

Chlamydia bacteria are obligate intracellular pathogens which can cause a variety of disease in humans and other vertebrate animals. To successfully complete its life cycle, Chlamydia must evade both intracellular innate immune responses and adaptive cytotoxic T cell responses. Here, we report on the role of the chlamydial lipooligosaccharide (LOS) in evading the immune response. Chlamydia infection is known to block the induction of apoptosis. However, when LOS synthesis was inhibited during Chlamydia trachomatis infection, HeLa cells regained susceptibility to apoptosis induction following staurosporine treatment.
KEYWORDS: Chlamydia, antigen processing, apoptosis, dendritic cells
ABSTRACT
Chlamydia bacteria are obligate intracellular pathogens which can cause a variety of disease in humans and other vertebrate animals. To successfully complete its life cycle, Chlamydia must evade both intracellular innate immune responses and adaptive cytotoxic T cell responses. Here, we report on the role of the chlamydial lipooligosaccharide (LOS) in evading the immune response. Chlamydia infection is known to block the induction of apoptosis. However, when LOS synthesis was inhibited during Chlamydia trachomatis infection, HeLa cells regained susceptibility to apoptosis induction following staurosporine treatment. Additionally, the delivery of purified LOS to the cytosol of cells increased the levels of the antiapoptotic protein survivin. An increase in survivin levels was also detected following C. trachomatis infection, which was reversed by blocking LOS synthesis. Interestingly, while intracellular delivery of lipopolysaccharide (LPS) derived from Escherichia coli was toxic to cells, LOS from C. trachomatis did not induce any appreciable cell death, suggesting that it does not activate pyroptosis. Chlamydial LOS was also a poor stimulator of maturation of bone marrow-derived dendritic cells compared to E. coli LPS. Previous work from our group indicated that LOS synthesis during infection was necessary to alter host cell antigen presentation. However, direct delivery of LOS to cells in the absence of infection did not alter antigenic peptide presentation. Taken together, these data suggest that chlamydial LOS, which is remarkably conserved across the genus Chlamydia, may act both directly and indirectly to allow the pathogen to evade the innate and adaptive immune responses of the host.
INTRODUCTION
Bacteria in the genus Chlamydia are obligate intracellular pathogens which can infect a variety of animals, leading to a diverse collection of serious diseases (1). In humans, Chlamydia trachomatis is the most commonly reported sexually transmitted bacterial pathogen, while infections of the eye are the leading cause of preventable blindness in the world (2, 3). Chlamydia pneumoniae commonly causes pneumonia in infected humans and can infect animals as well (4). Other Chlamydia spp. can cause zoonotic infections. Chlamydia abortus infects domestic ruminants, primarily sheep, and is a leading cause of “abortion storm,” which can be economically devastating to the agriculture industry and can cause abortion in humans (5). Chlamydia psittaci can infect a wide range of hosts and can lead to life-threatening pneumonia in humans (6, 7). While most chlamydial infections can be treated with antibiotics, resistance has been detected in Chlamydia suis (8). Lateral transfer of genes, at least in vitro, suggest that members of the genus Chlamydia have the ability to develop resistance to antibiotics (9). Therefore, understanding the biology of chlamydial infections is necessary for developing new therapies to combat infections.
Like other Gram-negative bacteria, Chlamydia generates a lipopolysaccharide (LPS) which decorates the outer bacteria cell membrane. However, the structure of the molecule is simple and consists of a trisaccharide of 3-deoxy-d-manno-oct-2-ulosonic residues and a penta-acyl lipid (10, 11). Because of its uncomplicated, short sugar chain, the molecule is often referred to as lipooligosaccharide (LOS). Remarkably, LOS is antigenically conserved among all chlamydial strains examined to date (12, 13). This stands in stark contrast to the high level of variation in LPS molecules from other bacteria. The reason for this high level of conservation in chlamydial LOS is unknown.
Obligately intracellular pathogens of vertebrate animals must avoid two elements of the host immune response: the innate intracellular immune response and the adaptive cytotoxic T cell response. In response to infection, host cells will often undergo some form of programmed cell death, be it apoptosis, pyroptosis, or necroptosis, to eliminate the infectious agent (14, 15). Innate immune sensors often respond to the presence of pathogen-associated molecular patterns (PAMPs) to begin triggering these cell death pathways (16, 17). Pathogen-generated proteins can also be degraded within host cells to short antigenic peptides, which can subsequently be presented to CD8+ cytotoxic T cells via the major histocompatibility complex (MHC) class I direct antigen presentation pathway (18–20). Upon recognition of the presented peptide, antigen-specific T cells kill the antigen-presenting cell. Unsurprisingly, intracellular pathogens have evolved numerous ways to evade these host immune responses (15, 21–23).
Chlamydia infection can prevent apoptosis of infected cells or at least delay cell death until infectious progeny are ready to be released from infected cells (24–27). The exact mechanism is unknown, though certain Inc proteins are necessary (27). In addition to preventing host cell death, Chlamydia infections also evade cytotoxic T cell responses (28, 29), possibly by manipulating the direct antigen presentation pathway (30–32). Recent work by our group found enhanced presentation of a model host antigen following infection with C. trachomatis, suggesting that skewing peptide presentation to favor host-derived peptides over pathogen-derived peptides may help Chlamydia evade the host immune response (33, 34). Interestingly, enhanced host peptide presentation was dependent on LOS synthesis during infection (34), as treatment with LPC-011, an inhibitor of bacterial LpxC (35), reversed the antigen presentation phenotype. This observation held true for a variety of Chlamydia spp. tested and suggested that altering MHC class I antigen presentation may be a conserved feature in different species of Chlamydia.
Here, we tested if LOS also has a role in preventing programmed cell death during infection. When LOS synthesis was prevented during infection, cells became susceptible to staurosporine-induced programmed cell death. We also transfected purified LOS into cells and found that not only is LOS far less toxic than Escherichia coli LPS, but also, LOS did not activate programmed cell death pathways. In fact, LOS was able to upregulate certain antiapoptotic proteins, such as survivin. We also measured dendritic-cell (DC) maturation in response to LOS exposure. LOS was far less potent at inducing maturation than E. coli LPS, as measured by the upregulation of DC cell surface markers necessary for T cell priming. However, LOS by itself did not enhance the presentation of a model host protein, as we have previously observed during infection. Taken together, these data suggest that LOS may protect Chlamydia from both innate, intracellular immune responses and adaptive cytotoxic T cell responses.
