

Abstract
Steroid hormones, including glucocorticoids and androgens, have potent actions to regulate many cellular processes within the liver. The steroid A-ring reductase, 5β-reductase (AKR1D1), is predominantly expressed in the liver, where it inactivates steroid hormones and, in addition, plays a crucial role in bile acid synthesis. However, the precise functional role of AKR1D1 to regulate steroid hormone action in vitro has not been demonstrated. We have therefore hypothesised that genetic manipulation of AKR1D1 has the potential to regulate glucocorticoid availability and action in human hepatocytes.
In both liver (HepG2) and non-liver cell (HEK293) lines, AKR1D1 over-expression increased glucocorticoid clearance with a concomitant decrease in the activation of the glucocorticoid receptor and the down-stream expression of glucocorticoid target genes. Conversely, knockdown of AKR1D1 using siRNA decreased glucocorticoid clearance and reduced the generation of 5β-reduced metabolites. In addition, the two 5α-reductase inhibitors finasteride and dutasteride failed to effectively inhibit AKR1D1 activity in either cell-free or hepatocellular systems.
Through manipulation of AKR1D1 expression and activity, we have demonstrated its potent ability to regulate glucocorticoid availability and receptor activation within human hepatoma cells. These data suggest that AKR1D1 may have an important role in regulating endogenous (and potentially exogenous) glucocorticoid action that may be of particular relevance to physiological and pathophysiological processes affecting the liver.
Keywords: Steroid hormones, 5β-reductase, AKR1D1, Glucocorticoids, GR
1. Introduction
Glucocorticoids are released in response to stress and play an important role in inflammation, cellular growth, development, body fluid homeostasis, carbohydrate, lipid, and protein metabolism. However, circulating glucocorticoid excess has been associated with a classical phenotype of adverse metabolic features including central obesity, insulin resistance, type 2 diabetes and hepatic triacylglycerol (TAG) accumulation. These effects are exemplified in patients with Cushing’s disease, which is characterised by increased glucocorticoid secretion due to pituitary adenoma autonomously secreting ACTH [1]. The majority of glucocorticoid actions are mediated through a classical steroid hormone pathway whereby upon glucocorticoid binding in the cytosol to its cognate glucocorticoid receptor (GR), there is dissociation from the associated heat shock protein complex and subsequent translocation to the nucleus where the glucocorticoid-GR complex binds to glucocorticoid response elements to alter gene transcription.
Importantly, and independent of circulating levels, glucocorticoid action is also regulated at a pre-receptor level within key target tissues, including the liver, by a series of enzymes that can either enhance their clearance or augment their action. In this regard, the 11β-HSD isozymes and the 5α-reductases (5αR) have been extensively described. 11β-HSD1 catalyses the conversion of inactive cortisone to active cortisol and global deletion of 11β-HSD1 has been associated with increased insulin sensitivity, protection from hepatic steatosis, reduced expression of lipolytic enzymes in adipose tissue and a beneficial metabolic phenotype in mice [2–5]. Furthermore, hepatic over-expression of 11β-HSD1 has been linked to increased hepatic TAG content in mice [6]. In humans, there is evidence of decreased hepatic 11β-HSD1 activity in obese patients [7,8]. Similarly, deletion of 5αR type 1 has been associated with increased hepatic lipid accumulation in mice [9,10], whilst, in urinary steroid profiles from obese patients, 5αR activity increases with indices of insulin resistance [11,12].
5β-reductase (AKR1D1) is a member of the aldo-keto-reductase (AKR) superfamily 1 of enzymes and is the first member of the 1D subfamily (AKR1D1), along with the rat (AKR1D2) and the rabbit (AKR1D3) 5β-reductase isoforms [13,14]. The role of AKR1D1 to regulate glucocorticoid (and other steroid hormone) action has not been examined in detail and there are currently no published data that have tried to assess its importance in regulating glucocorticoid action using appropriate in vitro cell systems. The enzyme encoded by this gene has a molecular weight of 37 kDa. It utilizes NADPH as an electron donor and catalyses a stereospecific irreversible double bond reduction between the 4th and the 5th carbon of the A-ring of steroids. It is highly expressed in the liver where it uses the C19-C27 steroids (including cortisol, cortisone, testosterone and androstenedione) as substrates to generate all 5β-reduced dihydrosteroid metabolites [15,16]. Both cortisol and cortisone are metabolized to 5β-dihydrocortisol and 5β-dihydrocortisone respectively by AKR1D1, and are then subsequently converted, in a non-rate limiting step, to their tetrahydro-metabolites (tetrahydrocortisol, 5β-THF and tetrahydrocortisone, 5β-THE) by 3α-hydroxysteroid dehydrogenases, AKR1C1, AKR1C2, AKR1C3, AKR1C4, which are all expressed in human liver [17]. Thus, enzymes from the same gene family work sequentially to produce tetrahydro-steroids.
The demonstration that AKR1D1 can alter glucocorticoid availability to impact upon cellular function is currently lacking, although several studies have characterised the ability of AKR1D1 to metabolize steroid hormones and bile acid (BA) intermediates [15,18,19] using purified recombinant protein. However, little is known about the ability of this enzyme to regulate steroid hormone availability using intact cellular systems. The aim of our study was therefore to demonstrate that in appropriate human cell models (notably liver derived cell lines where AKR1D1 is highly expressed), AKR1D1 is able to metabolize glucocorticoids and regulate GR activation and down-stream gene transcription.
2. Materials and methods
2.1. Cell culture
HepG2 cells were a gift from Dr Karl Morten and HEK293 cells from Prof. Anna Gloyn (University of Oxford). Both cell lines were cultured in Dulbecco’s Minimum Essential Medium (DMEM) (Zen Bio Inc, Durham, NC, USA), containing 4.5 g/L glucose, and supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin and 1% non-essential amino acids.
For all cell treatments, HepG2 and HEK293 cells were cultured in serum-free and phenol red-free media containing cortisone or cortisol (Sigma-Aldrich, Dorset, UK). Treatments were started 48 h after transfection with the constructs described below. The details of dose ranges and timing of treatment are presented in the results section.
2.2. Transfection studies
AKR1D1 over-expression studies were performed in 12-well Cell Bind plates (CORNING, Flintshire, UK). The pCMV6-XL4+AKR1D1 (Origene Technologies, Rockville, USA) construct was used and 1 μg DNA and 2 μL X-tremeGENE DNA transfection Reagent (Roche, Hertfordshire, UK) were diluted in 100 μL OPTIMEM serum-free media (Invitrogen, Paisley, UK). The mixture was vortexed and incubated at room temperature for 20 min and, subsequently, 100 μL was added to each well and cells incubated at 37 °C for 48 h prior to treatment.
For AKR1D1 knockdown studies, cells were plated in 24-well Cell Bind plates (CORNING, Flintshire, UK). AKR1D1 siRNA molecules were purchased from Invitrogen (Paisley, UK). 20 nmol of AKR1D1 siRNA was diluted in 25 μL OPTIMEM serum-free media (Invitrogen, Paisley, UK) and, in a separate tube, 2.5 μL Lipofectamine RNAiMAX (Invitrogen, Paisley, UK) was diluted in 25 μL OPTIMEM serum-free media. The contents of the two tubes were combined by gentle pipetting and incubated at room temperature for 5 min. 50 μL of the resulting transfection solution was added drop-wise to each well and cells incubated at 37 °C for 48 h prior to treatment.
