

Abstract
This review describes and summarizes the role of neuronal nitric oxide synthase (nNOS) on the central nervous system, particularly on brain regions such as the ventrolateral medulla (VLM) and the periaqueductal gray matter (PAG), and on blood vessels and the heart that are involved in the regulation and control of the cardiovascular system (CVS). Furthermore, we shall also review the functional aspects of nNOS during several physiological, pathophysiological, and clinical conditions such as exercise, pain, cerebral vascular accidents or stroke and hypertension. For example, during stroke, a cascade of molecular, neurochemical, and cellular changes occur that affect the nervous system as elicited by generation of free radicals and nitric oxide (NO) from vulnerable neurons, perioxide formation, superoxides, apoptosis, and the differential activation of three isoforms of nitric oxide synthases (NOSs), and can exert profound effects on the CVS. Neuronal NOS is one of the three isoforms of NOSs, the others being endothelial (eNOS) and inducible (iNOS) enzymes. Neuronal NOS is a critical homeostatic component of the CVS and plays an important role in regulation of different systems and disease process including nociception. The functional and physiological roles of NO and nNOS are described at the beginning of this review. We also elaborate the structure, gene, domain, and regulation of the nNOS protein. Both inhibitory and excitatory role of nNOS on the sympathetic autonomic nervous system (SANS) and parasympathetic autonomic nervous system (PANS) as mediated via different neurotransmitters/signal transduction processes will be explored, particularly its effects on the CVS. Because the VLM plays a crucial function in cardiovascular homeostatic mechanisms, the neuroanatomy and cardiovascular regulation of the VLM will be discussed in conjunction with the actions of nNOS. Thereafter, we shall discuss the up-to-date developments that are related to the interaction between nNOS and cardiovascular diseases such as hypertension and stroke. Finally, we shall focus on the role of nNOS, particularly within the PAG in cardiovascular regulation and neurotransmission during different types of pain stimulus. Overall, this review focuses on our current understanding of the nNOS protein, and provides further insights on how nNOS modulates, regulates, and controls cardiovascular function during both physiological activity such as exercise, and pathophysiological conditions such as stroke and hypertension.
Keywords: Ventrolateral Medulla, Periaqueductal Gray Matter, Nitric Oxide, nNOS, Nociception, Cardiovascular Regulation, Cerebral Vascular Accident, Hypertension, Stroke, Autonomic Nervous System, Nociception, nNOS Gene, nNOS Antagonist, Baroreceptor, Exercise Pressor Reflex, Arteriosclerosis, Thrombus, Endothelial Cells, Glutamate, GABA, Blood Pressure, Heart Rate, Exercise, Reactive Oxygen Species, Antioxidants, Hypothalamus
1. Introduction
Neuronal NOS is a very important member of a family of three enzymes known as nitric oxide synthases (NOSs) that catalyzes the production of NO from L-arginine, an α-amino acid that is used in the biosynthesis of urea, ornithine, agmantine, creatine, citrulline, and glutamate [1–4]. Neuronal NOS, also known as constitutive NOS (cNOS) or NOS1, is an isoenzyme because it is encoded by a different gene when compared to the other NOS isoforms. The gene that encodes nNOS is located on chromosome 12 in humans [2,5,6]. This review about nNOS is designed to elaborate the wide variety of roles, including its redox and antioxidant properties, played by nNOS within the cells, tissues, organs, peripheral nervous system, and the central nervous system, particularly the VLM and PAG that are directly and/or indirectly involved in the regulation, modulation, integration and control of the CVS in several physiological condition such as exercise and nociceptive pathways. Furthermore, we shall also summarize and review the functional aspects of nNOS during some pathophysiological and clinical conditions such as cerebral vascular accidents or stroke, hypertension and pain along with its antioxidant properties, the focus of this very important forum regarding nNOS. However, the functions, importance and clinical significance of nNOS will not be appreciated without a fundamental understanding of the role of NOSs and the actions of the final product, NO, on human and other mammalian systems, both in physiological and pathophysiological states. Thus, a brief summary of a) NOSs and b) NO effects on the CVS, cardiovascular functions and other disease processes will be presented below. Overall, this review focuses on the physiological actions and regulatory functions of nNOS along with its pathological significance and clinical implications with particular emphasis on different components of the CVS and cardiovascular diseases such as stroke and hypertension.
a). Isoforms of Nitric Oxide Synthases
All three NOSs catalyze the following reaction that results in the final production of NO (Knowles and Moncada, 1994):
However, all three NOSs isoforms play a role in catalyzing other leak and side reactions, such as superoxide production at the expense of nicotinamide adenine dinucleotide phosphate or commonly known as NADP+ while the reduced form of NADP+ is abbreviated as NADPH [2]. Because all the three different family members of NOSs are encoded by separate genes, they are also known as isoforms or isoenzymes of NOS: two of them belong to the constitutive NOS (cNOS) and the third one is inducible NOS or iNOS. Experiments with cloning the NOSs enzymes have discovered that cNOS includes both brain constitutive (NOS1) or commonly known as neuronal constitutive (nNOS) and endothelial constitutive (NOS3) or endothelial NOS (eNOS), while the third isoenzyme that is cloned is named as the inducible (NOS2) gene or inducible NOS or iNOS [7,8]. In order to elaborate further, nNOS has been cloned from chromosome 12 located within nervous tissues and skeletal muscle type II, and its major function has been initially thought to be cellular communication [7,8]. On the other hand, iNOS was cloned from chromosome 17, located within the immune and the CVS, and the major function was elucidated as immune defense against pathogens [7–9]. In addition, eNOS has been cloned from chromosome 7 within the endothelial cells with a very important function of causing vasodilation [7]. It should be noted that a drug called Ronopterin or VAS-203 or 4-amino-tetrahydrobiopterin (4-ABH4), an analogue of tetrahydrobiopterin (BH4), which is a cofactor of NOS, is a non-selective NOS antagonist that has been under research and development as a neuroprotective agent for the management of traumatic brain injury, such as stroke [1]. Finally, NOSs activity has also been demonstrated in several bacterial species that include some dangerous pathogens such as Bacillus anthracis and Staphylococcus aureus [10].