RESULTS
To determine if the synthesis of LOS during Chlamydia infection is necessary to prevent host cell apoptosis, we treated C. trachomatis-infected HeLa cells with LPC-011, an inhibitor of LOS synthesis. LPC-011 acts by inhibiting LpxC, an enzyme in the LOS synthesis pathway. Work by our group (33, 34) and others (35) has demonstrated that treatment with LPC-011 can inhibit LOS synthesis in a variety of cell types during infection with different Chlamydia species. Previous work has shown that Chlamydia infection protects infected cells from programmed cell death when the cells are treated with staurosporine, a nonspecific inducer of apoptosis (24). Treatment of HeLa cells with staurosporine resulted in an increase in DNA breaks, as measured by TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling) assay and changes to the DNA morphology (Fig. 1A) highlighted by increased Hoechst fluorescence at particular foci. Infection of HeLa cells with C. trachomatis L2 prevented changes to the DNA morphology following staurosporine treatment (Fig. 1B), indicating that during chlamydial infections, host cell apoptosis is prevented, as previously observed (24). However, when infected HeLa cells were treated with LPC-011 immediately after infection and apoptosis was subsequently induced with staurosporine, an increase in DNA damage foci in the nucleus was detected (Fig. 1B and C). The absence of LOS during infection induces chlamydial aberrancy, a developmental state that arises when chlamydiae are subjected to a variety of stressful culture conditions and fail to complete their replication cycle (34, 35). We therefore treated C. trachomatis L2-infected cells with ampicillin, which can induce an aberrancy-like state but does not have an effect on LOS production. When treated with ampicillin, infected cells remained resistant to staurosporine-induced apoptosis (Fig. 1B and C), indicating that the induction of aberrancy alone does not render host cells susceptible to induction of apoptosis. LPC-011 treatment alone had no effect on staurosporine-induced apoptosis in the absence of infection (Fig. 1D). These data suggest that LOS synthesis is necessary to prevent host cell programmed cell death during Chlamydia infection.
FIG 1.

Chlamydial LOS synthesis is necessary to prevent host cell programmed cell death. (A) HeLa cells were treated with staurosporine to induce nonspecific programmed cell death and DNA nicking, analyzed via the TUNEL assay (top) and via Hoechst staining (bottom) to detect changes to DNA nuclear morphology (bar, 10 μm). (B) C. trachomatis-infected HeLa cells were treated with either LPC-011 or ampicillin for the duration of the infection. At approximately 24 h postinfection, staurosporine was added, and 3 h later cells were analyzed for changes in DNA nuclear morphology. (C and D) A blinded observer quantified cells with changes to DNA nuclear morphology across five randomly selected fields for each treatment. Treatment with LPC-011 of infected cells resulted in a statistically significant increase in cells with changes to nuclear morphology consistent with the induction of programmed cell death compared to untreated or ampicillin-treated cells (**, P < 0.005; ***, P < 0.001; n.s, not significant).
We and others have detected LOS outside the chlamydial inclusion and present in the infected cell and neighboring host cells (36–39), which is due in part to the release of outer membrane vesicles during the infection process. As previously observed, LOS is detected as a diffuse staining outside the inclusion in infected HeLa cells (Fig. 2A), demonstrating that LOS is not restricted to the inclusion during infection and is positioned to interact with cellular components outside the inclusion and perhaps in neighboring cells. In order to directly study the effects of LOS on the apoptotic process, it is necessary to deliver LOS to the cytoplasm of cells, where it interacts with host factors, as would normally occur during an infection. We utilized the Amaxa Nucleofector to transfect cells with purified LOS. To confirm LOS localization to the intracellular compartments, we visualized LOS using scanning laser confocal microscopy to obtain cross-sectional views of cells. Cells were triply stained with anti-LOS antibody, DAPI (4′,6-diamidino-2-phenylindole), and fluorescent wheat germ agglutinin (WGA), which stains the plasma membrane. LOS was clearly visible within the cells following transfection, whereas it was bound primarily to the cell surface when cells were exposed to LOS but not transfected (Fig. 2B).
FIG 2.

Intracellularly delivered chlamydial LOS is not toxic and does not induce programmed cell death. (A) HeLa cells were infected with C. trachomatis L2 and treated with LPC-011 for 24 h or left untreated. Cells were then fixed and analyzed for LOS by staining with antibodies against LOS (green) or the inclusion membrane protein IncA (red) and appropriate secondary antibodies (bar, 10 μm). (B) JY cells were transfected with PBS or purified chlamydial LOS from C. trachomatis L2, allowed to adhere to poly-l-lysine-coated coverslips, and cultured for 2 h prior to confocal microscopy analysis. Fixed and permeabilized cells were analyzed for LOS (green), DNA (blue), or the plasma membrane (red), and sequential z-stacks were collected (bar, 10 μm). Transfected cells were compared to mock-transfected cells. Individual planes were analyzed to visualize the interior of the cell. (C) JY cells were transfected with equivalent doses of either chlamydial LOS or LPS derived from E. coli (from two different commercial sources). Three hours later, cells were analyzed for PI uptake to measure cell death. (D) JY cells were transfected with various doses of E. coli LPS, and cell death was measured by PI uptake. (E and F) JY cells were transfected with either chlamydial LOS or one of two doses of E. coli LPS, cultured for 3 h, and analyzed by Western blotting for the loss of procaspase 3 (E) or PARP (F). IB, immunoblot.
Intracellular LPS from Gram-negative bacteria such as E. coli is known to interact with inflammasomes to induce cell death via the pyroptosis pathway (40–42). To determine if intracellular LOS induced a phenotype similar to that induced by E. coli LPS, we compared cell death 2 h after transfection with either LPS or LOS. Exposure of JY cells to either LPS or LOS did not induce any measurable death, as measured by propidium iodide (PI) uptake (Fig. 2C). However, transfection with LPS resulted in rapid loss of viability, whereas LOS transfection did not induce any measurable increase in cell death compared to phosphate-buffered saline (PBS)-transfected controls (Fig. 2C). We then performed dose-response titration of LPS to determine the lowest level which could induce cell death equivalent to LOS (Fig. 2D). An approximately 1,000-fold reduction in LPS was necessary to lower levels of cellular death compared to that induced by LOS. We next examined if LOS could activate the apoptosis pathway by examining cell lysates for the loss of procaspase 3 and poly(ADP-ribose) polymerase 1 (PARP-1). LPS, but not LOS, transfection of JY cells resulted in loss of the native forms of both proteins, indicating the activation of programmed cell death pathways (Fig. 2E and F). Therefore, unlike LPS, LOS does not induce programmed cell death when delivered intracellularly.
To determine if LOS played an active role in preventing cellular death, we analyzed expression of apoptosis-related proteins using a 35-protein antibody array. Cells were transfected with LOS or PBS as a control and cultured for 3 h before a cell lysate was made. The lysate was then applied to the array, and each protein was analyzed and quantified. This experiment was repeated four times, and the average Z-score was calculated (Fig. 3A). The proteins survivin, XIAP, and cSMAC/Diablo had increased mean Z-score averages which were significantly different from zero (P < 0.05). We confirmed a significant increase in survivin levels by Western blot analysis following transfection of LOS into cells (Fig. 3B). LPS transfection did not increase levels of survivin. Next, we quantified the levels of survivin in cells following infection with C. trachomatis. Infection resulted in an increase in survivin levels similar to that in uninfected cells (P < 0.05), and treatment of infected cells with LPC-011 prevented an increase in survivin levels (Fig. 3C). These data indicate that the presence of LOS within cells can increase the levels of an antiapoptotic protein.