2.3. RNA extraction and gene expression
Total RNA was extracted from cells using the Tri-Reagent system (Sigma-Aldrich, Dorset, UK) and RNA concentration determined spec-trophotometrically at OD260 on a Nanodrop spectrophotometer (Thermo Scientific, Hemel Hempstead, UK). Reverse transcription was performed in a 20 μL volume; 1 μg of total RNA was incubated with 10x RT Buffer, 100 mM dNTP Mix, 10x RT Random Primers, 50U/μL MultiScribe Reverse Transcriptase and 20U/μL RNase Inhibitor. The reaction was performed under the following conditions; 25 °C for 10 min, 37 °C for 120 min and then terminated by heating to 85 °C for 5 min. All quantitative PCR experiments were conducted using an ABI 7900 H T sequence detection system (Perkin-Elmer Applied Biosystems, Warrington, UK). Reactions were performed in 6 μL volumes on 384-well plates in reaction buffer containing 3 μL of 2 x KlearKall Master Mix with standard ROX (LGC Genomics, Middlesex, UK). All primers (18SrRNA: Hs03003631_g1, AKR1D1: Hs00818881, SGK1: Hs00178612, DUSP1: Hs00610256_g1, GILZ: Hs00608272) were supplied by Thermo Fisher Scientific (Waltham, Massachusetts, USA) and reagents and all target genes were labelled with FAM. The reaction conditions were; 95 °C for 3 min, then 40 cycles of 95 °C for 3 s and 60 °C for 20 s. The Ct (dCt) of each sample using the following calculation (where E is reaction efficiency - determined from a standard curve): ΔCt = E[min Ct−sample Ct] using the 1/40 dilution from a standard curve generated from a pool of all cDNAs as the calibrator for all samples. The relative expression ratio was calculated using the following: Ratio= ΔCt[target]/ ΔCt[ref] and expression values were normalized to 18SrRNA [20].
2.4. Protein extraction and immunoblotting
Total protein was extracted from cells using RIPA buffer (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0) (Sigma-Aldrich, Dorset, UK), and protease inhibitor cocktail (ThermoFisher Scientific, Loughborough, UK). Protein concentrations were measured using a commercially available assay (Bio-Rad Laboratories Inc., Hercules, CA) according to the manufacturer’s protocol. Primary AKR1D1 (HPA057002, Atlas Antibodies AB, Bromma, Sweden), β-tubulin, (2146, Cell Signalling, Danvers, USA) and secondary antibodies (P044801–2, Agilent, Santa Clara, USA) were used at a dilution of 1/1000 (primary) and 1/2000 (secondary). Bands were visualised with ECL (Pierce Thermo Fisher Scientific) and ChemiDocXS imager (Biorad). Western blots were quantified by densitometry analysis using ImageJ (NIH, Bethesda, MD, http://rsb.info.nih.gov/ij), normalised to β-tubulin to correct for variability in gel loading.
2.5. Luciferase reporter assay
In order to determine GR activation, cells were plated in 12-well Cell Bind plates (CORNING, Flintshire, UK) and co-transfected with AKR1D1 overexpression vector (as described above) and GRE-reporter -a mixture of an inducible GRE-responsive firefly luciferase construct and a constitutively expressing renilla luciferase construct (QIAgen, Manchester, UK). Cell lysates were harvested in passive lysis buffer, and reporter activity was measured using the Luciferase Assay System (Promega) and an EnSpire Multimode plater reader (PerkinElmer, Massachusetts, USA). The data were presented as the ratio of firefly to renilla luciferase activity (Fluc/Rluc).
2.6. Cell-free system inhibitor studies
The construct encoding the AKR1D1 cDNA was provided by the Structural Genomic Consortium (SGC), University of Oxford, and was inserted into BL-21 Rosetta bacteria cells. Recombinant human AKR1D1 was expressed and purified as previously described [21].
For inhibitor cell-free studies, incremental amounts of finasteride or dutasteride (final concentration 0–500 μM) (Sigma-Aldrich, Dorset, UK) were tested using a substrate concentration of 40 μM, in the presence of AKR1D1 (final concentration 5 μM) in 100 mM potassium phosphate (pH 6.0), 20 μM NADPH (Sigma-Aldrich, Dorset, UK) and 4% acetonitrile, in a final volume of 100 μL at 37 °C. NADPH reduction was then measured using a BMG Labtech Flurostar fluorescence spectrophotometer (λ excitation = 340 nm, λ emission = 460 nm) for 35 min. The change in relative fluorescence units over 35 min in the absence of inhibitor was given a value of 100% enzyme activity.
2.7. Steroid hormone measurements
Cortisol and cortisone as well as their 5β-reduced metabolites 5β-tetrahydrocortisol (5β-THF) and 5β-tetrahydrocortisone (5β-THE) were analyzed by gas chromatography-mass spectrometry (GC–MS) using established methodology [22]. Steroids were extracted from cell media after addition of the internal standard, cortisol-d4. Briefly, transfected cells were incubated with cortisol (500 nM) or cortisone (200 nM) at different time points (2–24 h). 1 ml of media was collected, extracted by Sep Pak Columns (SPE) and samples were derivitized overnight to form methyloxime-trimethylsilylethers (MO-TMS). The final derivative was dissolved in 55 μL cyclohexane and transferred to an auto-sampler vial for gas chromatography mass spectrometry analysis using an Agilent 5973 instrument (www.agilent.com) in a selected ion monitoring mode.
2.8. Statistics
Data are presented as mean ± standard error unless otherwise stated. Two-tailed, paired t-tests were used to compare single treatments to control. For comparisons between control and different treatments, statistical analysis was performed using one-way analysis of variance (ANOVA) with Dunnett corrections. To compare mean differences between groups that had been split on multiple treatments, doses or times, two-way analysis of variance (ANOVA) with Sidak corrections was used. Statistical analysis on qPCR data was performed on mean relative expression ratio values (Ratio= ΔCt[target]/ ΔCt [20]. Data analysis was performed using Graphpad Prism software (Graphpad Software Inc, La Jolla, USA) and considered statistically significant at p < 0.05, unless otherwise stated.
3. Results
3.1. AKR1D1 regulates glucocorticoid clearance in both liver and non-liver cell lines
To define the functional role of AKR1D1 in vitro, HEK293 cells were transfected with either empty pCMV6-XL4 vector (EV) or AKR1D1 containing vector (Origene Technologies) for 48 h. Successful over-expression of AKR1D1 was confirmed using qPCR and immunoblotting (Fig. 1a and b).
Fig. 1.
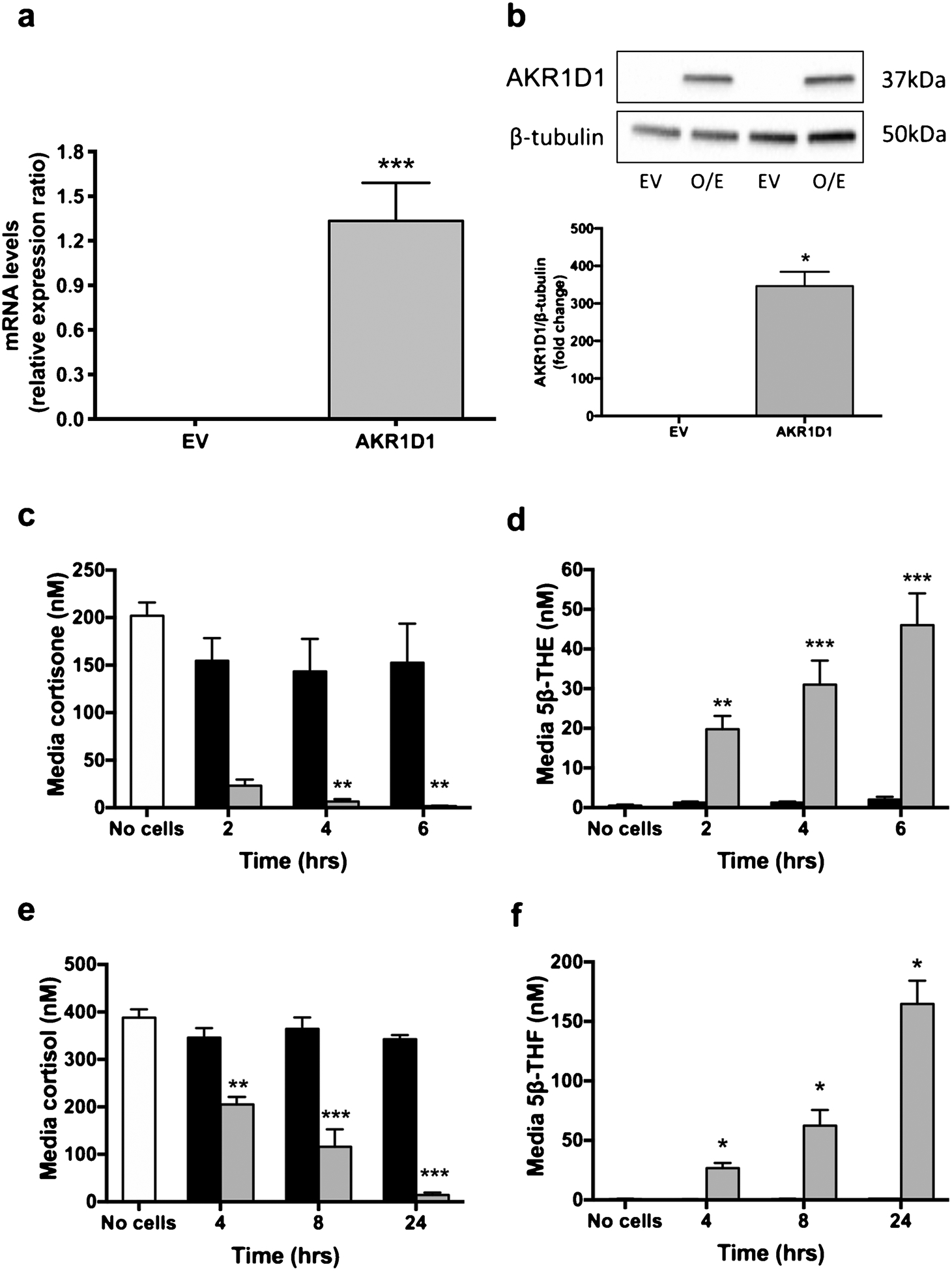
AKR1D1 expression, following over-expression in HEK293 cells, as measured by qPCR (a) and western blotting (b). AKR1D1 over-expression (grey bars) increases cortisone clearance (c) and tetrahydrocortisone generation [5β-THE] (d) in a time-dependent manner, compared to no cell controls (white bars) or vector only transfected cells (black bars). Similarly, in HEK293 cells, AKR1D1 over-expression (grey bars) increases cortisol clearance (e) as well as 5β-THF generation (f), in a time-dependent manner. qPCR data were normalised to 18SrRNA. Data are presented as mean ± se of n = 3 experiments, performed in triplicate, *p < 0.05, **p < 0.01, ***p < 0.001, compared to vector only transfected controls.
