

Abstract
Craniofacial muscle pain is highly prevalent in temporomandibular disorders but is difficult to treat. Enhanced understanding of neurobiology unique to craniofacial muscle pain should lead to the development of novel mechanism-based treatments. Herein, we review recent studies to summarize neural pathways of craniofacial muscle pain. Nociceptive afferents in craniofacial muscles are predominantly peptidergic afferents enriched with TRPV1. Signals from peripheral glutamate receptors converge onto TRPV1, leading to mechanical hyperalgesia. Further studies are needed to clarify whether hyperalgesic priming in nonpeptidergic afferents or repeated acid injections also affect craniofacial muscle pain. Within trigeminal ganglia, afferents innervating craniofacial muscles interact with surrounding satellite glia, which enhances the sensitivity of the inflamed neurons as well as nearby uninjured afferents, resulting in hyperalgesia and ectopic pain originating from adjacent orofacial tissues. Craniofacial muscle afferents project to a wide area within the trigeminal nucleus complex, and central sensitization of medullary dorsal horn neurons is a critical factor in muscle hyperalgesia related to ectopic pain and emotional stress. Second-order neurons project rostrally to pathways associated with affective pain, such as parabrachial nucleus and medial thalamic nucleus, as well as sensory-discriminative pain, such as ventral posteromedial thalamic nuclei. Abnormal endogenous pain modulation can also contribute to chronic muscle pain. Descending serotonergic circuits from the rostral ventromedial medulla facilitate activation of second-order neurons in the trigeminal nucleus complex, which leads to the maintenance of mechanical hyperalgesia of inflamed masseter muscle. Patients with temporomandibular disorders exhibit altered brain networks in widespread cortical and subcortical regions. Recent development of methods for neural circuit manipulation allows silencing of specific hyperactive neural circuits. Chemogenetic silencing of TRPV1-expressing afferents or rostral ventromedial medulla neurons attenuates hyperalgesia during masseter inflammation. It is likely, therefore, that further delineation of neural circuits mediating craniofacial muscle hyperalgesia potentially enhances treatment of chronic muscle pain conditions.
Keywords: temporomandibular disorder, orofacial muscle pain, primary afferents, trigeminal ganglia, descending pain modulation, chemogenetics
Introduction
Craniofacial muscle pain is associated with substantial morbidity and affects individuals through different conditions. Acute conditions, such as space abscesses, can cause muscle pain. Tension-type headaches are also likely to involve pericranial muscle pain. Temporomandibular disorders (TMDs) are the most prevalent conditions involving craniofacial muscle pain. In diagnostic criteria for TMD, masticatory muscle disorders include myalgia, tendonitis, myositis, and spasm (Schiffman et al. 2014). Myalgia is defined as “pain of muscle origin that is affected by jaw movement, function, or parafunction, and replication of this pain occurs with provocation testing of the masticatory muscles.” Since TMD occurrence is influenced by multiple factors, including jaw parafunction, psychosocial conditions, and genetic factors, it is important to understand in detail the peripheral and central neurobiological mechanisms underlying pain from the jaw muscles.
Recent studies suggest that the craniofacial system and the spinal system show distinct peripheral and central pain processing or different sensitivities. Acute inflammation induced by carrageenan produces greater and longer pressure-induced mechanical hyperalgesia in the masseter muscle than in the gastrocnemius muscle (Bagues et al. 2017). Trigeminal nociceptors uniquely contain a subset of afferents that bypass second-order neurons in the medullary dorsal horn and directly project to the parabrachial nucleus (PBN), which may enhance affective pain (Rodriguez et al. 2017). Furthermore, a calcitonin gene-related peptide (CGRP) receptor antagonist attenuates orofacial neuropathic pain but not spinal neuropathic pain (Michot et al. 2012). Therefore, the mechanisms of craniofacial pain are unique and cannot be considered identical to those of spinal pain.
In this review, we highlight recent studies focusing on the molecular and cellular mechanisms of craniofacial pain conditions, particularly muscle pain. A fuller understanding of the neurobiological mechanisms unique to craniofacial muscle pain should expose gaps in current knowledge and enhance developing tailored treatments. Selective targeting of molecules or their signaling associated with hyperalgesia will support the development of new analgesic strategies. The identification of critical neural pathways responsible for persistent pain is also important in light of recent developments in advanced methods for neural circuit manipulation.
Primary Afferents for Acute and Chronic Craniofacial Muscle Pain
Nociceptive inputs from the craniofacial muscles are carried by trigeminal nerves, the cell bodies of which are located in trigeminal ganglia (TG). Central terminals of trigeminal afferents terminate on the second-order neurons in the trigeminal sensory nucleus complex in the brainstem. Like many other chronic pain conditions, myalgia from TMD is not usually accompanied by obvious signs of injury or muscle inflammation, such as the facial swelling that is present in infectious conditions (e.g., submasseteric abscess). Therefore, sensitization of the central rather than peripheral nervous system is thought to make a more critical contribution to the maintenance of chronic TMD pain. Nonetheless, recent studies suggest that acute and chronic muscle pain conditions in patients with TMD are attributable to inputs from primary afferents: patients with chronic TMD myalgia show increased levels of cytokines in the masseter muscle (Louca Jounger et al. 2017). Algogenic substances, such as glutamate or serotonin, are increased in the masseter muscle following tooth clenching in patients with chronic TMD (Dawson et al. 2015). Injection of local anesthetics to active trigger points in masticatory muscles attenuates myofascial pain in the head and neck (Nouged et al. 2019), suggesting that primary afferent inputs from the masticatory muscles contribute to the occurrence of chronic pain in TMD. Thus, it is necessary to identify the peripheral molecular mechanisms to fully understand the pathophysiology and optimal treatment of pain associated with TMD.
Glutamate Receptor and TRP Channel Mechanisms in Peptidergic Afferents
Approximately a quarter of retrogradely labeled masseter afferents in TG are small- to medium-sized peptidergic afferents that express CGRP or substance P (SubP; Ambalavanar et al. 2003). Nonpeptidergic masseter afferents that bind isolectin B4 (IB4) are rare (5%; Ambalavanar et al. 2003). In TG, TRPV1 is predominantly expressed in peptidergic afferents (Joseph et al. 2019), and approximately 25% of masseter afferents express TRPV1 (Ro et al. 2009). Recent studies have offered convincing evidence for the TRPV1 contribution to craniofacial muscle pain. In rat models of masseter inflammation with complete Freund’s adjuvant (CFA), masseter injection of a TRPV1 inhibitor suppresses mechanical hyperalgesia. Inhibition of TRPV1 also attenuates spontaneous pain and ongoing pain following masseter inflammation (Wang et al. 2017; Wang, Brigoli, et al. 2018).