FIG 3.
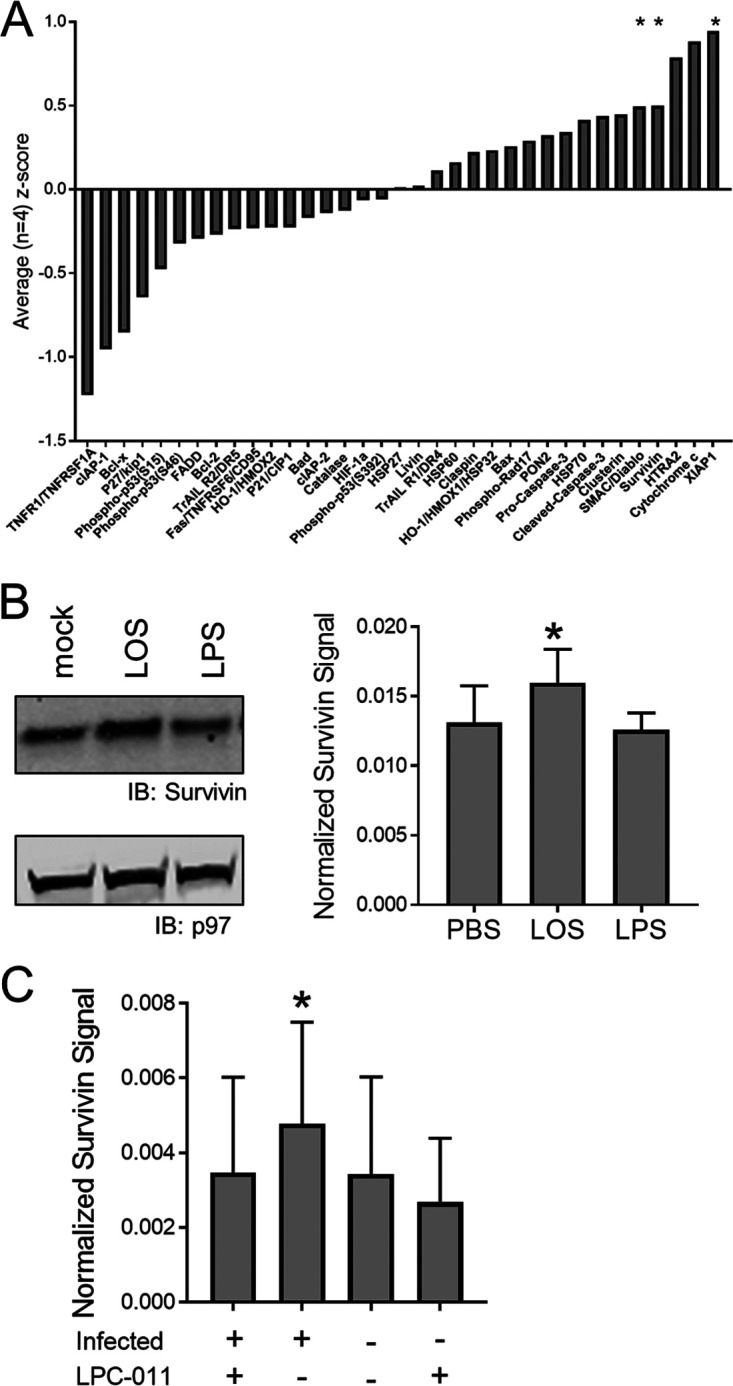
Chlamydial LOS increases survivin levels. (A) JY cells were transfected with PBS or chlamydial LOS and cultured for 3 h prior to analysis by apoptosis protein array. The fold change in protein levels of LOS-transfected cells compared to PBS controls for each protein was calculated. The average Z-score from four independent experiments was determined for each protein, and changes in the scores for survivin, cSMAC/Diablo, and XIAP were noted to be statistically significantly different from zero (*, P < 0.05). (B) The increase in survivin levels following LOS transfection was confirmed by Western blot analysis (*, P < 0.05; n = 5). (C) Survivin levels were quantified by Western blotting following C. trachomatis infection with or without LPC-011 treatment. Infected untreated cells had an increased level of survivin compared to all other cells (*, P < 0.05; n = 5).
Dendritic cells (DC) are important sentinel cells within the host that link innate and adaptive immune responses by acquiring and presenting antigen to T lymphocytes. DC respond to particular molecular signals which are not normally present during tissue homeostasis but whose presence signifies a danger to the host. LPS is one such danger signal: DC which sense LPS undergo the process of maturation in order to better activate T cells. Previous reports have demonstrated that Chlamydia infection can induce DC maturation, though less robustly than other bacterial products (43, 44). To determine if chlamydial LOS alone can induce DC maturation, we cultured DC from murine bone marrow precursors and measured a variety of cell surface markers by flow cytometry following exposure to various doses of LOS and LPS (Fig. 4). MHC class I levels were enhanced following exposure to LPS and remained unchanged at even the lowest doses of LPS tested (4 ng/ml). In contrast, 1,000-fold more LOS was needed for MHC class I to reach the plateaued levels observed following LPS treatment. Most other DC maturation markers showed a dose response to LPS treatment, with PD-L1 being the lone exception. PD-L1 expression reached a maximum at the lowest level of LPS tested, similar to MHC class I. In contrast, LOS treatment resulted in a modest increase above background for all markers tested, though the levels of LOS needed to equate to LPS treatment were 100-fold (MHC class II) to 1,000-fold (OX-40L and CD80) greater. PD-L1 and CD40 levels, while rising above the background levels of immature DC, were not as high as the expression level measured with lowest doses of LPS. Therefore, LOS is capable of inducing cell surface markers of DC maturation; however, it is ∼1,000-fold less potent than LPS.
FIG 4.

Chlamydial LOS is a poor stimulator of DC maturation. BMDC were derived from HLA-A2 transgenic mice on a C57BL/6 background by culturing bone marrow cells in GM-CSF for 10 days. Various doses of either E. coli LPS (black circles) or C. trachomatis L2 LOS (white circles) were added to cells, and cells were cultured overnight to induce DC maturation. Cells were stained for particular DC maturation markers, and the mean fluorescence intensity (MFI) for the population (for CD40, HLA-A2, OX40L, PD-L1, and I-Ab) or the percentage of cells positive for CD80 is indicated. Dotted lines represent background staining of immature BMDC. The results are representative of three independent experiments.