b). Nitric Oxide
Although this review will focus on the effects of nNOS on the CVS and nociception, a brief description of the role of NO on cardiovascular activity is warranted, because of the fact that all three NOSs mediate their effects and actions via the final product which is NO. However, exactly how much NO is produced by each of the NOS isoenzyme in physiological state or in a disease condition is unknown, but under inflammatory conditions in the brain, a molecule of iNOS isoenzyme can generate 100–7000 times more NO than one molecule of the nNOS isoform [11]. Acting via the second messenger cyclic GMP (cGMP), NO is a very important neuronal mediator of intracellular signal transduction processes and has a wide range of physiological, pathophysiological, and pharmacological actions on numerous systems of humans and mammals, including the cardiovascular and the nervous systems [11–17]. The literature has numerous research publications, reviews, book chapters, and books on NO, including reviews on target validation of nNOS in physiological homeostasis as well as in several disease states. These diseases include but not limited to stroke and cardiovascular dysfunction; neurodegenerative disorders (e.g., Alzheimer’s disease, Parkinson’s-like symptoms); epilepsy, cerebellar dysfunctions (e.g., essential tremor); autoimmune encephalomyelitis; brain, breast and lung cancers; systemic sclerosis; and skin lesions [18–23].
The brainstem in the medulla oblongata contains several critical nuclei that control cardiovascular functions via the SANS which is strongly influenced by NO and oxygen free radicals. Long-term NOSs inhibition and less NO production have age-dependent consequences on radical signaling along with antioxidant or detoxificant properties [24]. Sleep deprivation has also been shown to result in endothelial dysfunction in addition to the development of hypertension via the NO signaling pathway, that can be reversed by L-arginine supplementation which can be easily administered orally [18]. There is ample evidence that the NO pathway has a strong correlation with cardiovascular diseases, cognitive impairment, and dementia [19]. A randomized, double-blind, and placebo-controlled trial has been proposed to evaluate the feasibility of sodium nitrate that will increase systemic levels of NO precursors in plasma as part of a treatment protocol for acute congestive heart failure [25]. In the same line, a short-term increase in dietary nitrate that will increase the availability of NO has been shown to reduce carotid atherosclerosis and ischemic cardiovascular disease in older women [26]. Even a randomized controlled trial of platelet inhibition by Prasugrel® (a thienopyridine derivative, a platelet inhibitor and an irreversible antagonist of P2Y12 ADP receptors) in patients with unstable angina pectoris has shown significant improvement in cardiovascular function mediated via an increase in endothelial NO bioavailability [27]. Finally, a recent review describes the interaction between reactive oxygen species (ROS) production and antioxidant activity associated with atherosclerosis, hypertension, heart failure and myocardial infarction [20]. Thus, it can be summarized that NO plays a major role not only in physiological homeostasis but also has a critical relationship with many diseases associated with the cardiovascular and nervous systems such as stroke, hypertension and neurodegeneration.
2. Gene Structure, Organization, Binding Sites and Receptors of Neuronal Nitric Oxide Synthase
First discovered in 1990 after its neuronal isolation, nNOS has many applications in the development and homeostatic regulation of different mammalian systems [28]. From the onset of fetal development, nNOS plays a key role in the regulation of signaling and plasticity of neurons [28]. In order for the appropriate development to occur, nNOS is regulated physiologically according to the needs of the specific tissue and organ; it is regulated by phosphorylation and several phosphorylation sites for nNOS were already identified [29–31]. As a result, nNOS has been integrated in the evolutionary development of multiple vital enzymatic processes designed accordingly to specific neurons and cells [28]. Although the role of nNOS on the CVS has yet to be firmly established, numerous results from scientific medical research have reported the discovery of significant novel findings. These studies are specifically related to the biochemical properties, structural domain, and motif of nNOS, and the subsequent array of action of its final product, NO. This section will briefly summarize the role of the structure and domain of nNOS with particular emphasis on its effect on the CVS.
a). Domain and Motif of Neuronal Nitric Oxide Synthase
Although nNOS has been subdivided into multiple splicing variants like nNOSαa, nNOSαb, nNOSβ, and nNOSγ [32–34], we shall focus our review briefly on the general domain of the nNOS protein. The nNOS isoform exhibits a bidomain structure consisting of an N-terminal oxygenase domain and a C-terminal reductase domain [35]. The N-terminal domain bears binding sites for heme, BH4 and L-arginine, whereas the C-terminal domain has binding sites corresponding to flavin-adenine dinucleotide (FAD), flavin-mononucleotide (FMN), and NADPH. The two nNOS domains are connected by a CaM-binding sequence, also known as CaM-recognition site, and indicate the significance of CaM in affecting the catalytic activity by controlling the spatial orientation of these two regions as shown in Figure 1 [36,37]. Calmodulin is a sensor that resides within the intracellular space and regulates a multitude of different enzymes and ion channels, including the regulation or modulation of membrane transport proteins. Calmodulin transmits functional signals based on Ca2+ binding to CaM binding-motifs. It has been shown using cardiac magnetic resonance imaging (MRI) studies that nNOS plays a prominent role in modulating Ca2+ influx in the myocardial cells of the heart of mammals [38,39]. Furthermore, phosphorylation of nNOS at the SER847 sequence by CaM occurs in the hippocampal area of the rat brain following a transient focal ischemic attack [40]. Finally, it has been shown that increased intracranial pressure can induce a CaM-dependent protein kinase II (CaMPKII) mediated phosphorylation of nNOS at SER847 sequence in the rat hippocampus following an episode of subarachnoid hemorrhage. Tetrahydrobiopterin (BH4) couples the reductive oxygen activation to L-arginine oxidation [41]. The BH4 binding site and the active-site pocket that consists of heme and L-arginine binding sites are vital for nNOS dimerization [42]. In addition, two-hybrid studies with oxygenase and reductase domains, respectively, in yeast demonstrate that the association of nNOS into active dimers involves interactions between the reductase domains and between the reductase and oxygenase regions [43]. Altogether, the structure of the nNOS isoform itself has a profound correlation with cardiovascular disorders including cerebral ischemia or stroke. Figure 1 illustrates the complex bidomain structure of nNOS.