Following AKR1D1 over-expression, HEK293 cells were treated with cortisone (200 nM) (incubation time range 2–6 h) and media concentrations of cortisone and its metabolites, including 5β-THE, measured by GC–MS. There was no change in cell media cortisone concentration in the presence of empty vector (EV), however we observed a time-dependent increase in cortisone clearance in cells over-expressing AKR1D1, with a parallel increase in 5β-THE generation (Fig. 1c and d). Similar experiments were performed incubating transfected cells with cortisol rather than cortisone. Considering the lower catalytic efficiency (kcat) of AKR1D1 for cortisol, compared to cortisone, as shown by Chen et al. [15], cells were treated with higher concentrations of cortisol for a longer period of time (500 nM, 4–24 h incubation). There was no effect on cortisol clearance in HEK293 cells transfected with empty vector, whilst AKR1D1 over-expression significantly reduced cell media cortisol levels and increased 5β-tetrahydrocortisol (5β-THF) generation in a time-dependent manner (Fig. 1e and f).
In order to determine the impact of genetic manipulation of AKR1D1 expression in a functionally relevant context, a further set of experiments was performed in the human hepatoma cell line, HepG2 cells. As described above, HepG2 cells were transfected with either empty pCMV6-XL4 vector or AKR1D1 vector (Origene Technologies) and successful over-expression was confirmed using qPCR and western blotting assays (Fig. 2a and b).
Fig. 2.
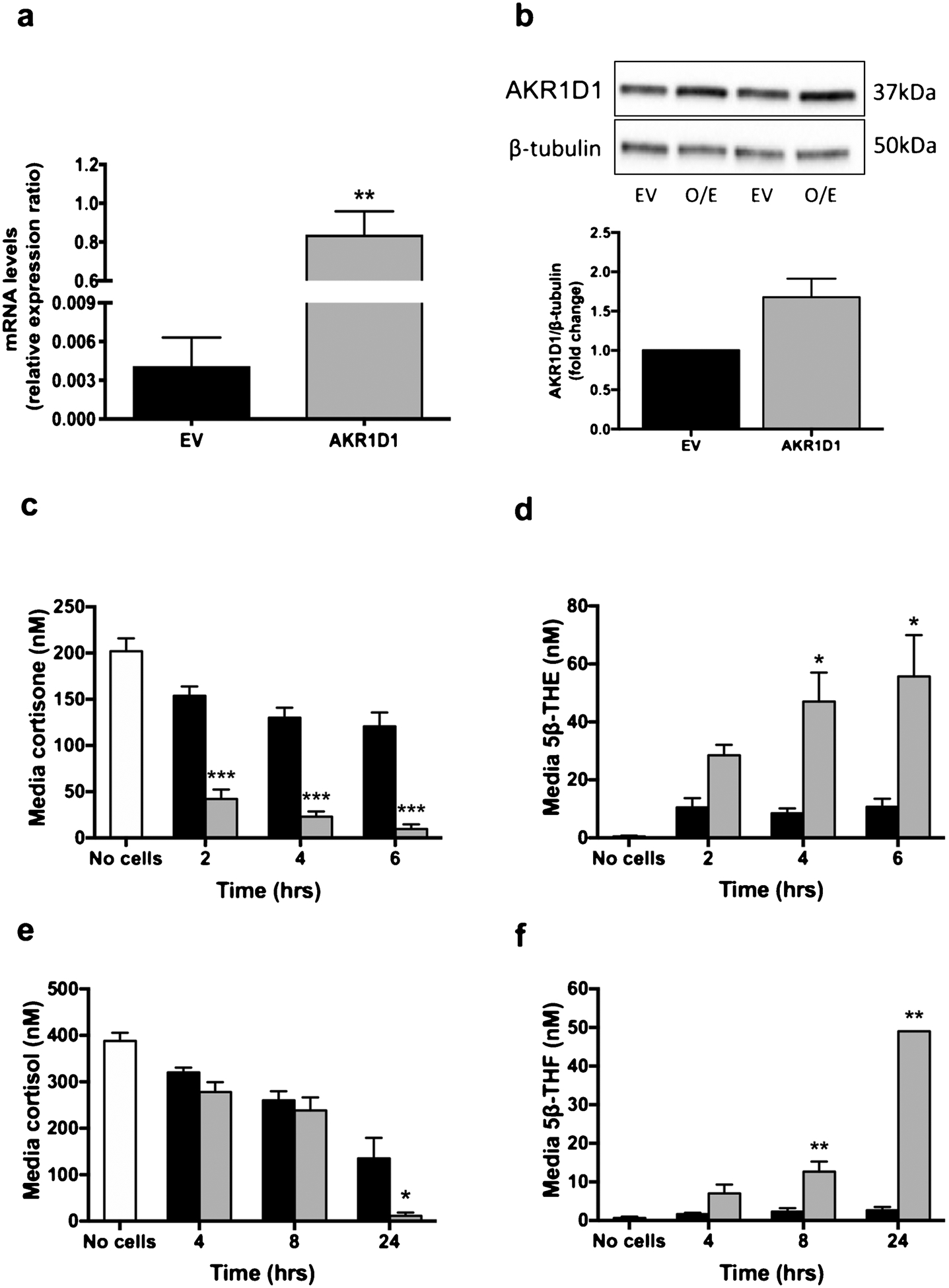
AKR1D1 expression, following over-expression in HepG2 cells, as measured by qPCR (a) and western blotting (b). AKR1D1 over-expression (grey bars) increases cortisone clearance (c) and tetrahydrocortisone generation [5β-THE] (d) in a time-dependent manner, in HepG2 cells, compared to no cell controls (white bars) or vector only transfected cells (black bars). Similarly, in HepG2 cells, AKR1D1 over-expression (grey bars) increases cortisol clearance (e) as well as 5β-THF generation (f), in a time-dependent manner. qPCR data were normalised to 18SrRNA. Data are presented as mean ± se of n = 3 experiments, performed in triplicate, *p < 0.05, **p < 0.01, ***p < 0.001, compared to vector only transfected controls.