We previously demonstrated enhanced MHC class I presentation of host cell peptides derived from defective proteins during chlamydial infections (33, 34). The synthesis of LOS by the infecting bacteria was necessary for this phenotype, as treatment with LPC-011 reversed the antigen presentation phenotype (Fig. 5A). To test if the presence of LOS was sufficient for enhanced antigen presentation, we transfected JY/SCRAP-SVG cells with LOS and treated them with the compound Shield-1, which stabilizes the model antigen SCRAP-SVG, allowing only defective proteins to be degraded and presented at the cell surface. The presence of transfected LOS did not enhance host peptide presentation (Fig. 5B). Therefore, the presence of LOS alone, at least at the levels tested here, is insufficient to enhance presentation of host peptides derived from defective proteins.
FIG 5.
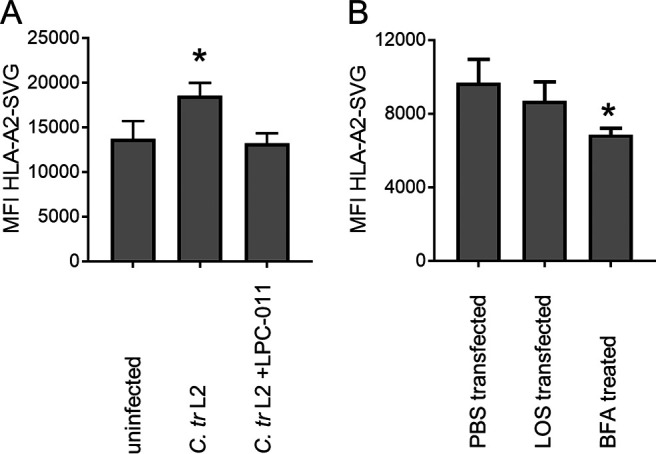
Intracellular LOS does not increase self-peptide antigen presentation. (A) JY/SCRAP-SVG cells were infected with C. trachomatis L2, treated with LPC-011, and cultured for 12 h prior to the addition of Shield-1. Cells were cultured for an additional 12 h and analyzed for HLA-A2-SVG peptide presentation by staining cells with the MAb RL15A and measuring the MFI using flow cytometry. (B) JY/SCRAP-SVG cells were transfected with different doses of LOS (or PBS controls) and cultured for 12 h in the presence of Shield-1 prior to staining with RL15A MAb to measure HLA-A2-SVG peptide levels (*, P < 0.05).
DISCUSSION
LOS is remarkably well conserved among Chlamydia species. This conservation is unexpected, especially when one considers other Gram-negative bacteria, which often have remarkably diverse glycan structures decorating the LPS molecules found on the bacterial outer membrane. Indeed, variation in what is referred to as the O chain can be used to serotype different strains of a single bacterial genus or species. For instance, there are currently 186 different recognized O antigens in E. coli (45). Diverse LPS structures also exist for Salmonella strains, which contain 54 known O antigens, and Vibrio species (193 different O-chain serogroups), and 20 known O-antigen structures are recognized in Pseudomonas aeruginosa (reviewed in reference 46). LPS diversity extends beyond infectious bacteria. A very abundant lineage of planktonic marine bacteria shows remarkable genetic plasticity at loci which are responsible for generating LPS (47, 48). Considering these examples, the antigenic similarity of Chlamydia LOS across members of the genus is striking.
While the mystery of LOS conservation remains unsolved, it has long been known that chlamydial LOS is far less toxic than the LPS molecules of other Gram-negative bacteria (49). Chlamydial LOS is known to be 10- to 100-fold less potent than LPS from other Gram-negative bacteria in stimulating tumor necrosis factor alpha (TNF-α) production in cell lines and immune cells such as DC (43, 50, 51). Recent work has demonstrated that LOS is at least 100-fold less potent than E. coli LPS at inducing endotoxic shock in mice (52), and in agreement with the results presented here, transfection of LOS into macrophages did not induce cell death, whereas LPS from E. coli was toxic (52). Reduced intracellular toxicity would of course be advantageous for an intracellular bacterium, and therefore, conservation of LOS may have evolved to ensure ongoing host cell survival.
Preventing, or at least delaying, host cell apoptosis is an important adaptive trait for chlamydiae. Resistance to apoptosis induction following Chlamydia infection was first reported over 2 decades ago (24). Apoptosis of cells is often delayed (25), presumably to allow Chlamydia to complete its developmental cycle, and if apoptosis is successfully induced early enough, the replication cycle is disrupted (53). Here, we report that LOS synthesis is necessary to convey apoptosis protection following infection. Furthermore, the presence of LOS in the cytoplasm can increase levels of anti-apoptotic proteins, suggesting a direct role for LOS in preventing apoptosis. This is reminiscent of a report by van Zandbergen et al. which showed that exposure of polymorphonuclear leukocytes (PMNs) to chlamydial LOS prevented apoptosis induction (54). Interestingly, LPS molecules from both E. coli and Salmonella enterica also prevented PMN apoptosis, though we found that introduction of LPS into the cytosol of JY cells quickly resulted in cell death. While more work will be needed to establish exactly how LOS can protect against apoptosis induction, it is intriguing to speculate that the conserved nature of LOS across the genus Chlamydia may serve to prevent premature host cell death.
Recent reports have suggested that chlamydial infections can trigger a second programmed cell death pathway, pyroptosis, if cells have functional inflammasomes (55, 56), particularly the noncanonical inflammasome (57). LPS can interact directly with noncanonical inflammasomes to trigger pyroptosis (40–42); however, transfection of chlamydial LOS into macrophages failed to induce pyroptosis (52). Yang et al. hypothesized that the penta-acyl structure of chlamydial LOS may fail to interact with inflammasomes (52). Though chlamydial LOS does not induce pyroptosis, chlamydial metabolites such as cyclic di-AMP have been shown to interact with STING and trigger pyroptosis (58).
Chlamydia must also evade adaptive immune responses, primarily cytotoxic-T-cell responses. CD8+ T cells are primed by DC which have captured, processed, and cross-presented exogenous antigens. In addition to cross-presenting antigens, DC undergo maturation when exposed to pathogen-associated molecular patterns such as LPS. Previous work has shown that LOS alone is a poor inducer of TNF-α expression by DC, but LOS-induced maturation was not measured (43). Here, we demonstrate that LOS-treated bone marrow-derived DC (BMDC) induced a partial maturation phenotype compared to LPS treatment. At least 1,000-fold less LPS than LOS was needed to induce equivalent levels of MHC class I expression. Furthermore, levels of T-cell-costimulatory molecules, such as CD80, OX-40L, and CD40, as well as the T-cell-inhibitory molecule PD-L1, on LOS-treated BMDC were reduced 100- to 1,000-fold compared to those seen with LPS. These data corroborate other work showing that exposure of DC to chlamydiae fails to induce robust DC maturation (44) and suggest that chlamydial LOS fails to trigger DC maturation during infection, reducing the ability of DC to prime cytotoxic T cell responses.
