

Abstract
Nephronophthisis (NPHP) is one of the renal ciliopathies and is also a cystic renal disorder with an autosomal recessive inheritance, which usually progresses to end-stage renal disease (ESRD). It affects children, adolescents, and young adults. In approximately 15% of cases, the features of a ciliopathy syndrome, which include liver fibrosis, skeletal anomalies, retinal abnormalities, and neurodevelopmental delay, will be present. We describe a case of a 2-year-old male child with ESRD on hemodialysis and a family record of a similar condition (his brother). The clinical features of this child are succinctly summarized. The genetic study was conducted using whole exome sequencing. TTC21B mutational variants were detected in our patient who exhibited nephrotic-range proteinuria, focal segmental glomerulosclerosis, and tubulointerstitial lesions that evolved to ESRD. Compound heterozygous mutations, c.626c > t (p.P209L) in exon 6 and c.450 g > a (p.W150Ter) in exon 5, were uncovered. These findings are in line with the description of autosomal recessive NPHP type 12. Both clinical and pathological diagnoses of NPHP are critical, bearing in mind ESRD as well as its related extrarenal defining features. Identification of the pathogenic variants in the TTC21B gene assisted in the successful proof of the clinical diagnosis NPHP12 as well as providing information for formal suitable prenatal counseling.
Keywords: ciliopathy, ESRD, nephronophthisis, TTC21B, whole exome sequencing
Introduction
Nephronophthisis (NPHP) is an autosomal recessive disorder, which proceeds to end-stage renal disease (ESRD) in early life. 1 In excess of 20 genes have already been stated as typical contributory genes of NPHP. Likewise, more than 40 genes have been linked to renal ciliopathies. 2 Such genes encode proteins located in primary cilia and connected structures. 3
The main cause of NPHP is often a dysfunction associated with primary cilia, which are usually nonmotile cilia in tissues. 4 Typically, the anticipated prevalence of NPHP fluctuates from 1:50,000 live-born in Finland and Canada to 1: 1,000,000 in the United States of America. 5
Roughly 80 to 90% of people with NPHP have isolated NPHP, whereas 10 to 20% have extrarenal manifestations that involve divergent disorders. 6 NPHP may be a multisystem disorder, which could influence the liver, eye, brain, and different organs. This may begin with prenatal dysplasia and advance to postnatal degeneration and fibrosis. 2
The TTC21B gene is located on chromosome 2q24.3. It is made up of 29 exons comprising 79.9kb of genomic DNA. The TTC21B gene encodes IFT139 protein, a part of the intraflagellar transport domain in the cilia. This domain is necessary for retrograde intraflagellar transport, which is depicted as a cause of NPHP. 7 As of now, 45 mutational variants inside TTC21B exons with a hotspot of c. 626C > T (p. P209L) have been uncovered. These variants are predominantly allocated in the European community and to North African but not Asian countries. 8
TTC21B genetic mutations are connected with many ciliopathies. TTC21B mutations may cause altered cilia structure and abnormal cell migration, assuming a hypomorphic status. 9 The p.P209L mutation has recently been assumed to play a role in microtubule network alterations. These alterations may influence cytoskeleton elements and disrupt podocyte assembly, thus reinforcing the assumption of its hypomorphic impact. 7 It additionally works as a modifier gene in the ciliopathy scale along with other genes eliciting ciliopathies disorders that affect the disease spectrum and progression. 7
Furthermore, TTC21B mutations can cause isolated NPHP, Jeune asphyxiating thoracic dystrophy, and Joubert syndrome. 7
Case Presentation
Our case is a 2-year-old male, the second sibling of a consanguineous union, full-term with normal development. His brother is 5 years old with a history marked by ESRD who was treated with regular hemodialysis since the age of 2.5 years; the etiology was obscure and the left kidney could not be visualized; otherwise, there was no family history of a comparable condition ( Fig. 1 ). Unfortunately, there was no genetic study or screening for urinary proteins previously directed to the parents or the brother. In our case, the mother underwent a four-dimensional ultrasound during pregnancy, which revealed right pelvic–ureteric junction obstruction. At the age of 8 months, abdominal ultrasonography showed right moderate hydronephrosis, and renal parenchymal echogenicity was equivalent to that of the liver with maintained corticomedullary differentiation (bilateral grade I nephropathy), although renal function was normal.