Patients with myofascial TMD show increased interstitial glutamate in the masseter muscle as compared with healthy subjects (Castrillon et al. 2010). Receptors for glutamates, such as N-methyl-D-aspartate (NMDA) receptors and metabotropic glutamate receptor 5 (mGluR5), are expressed in nociceptors, and their activation in muscle afferents leads to hyperalgesia (Lee, Saloman, et al. 2012; Chung, Lee, et al. 2015). Interestingly, mechanical hyperalgesia of the masseter muscle induced by glutamate receptor agonists is prevented by preemptive injection of TRPV1 antagonist into the masseter muscle (Lee, Saloman, et al. 2012; Chung, Lee, et al. 2015). NMDA increases PKC-dependent phosphorylation of TRPV1 serine 800 residue (TRPV1 S800) in dissociated trigeminal neurons, suggesting that TRPV1 S800 phosphorylation is a convergent pathway in the glutamate-induced regulation of TRPV1 (Lee, Chung, and Ro 2012). In a mouse line in which TRPV1 S801, an orthologue residue of rat TRPV1 S800, is mutated into alanine to prevent phosphorylation, mouse grimace scale (MGS) following masseteric CFA injection is attenuated as compared with wild-type mice, suggesting that TRPV1 S801 phosphorylation-dependent mechanisms contribute to CFA-induced spontaneous pain (Joseph et al. 2019).
TRPV1+ afferents are enriched with not only TRPV1 but other nociceptive molecules. TRPA1 is highly colocalized with TRPV1 and is expressed in approximately 10% of masseter afferents (Ro et al. 2009). Expression of TRPA1 and TRPV1 are upregulated in TG following masseter inflammation (Asgar et al. 2015; Chung et al. 2016). Inhibition of TRPA1 attenuates mechanical hyperalgesia, spontaneous pain, and ongoing pain during masseter inflammation (Asgar et al. 2015; Wang et al. 2017). Simultaneous pharmacologic inhibition of TRPV1 and TRPA1 additionally reduces ongoing pain (as assessed by conditioned place preference; Wang, Brigoli, et al. 2018). CGRP and SubP are upregulated following masseter inflammation, and CGRP receptor antagonists attenuate hyperalgesia from masseter inflammation (Ambalavanar et al. 2006). Furthermore, TRPV1+ afferents undergo broad changes in gene expression during masseter inflammation. Transcriptomic assay of TG after masseter inflammation showed upregulation of multiple pain-related genes that are known to be enriched in TRPV1+ afferents (e.g., Nav1.8, Tac1, PKCα; Chung et al. 2016). Therefore, the inhibition of the entire function of TRPV1+ afferents is more likely to produce greater analgesia for craniofacial muscle pain than can be produced by inhibiting individual molecules.
The importance of TRPV1+ afferents in craniofacial muscle pain was unequivocally demonstrated through the manipulation of TRPV1+ afferents. Chemical ablation of TRPV1+ afferents by injection of resiniferatoxin into the trigeminal subnucleus caudalis (Vc) or localized functional silencing of TRPV1-lineage afferent terminals within masseter muscle attenuates spontaneous pain and bite-evoked pain from masseter inflammation (Wang et al. 2017). Furthermore, ablation of neurokinin 1 receptor (NK1)–expressing second-order neurons in Vc attenuates spontaneous and bite-evoked pain from masseter inflammation (Wang et al. 2017). Since NK1 is a receptor for SubP and NK1+ neurons in Vc are projection neurons carrying capsaicin-evoked pain (Saito et al. 2017), these data support the notion that signaling through peptidergic afferents mediates hyperalgesia under craniofacial muscle inflammation. The roles of TRPV1+ afferents are depicted in Figure 1A.
Figure 1.

Potential mechanisms underlying muscle pain in peptidergic and nonpeptidergic afferents. (A) Sensitization of masseter afferents by glutamate receptor and TRP channel interaction in peptidergic afferents leads to mechanical hyperalgesia of masseter muscles and spontaneous pain under masseter inflammation. Glutamate receptor invokes PKC to phosphorylate TRPV1. TRPA1 is highly colocalized with TRPV1 and mediates masseter hyperalgesia. TRPV1+ afferents also mediate bite-evoked pain through unknown mechanisms, independent of TRPV1 or TRPA1. (B) IB4+ nonpeptidergic afferents mediate hyperalgesic priming. Exposure of muscle afferents to initial stimuli, such as interleukin 6 (IL6), glial cell line–derived neurotrophic factor (GDNF), and monocyte chemoattractant protein 1 (MCP1), leads to a priming state in nociceptors. In primed nociceptors, prostaglandin receptors (EP) are associated with exchange proteins activated by cAMP (Epac) and protein kinase Cε (PKCε), in addition to canonical protein kinase A (PKA) signaling. (C) Repeated acid injection into the muscle produces secondary hyperalgesia in skin through peptidergic afferents. Acid-sensing ion channel 3 (ASIC3), TRPV1, and Nav1.8 mediate hyperalgesia, which is independent of PKCε. Mechanisms elaborated in panels B and C are established in limb muscle. Further studies are needed to clarify whether hyperalgesic priming or repeated acid injections also affect craniofacial muscle pain.
Hyperalgesic Priming in Nonpeptidergic Afferents
Acute injury or inflammation induces long-lasting neuroplastic changes in nociceptor signaling, mainly involving protein kinase Cε (PKCε; Hucho et al. 2005). Although the pain provoked by the initial insult is transient, such neuroplastic changes prime nociceptors to be more susceptible to subsequent stimuli. Consequently, a second, modest stimulus by cytokines or inflammatory mediators (e.g., a low dose of prostaglandin E2 [PGE2]) provokes robust and prolonged hyperalgesia, which would not occur in the absence of the initial insult. This paradigm, so-called hyperalgesic priming, has been proposed as the mechanism underlying the transition of pain from acute to chronic at the level of primary afferents. In limb muscles, hyperalgesic priming is mediated by IB4+ nonpeptidergic nociceptive afferents, which contribute to mechanical hyperalgesia in limb muscles (assessed by pressure on the gastrocnemius muscle following inflammation or eccentric exercise; Alvarez et al. 2012; Fig. 1B).
Hyperalgesic priming could also underpin the chronic muscle pain associated with TMD. Multiple cytokines and inflammatory mediators are increased in the craniofacial muscles of patients with chronic TMD (Dawson et al. 2015; Louca Jounger et al. 2017), and such changes may occur repeatedly, for example, during repeated clenching bouts. In jaw muscles, multiple repeated injection of NGF produces increased mechanical sensitivity in the masseter but not the temporal muscle without affecting the frequency of referred pain (Exposto et al. 2018). Thus, despite the paucity of IB4+ afferents in masticatory muscles (Ambalavanar et al. 2003), the involvement of hyperalgesic priming in the muscle hyperalgesia observed in these studies cannot be excluded.