Despite significant levels of basal expression of AKR1D1 in untreated HepG2 cells, AKR1D1 over-expression increased cortisone clearance and 5β-THE generation (Fig. 2c and d). In addition, cortisol clearance and 5β-THF generation were both increased in a time-dependent manner (Fig. 2e and f). In addition to 5β-THF, cortisone (24 h, EV: 64.3 ± 6.9 vs. AKR1D1: 9.0 ± 4.0 nM, p < 0.001) as well as 5β-THE levels (24 h, EV: 25.3 ± 10.5 vs. AKR1D1: 94.0 ± 15.6 nM, p < 0.001) were detected, as a result of endogenous 11β-HSD2 (converting cortisol to cortisone) and AKR1D1 (generating 5β-THE) activity, respectively. AKR1D1 knockdown experiments were performed in HepG2 cells using siRNA and achieved a 91.8% reduction in mRNA expression (scrambled control: 0.86 ± 0.09 vs. AKR1D1 knockdown:0.07 ± 0.007, p < 0.001) (Fig. 3a) and a 1-fold reduction in AKR1D1 protein levels (Fig. 3b). Consistent with our over-expression data, AKR1D1 knockdown in HepG2 cells limited cortisone clearance (Fig. 3c) and significantly decreased 5β-THE generation (Fig. 3d).
Fig. 3.
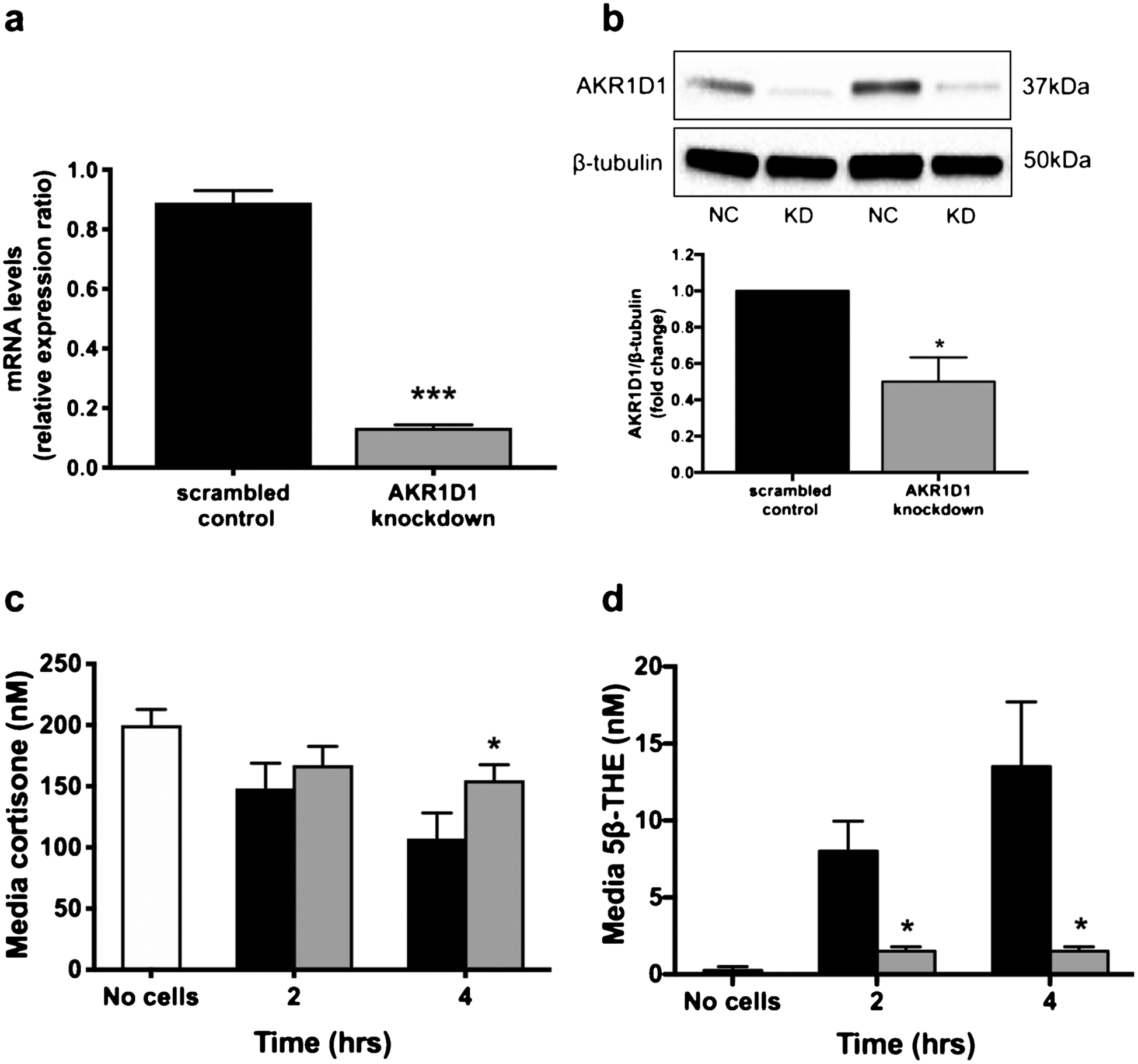
AKR1D1 expression, following transient knockdown in HepG2 cells, as measured by qPCR (a) and western blotting (b). Decreased AKR1D1 expression (grey bars) decreases cortisone clearance (c) and 5β-THE generation (d) in a time-dependent manner, in HepG2 cells. qPCR data were normalised to 18SrRNA. Data are presented as mean ± se of n = 3 experiments, performed in triplicate, *p < 0.05, **p < 0.01, ***p < 0.001, compared to vector only transfected controls.
3.2. AKR1D1 regulates GR receptor activation and gene transcription in vitro
In order to determine the functional cellular impact of altered pre-receptor glucocorticoid metabolism due to changes in AKR1D1 expression and activity on glucocorticoid receptor activation, dual transfection experiments were performed. HepG2 and HEK293 cells were transfected with both the AKR1D1 expressing vector and a commercially available GRE luciferase construct (QIAgen, Manchester, UK). In both cell lines, AKR1D1 over-expression decreased glucocorticoid receptor activation in the presence of cortisol (500 nM) (Fig. 4a), resulting, down-stream, to a decrease in mRNA expression of known glucocorticoid regulated genes (SGK1, DUSP1 and GILZ) (Fig. 4b and c).
Fig. 4.