Fig. 1.
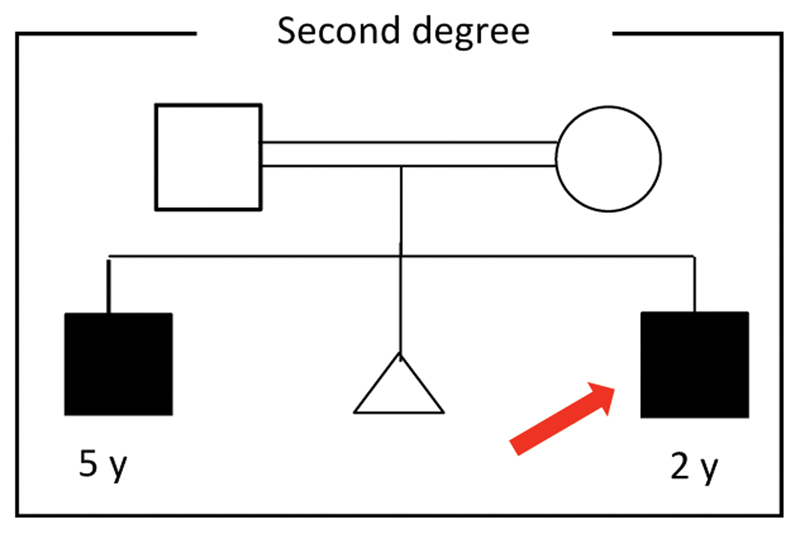
Family pedigree of the study case.
Afterward, at the age of 16 months, laboratory checks uncovered nephrotic-range proteinuria (10.5 gm/day), serum albumin of 3.4 g/dL, hemoglobin of 9 g/dL, and mean corpuscular volume of 72.14 fl. Renal function studies showed elevated serum creatinine (1.6 mg/dL) and blood urea nitrogen (21.1 mg/dL). The autoimmune profile was within the normal range, including serum complement, antinuclear antibody, antidouble-stranded DNA antibody, and antineutrophil cytoplasmic antibodies. Abdominal ultrasonography showed renal cortical echogenicity (greater than that of hepatic echogenicity) with a loss of corticomedullary differentiation for both kidneys (bilateral grade IV nephropathy) and right moderate hydronephrosis ( Table 1 ). Renal biopsy yielded 10 glomeruli under light microscopy, which included 7 globally sclerotic glomeruli with periglomerular fibrosis and 3 glomeruli showing segmental sclerosis. Marked tubular atrophy was seen along with intraluminal hyaline casts, and some tubules were dilated. Severe mixed tubulointerstitial inflammation and fibrosis were observed, which portrayed a state of FSGS with severe chronic tubulointerstitial nephritis ( Table 2 ). The condition of the patient deteriorated rapidly to ESRD with hypertension, and hemodialysis was started within 2 months. The two brothers had only renal manifestations, and no extrarenal abnormalities were detected.
Table 1. Summary of consecutive tests conducted for the patient.
| Age | 8 mo | 16 mo |
|---|---|---|
| Investigations | ||
| Renal function tests | ||
| BUN (mg/dL) | 8 | 21.1 |
| Creatinine (mg/dL) | 0.5 | 1.6 |
| Serum albumin (gm/dL) | Not performed | 3.4 |
| Complete blood picture | ||
| Hb | 10.5 g/dL | 9 g/dL |
| Platelets | 223,000/m 3 | 472,000/m 3 |
| WBCs | 6,600/m 3 | 8,000/m 3 |
| Urine analysis | ||
| Protein in urine | Absent | ++ |
| Protein/creatinine ratio | Not performed | 10.5 g/d |
| Abdominal ultrasonography | Bilateral grade 1 nephropathy | Bilateral grade 4 nephropathy |
| Renal biopsy | Not performed | Severe chronic tubulointerstitial nephritis (FSGS) |
Abbreviations: BUN, blood urea nitrogen; FSGS, focal segmental glomerulosclerosis; Hb, hemoglobin; WBC, white blood cells.