Figure 1:
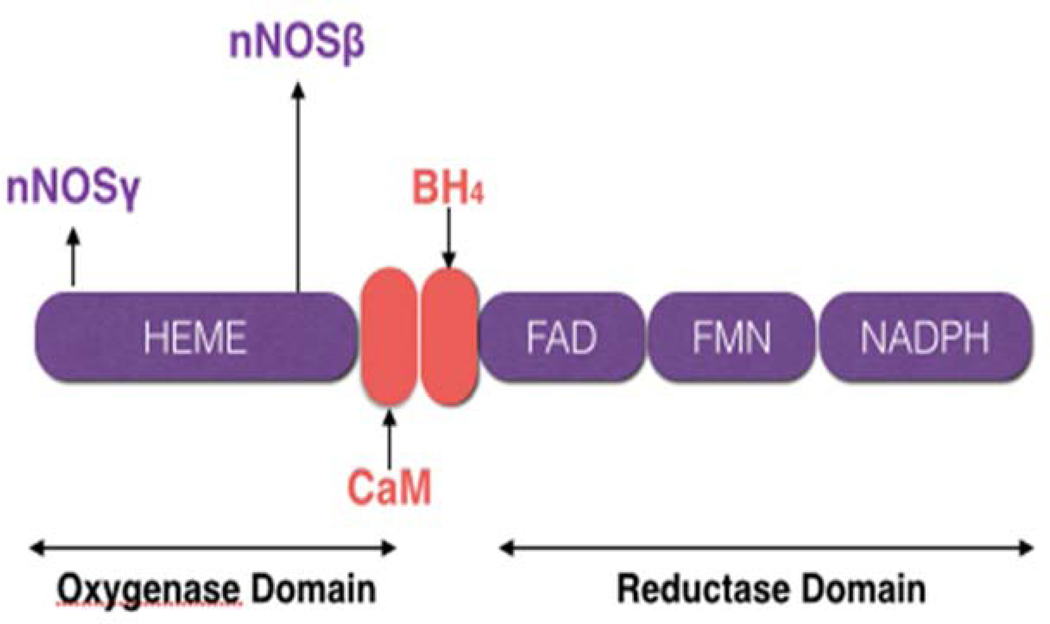
Bidomain structure of nNOS isoform and the splicing variants (nNOSβ and nNOSγ). The electrons donated by NADPH (nicotinamide adenine dinucleotide phosphate) to the reductase domain reach the oxygenase domain via two redox carriers, FAD (flavin-adenine dinucleotide) and FMN (flavin mononucleotide). In the oxygenase domain, the electrons interact with heme and BH 4 (tetrahydrobiopterin) to cause arginine oxidation to citrulline and NO (nitric oxide).
The location of nNOS on chromosome 12 holds tremendous clinical significance. For example, there is a significant difference between two species of rats (different only by one locus) in chromosome 12 and is associated with high contractility and lowered elasticity of resistance in the vasculature system during a rise in blood pressure, suggesting that within the two different congenital rat models, vascular remodeling and dysfunction can contribute to hypertension [5,44]. There is a paradoxical concept in the sense that both genetic subtypes are associated with the production of NO as well as regulation of vascular hypertension and are encoded within the same gene. Therefore, in order to further our knowledge about nNOS and its subsequent formation of NO would require future research on hypertension at the genetic and molecular levels.
b). Binding Sites
The binding sites located on the nNOS protein molecule are particularly examined for their individual as well as their combined abilities in order to further understand the physiology of the CVS as well as to elucidate the factors which contribute to cardiovascular and other diseases. For example, with regards to the designation of the BH4 binding site, it has been discovered that a pharmacological treatment with BH4 has shown to reverse pulmonary hypertension in a rat hypoxia model [45]. Similarly, the nNOS binding site for L-arginine is regarded with a greater interest. As people get older, peripheral arteries have a dwindling ability to dilate, due to endothelial dysfunction, particularly when such sequence of events within the vascular system furthers the risk of cardiovascular disease (CVD) with increasing age. L-arginine contributes to numerous physiological processes including nitrogen detoxification, immune-competency, growth hormone (GH) secretion, and insulin secretion and a considerable amount of attention has been placed on the ability of this amino acid to affect the overall vascular endothelial functions [46].
Reactive oxygen species play a large role in the pathogenesis of vascular diseases, and therefore, a careful consideration of nNOS’s relationship with NADPH cannot be underestimated. Scientists and researchers have been focused on the NADPH oxidase (NOX) family with reference to a large variety of ischemic and ROS-mediated vascular diseases. The NOX family (including seven isoforms of the NADPH oxidase catalytic unit) is quite unique as they also generate the ROS. The relevant enzymatic isoforms which are associated with vasculopathies are the NOX1, NOX2, and NOX4 of the NOX family [47]. Genetic mutations which are associated with a loss of NOX1 functionality have been shown to lower the occurrence of ROS-induced retinopathy in mouse models [47]. A reduction of ROS activity can lower the incidence of vascular injury. An actively working nNOS molecule moves charged electrons from NADPH, through the FAD and FMN (carboxy-terminal domain) to the heme (amino-terminal oxygenase domain; Figure 1). An essential cofactor BH4 binds the molecule oxygen and L-Arginine to the oxygenase domain. Ultimately at the amino-terminal oxygenase domain, these electrons are used to reduce and activate oxygen (O2) as well as to oxidize L-arginine to L-citrulline and NO [48] (Figure 1). Finally, by using electrons from NADPH to ultimately produce NO, the nNOS isoform plays the role of reducing the production of ROS and at the same time eliciting vasodilation and smooth muscle relaxation.