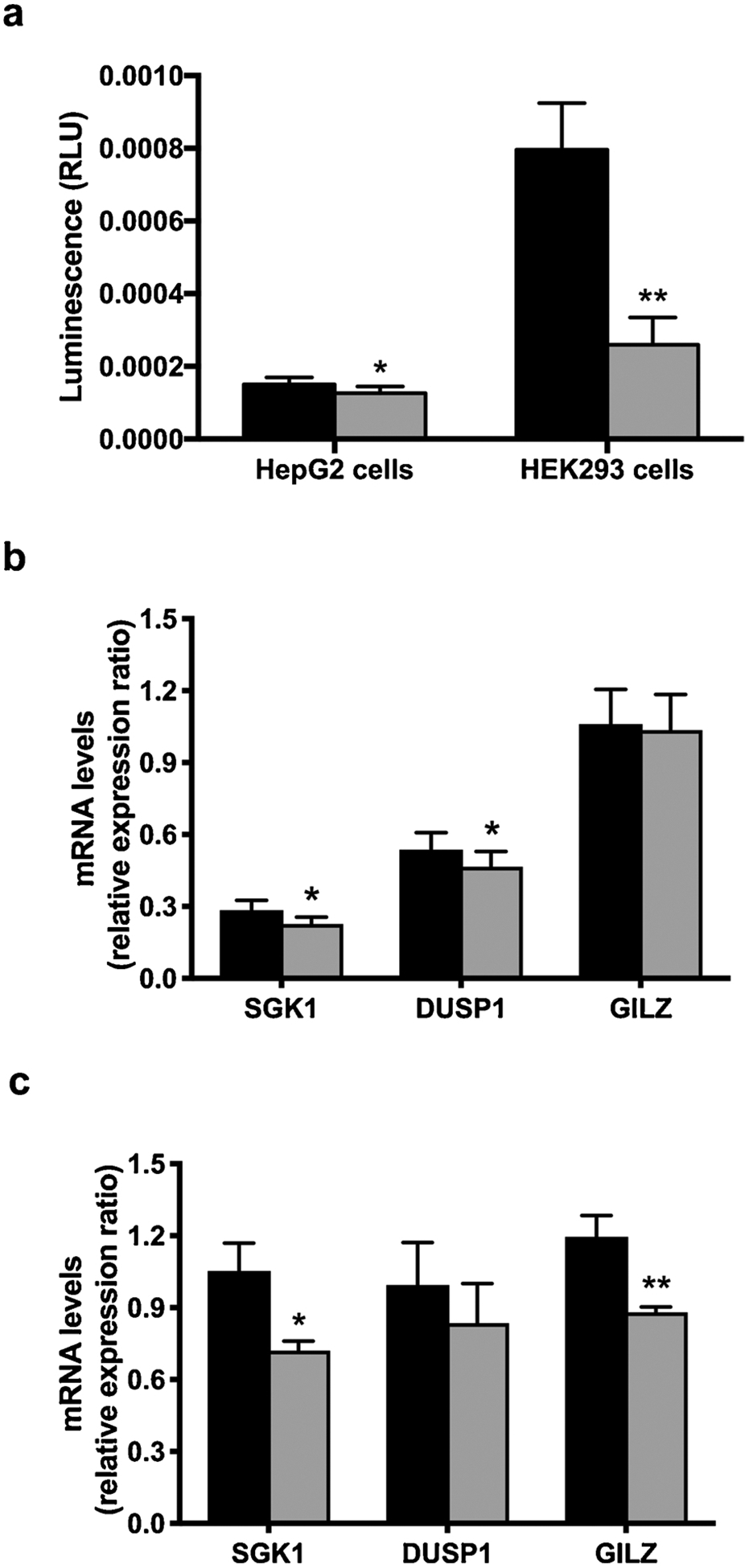
AKR1D1 over-expression (grey bars) significantly decreases activation of the glucocorticoid receptor in both HepG2 and HEK293 cells, as measured by activation of GRE-luciferase-reporter (a). In addition, AKR1D1 over-expression decreases the expression of the glucocorticoid receptor regulated genes SGK1, DUSP1 and GILZ in both HepG2 (b) and HEK293 (c) cells. qPCR data were normalised to 18SrRNA. Firefly luciferase activity was normalised to renilla luciferase. Data are presented as mean ± se of n = 7 and n = 3 experiments for HepG2 and HEK293 cells, respectively. All experiments were performed in triplicate, *p < 0.05, **p < 0.01, ***p < 0.001, compared to vector only transfected controls.
As previously mentioned, HepG2 cells also express steroid metabolising enzymes other than AKR1D1, including 11β-HSD2 and 5αR type 1. These enzymes all increase glucocorticoid clearance, leading to a further decrease in intracellular cortisol availability and may potentially explain the smaller effect size observed in HepG2, compared to HEK293 cells. In total, our data provide robust evidence that AKR1D1 is able regulate glucocorticoid receptor activation and consequent gene transcription through enhanced cortisol clearance.
3.3. Inhibition of AKR1D1
There are currently no specific inhibitors available targeting AKR1D1. In a previous study by Drury et al. [23], finasteride, a selective 5αR type 2 inhibitor, had a limited impact to inhibit AKR1D1 activity using testosterone as a substrate. Here, we explored the potential of finasteride (selective type 2 5αR inhibitor) and dutasteride (a non-selective type 1 and 2 5αR inhibitor), to block AKR1D1 activity using glucocorticoids as substrates, in both cell-free and in vitro systems. In order to determine the impact of 5αR inhibitors on AKR1D1 activity in cell-free systems, AKR1D1 recombinant human protein was generated and its activity was examined against incremental doses of either finasteride or dutasteride. Finasteride partially inhibited AKR1D1 recombinant protein activity in a dose-dependent manner, when cortisone and corticosterone were used as substrates. Dutasteride was less potent than finasteride and significantly reduced AKR1D1 activity only at the highest inhibitor concentrations (Fig. 5a and b). Neither finasteride nor dutasteride had any effect on AKR1D1 activity when cortisol was used as substrate (Fig. 5c). To complement our studies, we then explored the potential of these 5αR inhibitors to impair AKR1D1 activity using cell-based systems. HepG2 cells were treated with 200 nM cortisone for 8 h and 2 μM of finasteride and dutasteride (representing a 10-fold excess above substrate concentration). As expected, neither finasteride nor dutasteride had any impact upon AKR1D1 mRNA expression (Fig. 5d). Consistent with the cell-free system data, finasteride (but not dutasteride) was able to modestly decrease cortisone clearance (Fig. 5e). However, the reduction in the 5β-reduced metabolite, 5β-THE, failed to reach statistical significance (Fig. 5f) suggesting that the decrease in cortisone clearance may be mediated (at least in part) through the action of finasteride on other reductases. Low levels of 5α-THF were detected in the culture media (indicative of cortisone to cortisol conversion and subsequent 5α-reduction to generate 5α-THF). Both finasteride and dutasteride decreased 5α-THF generation (vehicle:31.00 ± 23.07 vs. finasteride: 9.33 ± 6.0 vs. dutasteride:2.33 ± 2.33 nM), consistent with their well-established inhibitory action upon 5αR isoforms, putatively contributing to cortisone clearance through this mechanism. It should be noted that, by contrast, the Ki values of finasteride and dutasteride for 5αR isozymes are in the low nanomolar range [24,25]. Thus, the modest effects observed with these inhibitors on AKR1D1 activity are unlikely to affect the pharmacological profile of these inhibitors.
Fig. 5.
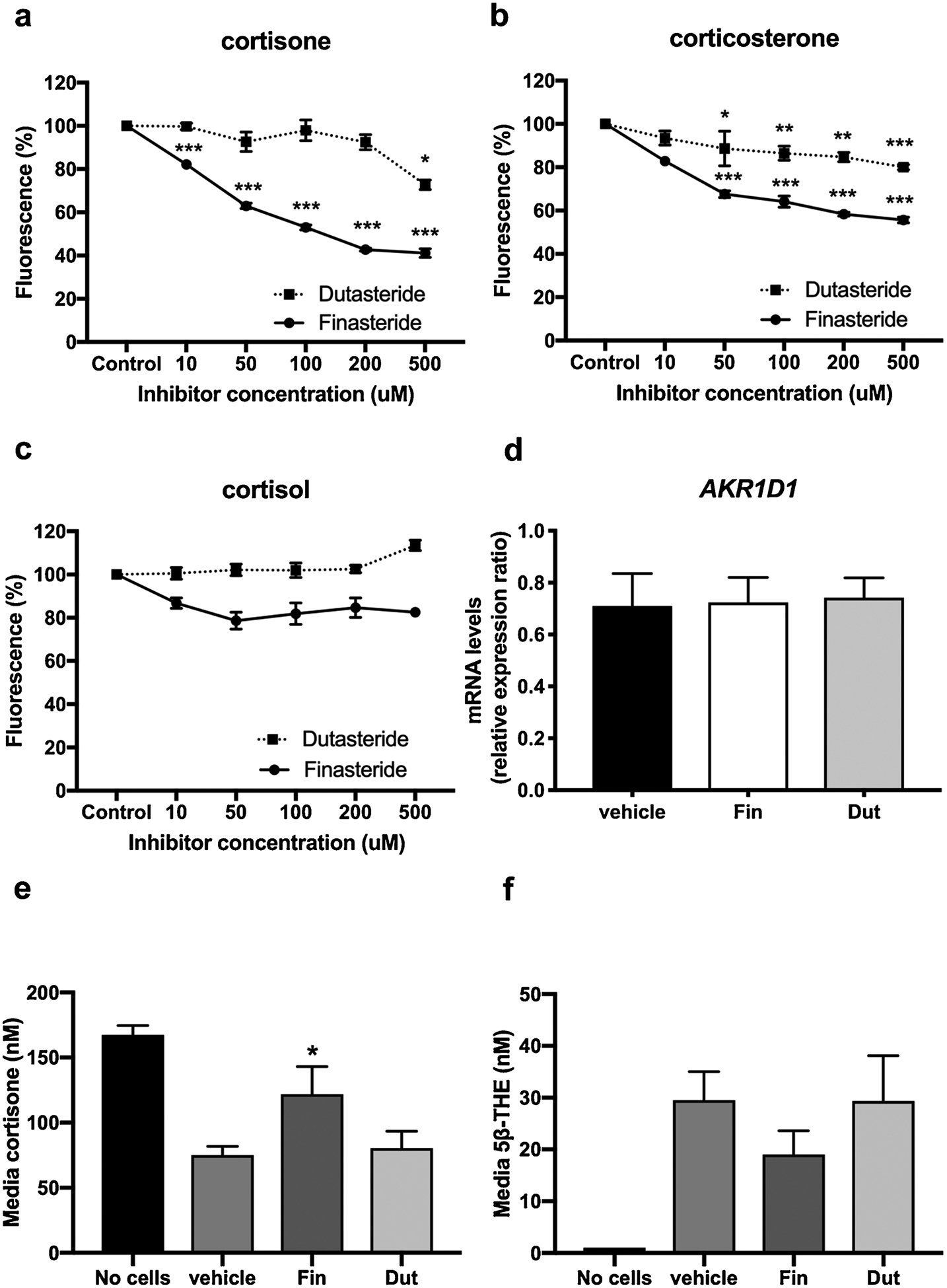
AKR1D1 inhibition by finasteride and dutasteride using cortisone (a), cortisol (b) and corticosterone (c) as substrates in a cell free system using recombinant human protein, as measured by NADPH reduction for 35 min. Data are expressed as % enzyme activity of n = 3 replicates. Effect of finasteride and dutasteride on AKR1D1 mRNA expression (d). Effect of finasteride and dutasteride on AKR1D1 activity, as measured by cortisone clearance (e) and 5β-THE generation (f), in HepG2 cells. qPCR data were normalised to 18SrRNA. Data are expressed as mean ± se of n = 3 experiments (*p < 0.05, **p < 0.01, ***p < 0.001, compared to control).