Note: Bold text refers to increased values with illness progression.
Table 2. Histological and morphological findings described by the renal biopsy.
| Histological abnormalities | |
|---|---|
| Gross | LM: single fragmented core measured ∼0.8 cm EM: single core measured 0.8 cm |
| Light microscopy |
Sections examined revealed corticomedullary renal tissue core containing
10
glomeruli.
• 7/10 glomeruli are globally sclerosed with periglomerular fibrosis • 3/10 of glomeruli show segmental sclerosis • Marked tubular atrophy is seen together with intraluminal hyaline casts; some tubules are dilated • Severe mixed tubulointerstitial inflammation and fibrosis were found • Thick-walled blood vessels are noted |
| Immunostaining | • lgG, lgM, lgA: focal mesangial (1+) |
Abbreviations: EM, electron microscopy; Ig, immunoglobulin; LM, light microscopy.
Methods
Informed consent was obtained from the parents of the children for blood collection and molecular testing. The research paper was endorsed by the local ethical committee of Menoufia University. The genetic analysis was performed in the form of whole exome sequencing (WES). RNA picks up halts aligned with approximately 60 MB of the human exon. These cover more than 99% of the regions in Reference Sequence, GENCODE, and the Consensus Coding Sequence catalogs. It is employed to enrich regions of relevance on genomic DNA using Agilent's SureSelect Human All Exon V6 kit. Typically, the created target is sequenced on an Illumina platform to achieve a depth of approximately 100 × . Typically, approximately 97% of the intended bases are counted in more than 10 times. The end-to-end in-house bioinformatics pipeline (including base call out, reads alliance to GRCh37/hg19 genetic assembly, riddling out of inferiority readings, and other artifacts) is followed by variant detection. All mutational variations registered in ClinVar or The Human Gene Mutation databases were recorded. The genetic variation with a minor allele frequency of <1% recorded in the Genome Aggregation database is reported as well. The coding exons plus about 20 adjacent intronic bases were examined. The uncovered mutational variations are evaluated for pathogenicity. Also, they are categorized into five classes as stated by the American College of Medical Genetics and Genomics (ACMG) recommendations. 10 Typically, the genetic variations assumed to cause disease are reported. Those genetic variants assumed to be benign or likely benign are not reported.
Results
Pathogenic TTC21B mutations were identified in our case. A compound heterozygous pathogenic variant c.626C > T (p.P209L) and a likely heterozygous likely pathogenic variant c.450G > A (p.W150Ter) were identified in exons 6 and 5, respectively.
The p.P209L is a missense variant with Proline to Leucine change at position 209 (NM_024753.4:c.626C > T). The outcome is matched with the diagnosis of autosomal recessive NPHP type 12, as reported by Davis et al 7 and Bullich et al. 11 This genetic variation is recorded as pathogenic by ClinVar (research, Variation ID: 30935). It is also delegated as a pathogenic variant (class 1) as indicated by the ACMG guidelines. 10
The p.W150Ter variant creates a premature stop codon (NM_024753.4:c.450G > A). The p.W150Ter has not been recently portrayed in other literature and not reported in ClinVar. ACMG classifies it as a likely pathogenic (class 2) variant. 10
No other variants were found in other NPHP-related genes, and no mutations were uncovered in other hereditary nephrotic syndrome-related genes in our case.