c). Advanced Structure of Neuronal Nitric Oxide Synthase
Neuronal NOS protein consists of 1434 amino acids and a molecular weight of 161 kDa [49]. Multiple mRNA transcripts of nNOS can be generated via alternate promoter usage, alternative splicing, and cassette insertions/deletions. In addition, some transcripts are directly translated into the nNOS protein isoform with modified structure and/or functions [50]. The full-length transcript (without excisional mRNA codon removal) translation initiates in exon 2 results in a 161 kDa protein designated as nNOSα while deletion of exon 2 in stem cells forms two shorter splice variants, nNOSβ (136 kDa) and nNOSγ (125kDa) [33,34]. Figure 2 illustrates the different splice variants of the nNOS family.
Figure 2. Difference between nNOS α and nNOS β splice variants.
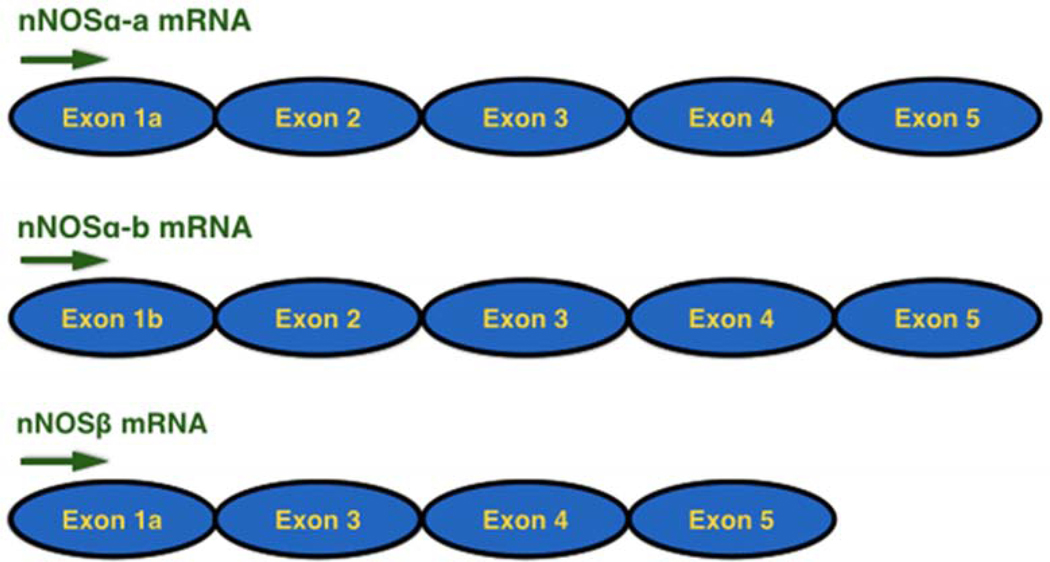
Serine, Threonine, Arginine), Tetrahydrobiopterin, Calmodulin, Flavin mononucleotide, Flavin adenine dinucleotide, and Nicotinamide adenine dinucleotide phosphate are common structural components present in all the splice variants of nNOS (neuronal nitric oxide synthase). The arrows indicate the direction of phosphorylation at the respective sites on NOS (nitric oxide synthase) activity.
Tetrahydrobiopterin (BH4) couples the reductive oxygen activation to L-arginine oxidation [41]. The BH4 binding site and the active-site pocket that consists of heme and L-arginine binding sites are vital for nNOS dimerization [42]. As mentioned above, the association of nNOS into active dimers involves interactions between the reductase domains and between the reductase and oxygenase regions [43]. The CaM sequence stimulates nNOS catalytic activity and electron transfer into its flavin and heme centers [51]. Neuronal NOS contains PDZ (PSD-95, discs-large and zona occludens-1) domains which consist of a consensus sequence of 90 amino acids that mediate protein-protein interactions via localization of nNOS to appropriate sites in the neuron [52–54]. As mentioned previously, there are three specific and distinct forms of NOS: iNOS, eNOS and nNOS, and all of these isoenzymes are expressed in cardiac myocytes with some variations. For example, the activity of iNOS has been detected in a variety of cell types, including endocardial and endothelial cells, vascular smooth muscle cells, fibroblasts, and neonatal and adult cardiac myocytes from several species [55].
d). Receptors Associated with Neuronal Nitric Oxide Synthase
Neuronal NOS is located at the postsynaptic membrane and attached to the N-terminal PSD/Discs-large/ZO-1 homologous (PDZ)-binding domain, which is subsequently attached to the N-methyl-D-aspartate (NMDA) subtype of the glutamate receptor family or NMDAR [28]. The NMDAR is driven by an influx of Ca2+ from the synaptic cleft, which will also propel the production of NO for signal potentiation [28]. Calcium is the primary regulator of nNOS transcription and activity; Ca2+ binding to CaM results in an electron transfer from the C-terminal (flavin domain) to the N-terminal (heme domain) by the displacement of an autoinhibitory insert, resulting in the biosynthesis of NO [56]. Neuronal NOS is transported to the Ca2+ handling proteins, such as the NMDAR-Ca2+ channel associations [57]. The crucial relationship between nNOS and NMDAR has been described through observations regarding homocysteine, which activates NMDAR in the central nervous system [58,59]. Homocysteine disrupts the production and activity of the NO molecule. Because of the fact that NO functions to decrease vascular smooth muscle cell propagation, its suppression by homocysteine could promote myointimal hyperplasia and narrowing of a blood vessel. Therefore, high homocysteine levels are considered a risk factor for cardiovascular diseases. This relationship demonstrates the vital role that nNOS plays with the NMDAR and the Ca2+ channels; further elaborating its essential role in the homeostasis of multiple organ systems, including the CVS, and their interactions with each other. Figure 3 is a schematic illustration of such nNOS and NMDAR interactions.