4. Discussion
In this study, we demonstrated, for the first time, that manipulation of AKR1D1 expression and activity is able to regulate glucocorticoid availability in liver and non-liver cell lines. AKR1D1 over-expression leads to increased glucocorticoid clearance and decreased GR activation, whilst AKR1D1 knockdown is able to decrease glucocorticoid clearance in human hepatocytes. In addition, we showed that finasteride and dutasteride, two 5αR inhibitors, failed to prevent AKR1D1-induced glucocorticoid clearance, in either cell-free or in vitro cellular systems.
The crystal structure and enzymology of human AKR1D1 has been extensively characterized [14,26]. AKR1D1 has broad substrate specificity [15]. Disease-causing missense mutations lead to decreased functional activity and when they occur in evolutionary conserved amino acids, enzyme stability is impaired [15,27]. Patients with mutations in AKR1D1 present with a rare and potentially severe form of neonatal cholestasis and can be treated effectively with bile acid replacement [28]. However, survival into adulthood in the absence of treatment is reported. The role of AKR1D1 in steroid hormone metabolism in vitro and in vivo has not been explored in detail. The urinary steroid profile in a single patient with a missense mutation in AKR1D1 (662C > T resulting in a Pro198Leu substitution) provided the first in vivo evidence of the ability of AKR1D1 to alter steroid hormone availability with the potential to alter glucocorticoid availability and action. In this patient, there was a marked reduction of all 5β-reduced steroid hormone metabolites, and in particular, an absence of 5β-tetrahydrocortisol [29]. There was a partial compensatory increase in 5α-reduced metabolites.
Our findings in human hepatoma cells suggest that AKR1D1 expression may act as a protective mechanism against glucocorticoid excess by not only reducing cortisol within the liver, but also limiting the generation of cortisol from cortisone [through enhanced clearance of substrate (cortisone) for 11β-HSD1]. In this regard, there are important parallels with the isozymes of 11β-HSD and 5αR. Genetic and pharmacological manipulation of these systems alters steroid hormone availability in a tissue-specific manner that is independent of circulating steroid hormone levels. In addition, changes in activity and expression of 11β-HSDs and 5αRs have been linked to the development of metabolic diseases including NAFLD, T2DM and insulin resistance [12,30,31]. Furthermore, clinical studies using pharmacological inhibitors have shown improvements in clinical phenotype with selective 11β-HSD1 inhibitors [32–34] and a detrimental effect of 5αR inhibitors [35,36] putatively through decreasing and increasing tissue-specific cortisol availability, respectively.
In this study, we have focused exclusively on the role of AKR1D1 to metabolize endogenous glucocorticoids. Whilst this is clearly important in understanding its physiological role, there is a broader context in that 2–3% of the population of the UK and USA are prescribed therapeutic glucocorticoids, including dexamethasone, prednisone and prednisolone [37,38]. It is well established that synthetic glucocorticoids are poorly metabolized within human tissues, and demonstrate poor cortisol binding globulin affinity and slower plasma removal rates, thus having more prolonged action when compared to endogenous glucocorticoids [39–41]. However, several studies have shown that synthetic glucocorticoids can also be inactivated through a variety of metabolic pathways, including oxidation or reduction by CYP3A4, isoforms of 11β-HSD and 5αR [42–45]. In this regard, the ability of AKR1D1 is entirely unexplored, but is likely to be of equal importance. In addition, little is currently known about the ability of glucocorticoids in the regulation of expression and activity of AKR1D1.
AKR1D1 is very highly expressed in human liver and based upon the observations outlined above, with respect to other steroid hormone pre-receptor metabolising enzymes, it is entirely plausible that AKR1D1 could represent a critical regulatory step in the control of metabolic phenotype within the liver. There is an established body of evidence highlighting the role of the GR in the regulation of carbohydrate metabolism; glucocorticoids perform their metabolic role through activation of the GR and regulate the expression of major gluconeogenic enzymes such as PCK1, G6Pase and tyrosine aminotransferase [46]. Furthermore, hepatocyte specific GR knockout mice exhibit hypoglycaemia after prolonged fasting due to impaired gluconeogenic enzyme expression [47]. Glucocorticoids also regulate lipid metabolism in a variety of tissues including adipose tissue and the liver [48–51]. Rodent studies including primary cultures of rat hepatocytes have demonstrated increased activity of hepatic acetyl-CoA carboxylase (rate limiting step of DNL) and fatty acid synthase after glucocorticoid treatment [52] as well as increased lipogenesis after glucocorticoid treatment in the presence of insulin [53]. Consistent with this, cortisol treatment has been reported to increase lipogenesis following insulin stimulation in primary human hepatocytes [51]. Moreover, decreased cortisol clearance via 5αR1 deletion has also been associated with hepatic steatosis in mice [9]. Taken together, these observations demonstrate not only the potent ability of glucocorticoids to manipulate metabolic phenotype in the liver, but also the critical role that pre-receptor regulation can play.
Specifically, with regards to AKR1D1, there are currently no published data on genetically modified animal models that might help aid our understanding of the true role of AKR1D1 in vivo and there are very limited clinical data. A single paper has suggested that there is decreased expression in liver biopsies from patients with diabetes when compared against non-diabetic controls [54]. An additional study that examined AKR1D1 activity using urinary steroid metabolites suggested that increased activity was associated with increased hepatic lipid content [55].
The ability of AKR1D1 to regulate cytosolic availability of a variety of steroid hormones in human hepatocytes indicates the presence of an intracellular complex system for steroid hormone metabolism. However, in addition, AKR1D1 performs a fundamental step in BA synthesis, as it is involved in an intermediate step in both the classic and alternative BA synthesis pathway, that lead to the generation of the primary BAs cholic acid and chenodeoxycholic acid. BAs are now well established as important signalling molecules, acting locally as well as in distant tissues, and are key regulators of systemic metabolism [56], and therefore the role that AKR1D1 plays in BA synthesis adds a further layer of complexity to the mechanisms that may underpin the role of this enzyme to regulate metabolic phenotype.
Although the important role of AKR1D1 in steroid hormone metabolism is now evident, to date, no specific pharmacological inhibitors for AKR1D1 are available. A previous study by Drury et al. 2009 reported that finasteride, a specific 5αR type 2 inhibitor, partially inhibited AKR1D1 activity [23]. Our cell-based studies have now provided evidence to suggest that finasteride is able to impact a modest inhibitory action on AKR1D1 (but much less so with the non-selective inhibitor, dutasteride). However, considering that cortisone can also be metabolized by 5αRs [57,58], it is possible the inhibition of cortisone clearance that we observed could have been mediated (at least in part), by the ability of finasteride to inhibit 5αR activity. The development of potent and specific inhibitors of AKR1D1 activity will undoubtedly enhance our ability to dissect the role of AKR1D1 cellular function and may also have therapeutic potential in metabolic disease, but this remains to be formally evaluated.
