of Clinical Investigation 99: 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Busse W, Corren J, Lanier BQ, et al. (2001) Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. The Journal of Allergy and Clinical Immunology 108(2): 184–190. [DOI] [PubMed] [Google Scholar]
- 64. Solèr M, Matz J, Townley R, et al. (2001) The anti-IgE antibody omalizumab reduces exacerbations and steroid requirement in allergic asthmatics. European Respiratory Journal 18(2): 254–261. [DOI] [PubMed] [Google Scholar]
- 65. Buhl R, Solèr M, Matz J, et al. (2002) Omalizumab provides long-term control in patients with moderate-to-severe allergic asthma. European Respiratory Journal 20(1): 73–78. [DOI] [PubMed] [Google Scholar]
- 66. Hoshino M, Ohtawa J. (2012) Effects of adding omalizumab, an anti-immunoglobulin E antibody, on airway wall thickening in asthma. Respiration 83(6): 520–528. [DOI] [PubMed] [Google Scholar]
- 67. Lowe PJ, Renard D. (2011) Omalizumab decreases IgE production in patients with allergic (IgE-mediated) asthma; PKPD analysis of a biomarker, total IgE. British Journal of Clinical Pharmacology 72(2): 306–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kroegel C, Foerster M, Lerche K, et al. (2015) Long-term efficacy of omalizumab (OMA) for patients with severe allergic asthma (SAA). Clinical assessment and relationship to serum IgE concentrations. European Respiratory Journal 46: PA2561. [Google Scholar]
- 69. Lin CH, Cheng SL. (2016) A review of omalizumab for the management of severe asthma. Drug Design, Development and Therapy 10: 2369–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chan MA, Gigliotti NM, Dotson AL, et al. (2013) Omalizumab may decrease IgE synthesis by targeting membrane IgE+ human B cells. Clinical and Translational Allergy 3: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Maggi L, Rossettini B, Montaini G, et al. (2018) Omalizumab dampens type 2 inflammation in a group of long-term treated asthma patients and detaches IgE from FcεRI. European Journal of Immunology 48(12): 2005–2014. [DOI] [PubMed] [Google Scholar]
- 72. Teach SJ, Gill MA, Togias A, et al. (2015) Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. The Journal of Allergy and Clinical Immunology 136(6): 1476–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Labrador-Horrillo M, Ferrer M. (2015) Profile of omalizumab in the treatment of chronic spontaneous urticaria. Drug Design, Development and Therapy 9: 4909–4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gevaert P, Calus L, Van Zele T, et al. (2013) Omalizumab is effective in allergic and nonallergic patients with nasal polyps and asthma. The Journal of Allergy and Clinical Immunology 131(1): 110–116. [DOI] [PubMed] [Google Scholar]
- 75. Bousquet J, Cabrera P, Berkman N, et al. (2005) The effect of treatment with omalizumab, an anti-IgE antibody, on asthma exacerbations and emergency medical visits in patients with severe persistent asthma. Allergy 60(3): 302–308. [DOI] [PubMed] [Google Scholar]
- 76. Brusselle G, Michils A, Louis R, et al. (2009) ‘Real-life’ effectiveness of omalizumab in patients with severe persistent allergic asthma: The PERSIST study. Respiratory Medicine 103(11): 1633–1642. [DOI] [PubMed] [Google Scholar]
- 77. Braunstahl GJ, Chen CW, Maykut R, et al. (2013) The eXpeRience registry: The ‘real-world’ effectiveness of omalizumab in allergic asthma. Respiratory Medicine 107(8): 1141–1151. [DOI] [PubMed] [Google Scholar]
- 78. Kirchnerová OR, Valena T, Novosad J, et al. (2019) Real-world effectiveness and safety of omalizumab in patients with uncontrolled severe allergic asthma from the Czech Republic. Postepy Dermatol Alergol 36(1): 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bystroň J, Hutyrová B. (2016) Vyhodnocení účinnosti léčby anti-IgE monoklonální protilátkou nejen u těžkého alergického astmatu. Klinická Farmakologie A Farmacie 30: 14–18. [Google Scholar]