Figure 3. Negative feedback loop for nNOS control of Ca2+ entry through NMDA receptor.
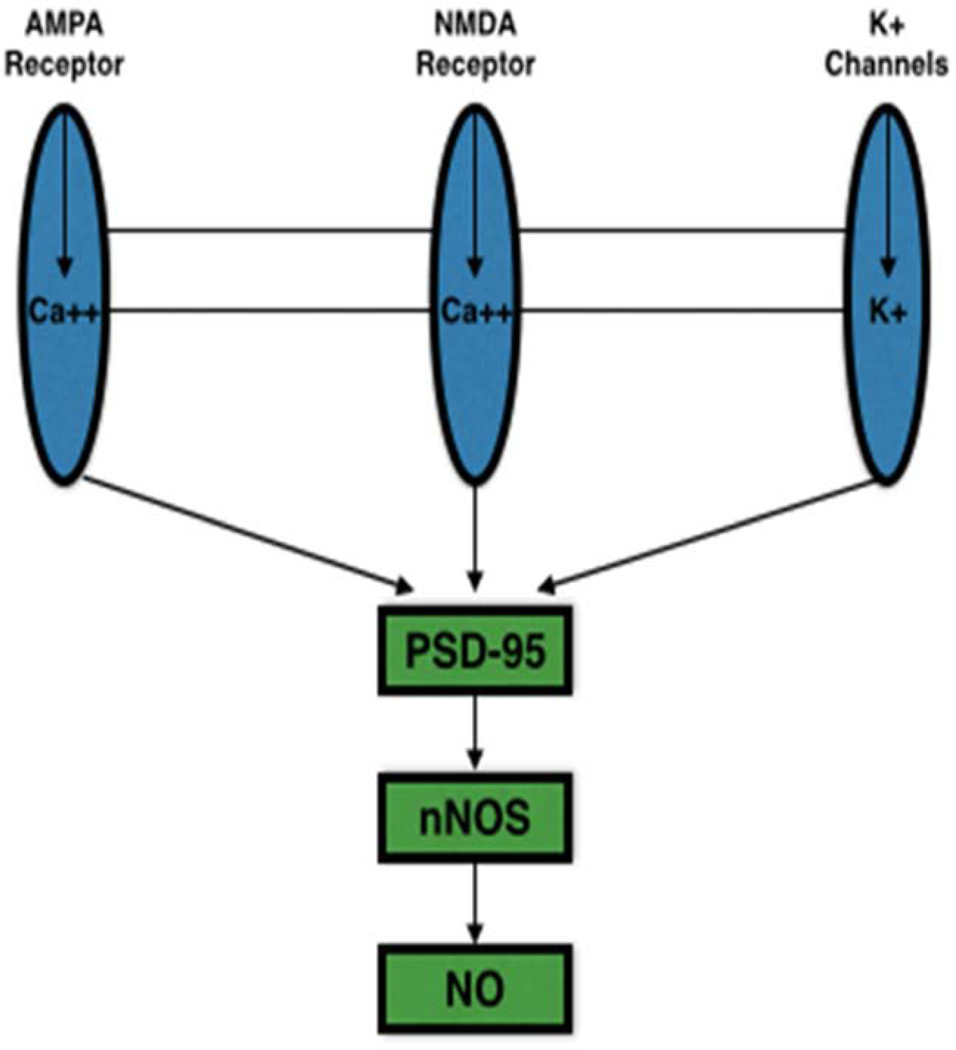
The entry of Ca2+ into the cell is coupled to nNOS (neuronal nitric oxide synthase) through the PDZ domain, PSD-95. As the level of Ca2+ in the cell increases, nNOS-derived NO (nitric oxide) acts on glutamate receptors (Both NMDA (N-methyl-D-aspartate) and AMPA) in order to inhibit further entry of Ca2+ by S-nitrosylation reaction.
e). Regulation of Neuronal Nitric Oxide Synthase
There are a number of molecular regulators of nNOS that are active within the human body. For example, the heat shock protein 90 (HSP90) stimulates nNOS activity by increasing its binding to Ca2+/CaM [60]. HSP90 is upregulated with increased nNOS expression. On the other hand, caveolae 1 (Cav-1) and caveolae 3 (Cav-3) proteins are downregulated by an increase in nNOS expression [61]. Caveolae are invaginations of the plasma membrane which are rich in cholesterol and sphingolipids. There are three isoforms of caveolins or the structural proteins of caveolae. For example, Cav-1 functions to generate caveolae in non-muscle cell types and provides for membrane localization of Cav-2. Then, Cav-3 is needed to form caveolae in skeletal, cardiac, and smooth muscle cells [62]. Studies using a model of genetically-engineered mice which lack caveolin genes have given researchers the ability to properly understand the importance of each distinct NOS isoforms which may contribute to the pathophysiology of many human diseases. It has been shown that Cav-1 KO (gene knockout) mice develop a severe type of cardiomyopathy, which shortens their lifespans significantly [62]. However, Cav-3 is actually predominant in cardiac myocytes and in the heart of a Cav-3 KO mouse; levels of cyclic AMP (cAMP) and the β-adrenergic second messenger signaling mechanism in association with ATP production are inhibited or decreased [62]. The Cav-1 KO mice also have an overactive production of all NOSs due to the previously mentioned inverse relationship between Cav-1 and nNOS activity. A change in NOSs activity in Cav-1 KO mice can be linked to the development of cardiopulmonary pathologies due to its critical roles on cardiovascular homeostasis, vascular tone, vascular remodeling, platelet aggregation, and angiogenesis [62]. Another regulator of nNOS is HSP70, which has been shown to be generated within the rostral ventrolateral medulla (RVLM) neurons resulting in an overexpression of the nNOS protein [63,64]. This may explain the neuroprotection during toxic septic shock as a result of endotoxins in the blood. In addition, the mechanisms underlying neuroprotection involve an enhancement of protein kinase G (PKG) signaling pathway and inhibition of the nNOS/peroxynitrite pathway that prevents cardiovascular depression [63,64]. In addition, evidence suggests that the autoinhibitory domain physiologically modulates nNOS activity wherein nNOS functions as structural element that is displaced upon calmodulin binding, resulting in loss of inhibition of NO synthesis [65]. Oxidative stress can induce phosphorylation of neuronal NOS in cardiomyocytes through AMP-activated protein kinase (AMPK) [66]. Finally, a novel protein that is named Protein Inhibitor of Neuronal Nitric-oxide Synthase (PIN) has been shown to bind to a 17-Amino Acid Residue Fragment of the nNOS and it provides further insights into the nNOS inhibition mechanism by PIN [67].