In conclusion, we have shown that AKR1D1 regulates glucocorticoid availability and GR activation and therefore represents a key pre-receptor regulator of hormonal action in human liver. However, there are additional questions that need to be addressed including dissecting the broader role of AKR1D1 in an in vivo context in both rodents and humans.
Acknowledgments
This work was supported by the Medical Research Council, UK (programme grant awarded to JWT, ref. MR/P011462/1); NIHR Oxford Biomedical Research Centre (Principal investigator award to JWT) based at Oxford University Hospitals NHS Trust and University of Oxford; British Heart Foundation (senior fellowship awarded to LH ref. FS/15/56/31645); NIHR Birmingham Biomedical Research Centre (Principal investigator award to WA); JED is supported by NIHR Oxford Biomedical Research Centre, Oxford UK. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health or the National Institute of Environmental Health.
Financial support
Medical Research Council (programme grant to JWT ref. MR/P011462/1); NIHR Oxford Biomedical Research Centre (principal investigator award to JWT); British Heart Foundation (senior fellowship to LH ref. FS/15/56/31645); National Institute of Environmental Health Sciences (P30-ES013508 awarded to TMP).
Footnotes
Disclosure summary
Nothing to declare. TMP is founder of Penzymes and is a consultant for Tokai and Sage Pharmaceutics and is a recipient of a Sponsored Research Agreement from Forendo.
References
- [1].Wang M, The role of glucocorticoid action in the pathophysiology of the Metabolic Syndrome, Nutr. Metab. (Lond.) 2 (2005) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Morton NM, Holmes MC, Fiévet C, Staels B, Tailleux a, Mullins JJ, Seckl JR, Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice, J. Biol. Chem 276 (2001) 41293–41300. [DOI] [PubMed] [Google Scholar]
- [3].Morgan SA, McCabe EL, Gathercole LL, Hassan-Smith ZK, Larner DP,Bujalska IJ, Stewart PM, Tomlinson JW, Lavery GG, 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess, Proc. Natl. Acad. Sci 111 (2014) E2482–E2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang Y, Yan C, Liu L, Wang W, Du H, Fan W, Lutfy K, Jiang M, Friedman TC,Liu Y, 11β-Hydroxysteroid dehydrogenase type 1 shRNA ameliorates glucocorticoid-induced insulin resistance and lipolysis in mouse abdominal adipose tissue, AJP Endocrinol. Metab 308 (2015) E84–E95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C,Walker BR, Flier JS, Mullins JJ, Seckl JR, Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11β-hydroxysteroid dehydrogenase type 1-deficient mice, Diabetes (2004). [DOI] [PubMed] [Google Scholar]
- [6].Paterson JM, Morton NM, Fievet C, Kenyon CJ, Holmes MC, Staels B, Seckl JR, Mullins JJ, Metabolic syndrome without obesity: hepatic over-expression of 11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice, Proc. Natl. Acad. Sci. U. S. A 101 (2004) 7088–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stewart PM, Boulton A, Kumar S, Clark PM, Shackleton CH, Cortisol metabolism in human obesity: impaired cortisone-cortisol conversion in subjects with central adiposity, J. Clin. Endocrinol. Metab 84 (1999) 1022–1027. [DOI] [PubMed] [Google Scholar]
- [8].Rask E, Walker BR, Söderberg S, Livingstone DEW, Eliasson M, Johnson O,Andrew R, Olsson T, Tissue-specific changes in peripheral cortisol metabolism in obese women: increased adipose 11beta-hydroxysteroid dehydrogenase type 1 activity, J. Clin. Endocrinol. Metab 87 (2002) 3330–3336. [DOI] [PubMed] [Google Scholar]
- [9].Dowman JK, Hopkins LJ, Reynolds GM, Armstrong MJ, Nasiri M, Nikolaou N, Van Houten ELAF, Visser JA, Morgan SA, Lavery GG, Oprescu A,Hübscher SG, Newsome PN, Tomlinson JW, Loss of 5α-reductase type 1 accelerates the development of hepatic steatosis but protects against hepatocellular carcinoma in male mice, Endocrinology 154 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Livingstone DEW, Barat P, Di Rollo EM, Rees GA, Weldin BA, Rog-Zielinska EA, MacFarlane DP, Walker BR, Andrew R, 5α-reductase type 1 deficiency or inhibition predisposes to insulin resistance, hepatic steatosis and liver fibrosis in rodents, Diabetes 64 (2015). [DOI] [PubMed] [Google Scholar]
- [11].Andrew R, Phillips DIW, Walker BR, Obesity and gender influence cortisol secretion and metabolism in man, J. Clin. Endocrinol. Metab 83 (1998) 1806–1809. [DOI] [PubMed] [Google Scholar]
- [12].Tomlinson JW, Finney J, Gay C, Hughes BA, Hughes SV, Stewart PM, Impaired glucose tolerance and insulin resistance are associated with increased adipose 11beta-hydroxysteroid dehydrogenase type 1 expression and elevated hepatic 5alpha-reductase activity, Diabetes 57 (2008) 2652–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Onishi Y, Noshiro M, Shimosato T, Okuda K, Molecular cloning and sequence analysis of cDNA encoding δ4-3-ketosteroid 5β-reductase of rat liver, FEBS Lett. 283 (1991) 215–218. [DOI] [PubMed] [Google Scholar]
- [14].Faucher F, Cantin L, Luu-The V, Labrie F, Breton R, Crystal structures of human Δ4–3-ketosteroid 5β-reductase (AKR1D1) reveal the presence of an alternative binding site responsible for substrate inhibition, Biochemistry 47 (2008) 13537–13546. [DOI] [PubMed] [Google Scholar]
- [15].Chen M, Drury JE, Penning TM, Substrate specificity and inhibitor analyses of human steroid 5β-reductase (AKR1D1), Steroids 76 (2011) 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jin Y, Chen M, Penning TMM, Rate of steroid double-bond reduction catalysed by the human steroid 5β-reductase (AKR1D1) is sensitive to steroid structure: implications for steroid metabolism and bile acid synthesis, Biochem. J 462 (2014) 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M,Palackal N, Ratnam K, Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones, Biochem. J 351 (2000) 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen M, Penning TM, 5β-reduced steroids and human Δ4–3-ketosteroid 5β-reductase (AKR1D1), Steroids 83 (2014) 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rižner TL, Penning TM, Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism, Steroids 79 (2014) 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pfaffl MW, A new mathematical model for relative quantification in real-time RTPCR, Nucleic Acids Res. 29 (2001) e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kavanagh KL, Guo K, Dunford JE, Wu X, Knapp S, Ebetino FH, Rogers MJ, Russell RGG, Oppermann U, The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs, Proc. Natl. Acad. Sci 103 (2006) 7829–7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shackleton CHL, Profiling steroid hormones and urinary steroids, J. Chromatogr. B Biomed. Sci. Appl 379 (1986) 91–156. [DOI] [PubMed] [Google Scholar]
- [23].Drury JE, Di Costanzo L, Penning TM, Christianson DW, Inhibition of human steroid 5β-reductase (AKR1D1) by finasteride and structure of the enzyme-inhibitor complex, J. Biol. Chem 284 (2009) 19786–19790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jenkins EP, Andersson S, Imperato-McGinley J, Wilson JD, Russell DW, Genetic and pharmacological evidence for more than one human steroid 5 alpha-reductase, J. Clin. Invest 89 (1992) 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tindall DJ, Rittmaster RS, The rationale for inhibiting 5alpha-reductase isoenzymes in the prevention and treatment of prostate cancer, J. Urol 179 (2008) 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Di Costanzo L, Drury JE, Penning TM, Christianson DW, Crystal structure of human liver Δ4–3-ketosteroid 5β-reductase (AKR1D1) and implications for substrate binding and catalysis, J. Biol. Chem 283 (2008) 16830–16839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mindnich R, Drury JE, Penning TM, The effect of disease associated point mutations on 5β-reductase (AKR1D1) enzyme function, Chem. Biol. Interact (2011) 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Clayton PT, Disorders of bile acid synthesis, J. Inherit. Metab. Dis (2011), 10.1007/s10545-010-9259-3. [DOI] [PubMed] [Google Scholar]