Long-lasting Hyperalgesia by Repeated Acid Injection in Peptidergic Afferents
Ischemic acidosis in muscle is relevant to muscle pain. Although mechanical hyperalgesia of the hindpaw produced by a single injection of acid (pH 4) into the gastrocnemius muscle of rats is transient and disappears within hours, a second injection after 5 d leads to hyperalgesia that lasts for several days (Sluka et al. 2001; Chen et al. 2014; Gong et al. 2016). The repeated injection of acid has been used as a rodent model of prolonged hyperalgesia. Although this model appears to be similar to the aforementioned hyperalgesic priming, its underlying mechanisms are distinct. In contrast to hyperalgesic priming occurring in IB4+ afferents, hyperalgesia by repeated acid injection occurs in IB4-negative afferents and depends on TRPV1, acid-sensing ion channel 3, and Nav1.8 but not PKCε (Chen et al. 2014; Fig. 1C).
Two injections of acid into the rat masseter muscle have been shown to induce prolonged mechanical hyperalgesia (Lund et al. 2010). In this study, mechanical sensitivity was assessed on masseter muscles with a 15-g von Frey filament, which is close to the cutaneous mechanical threshold; therefore, mechanical hyperalgesia is likely to represent secondary hyperalgesia derived from the skin overlying the masseter muscle. By contrast, repeated injection of acid into the masseter muscle did not produce mechanical hyperalgesia when assessed with greater pressure (>100-g baseline threshold) on the masseter muscle (Ambalavanar et al. 2007). Therefore, it is unclear whether repeated acid injection or hyperalgesic priming actually mediates hyperalgesia from the masseter muscles. In contrast to rodent limb muscles, repeated acid injection into the masseter muscle produces transient muscle pain but does not cause prolonged muscle hyperalgesia or functional pain in humans (Castrillon et al. 2013; Louca Jounger et al. 2019).
Intraganglionic Mechanisms
Molecular and cellular mechanisms of complex dynamic interactions within TG have been thoroughly reviewed elsewhere (Shinoda et al. 2019). Multiple layers of interaction occur within TG in the presence of nerve injury or peripheral inflammation. Afferents projecting to the site of injury or inflammation interact with satellite glia (SG) surrounding the given neurons, which enhances the sensitivity of the injured neurons. Injured neurons and surrounding SG can sensitize the adjacent afferents projecting to uninjured tissue, which induces pain from an ectopic site. Such spreading can occur across neurons projected to different dermatomes (e.g., mandibular area to maxillary area) or different tissues (e.g., tooth pulp to masseter muscle). Here, we discuss recent studies on craniofacial muscle pain, as diagrammed in Figure 2.
Figure 2.
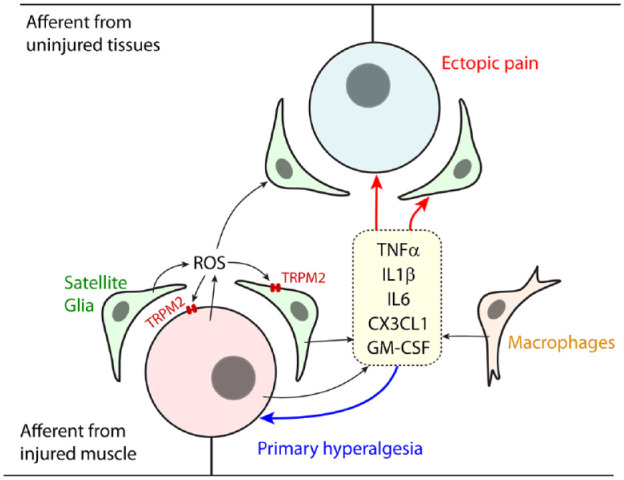
Schematic of intraganglionic mechanisms of craniofacial muscle pain. Nociceptive inputs from injured or inflamed craniofacial muscle produce extensive changes in gene expression in primary afferents and satellite glia within trigeminal ganglia. Reactive oxygen species (ROS) are increased, which may enhance genetic and functional changes. Primary afferents, satellite glia, and macrophages produce an array of cytokines and chemokines, such as tumor necrosis factor α (TNFα), interleukin 1β (IL1β), interleukin 6 (IL6), C-X3-C motif chemokine ligand 1 (CX3CL1; fractalkine), and granulocyte-macrophage colony-stimulating factor (GM-CSF). These factors sensitize injured afferents to enhance hyperalgesia (primary hyperalgesia). These sensitizing factors propagate into adjacent neurons from uninjured tissues, producing ectopic pain.
CFA-induced inflammation of the masseter muscle produces changes in approximately 17% of all genes within the TG, including multiple cytokines, ion channels, G protein–coupled receptors, and neurotrophic factors (Chung et al. 2016). In this study, gene changes should reflect the net alteration of transcriptomes from not only neurons but also SG and immune cells within the TG. Masseter inflammation or restraint stress induces the activation and proliferation of SG with upregulation of glial fibrillary acidic protein, a marker of SG (Zhao et al. 2015; Chung et al. 2016). Masseter inflammation also causes increased reactive oxygen species with proinflammatory cytokines, such as interleukin 1β or tumor necrosis factor α, in the TG (Chung, Asgar, et al. 2015). These factors may be derived from SG or macrophages. Indeed, sustained mouth opening in mice induces increased infiltration of macrophages and upregulation of cytokines within TG (Hawkins and Durham 2016; Wang, Shi, et al. 2018). Such changes in intraganglionic cytokines or chemokines following jaw muscle inflammation or injury should produce primary hyperalgesia. For example, temporal muscle afferents release fractalkine to activate the surrounding SG through C-X3-C motif chemokine receptor 1, which mediates mechanical hyperalgesia from the temporal muscle (Cairns et al. 2017). Increases in ganglionic cytokines following muscle inflammation can induce ectopic pain from other regions. Likewise, inflammation of other tissues may propagate into the jaw muscles via intraganglionic mechanisms. Temporomandibular joint inflammation increases SG activity and the level of tumor necrosis factor α in the TG, which mediates mechanical hyperalgesia of the masseter muscle without spreading inflammation to the muscle (Ito et al. 2018). Therefore, intraganglionic mechanisms involving SG and immune cells can produce craniofacial muscle hyperalgesia and ectopic pain. Intraganglionic spreading of sensitizing signals raises the possibility that aforementioned roles of peptidergic and nonpeptidergic afferents are not completely segregated along ascending pain pathways. In this regard, mechanical hyperalgesia induced by masseter injection of capsaicin, which activates peptidergic afferents, can be attenuated by an agonist of δ-opioid receptor or by an inhibitor of G protein–coupled inwardly rectifying potassium channels, both of which are expressed in nonpeptidergic afferents (Chung et al. 2014).