In addition to inhibiting the priming of CD8+ T cell responses, we have demonstrated a role for LOS in infected cells to alter direct MHC class I antigen presentation. Chlamydia infection of cells resulted in enhanced MHC class I presentation of a host-derived peptide originating from a model substrate, more specifically, when the peptide was derived from inherently defective forms of the model protein, referred to as a DRiP. We hypothesize that when self-peptide presentation is enhanced, there is a corresponding decrease in chlamydial peptides being presented, thus masking the cell from cytotoxic T cell responses. When LOS synthesis was inhibited by treating with LPC-011, self-antigen presentation returned to normal. However, the data presented here suggest that this antigen presentation phenotype is not the direct result of LOS interactions with host cell components, as transfection of LOS into cells failed to recapitulate the alteration in DRiP presentation. We hypothesize that the presence of LOS during infection allows other chlamydia-derived factors to interact with the host cell in order to skew peptide presentation.
In summary, chlamydial LOS appears to have many direct and indirect functions to allow Chlamydia to successfully evade both innate intracellular immune responses and cytotoxic T cell responses. LOS may therefore be conserved across the genus to allow Chlamydia spp. to successfully evade host immune responses.
MATERIALS AND METHODS
Cells, bacteria, and infections.
JY and JY/SCRAP-SVG cells were grown in RPMI 1640 supplemented with 7.5% fetal calf serum (FCS) with HEPES and Glutamax (all from Thermo Fisher) as previously described (33). HeLa cells were grown in Dulbecco modified Eagle medium (DMEM) with 7.5% FCS and Glutamax. All cells were cultured at 37°C in 5% CO2. C. trachomatis strain L2-434 (serovar L2) transformed with mCherry plasmid was a gift from Robert J. Suchland, University of Washington. Infection of JY cells was done as previously described (33). Briefly, a total of 5 × 105 cells were centrifuged at a relative centrifugal force (RCF) of 100 for 5 min and plated in 0.5 ml medium in a 24-well plate. C. trachomatis was added at a multiplicity of infection (MOI) of 10, and the plate was spun at an RCF of 980 for 1 h. Medium was then added to directly to the wells, and the plates were returned to the incubator. HeLa cells (5 × 104 cells) were seeded on a coverslip in a 24-well plate. After 24 h, C. trachomatis was added to an MOI of 10 to 20, and the plate was spun at an RCF of 980 for 1 h. The plates were returned to the incubator and cultured for up to 48 h.
Antibodies and reagents.
The anti-chlamydial LOS antibody EVI-HI and anti-chlamydial IncA clone 3H7 have been described (59). Alexa Fluor 647-coupled RL15A has been described (33, 60). The following antibodies were from BioLegend and used to monitor DC maturation: phycoerythrin (PE)-conjugated anti-CD80 (clone 16-10A1), allophycocyanin (APC)-conjugated anti-OX-40L (clone RM134L), APC–anti-HLA-A2 (clone BB7.2), fluorescein isothiocyanate (FITC)-conjugated anti-mouse I-Ab (clone AF6-120.1), APC–anti-PD-L1 (clone 10F.9G2), FITC–anti-CD40 (clone HM40-3), and Alexa Fluor 488–anti-mouse CD11c (clone N418). The secondary antibodies goat anti-mIgG2a–tetramethyl rhodamine isocyanate (TRITC) and goat anti-mIgG1–FITC were from Southern Biotech. The LpxC inhibitor LPC-011 was a kind gift from Pei Zhou and Raphael Valdivia from Duke University. The Proteome Profiler human apoptosis array kit was from R&D. Rabbit polyclonal antibodies specific for caspase 3, PARP, and survivin were all from Cell Signaling and were diluted 1:1,000 for Western blot analysis. The β-actin rabbit polyclonal antibody was from Bethyl Laboratories and was used at a dilution of 1:2,000. The p97-specific monoclonal antibody (MAb) was from Fitzgerald (clone 58.13.3) and was used at a dilution of 1:5,000. LOS (Chlamydia trachomatis serovar L2 LPS) was from Glycobiotech (Germany). LPS was from either Sigma (E. coli serotype O111:B4) or Molecular Probes (LPS from E. coli serotype O55:B5 coupled to Alexa Fluor 488). Both LPS and LOS were resuspended in PBS to a concentration of 1 mg/ml. PI, used at 1 mg/ml, was from Invitrogen.
Transfection.
JY cells were transfected with indicated doses of LOS or LPS using the Amaxa Nucleofector 96-well shuttle system (Lonza). For each transfection, ∼ 5 × 105 cells were harvested by centrifugation at an RCF of 100 and resuspended in 20 μl of Amaxa solution SF and mixed with 2 μl of either LPS or LOS solution to yield the required amounts. Cells were then transfected using program FF-120, incubated at 37°C for 5 min, harvested, placed in prewarmed complete medium, and cultured for 3 to 4 h prior to analysis.
Fluorescence and confocal microscopy.
Images were collected on a Leica DML fluorescence microscope fitted with a Retiga 2000R camera and processed with QCapture Pro 6.0 software (Q Imaging). HeLa cells were grown to ∼50% confluence on glass coverslips and subsequently infected with C. trachomatis at an MOI of 10. After 24 h, cells were treated with staurosporine at 1 μM for 4 h and fixed with 4% paraformaldehyde (PFA). TUNEL staining was performed using the TACS 2 TdT Fluorescein Kit (Trevigen) following the manufacturer's instructions. For examination of DNA morphology, nuclei were stained with Hoechst 34580 (2 μg/ml; Invitrogen) and examined under a 20× objective. The percent apoptotic cells was determined by examining five random fields (approximately 200 cells in total) in each treatment. Cells were scored by a blinded observer. The percent apoptotic cells was calculated. For examination of LOS and IncA distribution, cells were infected and either treated with LPC-011 for 24 h or left untreated. Cells were then stained with anti-chlamydial LOS MAb EVI-HI (IgG2A) and anti-IncA MAb 3H7 (IgG1), followed by the appropriate goat anti-mouse IgG isotype secondary antibodies. For confocal microscopy analysis of LOS-transfected cells, JY cells transfected with LOS were cultured on poly-d-lysine-coated coverslips for 2 h at 37°C. Cell membranes were then stained with 5 μg/ml WGA (Alexa Fluor 555 conjugate; Invitrogen) in PBS at 37°C for 10 min. After being washed, cells were fixed with 4% PFA and permeabilized with 0.2% Triton X-100 (EMD). Cells were then stained with anti-chlamydial LOS antibody. Vectashield (Vector Laboratories) containing 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) was used to stain DNA. Images were collected with a Zeiss LSM 780 NLO confocal microscope.
Flow cytometry.