- [29].Palermo M, Marazzi MG, Hughes B.a., Stewart PM, Clayton PT,Shackleton CHL, Human Δ4–3-oxosteroid 5β-reductase (AKR1D1) deficiency and steroid metabolism, Steroids 73 (2008) 417–423. [DOI] [PubMed] [Google Scholar]
- [30].Ahmed A, Rabbitt E, Brady T, Brown C, Guest P, Bujalska IJ, Doig C,Newsome PN, Hubscher S, Elias E, Adams DH, Tomlinson JW, Stewart PM, A switch in hepatic cortisol metabolism across the spectrum of non alcoholic fatty liver disease, PLoS One 7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Andrews RC, Herlihy O, Livingstone DEW, Andrew R, Walker BR, Abnormal cortisol metabolism and tissue sensitivity to cortisol in patients with glucose intolerance, J. Clin. Endocrinol. Metab 87 (2002) 5587–5593. [DOI] [PubMed] [Google Scholar]
- [32].Feig PU, Shah S, Hermanowski-Vosatka A, Plotkin D, Springer MS, Donahue S,Thach C, Klein EJ, Lai E, Kaufman KD, Effects of an 11beta-hydroxysteroid dehydrogenase type 1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome, Diabetes Obes. Metab 13 (2011) 498–504. [DOI] [PubMed] [Google Scholar]
- [33].Rosenstock J, Banarer S, VA F, SE I, Sun W, Yao W, Hollis G, Flores R, Levy R,WV W, JR S, Huber R, The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy, Diabetes Care 33 (2010) 1516–1522 7p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shah S, Hermanowski-Vosatka A, Gibson K, Ruck RA, Jia G, Zhang J,Hwang PMT, Ryan NW, Langdon RB, Feig PU, Efficacy and safety of the selective 11β-HSD-1 inhibitors MK-0736 and MK-0916 in overweight and obese patients with hypertension, J. Am. Soc. Hypertens 5 (2011) 166–176. [DOI] [PubMed] [Google Scholar]
- [35].Hazlehurst JM, Oprescu AI, Nikolaou N, Di Guida R, Grinbergs AEK, Davies NP, Flintham RB, Armstrong MJ, Taylor AE, Hughes BA, Yu J,Hodson L, Dunn WB, Tomlinson JW, Dual-5α-reductase inhibition promotes hepatic lipid accumulation in man, J. Clin. Endocrinol. Metab 101 (2016) 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Upreti R, Hughes KA, Livingstone DEW, Gray CD, Minns FC, Macfarlane DP,Marshall I, Stewart LH, Walker BR, Andrew R, 5alpha-reductase type 1 modulates insulin sensitivity in men, J. Clin. Endocrinol. Metab 99 (2014) E1397–E1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wooldridge JE, Anderson CM, Perry MC, Corticosteroids in advanced cancer, Oncology (Williston Park) (2001). [PubMed] [Google Scholar]
- [38].van Staa TP, Leufkens HGM, Abenhaim L, Begaud B, Zhang B, Cooper C, Use of oral corticosteroids in the United Kingdom, Qjm 93 (2000) 105–111. [DOI] [PubMed] [Google Scholar]
- [39].Araki Y, Yokota O, Kato T, Kashima M, Miyazaki T, Dynamics of synthetic corticosteroids in man, in: Pincus G, Nakao T, Tait JF (Eds.), Steroid Dyn, 1965, pp. 463–480. [Google Scholar]
- [40].Fell PJ, Kinetic studies of cortisol and synthetic corticosteroids in man, Clin. Endocrinol. (Oxf.) 1 (1972) 65–72. [DOI] [PubMed] [Google Scholar]
- [41].Sandberg AA, Slaunwhite W JR., Differences in metabolism of prednisolone-C14 and cortisol-C14, J. Clin. Endocrinol. Metab 17 (1957) 1040–1050. [DOI] [PubMed] [Google Scholar]
- [42].Rodchenkov GM, Vedenin AN, Uralets VP, Semenov VA, Characterization of prednisone, prednisolone and their metabolites by gas chromatography-mass spectrometry, J. Chromatogr. B Biomed. Sci. Appl 565 (1991) 45–51. [DOI] [PubMed] [Google Scholar]
- [43].Siebe H, Baude G, Lichtenstein I, Wang D, Bühler H, Hoyer G-A, Hierholzer K, Metabolism of dexamethasone: sites and activity in mammalian tissues, Kidney Blood Press. Res (1993). [DOI] [PubMed] [Google Scholar]
- [44].Best R, Nelson SM, Walker BR, Dexamethasone and 11-dehydrodexamethasone as tools to investigate the isozymes of 11β-hydroxysteroid dehydogenase in vitro and in vivo, J. Endocrinol (1997). [DOI] [PubMed] [Google Scholar]
- [45].Renner E, Horber FF, Jost G, Frey BM, Frey FJ, Effect of liver function on the metabolism of prednisone and prednisolone in humans, Gastroenterology (1986). [DOI] [PubMed] [Google Scholar]
- [46].Moraitis AG, Block T, Nguyen D, Belanoff JK, The role of glucocorticoid receptors in metabolic syndrome and psychiatric illness, J. Steroid Biochem. Mol. Biol 165 (2017) 114–120. [DOI] [PubMed] [Google Scholar]
- [47].Opherk C, Tronche F, Kellendonk C, Kohlmüller D, Schulze A, Schmid W,Schütz G, Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus, Mol. Endocrinol 18 (2004) 1346–1353. [DOI] [PubMed] [Google Scholar]
- [48].Slavin BG, Ong JM, Kern PA, Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes, J. Lipid Res 35 (1994) 1535–1541. [PubMed] [Google Scholar]
- [49].Gathercole LL, Morgan SA, Bujalska IJ, Hauton D, Stewart PM,Tomlinson JW, Regulation of lipogenesis by glucocorticoids and insulin in human adipose tissue, PLoS One 6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Xu C, He J, Jiang H, Zu L, Zhai W, Pu S, Xu G, Direct effect of glucocorticoids on lipolysis in adipocytes, Mol. Endocrinol 23 (2009) 1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nasiri M, Nikolaou N, Parajes S, Krone NP, Valsamakis G, Mastorakos G,Hughes B, Taylor A, Bujalska IJ, Gathercole LL, Tomlinson JW, 5α-reductase type 2 regulates glucocorticoid action and metabolic phenotype in human hepatocytes, Endocrinology 156 (2015) 2863–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Diamant S, Shafrir E, Modulation of the activity of insulin-dependent enzymes of lipogenesis by glucocorticoids, Eur. J. Biochem 53 (1975) 541–546. [DOI] [PubMed] [Google Scholar]
- [53].Amatruda JM, Danahy SA, Chang CL, The effects of glucocorticoids on insulin-stimulated lipogenesis in primary cultures of rat hepatocytes, Biochem. J 212 (1983) 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Valanejad L, Ghareeb M, Shiffka S, Nadolny C, Chen Y, Guo L, Verma R, You S,Akhlaghi F, Deng R, Dysregulation of Δ4–3-oxosteroid 5β-reductase in diabetic patients: implications and mechanisms, Mol. Cell. Endocrinol 470 (2018) 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Westerbacka J, Yki-Järvinen H, Vehkavaara S, Häkkinen AM, Andrew R,Wake DJ, Seckl JR, Walker BR, Body fat distribution and cortisol metabolism in healthy men: enhanced 5β-reductase and lower cortisol/cortisone metabolite ratios in men with fatty liver, J. Clin. Endocrinol. Metab 88 (2003) 4924–4931. [DOI] [PubMed] [Google Scholar]
- [56].Li T, Chiang JYL, Bile acid signaling in metabolic disease and drug therapy, Pharmacol. Rev 66 (2014) 948–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].McGuire JS, Tomkins GM, The heterogeneity of Δ4–3-ketosteroid reductases (5α),J. Biol. Chem 235 (1960) 1634–1638. [Google Scholar]
- [58].Gold NI, Crigler JF, Suggested in vivo irreversible metabolism of labelled cortisol during distribution: effects on kinetics measurements and role of modes and routes of administration, J. Clin. Endocrinol. Metab 34 (1972) 1025–1038. [DOI] [PubMed] [Google Scholar]