As mentioned above, the generation of NO is linked to the NMDAR, which under normal physiological process will limit NO production to small amounts. Unfortunately, pathological processes such as hypertension (HTN) and cerebrovascular accidents (CVA) may damage or alter this delicate process resulting in an abundance of NO generation [28]. Nitric oxide neurotoxicity can thus be exacerbated by oxygen free radicals and other ROS that arise from post injury status resulting in further degradation of proteinaceous material. Although caustic, this inflammatory phenomenon does not occur in neurons that possess nNOS possibly due to proximity interactions with phosphofructokinase (PFK1), which would protect the neurons through regulation of glycolysis. It has been shown that nNOS expression increases with CVA, and also causes changes in the homeostatic regulation of heart rate, blood pressure, myocardial contractility, and the autonomic nervous system (ANS); both physiologically and during times of stress/activity/exercise [68–71]. In summary, we have briefly described the structure, domain, location, motif, receptors involved, and regulation of nNOS, and its relationship with certain cardiovascular activities. The following section 3 will further elaborate on specific roles of nNOS on the CVS in both physiological and pathological states.
3. Role of Neuronal Nitric Oxide Synthase on Cardiovascular Functions in Physiological and Pathological States
Cardiovascular diseases are the leading cause of morbidity and mortality in this entire world, with myocardial infarction and stroke being the first and the third killer diseases, respectively, in the United States of America [72]. The time and money spent, the research performed on myocardial cells and the heart itself, and the plethora of studies done on regulatory mechanisms of cardiovascular function in both physiological and pathological situations as described in the medical literature are extraordinarily enormous. Among all the numerous receptors, various neurotransmitters, neural pathways and signaling mechanisms that are involved with the CVS, NO is a very important determinant for maintaining basal vascular tone; preventing platelet activation and aggregating; limiting the adhesion of white blood cells to the endothelial wall of the vasculature; controlling myocardial contractility; and mediating, modulating and integrating the regulation of the CVS [15,16,22,73]. In addition, NO has been identified as a key player in the pathogenesis of common cardiovascular diseases that include hypotensive shock, development of essential hypertension, and formation of atherosclerosis [74,75]. On October 12, 1998, the Nobel Prize Assembly awarded the Nobel Prize in Medicine to three scientists, Robert Furchgott, Louis Ignarro, and Ferid Murad, for their discoveries that NO is a key signaling molecule in the CVS. In the following paragraphs, we shall discuss the past and current literature regarding the role of nNOS on many aspects of the CVS such as cardiac myocytes, the heart, the vasculature, the different regions of the brain, several cardiovascular reflexes, and on cardiovascular regulatory mechanisms, i.e., the SANS and the PANS.
a). Cardiac Myocytes and Cardiac Functions: Role of Neuronal Nitric Oxide Synthase
An area of concern, as it pertains to the viability of the mammalian organism, is the CVS and the effects of nNOS on its regulation. Neuronal NOS, throughout the years of research, has been expanded from an identifying term used for just one protein to a family of variants consisting of nNOSαa, nNOSαb, nNOSβ and nNOSγ that are present on all muscle cell types including cardiac myocytes [68–71]. A study has identified that nNOS as well as Ca2+-CaM-dependent CaMPKII and NOX2 are important mediators in mechanical and chemical transduction processes that occur during cardiac contraction, and may provide new therapeutic approaches for treating stress-induced Ca2+ dysregulation, arrhythmias, and cardiomyopathy [76,77]. Cardiac muscle cells are regulated by several mechanisms: intrinsically via the automaticity of the sinoatrial node, atrioventricular node, and the purkinje fibers via L and T types of Ca2+ channels and extrinsically via the SANS and the PANS, the two divisions of the autonomic nervous system or ANS. Neuronal NOS is present inside the sarcoplasmic reticulum (SR) of cardiac myocytes (Figure 4) and allows more Ca2+ to exit the SR and resulting in positive ionotropic and dromotropic effects [78–80]. Neuronal NOS also increases phospholamban protein levels which inhibit SERCA and ultimately result in a decreased SR Ca2+ concentration [79]. Inhibition of SERCA and a simultaneous augmentation of phospholamban levels have negative ionotropic and dromotropic effects on cardiac myocytes. An increase or a decrease in nNOS activity in the regulation of intrinsic activity of cardiac muscle cells defines the positive or negative ionotropic and dromotropic effect, respectively.
Figure 4: Effect of nNOS on calcium handling.