Central Mechanisms for Craniofacial Muscle Pain
Central Sensitization of Second-Order Neurons in the Trigeminal Nucleus Complex
Nociceptive afferent inputs from TG terminate on second-order neurons in the trigeminal nucleus complex (TNC) and upper cervical spinal cord (Chichorro et al. 2017). Multiple preclinical studies have established that sensitization of medullary dorsal horn neurons through NMDA receptors, glial cells, neuropeptides, and multiple cytokines and neurotrophins underpins orofacial hyperalgesia, and recent detailed reviews on this topic are available (Chichorro et al. 2017; Shinoda et al. 2019). Sensitization of medullary dorsal horn neurons involving NMDA receptor and glial cells within Vc also plays an important role in craniofacial muscle hyperalgesia (Ren and Dubner 2011; Lin et al. 2019). In contrast to cutaneous hyperalgesia, muscle hyperalgesia involves sensitization of neurons in not only Vc and upper cervical spinal cord but also the transition zone between Vc and the interpolaris (Vc/Vi; Ren and Dubner 2011). Vc/Vi receives autonomic and vagal inputs, suggesting that somatoautonomic and somatovisceral integration occurs in this area (Ikeda et al. 2003). In addition, TNC receives convergent inputs from various orofacial tissues. The sensitization of TNC neurons induced by the injury of one tissue can produce hyperalgesia in an uninjured tissue. For example, pulpitis induces sensitization of Vc and Vc/Vi neurons, which produces masseter hyperalgesia (Shimizu et al. 2014). Sensitization of TNC neurons also mediates masseter hyperalgesia associated with stress: Forced swim stress increases Fos expression in Vc and Vc/Vi induced by masseter injection of formalin (Nakatani et al. 2018). Restraint stress induces masseter mechanical hyperalgesia through the NMDA receptor and neuronal nitric oxide synthase in Vc (Lin et al. 2019). Therefore, TNC is a critical relay station in which somatosensory input is integrated with multiple factors to modulate pain from craniofacial muscles.
Rostral projections of second-order neurons from Vc mainly terminate in PBN, medial thalamic nuclei, and ventral posteromedial thalamic nuclei (Saito et al. 2017). Masseter inflammation leads to the activation of Vc and Vc/Vi neurons that project primarily to the PBN and the nucleus submedius of the thalamus (Ikeda et al. 2003). Neurons in PBN then project to multiple brain regions involved in pain processing, including central amygdala, hypothalamus, and PAG (Kucyi et al. 2014), suggesting a role of PBN in affective pain and descending pain modulation (Chiang et al. 2019). VPM project to the somatosensory cortex and are involved in sensory discriminative aspects of pain, whereas medial thalamic nuclei project to the anterior cingulate cortex (ACC) and mediate affective aspects of pain (Iwata et al. 2011). Since proportions of Vc neurons projecting to VPM and PBN are altered by trigeminal neuropathy (Okada et al. 2019), it is possible that craniofacial muscle pain or TMD involves plasticity of ascending pain pathways. Neural pathways relevant to craniofacial muscle pain are depicted in Figure 3.
Figure 3.
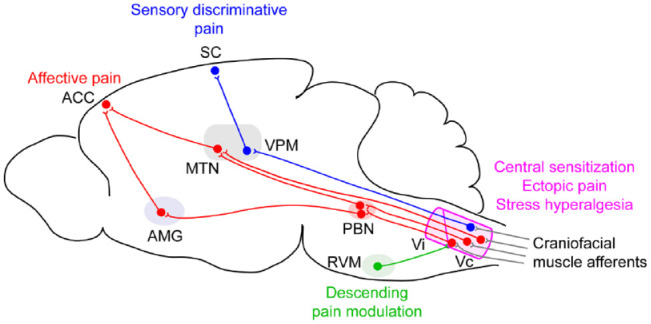
Schematic of central neural circuits of craniofacial muscle pain. Central sensitization in the trigeminal nucleus complex, especially the subnucleus caudalis (Vc) and transition zone of the Vc and subnucleus interpolaris (Vc/Vi), is critical for amplification of pain, ectopic hyperalgesia, and stress-induced hyperalgesia. Vc receives descending pain modulation inputs from rostral ventromedial medulla (RVM). Vc neurons are mainly projected to ventral posteromedial thalamic nuclei (VPM) and the parabrachial nucleus (PBN). VPM neurons project to the somatosensory cortex and mediate sensory discriminative aspects of pain. Medial thalamic nuclei (MTN) directly project to the anterior cingulate cortex (ACC), which contributes to affective pain. PBN widely projects to pain-related regions, including amygdala (AMG), which projects to ACC and mediates affective components of pain.
Descending Pain Modulation
Second-order neurons in the TNC receive descending inputs from several regions of the central nervous system, such as the rostral ventromedial medulla (RVM) or locus coeruleus, which modulates nociceptive input and transmission in the TNC. The RVM and the locus coeruleus receive inputs from the amygdala and PAG. Such descending modulation can be either inhibitory or facilitatory. Imbalance of inhibitory and facilitatory modulation in favor of facilitation under tissue or nerve injury can lead to chronic pain. Different regions of the PAG are involved in inhibitory and facilitatory regulation following the injection of hypertonic saline into rat gastrocnemius muscle. Lesioning of the dorsolateral PAG attenuates mechanical hyperalgesia, whereas ventrolateral PAG attenuates thermal hypoalgesia from the hindpaw (Lei et al. 2014). The RVM also mediates inhibition and facilitation. Descending facilitation from the RVM is mainly driven by descending projection of serotonergic neurons onto second-order neurons in Vc and Vc/Vi (Sugiyo et al. 2005; Okubo et al. 2013). CFA-induced masseter mechanical hyperalgesia is relieved by the lesioning of RVM or depletion of serotonin in RVM neurons (Sugiyo et al. 2005; Chai et al. 2012).
In humans, endogenous pain modulation is assessed by temporal summation of pain (facilitatory regulation) or conditioned pain modulation (inhibitory regulation). Functional pain conditions involving fibromyalgia, headache, or TMD are commonly suggested to accompany abnormal endogenous pain modulation (e.g., enhanced facilitation or impaired inhibition). Reports of endogenous pain modulation in patients with TMD are equivocal, as has recently been demonstrated (Moana-Filho et al. 2018). However, meta-analyses, including 22 studies involving patients with 3 nonparoxysmal orofacial pain conditions (TMD, burning mouth syndrome, and persistent dentoalveolar pain disorders), showed that pain facilitation is modestly enhanced in the patients experiencing pain as compared with those who were pain-free. By contrast, a trend whereby pain inhibition was impaired in patients with pain was observed but did not reach statistical significance due to considerable heterogeneity of the studies (Moana-Filho et al. 2018). These results suggest that abnormal endogenous pain modulation can contribute to central mechanisms underlying chronic muscle pain in patients with TMD. Therefore, pharmacologic or nonpharmacologic methods aimed at altering endogenous pain modulation may be viable options as potential therapies for chronic muscle pain.