Discussion
Ciliary dysfunction contributes to a wide scope of overlapping phenotypes, named as ciliopathies. This also is accentuated by genomic overlap. The causal genes can also give rise to modifying alleles, resulting in clinically defined phenotypes. Typically, this variability, which is noticed in glomerular disorders and cystic renal diseases, reinforces the statement that genetic variations in many genes coding functional protein elements that are brought together in common trails may impact clinical outcome ( Table 3 ). 3 6 12
Table 3. Genetic classification of NPHP and its related disorders.
| HGNC gene symbol | NPHP type | Disorders associated with mutations |
|---|---|---|
| NPHP1 | 1 | NPHP/SLSN/JBTS |
| INVS | 2 | NPHP/SLSN (including infantile NPHP) situs inversus |
| NPHP3 | 3 | NPHP/SLSN/MKS (including infantile NPHP) |
| NPHP4 | 4 | NPHP/SLSN |
| IQCB1 | 5 | SLSN/LCA |
| CEP290 | 6 | JBTS/BBS/MKS/LCA/SLSN |
| GLIS2 | 7 | NPHP |
| RPGRIP1L | 8 | JBTS/MKS |
| NEK8 | 9 | NPHP (including infantile NPHP) |
| SDCCAG8 | 10 | SLSN/BBS |
| TMEM67 | 11 | NPHP/MKS/JBTS/COACH syndrome |
| TTC21B | 12 | NPHP/JBTS |
| WDR19 | 13 | NPHP/JBTS |
| ZNF423 | 14 | JBTS |
| CEP164 | 15 | NPHP/SLSN/JBTS |
| ANKS6 | 16 | NPHP |
| IFT172 | 17 | NPHP/Jeune/Mainzer–Saldino syndrome |
| CEP83 | 18 | NPHP (including infantile NPHP) |
| DCDC2 | 19 | NPHP/liver fibrosis |
| MAPKBP1 | 20 | NPHP |
Abbreviations: BBS, Bardet–Biedl syndrome; COACH, cerebellar vermis hypo/aplasia, oligophrenia (mental retardation), ataxia, ocular coloboma, and hepatic fibrosis; JBTS, Joubert syndrome; LCA, Leber congenital amaurosis; MKS, Meckel syndrome; NPHP, nephronophthisis; SLNS, Senior–Løken syndrome.
Our case presented with nephrotic proteinuria, ESRD, and hypertension, and showed focal segmental glomerulosclerosis with tubulointerstitial lesions (e.g., interstitial fibrosis and marked tubular atrophy) on pathological evaluation. In addition, his brother experienced ESRD and is on hemodialysis. The WES for our case revealed compound heterozygous TTC21B variants, c.450G > A (p.W150Ter) and c.626C > T (p.P209L) in exons 5 and 6, respectively. Pathogenic variants in the TTC21B gene are associated with NPHP type 12. Other mutations that involve TTC21B by earlier reports are outlined in Table 4 .
Table 4. Mutations encountered in the TTC21B gene .
| TTC21B mutations | |
|---|---|
| 1 | c.626C > T (p.P209L) |
| 2 | c.1276C > G (p.H426D) |
| 3 | c.152–2A > G (Splice site) |
| 4 | c.3605 T > C (p.L1202P) |
| 5 | c.1654–7delTGTC (p.C552fsX1) |
| 6 | c.448 T > C (p.W105R) |
| 7 | c.3264–3C > G (splice site) |
| 8 | c.2758–2A > G (splice site) |
| 9 | c.1231C > T (R411X) |
| 10 | c.2384 T > C (L795P) |
| 11 | c.1656 T > A (C552X) |
| 12 | c.448 T > C (W150R) |
| 13 | c.1552 T > C (p.C518R) |
| 14 | c.1456dupA (p.R486KfsX22) |
| 15 | c.2211 + 3 A > G |
Both brothers had renal manifestation with no extrarenal abnormalities detected, which is consistent with the fact that 80% of patients possess isolated NPHP, whereas 20% of patients present with other ciliopathy phenotypes in addition to NPHP. 2
The p.P209L is a hotspot mutation, which is recognized as homozygous in 14 families and heterozygous in 5 families, with a diagnosis of NPHP or focal segmental glomerulosclerosis. 8 Almost all of these 14 families with homozygous p.P209L mutation had similar genotypes. They presented with hypertension, late-onset proteinuria, and ESRD between 15 and 32 years of age. 9 11 Curiously, the eight families with homozygous/heterozygous splicing and small deletions/insertions in TTC21B appear to have a more extreme phenotype than those with homozygous p.P209L mutations. Their clinical features included early-onset proteinuria and ESRD in the first 8 years of age 8 ; this is the circumstance in our patient, who had a heterozygous mutation with early-onset proteinuria and ESRD at 16 months of age.