- 80. Deschildre A, Marguet C, Langlois C, et al. (2015) Real-life long-term omalizumab therapy in children with severe allergic asthma. European Respiratory Journal 46(3): 856–859. [DOI] [PubMed] [Google Scholar]
- 81. Deschildre A, Marguet C, Salleron J, et al. (2013) Add-on omalizumab in children with severe allergic asthma: A 1-year real life survey. European Respiratory Journal 42: 1224–1233. [DOI] [PubMed] [Google Scholar]
- 82. Licari A, Castagnoli R, Denicolo C, et al. (2017) Omalizumab in children with severe allergic asthma: The Italian real-life experience. Current Respiratory Medicine Reviews 13(1): 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Humbert M, Taillé C, Mala L, et al. (2018) Omalizumab effectiveness in patients with severe allergic asthma according to blood eosinophil count: The STELLAIR study. European Respiratory Journal 51(5): 1702523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Casale TB, Luskin AT, Busse W, et al. (2019) Omalizumab effectiveness by biomarker status in patients with asthma: Evidence from PROSPERO, a prospective real-world study. The Journal of Allergy and Clinical Immunology: In Practice 7(1): 156–164.e1. [DOI] [PubMed] [Google Scholar]
- 85. Bousquet J, Rabe K, Humbert M, et al. (2007) Predicting and evaluating response to omalizumab in patients with severe allergic asthma. Respiratory Medicine 101(7): 1483–1492. [DOI] [PubMed] [Google Scholar]
- 86. Durrani SR, Viswanathan RK, Busse WW. (2011) What effect does asthma treatment have on airway remodeling? Current perspectives. The Journal of Allergy and Clinical Immunology 128(3): 439–448, quiz 449. [DOI] [PubMed] [Google Scholar]
- 87. Rabe KF, Calhoun WJ, Smith N, et al. (2011) Can anti-IgE therapy prevent airway remodeling in allergic asthma. Allergy 66(9): 1142–1151. [DOI] [PubMed] [Google Scholar]
- 88. Samitas K, Delimpoura V, Zervas E, et al. (2015) Anti-IgE treatment, airway inflammation and remodelling in severe allergic asthma: Current knowledge and future perspectives. The European Respiratory Review 24(138): 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Krčmová I, Novosad J, Malá E, et al. (2018) Small, prospective, observational, pilot study in severe asthmatic patients after omalizumab treatment discontinuation. Clinical Therapeutics 40: 1942–1953. [DOI] [PubMed] [Google Scholar]
- 90. Nopp A, Johansson SGO, Adédoyin J, et al. (2010) After 6 years with Xolair; a 3-year withdrawal follow-up. Allergy 65(1): 56–60. [DOI] [PubMed] [Google Scholar]
- 91. Nopp A, Johansson SGO, Ankerst J, et al. (2007) CD-sens and clinical changes during withdrawal of Xolair after 6 years of treatment. Allergy 62(10): 1175–1181. [DOI] [PubMed] [Google Scholar]
- 92. Kupryś-Lipińska I, Kuna P. (2014) Loss of asthma control after cessation of omalizumab treatment: Real life data. Postepy Dermatol Alergol 31(1): 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Molimard M, Mala L, Bourdeix I, et al. (2014) Observational study in severe asthmatic patients after discontinuation of omalizumab for good asthma control. Respiratory Medicine 108(4): 571–576. [DOI] [PubMed] [Google Scholar]
- 94. Slavin RG, Ferioli C, Tannenbaum SJ, et al. (2009) Asthma symptom re-emergence after omalizumab withdrawal correlates well with increasing IgE and decreasing pharmacokinetic concentrations. The Journal of Allergy and Clinical Immunology 123(1): 107–113. [DOI] [PubMed] [Google Scholar]
- 95. Busse W, Trazskoma B, Omachi T, et al. (2014) Evaluating omalizumab persistency of response after long-term therapy (XPORT). European Respiratory Journal 44: 3485. [Google Scholar]
- 96. Ledford D, Busse WW, Trzaskoma B, et al. (2017) A randomized multicenter study evaluating Xolair persistence of response after long-term therapy. The Journal of Allergy and Clinical Immunology 140(1): 162–169. [DOI] [PubMed] [Google Scholar]