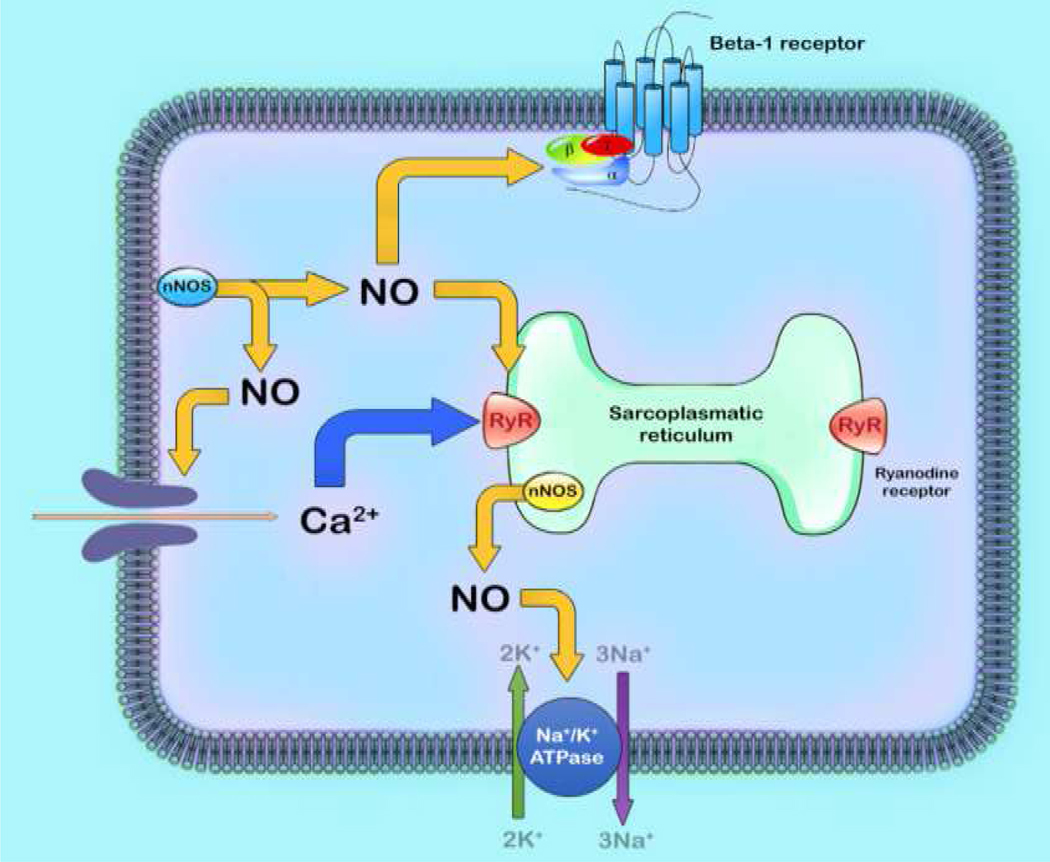
The process of excitation-contraction coupling begins with the entry of calcium (Ca2+) into the cell via the calcium current that triggers the further release of Ca2+ from sarcoplasmic reticulum. This stimulation of intracellular Ca2+ initiates and propagates contraction. Neuronal nitric oxide synthase (nNOS) have significant effects on myocyte contraction under basal conditions and via Beta-1 adrenergic receptor stimulation.
Neuronal NOS is involved in the control of cardiac muscle contraction. Inhibition of nNOS activity via gene disruption or by acute pharmacological blockade enhances basal cardiac contractility at physiological temperatures and over a wide range of heart rates [81,82]. An increase in left ventricular (LV) ejection fraction has been observed in nNOS−/− mice compared with wild type nNOS littermates, without any significant change in heart rate or blood pressure between the two strains [83]. In isolated LV myocytes, nNOS inhibition results in a Ca2+-mediated positive ionotropism [81,83].
In addition, the nNOS-derived NO increases the activity of Na+-K+-ATPase that indirectly affects the Ca2+ fluxes via an action on intracellular sodium (Na+) levels, resulting in a change in the activity of the Na+-Ca2+ exchanger [84]. Neuronal NOS-induced production of NO inhibits the L-type Ca2+ current via the NO-cGMP pathway as a result of attenuating guanylate cyclase-derived increases in peak Ca2+ current, and PKG phosphorylation elicits a decrease in the basal levels [85]. It is possible that this mechanism also involves a change in the redox state of the Ca2+ channel. In addition, nNOS-derived NO exerts a negative feedback effect on Ca2+ handling as the increase in intracellular Ca2+-mediated stimulation of nNOS synthesis and subsequent generation of NO further inhibits Ca2+ fluxes. Finally, nNOS plays a role in the regulation of proteins involved in Ca2+ handling via the Ca2+ channels that are linked to the ryanodine receptors [86]. Thus, in the myocardium, nNOS activity is regulated by beat-to-beat variations in intracellular Ca2+ levels [83].
The roles of oxidants and reactive oxygen species (ROS), including nNOS, on Ca2+ and Na+ movements in healthy and diseased cardiac myocytes and other pathological conditions such as cardiac ischemia-reperfusion injury have been well established. A review in the literature describes the protective effects of nNOS after ischemia-reperfusion injury, with particular emphasis on the subcellular localization of nNOS at the molecular level and the subsequent targets of its action [87]. Ischemic postconditioning can provide protection of the heart following an ischemia-reperfusion injury to the heart mediated via nNOS located within the SR and mitochondria [88]. The L-type Ca2+ channel activity and myofilament Ca2+ sensitivity in cardiac myocytes can be modulated by the nNOS protein in hypertensive rats [89]. The deleterious effects of chronic ethanol consumption on the levels of Brain Natriuretic Peptide (BNP) and subsequent expression of the nNOS isoform within cardiac myocytes contribute to worsening of cardiac remodeling and may lead to progression of cardiomyopathy [90]. Neuronal NOS decreases myocardial contractility via the inhibition of the L-type Ca2+ current amplitude and the intrinsic Ca2+ transients, and both mitochondria and xanthine oxidoreductase have been identified as potential targets for nNOS in cardiac disease animal models [82].