Cells were harvested and resuspended in Hanks balanced salt solution (HBSS) supplemented with 0.1% bovine serum albumin (BSA). Cells (105 to 2 × 105) were distributed to 96-well plates to which was added FcBlocking agent (BioLegend). After a 15-min incubation at 4°C, the plate was spun at 2,000 rpm, and cells were incubated with the indicated antibody at 4°C for 30 min. Cells were then washed in HBSS-BSA and resuspended in ∼100 μl of HBSS-BSA for immediate analysis on a BD Accuri C6 flow cytometer, and analysis was completed using BD CSampler analysis software. A minimum of 10,000 live cells were analyzed for each experiment. For propidium iodide (PI) uptake analysis, cells were incubated with PI at 5 μg/ml for 5 min at room temperature prior to analysis.
Apoptosis array and Western blotting.
The Proteome Profiler human apoptosis array kit was used following the manufacturer’s suggested guidelines. Briefly, cells transfected with PBS or LOS were cultured for 3 h, washed in PBS, and lysed at 107 cells/ml in the provided lysis buffer. Following centrifugation, 250 μl of cell lysate was added to the preblocked array membrane and gently rocked overnight at 4°C. Membranes were washed, biotinylated detection antibody cocktail was added to each membrane, and membranes were gently rocked for 1 h, followed by washing of the membrane and the addition of streptavidin-horseradish peroxidase (HRP). The membranes were scanned with an Azure c600 imager. For Western blot analysis, cells were resuspended in lysis buffer (PBS with 0.5% Triton X-100 and Roche Complete protease inhibitor) at 107 cells/ml and lysed on ice for 30 min. The lysate was centrifuged for 5 min, and the resulting supernatant was mixed with lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) and 100 μM dithiothreitol (DTT) and boiled for 20 min. Samples were resolved by gel electrophoresis on 4% to 12% bis-Tris polyacrylamide Bolt gels (Invitrogen) and transferred to nitrocellulose membranes using the Invitrogen iBlot 2 transfer apparatus. Membranes were blocked in a 4% milk–TBS-T (1 mM Tris [pH 7.5], 100 mM NaCl, 1% Tween 20) solution for 1 h and then exposed to primary antibody diluted in 0.5% milk–TBS-T overnight with gentle rocking. After a single washing in TBS-T, the appropriate secondary antibodies diluted in 0.5% milk–TBS-T were added to the membrane and incubated for 1 h at room temperature with gentle rocking. Membranes were washed in excess TBS-T followed by deionized water and visualized using an Odyssey SA infrared imaging system (Li-Cor). Quantitation of both Western blots and apoptosis arrays was done using Image Studio software (Li-Cor).
BMDC culture.
HLA-A2 transgenic mice on a C57BL/6 background (strain 003475; Jackson Laboratories) were sacrificed, and femurs were dissected and rinsed in 70% ethanol (EtOH). Bone marrow from femurs was flushed into complete medium (RPMI 1640 plus 7.5% fetal calf serum, HEPES, and Glutamax, supplemented with penicillin, streptomycin, and gentamicin) using a 23-gauge needle. Marrow was gently pipetted to create a single-cell suspension and then depleted of erythrocytes using ammonium-chloride-potassium (ACK) lysing buffer (VWR). Cells were resuspended to 107/ml in BMDC culture medium (RPMI 1640 supplemented with 7.5% fetal calf serum, HEPES, Glutamax, penicillin, streptomycin, gentamicin, and 20 ng/ml granulocyte-macrophage colony-stimulating factor [GM-CSF] from eBioscience). Two milliliters of prewarmed BMDC culture medium was added to each well of a 24-well plate, 50 μl of cells were then slowly dispensed into the middle of the well, and cells were incubated at 37°C in 5% CO2 for 4 days, at which time 500 μl of medium was carefully removed and discarded and 500 μl of warm BMDC medium was added to the plates. This was repeated again on day 7 and day 9 after cultures were established. On day 10, LPS or LOS diluted in PBS was added to each well. Cells were cultured overnight and harvested by pipetting the following day for analysis of maturation markers.
Statistics.
All two-way Student's t test analysis was completed using GraphPad Prism. For analysis of the apoptosis array, densitometry values for each protein were used as raw data. Levels of each protein in LOS-treated cells were divided by the values for control cells. The resulting ratios for each protein were averaged, and the average value was subtracted from each individual protein’s value. The difference between an individual protein and the average change for every protein was divided by the standard deviation for all ratios and reported as a Z-score. The Z-scores from 4 independent experiments were subsequently averaged, and statistical significance differing from a score of zero was calculated using GraphPad Prism.
ACKNOWLEDGMENTS
We thank Ali Al-Fotis for technical help and scoring microscopy images. We thank Pei Zhou and Raphael Valdivia from Duke University for the gift of LPC-011, which was supported by a grant from NIH AI094475 awarded to Pei Zhou.
This work was made possible by NIH grants R21AI121752 and R01AI130059 (to B.P.D.) and R21AI088540 (to D.D.R.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Wheelhouse N, Longbottom D. 2012. Endemic and emerging chlamydial infections of animals and their zoonotic implications. Transbound Emerg Dis 59:283–291. doi: 10.1111/j.1865-1682.2011.01274.x. [DOI] [PubMed] [Google Scholar]
- 2.Gottlieb SL, Martin DH, Xu F, Byrne GI, Brunham RC. 2010. Summary: the natural history and immunobiology of Chlamydia trachomatis genital infection and implications for chlamydia control. J Infect Dis 201:190–S204. doi: 10.1086/652401. [DOI] [PubMed] [Google Scholar]
- 3.Resnikoff S, Pascolini D, Etya'ale D, Kocur I, Pararajasegaram R, Pokharel GP, Mariotti SP. 2004. Global data on visual impairment in the year 2002. Bull World Health Organ 82:844–851. [PMC free article] [PubMed] [Google Scholar]
- 4.Roulis E, Polkinghorne A, Timms P. 2013. Chlamydia pneumoniae: modern insights into an ancient pathogen. Trends Microbiol 21:120–128. doi: 10.1016/j.tim.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Pospischil A, Thoma R, Hilbe M, Grest P, Gebbers JO. 2002. Abortion in woman caused by caprine Chlamydophila abortus (Chlamydia psittaci serovar 1). Swiss Med Wkly 132:64–66. [DOI] [PubMed] [Google Scholar]
- 6.Knittler MR, Sachse K. 2015. Chlamydia psittaci: update on an underestimated zoonotic agent. Pathog Dis 73:1–15. doi: 10.1093/femspd/ftu007. [DOI] [PubMed] [Google Scholar]
- 7.Lagae S, Kalmar I, Laroucau K, Vorimore F, Vanrompay D. 2014. Emerging Chlamydia psittaci infections in chickens and examination of transmission to humans. J Med Microbiol 63:399–407. doi: 10.1099/jmm.0.064675-0. [DOI] [PubMed] [Google Scholar]