Maladaptive Changes of Pain Circuits in the Brain
A study with positive emission tomography in humans showed that pain from the masseter muscle is represented in the bilateral dorsal-posterior insula, ACC, prefrontal cortex, and right posterior parietal cortex but not in the somatosensory cortex, suggesting a strong affective component of pain from masseter muscle (Kupers et al. 2004). Functional magnetic resonance imaging studies in patients with TMD indicate widespread changes in brain networks: patients with TMD show increased resting functional connectivity between the left anterior insular cortex and pregenual ACC (Ichesco et al. 2012) and between the medial prefrontal cortex and the posterior cingulate cortex/precuneus, medial thalamus, or PAG (Kucyi et al. 2014), suggesting the importance of the affective and cognitive aspects of pain and the descending regulatory system. A recent study indicated a role of striatum. Patients with TMD show decreased corticostriatal functional connectivity, for example, between ventral striatum and ventral frontal cortices (ACC or anterior insula), which might underpin altered motor control, pain processing, and cognition (He et al. 2018). A meta-analysis study suggested that these altered brain networks in patients with TMD are largely in common with those in patients with trigeminal neuropathy (e.g., changes in ACC, insular cortex, prefrontal cortex, and basal ganglia) with some distinct components (e.g., involvement of distinct regions of insular cortex and less involvement of thalamocortical connectivity in TMD; Lin 2014).
Functional connectivity reflects only temporal synchronization of blood flow changes between 2 areas, regardless of their direct structural and functional neural connection; therefore, actual contributions of the identified functional connectivity to pain are hard to interpret. Despite this limitation, these studies provide useful information allowing us to infer functional changes among multiple regions involved in pain transmission and modulation in patients with TMD. These human data provide an excellent starting point for reverse translational approaches to determine detailed neural circuit mechanisms of craniofacial muscle pain in the brains of experimental animals. For example, a recent study showed that enhanced synaptic transmission in ACC, as evaluated by stimulating the medial thalamus, was shown to maintain long-lasting masseter hyperalgesia induced by occlusal interference in rats (Xu et al. 2019).
Neural Circuit Manipulations as Potential Therapeutics for Craniofacial Muscle Pain
Recent developments in circuit-manipulating methods, including optogenetic or chemogenetic tools, are considered potentially beneficial for the treatment of chronic pain conditions (Iyer et al. 2016). These systems may be useful in manipulating the activity of a subset of neurons in the peripheral and central nervous systems to activate inhibitory circuits or to silence hyperactive circuits. Since infection with adeno-associated virus (AAV) is approved by the Food and Drug Administration, virally delivered genetic tools in well-defined circuits may be an option for improved treatment of chronic pain in the future. In mice, chemogenetic silencing of TRPV1+ afferents with inhibitory designer receptors exclusively activated by designer drugs (DREADD) receptor hM4Di, an engineered human M4 muscarinic receptor coupled with inhibitory G protein, relieves spontaneous and bite-evoked pain following masseter inflammation (Fig. 4). In comparison, bite-evoked pain can be inhibited by silencing TRPV1+ afferents but not by inhibition of TRPV1 or TRPA1 (Wang et al. 2017; Wang, Brigoli, et al. 2018; Joseph et al. 2019). Silencing of the peptidergic afferents should reduce nociceptive inputs from craniofacial muscles and reduce neurotransmission into the Vc. At the same time, neurogenic release of algogenic substances (e.g., glutamate, SubP) into muscle can be reduced, which will further suppress nociceptor activation and sensitization. In contrast to the silencing of cutaneous TRPV1+ afferents, localized silencing of masseter afferents is unlikely to be accompanied by adverse effects, such as loss of heat sensation.
Figure 4.
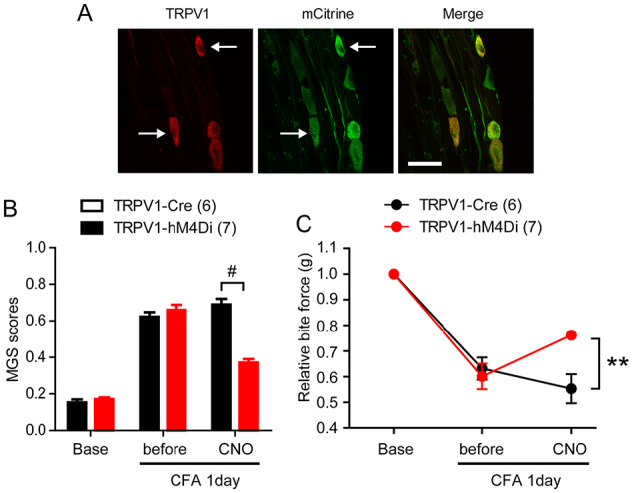
Chemogenetic inhibition of TRPV1-lineage afferents attenuates spontaneous and bite-evoked pain during masseter inflammation in mice. (A) TRPV1-Cre mice were crossed with R26-hM4Di mice to generate TRPV1-hM4Di mice. hM4Di is a Gi-coupled DREADD receptor that can be activated by clozapine-N-oxide (CNO). hM4Di is fused with mCitrine, which serves as a marker of hM4Di expression. Trigeminal ganglia of TRPV1-hM4Di mice were immunolabeled with anti-TRPV1 or anti-GFP. Arrows indicate neurons coexpressing TRPV1 and mCitrine. (B, C) Averaged mouse grimace scale (MGS) scores and bite force evaluated before and after bilateral masseter injection of CNO 1 d following bilateral masseter injection of complete Freund’s adjuvant (CFA) in TRPV1-hM4Di and TRPV1-Cre mice. Values are presented as mean ± SEM. **P < 0.001 and #P < 0.0001 in Bonferroni post hoc test following 2-way analysis of variance. (Reproduced from Wang et al. 2017 with permission.)
The chemogenetic approach is potentially useful for manipulating central circuits as well. For example, chemogenetic silencing of RVM neurons attenuated spontaneous and bite-evoked pain from masseter inflammation (Fig. 5). Inhibitory DREADD receptor hM4Di fused with mCherry or green fluorescence protein (GFP) control was expressed in RVM neurons by microinjection of AAV5. Fluorescent reporter proteins were robustly expressed in RVM neurons. GFP+ or mCherry+ axons were densely projected to the medullary dorsal horn, which was located close to the TRPV1+ central terminals of the primary afferents. Mice expressing hM4Di or GFP showed comparable baseline MGS and bite force. At 1 d after CFA injection into bilateral masseter muscles, both groups showed similar increases in MGS and decreases in bite force. Upon systemic injection of DREADD agonist compound 21, MGS was reduced and bite force elevated in the hM4Di group but not the GFP group. These results suggest that functional inhibition of the descending projection from the RVM to Vc reduces net facilitatory effects on spontaneous and bite-evoked pain, as well as mechanical hyperalgesia, following masseter inflammation. This approach can be useful for silencing other excitatory circuits that are involved in masseter pain. For example, the formalin-induced masseter pain response is alleviated by the chemogenetic silencing of ventroposterior thalamic nuclei in rats (Strand et al. 2018).