This stresses the significance of studying these uncommon and new genetic variations that could have an important clinical role in many cases where known and common genetic variants are absent. The incidence of consanguineous relationships is high in Arab residents. 13 This subsequently expands the occurrence of autosomal recessive disorders, some of which are incredibly uncommon and for which causative gene variations have never been distinguished. There is a requirement for a record of the mutations identified in every affected family with uncommon phenotypes. This will enable physicians to conduct proper genetic counseling and cost-effective genetic testing.
Conclusion
There are currently in excess of 20 genes that, when mutated, may lead to NPHP. Typically, mutational variation in TTC21B should be considered when renal biopsy reveals focal segmental glomerulosclerosis and tubulointerstitial lesions. This is particularly important in familial cases. Furthermore, ongoing research should be coordinated to uncover new mutations that may reveal the disease in many undiscovered circumstances.
Funding Statement
Funding None.
Footnotes
Conflict of Interest None declared.
References
- 1.Halbritter J, Porath J D, Diaz K A et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet. 2013;132(08):865–884. doi: 10.1007/s00439-013-1297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Srivastava S, Molinari E, Raman S, Sayer J A. Many genes-one disease? Genetics of nephronophthisis (NPHP) and NPHP-associated disorders. Front Pediatr. 2018;5:287. doi: 10.3389/fped.2017.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arts H H, Knoers N V. Current insights into renal ciliopathies: what can genetics teach us? Pediatr Nephrol. 2013;28(06):863–874. doi: 10.1007/s00467-012-2259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirk R. Genetics: Causative genes identified in rare renal ciliopathies. Nat Rev Nephrol. 2014;10(02):64. doi: 10.1038/nrneph.2013.261. [DOI] [PubMed] [Google Scholar]
- 5.Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20(01):23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolf M T. Nephronophthisis and related syndromes. Curr Opin Pediatr. 2015;27(02):201–211. doi: 10.1097/MOP.0000000000000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis E E, Zhang Q, Liu Q et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43(03):189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H, Su B, Liu X, Xiao H, Ding J, Yao Y. Mutations in TTC21B cause different phenotypes in two childhood cases in China. Nephrology (Carlton) 2018;23(04):371–376. doi: 10.1111/nep.13008. [DOI] [PubMed] [Google Scholar]
- 9.Huynh Cong E, Bizet A A, Boyer O et al. A homozygous missense mutation in the ciliary gene TTC21B causes familial FSGS. J Am Soc Nephrol. 2014;25(11):2435–2443. doi: 10.1681/ASN.2013101126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(05):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bullich G, Vargas I, Trujillano D et al. Contribution of the TTC21B gene to glomerular and cystic kidney diseases. Nephrol Dial Transplant. 2017;32(01):151–156. doi: 10.1093/ndt/gfv453. [DOI] [PubMed] [Google Scholar]
- 12.Slaats G G, Giles R H. Are renal ciliopathies (replication) stressed out? Trends Cell Biol. 2015;25(06):317–319. doi: 10.1016/j.tcb.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Hamamy H, Bittles A H. Genetic clinics in Arab communities: meeting individual, family and community needs. Public Health Genomics. 2009;12(01):30–40. doi: 10.1159/000153428. [DOI] [PubMed] [Google Scholar]