- 8.Dugan J, Andersen AA, Rockey DD. 2007. Functional characterization of IScs605, an insertion element carried by tetracycline-resistant Chlamydia suis. Microbiology 153:71–79. doi: 10.1099/mic.0.29253-0. [DOI] [PubMed] [Google Scholar]
- 9.Suchland RJ, Sandoz KM, Jeffrey BM, Stamm WE, Rockey DD. 2009. Horizontal transfer of tetracycline resistance among Chlamydia spp. in vitro. Antimicrob Agents Chemother 53:4604–4611. doi: 10.1128/AAC.00477-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rund S, Lindner B, Brade H, Holst O. 1999. Structural analysis of the lipopolysaccharide from Chlamydia trachomatis serotype L2. J Biol Chem 274:16819–16824. doi: 10.1074/jbc.274.24.16819. [DOI] [PubMed] [Google Scholar]
- 11.Rund S, Lindner B, Brade H, Holst O. 2000. Structural analysis of the lipopolysaccharide from Chlamydophila psittaci strain 6BC. Eur J Biochem 267:5717–5726. doi: 10.1046/j.1432-1327.2000.01635.x. [DOI] [PubMed] [Google Scholar]
- 12.Brade H, Brade L, Nano FE. 1987. Chemical and serological investigations on the genus-specific lipopolysaccharide epitope of Chlamydia. Proc Natl Acad Sci U S A 84:2508–2512. doi: 10.1073/pnas.84.8.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nurminen M, Leinonen M, Saikku P, Makela P. 1983. The genus-specific antigen of Chlamydia: resemblance to the lipopolysaccharide of enteric bacteria. Science 220:1279–1281. doi: 10.1126/science.6344216. [DOI] [PubMed] [Google Scholar]
- 14.Jorgensen I, Miao EA. 2015. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev 265:130–142. doi: 10.1111/imr.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamkanfi M, Dixit VM. 2010. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8:44–54. doi: 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Charles A, Janeway J, Medzhitov R. 2002. Innate immune recognition. Annu Rev Immunol 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 17.Chow KT, Gale M Jr, Loo Y-M. 2018. RIG-I and other RNA sensors in antiviral immunity. Annu Rev Immunol 36:667–694. doi: 10.1146/annurev-immunol-042617-053309. [DOI] [PubMed] [Google Scholar]
- 18.York IA, Rock KL. 1996. Antigen processing and presentation by the class I major histocompatibility complex. Annu Rev Immunol 14:369–396. doi: 10.1146/annurev.immunol.14.1.369. [DOI] [PubMed] [Google Scholar]
- 19.Yewdell JW. 2007. Plumbing the sources of endogenous MHC class I peptide ligands. Curr Opin Immunol 19:79–86. doi: 10.1016/j.coi.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 20.van Endert P. 2011. Providing ligands for MHC class I molecules. Cell Mol Life Sci 68:1467–1469. doi: 10.1007/s00018-011-0654-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yewdell JW, Hill AB. 2002. Viral interference with antigen presentation. Nature Immunol 3:1019–1025. doi: 10.1038/ni1102-1019. [DOI] [PubMed] [Google Scholar]
- 22.Hansen TH, Bouvier M. 2009. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol 9:503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- 23.Ulland TK, Ferguson PJ, Sutterwala FS. 2015. Evasion of inflammasome activation by microbial pathogens. J Clin Invest 125:469–477. doi: 10.1172/JCI75254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in Chlamydia-infected cells: blockade of mitochondrial dytochrome c release and caspase activation. J Exp Med 187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsuo J, Haga S, Hashimoto K, Okubo T, Ozawa T, Ozaki M, Yamaguchi H. 2019. Activation of caspase-3 during Chlamydia trachomatis-induced apoptosis at a late stage. Can J Microbiol 65:135–143. doi: 10.1139/cjm-2018-0408. [DOI] [PubMed] [Google Scholar]
- 26.Ying S, Fischer SF, Pettengill M, Conte D, Paschen SA, Ojcius DM, Häcker G. 2006. Characterization of host cell death induced by Chlamydia trachomatis. Infect Immun 74:6057–6066. doi: 10.1128/IAI.00760-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weber MM, Lam JL, Dooley CA, Noriea NF, Hansen BT, Hoyt FH, Carmody AB, Sturdevant GL, Hackstadt T. 2017. Absence of specific Chlamydia trachomatis inclusion membrane proteins triggers premature inclusion membrane lysis and host cell death. Cell Rep 19:1406–1417. doi: 10.1016/j.celrep.2017.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ibana J, Myers L, Porretta C, Lewis M, Taylor S, Martin D, Quayle A. 2012. The major CD8 T cell effector memory subset in the normal and Chlamydia trachomatis-infected human endocervix is low in perforin. BMC Immunol 13:66. doi: 10.1186/1471-2172-13-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fankhauser SC, Starnbach MN. 2014. PD-L1 limits the mucosal CD8+ T cell response to Chlamydia trachomatis. J Immunol 192:1079–1090. doi: 10.4049/jimmunol.1301657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong G, Liu L, Fan T, Fan P, Ji H. 2000. Degradation of transcription factor Rfx5 during the inhibition of both constitutive and interferon γ–inducible major histocompatibility complex class I expression in chlamydia-infected cells. J Exp Med 191:1525–1534. doi: 10.1084/jem.191.9.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ibana JA, Schust DJ, Sugimoto J, Nagamatsu T, Greene SJ, Quayle AJ. 2011. Chlamydia trachomatis immune evasion via downregulation of MHC class I surface expression involves direct and indirect mechanisms. Infect Dis Obstet Gynecol 2011:420905. doi: 10.1155/2011/420905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caspar-Bauguil S, Puissant B, Nazzal D, Lefevre JC, Thomsen M, Salvayre R, Benoist H. 2000. Chlamydia pneumoniae induces interleukin-10 production that down-regulates major histocompatibility complex class I expression. J Infect Dis 182:1394–1401. doi: 10.1086/315856. [DOI] [PubMed] [Google Scholar]
- 33.Cram ED, Simmons RS, Palmer AL, Hildebrand WH, Rockey DD, Dolan BP. 2016. Enhanced direct major histocompatibility complex class I self-antigen presentation induced by Chlamydia infection. Infect Immun 84:480–490. doi: 10.1128/IAI.01254-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cram ED, Rockey DD, Dolan BP. 2017. Chlamydia spp. development is differentially altered by treatment with the LpxC inhibitor LPC-011. BMC Microbiol 17:98. doi: 10.1186/s12866-017-0992-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen BD, Cunningham D, Liang X, Chen X, Toone EJ, Raetz CRH, Zhou P, Valdivia RH. 2011. Lipooligosaccharide is required for the generation of infectious elementary bodies in Chlamydia trachomatis. Proc Natl Acad Sci U S A 108:10284–10289. doi: 10.1073/pnas.1107478108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campbell S, Richmond SJ, Yates PS, Storey CC. 1994. Lipopolysaccharide in cells infected by Chlamydia trachomatis. Microbiology 140:1995–2002. doi: 10.1099/13500872-140-8-1995. [DOI] [PubMed] [Google Scholar]