Figure 5.
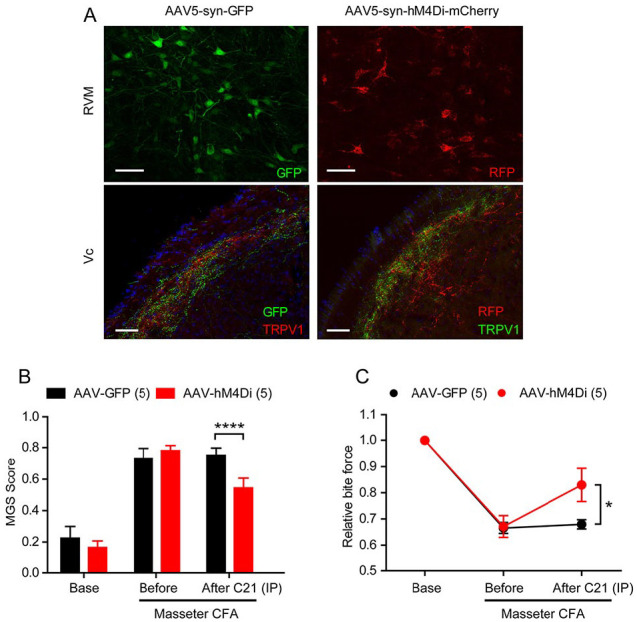
Chemogenetic inhibition of rostral ventromedial medulla (RVM) neurons attenuates spontaneous and bite-evoked pain during masseter inflammation in mice. (A) Adeno-associated virus 5 encoding green fluorescence protein (GFP) or hM4Di fused with mCherry under a neuron-specific promoter synapsin (AAV5-Syn-GFP or AAV5-syn-hM4Di-mCherry) was stereotaxically injected into RVM. Immunohistochemical labeling of GFP or red fluorescent protein (RFP) in RVM (top) or GFP and TRPV1 in trigeminal nucleus caudalis (Vc; bottom) as indicated. Scale bar, 50 µm. (B, C) Four weeks after injection of AAV5-syn-GFP or AAV5-syn-hM4Di, mouse grimace scale (MGS) and bite force were measured before and 1 d after injecting complete Freund’s adjuvant (CFA) into bilateral masseter muscles. Compound21 (0.2 mg; nonhydrolyzable analogue of clozapine-N-oxide) was injected intraperitoneally (IP) to activate hM4Di for neuronal silencing, which was followed by measuring MGS and bite force 30 min to 1 h after injection. *P < 0.05 and ****P < 0.0001 in Bonferroni post hoc analysis following 2-way repeated measure analysis of variance. n = 5 in each group. Values are presented as mean ± SEM.
Conclusion
Chronic craniofacial muscle pain associated with conditions such as TMD involves multiple peripheral and central mechanisms. We discussed the potential roles of glutamate receptors and TRP interactions, hyperalgesic priming in primary afferents, and intraganglionic spreading mechanisms in TG. We also discussed the contribution of altered brain pain pathways, including descending pain modulation. These areas are likely to provide potential major neural pathways associated with craniofacial muscle pain. Identifying major neural circuits is important for enhancing our ability to treat intractable chronic pain conditions through pharmacologic or circuit-manipulating approaches in the future. Molecular and circuitry mechanisms in brain regions associated with chronic muscle pain, particularly the affective components of pain, require further delineation. Enhanced understanding of peripheral and central circuits in comorbid pain conditions, sexual dimorphism, and stress-induced hyperalgesia will also be critical for improving our ability to treat chronic muscle pain conditions.
Author Contributions
M.K. Chung, contributed to conception, design, data analysis, and interpretation, drafted the manuscript; S. Wang, contributed to conception, data acquisition, and analysis, critically revised the manuscript; J. Yang, contributed to conception and data acquisition, critically revised the manuscript; I. Alshanqiti, contributed to conception, drafted the manuscript; F. Wei, contributed to conception, design, data analysis, and interpretation, critically revised the manuscript; J.Y. Ro, contributed to conception, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
This study was supported by the National Institute of Dental and Craniofacial Research of the National Institutes of Health grants DE023846 (M.K.C.), DE027731 (M.K.C. and F.W.), and DE016 062 (J.Y.R.).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
ORCID iD: M.K. Chung  https://orcid.org/0000-0001-7637-1148
https://orcid.org/0000-0001-7637-1148
References
- Alvarez P, Gear RW, Green PG, Levine JD. 2012. IB4-saporin attenuates acute and eliminates chronic muscle pain in the rat. Exp Neurol. 233(2):859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambalavanar R, Moritani M, Haines A, Hilton T, Dessem D. 2003. Chemical phenotypes of muscle and cutaneous afferent neurons in the rat trigeminal ganglion. J Comp Neurol. 460(2):167–179. [DOI] [PubMed] [Google Scholar]
- Ambalavanar R, Moritani M, Moutanni A, Gangula P, Yallampalli C, Dessem D. 2006. Deep tissue inflammation upregulates neuropeptides and evokes nociceptive behaviors which are modulated by a neuropeptide antagonist. Pain. 120(1–2):53–68. [DOI] [PubMed] [Google Scholar]
- Ambalavanar R, Yallampalli C, Yallampalli U, Dessem D. 2007. Injection of adjuvant but not acidic saline into craniofacial muscle evokes nociceptive behaviors and neuropeptide expression. Neuroscience. 149(3):650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgar J, Zhang Y, Saloman JL, Wang S, Chung MK, Ro JY. 2015. The role of TRPA1 in muscle pain and mechanical hypersensitivity under inflammatory conditions in rats. Neuroscience. 310:206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagues A, Martin-Fontelles MI, Esteban-Hernandez J, Sanchez-Robles EM. 2017. Characterization of the nociceptive effect of carrageenan: masseter versus gastrocnemius. Muscle Nerve. 56(4):804–813. [DOI] [PubMed] [Google Scholar]
- Cairns BE, O’Brien M, Dong XD, Gazerani P. 2017. Elevated fractalkine (CX3CL1) levels in the trigeminal ganglion mechanically sensitize temporalis muscle nociceptors. Mol Neurobiol. 54(5):3695–3706. [DOI] [PubMed] [Google Scholar]
- Castrillon EE, Cairns B, List T, Svensson P, Ernberg M. 2013. Acidic saline-induced pain as a model for experimental masseter myalgia in healthy subjects. Eur J Pain. 17(10):1438–1446. [DOI] [PubMed] [Google Scholar]
- Castrillon EE, Ernberg M, Cairns BE, Wang K, Sessle BJ, Arendt-Nielsen L, Svensson P. 2010. Interstitial glutamate concentration is elevated in the masseter muscle of myofascial temporomandibular disorder patients. J Orofac Pain. 24(4):350–360. [PubMed] [Google Scholar]
- Chai B, Guo W, Wei F, Dubner R, Ren K. 2012. Trigeminal-rostral ventromedial medulla circuitry is involved in orofacial hyperalgesia contralateral to tissue injury. Mol Pain. 8:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WN, Lee CH, Lin SH, Wong CW, Sun WH, Wood JN, Chen CC. 2014. Roles of ASIC3, TRPV1, and NaV1.8 in the transition from acute to chronic pain in a mouse model of fibromyalgia. Mol Pain. 10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang MC, Bowen A, Schier LA, Tupone D, Uddin O, Heinricher MM. 2019. Parabrachial complex: a hub for pain and aversion. J Neurosci. 39(42):8225–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichorro JG, Porreca F, Sessle B. 2017. Mechanisms of craniofacial pain. Cephalalgia. 37(7):613–626. [DOI] [PubMed] [Google Scholar]
- Chung MK, Asgar J, Lee J, Shim MS, Dumler C, Ro JY. 2015. The role of TRPM2 in hydrogen peroxide-induced expression of inflammatory cytokine and chemokine in rat trigeminal ganglia. Neuroscience. 297:160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MK, Asgar J, Park J, Ro JY. 2016. Transcriptome analysis of trigeminal ganglia following masseter muscle inflammation in rats. Mol Pain. 12:1744806916668526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MK, Cho YS, Bae YC, Lee J, Zhang X, Ro JY. 2014. Peripheral G protein–coupled inwardly rectifying potassium channels are involved in δ-opioid receptor-mediated anti-hyperalgesia in rat masseter muscle. Eur J Pain. 18(1):29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MK, Lee J, Joseph J, Saloman J, Ro JY. 2015. Peripheral group I metabotropic glutamate receptor activation leads to muscle mechanical hyperalgesia through TRPV1 phosphorylation in the rat. J Pain. 16(1):67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson A, Ghafouri B, Gerdle B, List T, Svensson P, Ernberg M. 2015. Effects of experimental tooth clenching on pain and intramuscular release of 5-HT and glutamate in patients with myofascial TMD. Clin J Pain. 31(8):740–749. [DOI] [PubMed] [Google Scholar]
- Exposto FG, Masuda M, Castrillon EE, Svensson P. 2018. Effects of nerve growth factor experimentally-induced craniofacial muscle sensitization on referred pain frequency and number of headache days: a double-blind, randomized placebo-controlled study. Cephalalgia. 38(14):2006–2016. [DOI] [PubMed] [Google Scholar]
- Gong WY, Abdelhamid RE, Carvalho CS, Sluka KA. 2016. Resident macrophages in muscle contribute to development of hyperalgesia in a mouse model of noninflammatory muscle pain. J Pain. 17(10):1081–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins JL, Durham PL. 2016. Prolonged jaw opening promotes nociception and enhanced cytokine expression. J Oral Facial Pain Headache. 30(1):34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Li F, Gu T, Ma H, Li X, Zou S, Huang X, Lui S, Gong Q, Chen S. 2018. Reduced corticostriatal functional connectivity in temporomandibular disorders. Hum Brain Mapp. 39(6):2563–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucho TB, Dina OA, Levine JD. 2005. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4(+) neuron-specific mechanism. J Neurosci. 25(26):6119–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichesco E, Quintero A, Clauw DJ, Peltier S, Sundgren PM, Gerstner GE, Schmidt-Wilcke T. 2012. Altered functional connectivity between the insula and the cingulate cortex in patients with temporomandibular disorder: a pilot study. Headache. 52(3):441–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T, Terayama R, Jue SS, Sugiyo S, Dubner R, Ren K. 2003. Differential rostral projections of caudal brainstem neurons receiving trigeminal input after masseter inflammation. J Comp Neurol. 465(2):220–233. [DOI] [PubMed] [Google Scholar]
- Ito R, Shinoda M, Honda K, Urata K, Lee J, Maruno M, Soma K, Okada S, Gionhaku N, Iwata K. 2018. Tumor necrosis factor alpha signaling in trigeminal ganglion contributes to mechanical hypersensitivity in masseter muscle during temporomandibular joint inflammation. J Oral Facial Pain Headache. 32(1):75–83. [DOI] [PubMed] [Google Scholar]
- Iwata K, Miyachi S, Imanishi M, Tsuboi Y, Kitagawa J, Teramoto K, Hitomi S, Shinoda M, Kondo M, Takada M. 2011. Ascending multisynaptic pathways from the trigeminal ganglion to the anterior cingulate cortex. Exp Neurol. 227(1):69–78. [DOI] [PubMed] [Google Scholar]
- Iyer SM, Vesuna S, Ramakrishnan C, Huynh K, Young S, Berndt A, Lee SY, Gorini CJ, Deisseroth K, Delp SL. 2016. Optogenetic and chemogenetic strategies for sustained inhibition of pain. Sci Rep. 6:30570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph J, Qu L, Wang S, Kim M, Bennett D, Ro J, Caterina M, Chung MK. 2019. Phosphorylation of TRPV1 S801 contributes to modality-specific hyperalgesia in mice. J Neurosci. 39(50):9954–9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucyi A, Moayedi M, Weissman-Fogel I, Goldberg MB, Freeman BV, Tenenbaum HC, Davis KD. 2014. Enhanced medial prefrontal-default mode network functional connectivity in chronic pain and its association with pain rumination. J Neurosci. 34(11):3969–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupers RC, Svensson P, Jensen TS. 2004. Central representation of muscle pain and mechanical hyperesthesia in the orofacial region: a positron emission tomography study. Pain. 108(3):284–293. [DOI] [PubMed] [Google Scholar]
- Lee J, Chung MK, Ro JY. 2012. Activation of NMDA receptors leads to phosphorylation of TRPV1 S800 by protein kinase C and A-Kinase anchoring protein 150 in rat trigeminal ganglia. Biochem Biophys Res Commun. 424(2):358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Saloman JL, Weiland G, Auh QS, Chung MK, Ro JY. 2012. Functional interactions between NMDA receptors and TRPV1 in trigeminal sensory neurons mediate mechanical hyperalgesia in the rat masseter muscle. Pain. 153(7):1514–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J, Sun T, Lumb BM, You HJ. 2014. Roles of the periaqueductal gray in descending facilitatory and inhibitory controls of intramuscular hypertonic saline induced muscle nociception. Exp Neurol. 257:88–94. [DOI] [PubMed] [Google Scholar]