- 37.Karimi ST, Schloemer RH, Wilde CE. 1989. Accumulation of chlamydial lipopolysaccharide antigen in the plasma membranes of infected cells. Infect Immun 57:1780–1785. doi: 10.1128/IAI.57.6.1780-1785.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown WJ, Skeiky YAW, Probst P, Rockey DD. 2002. Chlamydial antigens colocalize within IncA-laden fibers extending from the inclusion membrane into the host cytosol. Infect Immun 70:5860–5864. doi: 10.1128/iai.70.10.5860-5864.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richmond SJ, Stirling P. 1981. Localization of chlamydial group antigen in McCoy cell monolayers infected with Chlamydia trachomatis or Chlamydia psittaci. Infect Immun 34:561–570. doi: 10.1128/IAI.34.2.561-570.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. 2013. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszyński A, Forsberg LS, Carlson RW, Dixit VM. 2013. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 42.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. 2014. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 43.Prebeck S, Brade H, Kirschning CJ, da Costa CP, Dürr S, Wagner H, Miethke T. 2003. The Gram-negative bacterium Chlamydia trachomatis L2 stimulates tumor necrosis factor secretion by innate immune cells independently of its endotoxin. Microbes Infect 5:463–470. doi: 10.1016/S1286-4579(03)00063-7. [DOI] [PubMed] [Google Scholar]
- 44.Prebeck S, Kirschning C, Dürr S, da Costa C, Donath B, Brand K, Redecke V, Wagner H, Miethke T. 2001. Predominant role of Toll-like receptor 2 versus 4 in Chlamydia pneumoniae-induced activation of dendritic cells. J Immunol 167:3316–3323. doi: 10.4049/jimmunol.167.6.3316. [DOI] [PubMed] [Google Scholar]
- 45.Scheutz F, Cheasty T, Woodward D, Smith HR. 2004. Designation of O174 and O175 to temporary O groups OX3 and OX7, and six new E. coli O groups that include verocytotoxin-producing E. coli (VTEC): O176, O177, O178, O179, O180 and O181. APMIS 112:569–584. doi: 10.1111/j.1600-0463.2004.apm1120903.x. [DOI] [PubMed] [Google Scholar]
- 46.Samuel G, Reeves P. 2003. Biosynthesis of O-antigens: genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydrate Res 338:2503–2519. doi: 10.1016/j.carres.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 47.Wilhelm LJ, Tripp HJ, Givan SA, Smith DP, Giovannoni SJ. 2007. Natural variation in SAR11 marine bacterioplankton genomes inferred from metagenomic data. Biol Direct 2:27. doi: 10.1186/1745-6150-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grote J, Thrash JC, Huggett MJ, Landry ZC, Carini P, Giovannoni SJ, Rappé MS. 2012. Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 3:e00252-12. doi: 10.1128/mBio.00252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brade L, Schramek S, Schade U, Brade H. 1986. Chemical, biological, and immunochemical properties of the Chlamydia psittaci lipopolysaccharide. Infect Immun 54:568–574. doi: 10.1128/IAI.54.2.568-574.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ingalls RR, Rice PA, Qureshi N, Takayama K, Lin JS, Golenbock DT. 1995. The inflammatory cytokine response to Chlamydia trachomatis infection is endotoxin mediated. Infect Immun 63:3125–3130. doi: 10.1128/IAI.63.8.3125-3130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heine H, Müller-Loennies S, Brade L, Lindner B, Brade H. 2003. Endotoxic activity and chemical structure of lipopolysaccharides from Chlamydia trachomatis serotypes E and L2 and Chlamydophila psittaci 6BC. Eur J Biochem 270:440–450. doi: 10.1046/j.1432-1033.2003.03392.x. [DOI] [PubMed] [Google Scholar]
- 52.Yang C, Briones M, Chiou J, Lei L, Patton MJ, Ma L, McClarty G, Caldwell HD. 2019. Chlamydia trachomatis lipopolysaccharide evades the canonical and noncanonical inflammatory pathways to subvert innate immunity. mBio 10:e00595-19. doi: 10.1128/mBio.00595-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ying S, Pettengill M, Latham ER, Walch A, Ojcius DM, Häcker G. 2008. Premature apoptosis of Chlamydia-infected cells disrupts chlamydial development. J Infect Dis 198:1536–1544. doi: 10.1086/592755. [DOI] [PubMed] [Google Scholar]
- 54.van Zandbergen G, Gieffers J, Kothe H, Rupp J, Bollinger A, Aga E, Klinger M, Brade H, Dalhoff K, Maass M, Solbach W, Laskay T. 2004. Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J Immunol 172:1768–1776. doi: 10.4049/jimmunol.172.3.1768. [DOI] [PubMed] [Google Scholar]
- 55.Finethy R, Jorgensen I, Haldar AK, de Zoete MR, Strowig T, Flavell RA, Yamamoto M, Nagarajan UM, Miao EA, Coers J. 2015. Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in Chlamydia-infected macrophages. Infect Immun 83:4740–4749. doi: 10.1128/IAI.00856-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen L, Liu X, Yu X, Ren R, Wang C, Zhao R, Meng G, Li S, Zhou X. 2017. Chlamydia muridarum infection of macrophages stimulates IL-1b secretion and cell death via activation of caspase-1 in an RIP3-independent manner. BioMed Res Int 2017:1–8. doi: 10.1155/2017/1592365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Allen J, Gyorke CE, Tripathy MK, Zhang Y, Lovett A, Montgomery SA, Nagarajan UM. 2019. Caspase-11 contributes to oviduct pathology during genital Chlamydia infection in mice. Infect Immun 87:e00262-19. doi: 10.1128/IAI.00262-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Webster SJ, Brode S, Ellis L, Fitzmaurice TJ, Elder MJ, Gekara NO, Tourlomousis P, Bryant C, Clare S, Chee R, Gaston HJS, Goodall JC. 2017. Detection of a microbial metabolite by STING regulates inflammasome activation in response to Chlamydia trachomatis infection. PLoS Pathog 13:e1006383. doi: 10.1371/journal.ppat.1006383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suchland RJ, Jeffrey BM, Xia M, Bhatia A, Chu HG, Rockey DD, Stamm WE. 2008. Identification of concomitant infection with Chlamydia trachomatis IncA-negative mutant and wild-type strains by genomic, transcriptional, and biological characterizations. Infect Immun 76:5438–5446. doi: 10.1128/IAI.00984-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim S, Li L, McMurtrey CP, Hildebrand WH, Weidanz JA, Gillanders WE, Diamond MS, Hansen TH. 2010. Single-chain HLA-A2 MHC trimers that incorporate an immundominant peptide elicit protective T cell immunity against lethal West Nile virus infection. J Immunol 184:4423–4430. doi: 10.4049/jimmunol.0903955. [DOI] [PMC free article] [PubMed] [Google Scholar]