- Lin CS. 2014. Brain signature of chronic orofacial pain: a systematic review and meta-analysis on neuroimaging research of trigeminal neuropathic pain and temporomandibular joint disorders. PLoS One. 9(4):e94300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Zhao Y, Cheng B, Zhao H, Miao L, Li Q, Chen Y, Zhang M. 2019. NMDAR and JNK activation in the spinal trigeminal nucleus caudalis contributes to masseter hyperalgesia induced by stress. Front Cell Neurosci. 13:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louca Jounger S, Christidis N, Svensson P, List T, Ernberg M. 2017. Increased levels of intramuscular cytokines in patients with jaw muscle pain. J Headache Pain. 18(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louca Jounger S, Eriksson N, Lindskog H, Oscarsson A, Simonsson V, Ernberg M, Christidis N. 2019. Repeated buffered acidic saline infusion in the human masseter muscle as a putative experimental pain model. Sci Rep. 9(1):15474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JP, Sadeghi S, Athanassiadis T, Caram Salas N, Auclair F, Thivierge B, Arsenault I, Rompre P, Westberg KG, Kolta A. 2010. Assessment of the potential role of muscle spindle mechanoreceptor afferents in chronic muscle pain in the rat masseter muscle. PLoS One. 5(6):e11131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michot B, Bourgoin S, Viguier F, Hamon M, Kayser V. 2012. Differential effects of calcitonin gene-related peptide receptor blockade by olcegepant on mechanical allodynia induced by ligation of the infraorbital nerve vs the sciatic nerve in the rat. Pain. 153(9):1939–1948. [DOI] [PubMed] [Google Scholar]
- Moana-Filho EJ, Herrero Babiloni A, Theis-Mahon NR. 2018. Endogenous pain modulation in chronic orofacial pain: a systematic review and meta-analysis. Pain. 159(8):1441–1455. [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Kurose M, Shimizu S, Hasegawa M, Ikeda N, Yamamura K, Takagi R, Okamoto K. 2018. Inhibitory effects of fluoxetine, an antidepressant drug, on masseter muscle nociception at the trigeminal subnucleus caudalis and upper cervical spinal cord regions in a rat model of psychophysical stress. Exp Brain Res. 236(8):2209–2221. [DOI] [PubMed] [Google Scholar]
- Nouged E, Dajani J, Ku B, Al-Eryani K, Padilla M, Enciso R. 2019. Local anesthetic injections for the short-term treatment of head and neck myofascial pain syndrome: a systematic review with meta-analysis. J Oral Facial Pain Headache. 33(2):183–198. [DOI] [PubMed] [Google Scholar]
- Okada S, Katagiri A, Saito H, Lee J, Ohara K, Iinuma T, Bereiter DA, Iwata K. 2019. Differential activation of ascending noxious pathways associated with trigeminal nerve injury. Pain. 160(6):1342–1360. [DOI] [PubMed] [Google Scholar]
- Okubo M, Castro A, Guo W, Zou S, Ren K, Wei F, Keller A, Dubner R. 2013. Transition to persistent orofacial pain after nerve injury involves supraspinal serotonin mechanisms. J Neurosci. 33(12):5152–5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Dubner R. 2011. The role of trigeminal interpolaris-caudalis transition zone in persistent orofacial pain. Int Rev Neurobiol. 97:207–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro JY, Lee JS, Zhang Y. 2009. Activation of TRPV1 and TRPA1 leads to muscle nociception and mechanical hyperalgesia. Pain. 144(3):270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez E, Sakurai K, Xu J, Chen Y, Toda K, Zhao S, Han BX, Ryu D, Yin H, Liedtke W, et al. 2017. A craniofacial-specific monosynaptic circuit enables heightened affective pain. Nat Neurosci. 20(12):1734–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H, Katagiri A, Okada S, Mikuzuki L, Kubo A, Suzuki T, Ohara K, Lee J, Gionhaku N, Iinuma T, et al. 2017. Ascending projections of nociceptive neurons from trigeminal subnucleus caudalis: a population approach. Exp Neurol. 293:124–136. [DOI] [PubMed] [Google Scholar]
- Schiffman E, Ohrbach R, Truelove E, Look J, Anderson G, Goulet JP, List T, Svensson P, Gonzalez Y, Lobbezoo F, et al. 2014. Diagnostic criteria for temporomandibular disorders (DC/TMD) for clinical and research applications: recommendations of the international RDC/TMD consortium network and orofacial pain special interest group. J Oral Facial Pain Headache. 28(1):6–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K, Matsumoto K, Noma N, Matsuura S, Ohara K, Komiya H, Watase T, Ogiso B, Tsuboi Y, Shinoda M, et al. 2014. Involvement of trigeminal tra nsition zone and laminated subnucleus caudalis in masseter muscle hypersensitivity associated with tooth inflammation. PLoS One. 9(10):e109168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda M, Kubo A, Hayashi Y, Iwata K. 2019. Peripheral and central mechanisms of persistent orofacial pain. Front Neurosci. 13:1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluka KA, Kalra A, Moore SA. 2001. Unilateral intramuscular injections of acidic saline produce a bilateral, long-lasting hyperalgesia. Muscle Nerve. 24(1):37–46. [DOI] [PubMed] [Google Scholar]
- Strand J, Stinson C, Bellinger LL, Peng Y, Kramer PR. 2018. G(i) protein functions in thalamic neurons to decrease orofacial nociceptive response. Brain Res. 1694:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyo S, Takemura M, Dubner R, Ren K. 2005. Trigeminal transition zone/rostral ventromedial medulla connections and facilitation of orofacial hyperalgesia after masseter inflammation in rats. J Comp Neurol. 493(4):510–523. [DOI] [PubMed] [Google Scholar]
- Wang GYF, Shi XQ, Wu W, Gueorguieva M, Yang M, Zhang J. 2018. Sustained and repeated mouth opening leads to development of painful temporomandibular disorders involving macrophage/microglia activation in mice. Pain. 159(7):1277–1288. [DOI] [PubMed] [Google Scholar]
- Wang S, Brigoli B, Lim J, Karley A, Chung MK. 2018. Roles of TRPV1 and TRPA1 in spontaneous pain from inflamed masseter muscle. Neuroscience. 384:290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Lim J, Joseph J, Wei F, Ro JY, Chung MK. 2017. Spontaneous and bite-evoked muscle pain are mediated by a common nociceptive pathway with differential contribution by TRPV1. J Pain. 18(11):1333–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XX, Cao Y, Mo SY, Liu Y, Xie QF. 2019. ACC plasticity maintains masseter hyperalgesia caused by occlusal interference. J Dent Res. 98(5):589–596. [DOI] [PubMed] [Google Scholar]
- Zhao YJ, Liu Y, Zhao YH, Li Q, Zhang M, Chen YJ. 2015. Activation of satellite glial cells in the trigeminal ganglion contributes to masseter mechanical allodynia induced by restraint stress in rats. Neurosci Lett. 602:150–155. [DOI] [PubMed] [Google Scholar]