On the other hand, both the basal tone and positive inotropic responses mediated via β−1 adrenergic receptor stimulation with low and high concentrations of isoproterenol are not mediated by nNOS in the LV myocytes [82]. However, one study reports a reduction in the inotropic response in nNOS−/− myocytes at room temperature [86]. It has been shown that the anti-hypertrophic and antioxidant effects of β3-adrenergic stimulation in myocytes require differential expression of nNOS phosphorylation [91]. The literature agrees that nNOS is solely responsible for the NO-mediated autocrine regulation of myocardial contraction and relaxation [92]. In addition, studies using nNOS inhibitors support the involvement of NO in cardiac arrhythmias where direct stimulation of the vagus nerve in rabbits promotes NO release via the nNOS protein within the ventricles of the heart [93,94]. NO release due to nNOS stimulation within the LV due to vagal nerve stimulation has antiarrhythmic effects in the heart [94]. Finally, an atrial-specific upregulation of micro RNA-31 in human atrial fibrillation is a key mechanism that results in the loss of atrial dystrophin and nNOS proteins which may lead to the electrical phenotype induced by atrial fibrillation [95]. Overall, we summarize that nNOS has very important and serious implications on cardiac myocytes and cardiac functions in both physiological and pathological situations such as cardiac remodeling and heart failure, and is evolving into a potential therapeutic tool for pharmacological interventions in many disease states.
b). Vasculature and Neuronal Nitric Oxide Synthase
Although the eNOS protein is the predominant regulator of arterial blood pressure via vasodilatation, nNOS is also present on endothelial cells and significantly contributes to the modulation of vascular perfusion [96,97]. Interestingly, other studies report that nNOS is expressed in endothelial cells and participates in the endothelial-dependent control of vascular relaxation [98,99]. Smooth muscle is found throughout the mammalian vasculature imbedded in the tunica media of most arterial circulatory system and some of the venous system. Homeostatic mechanisms at the autocrine and paracrine level result in an immediate vasodilatory effect during times of increased demand or increased blood pressure as a direct consequence of NO that increases the diameter of the vascular lumen, and thereby, decreasing vascular resistance by a 4-fold. Since 2009, emerging data propose that eNOS and nNOS have their separate distinctive roles in the physiologic local regulation of human microvascular tone in vivo that triggered further studies regarding the differential contribution of nNOS and eNOS in vascular regulation as observed in several human diseases [100–104]. In addition, a study using RT-PCR confirms the endothelial expression of both nNOS and eNOS, and that the differential distribution of NOS isoforms in brain blood vessels might explain their separate modes of activation [105]. In a disease state like essential hypertension, it has been shown that impaired mental stress-induced nNOS-mediated vasodilatation directly correlates with the increased sympathetic activity in hypertensive patients [101]. Thus, nNOS has serious consequences on the human circulatory system that is regulated by the brain.
On the other hand, nNOS plays a critical role in the transcription of regulatory systems within the endothelial cells during ocular hemo-vasculogenesis, vasculogenesis, and overall angiogenesis [102]. Neuronal NOS also regulates the dynamics of renal blood flow via acetylcholine-induced vasodilation or by norepinephrine/angiotensin II elicited renal vasoconstriction [106]. Furthermore, a study demonstrates that the nNOS isoform is a critical regulator of renal hemodynamics in the newborn piglet kidney [103]. It is worthy to note that the protein expression of nNOS is maximal within the trigeminal vascular system and other brain structures related to pain-related circulatory changes in rats [107]. Moreover, the nNOS-derived NO-cyclic GMP pathway has the potential to be an important target for the treatment of renal circulatory dysfunction, including chronic kidney failure and uremia [108]. The heat-shock protein HSP-90 has been identified as a signaling mediator of nNOS-dependent vasodilatory response in mesenteric arteries, observed in portal hypertensive states [109]. Indeed, local nNOS-derived release of NO is a critical regulator of basal blood flow in the human coronary vascular bed; thereby suggesting that nNOS has its own distinctive role in the physiological regulation of human coronary vascular tone in vivo [110]. It has been previously shown that nNOS predominantly increases production of NO during decreased periarteriolar oxygen tension, resulting in vasodilation as seen during a state of low blood pressure [111]. Another study indicates that nNOS inhibition by pharmacological interventions can improve, at least in part, the hippocampal dysfunction after an attack of brain ischemia [105]. Overall, we summarize the role of nNOS protein on the vasculature during both physiological state and pathological situations that can lead to further development of strategies in the treatment protocol of vascular disorders such as ischemia.
c). Sympathetic Autonomic Nervous System and Parasympathetic Autonomic Nervous System: Role of Neuronal Nitric Oxide Synthase
Both the SANS and the PANS have significant roles in the regulation, integration, and modulation of cardiovascular functions during physiological homeostasis as well as throughout pathophysiological situations like myocardial infarction or stroke. The ANS of the heart is divided into intrinsic and extrinsic divisions; additionally, the extrinsic ANS of the heart contains nerves that regulate communication between the central nervous system (CNS) and the heart while the intrinsic division of the cardiac ANS contains autonomic nerves that are present within the pericardial cavity and within the heart [112]. Many processes such as cellular properties of myocytes, the hemodynamics, blood pressure, and heart rate are regulated by the ANS [112]. Parasympathetic interaction in the CVS stimulates acetylcholine release which can be counteracted with sympathetic epinephrine release; additionally, cholinergic stimulus plays an important predominant role in cardiovascular protection against heart failure and ventricular arrhythmias [113,114]. For example, electrophysiological modulation of the cardiac cycle by the ANS is crucial, especially in circumstances such as atrial fibrillation where activation of parasympathetic or antiarrhythmic cycles and inhibition of sympathetic or arrhythmic periods are crucial for keeping a patient alive [113,114]. Activation of the PANS within the atria is antiarrhythmic while SANS activation is proarrhythmic in both the atria and ventricles. The origin of both the SANS and PANS is within the brainstem or the CNS and most of the studies done on nNOS that regulates cardiovascular function and activity via SANS and PANS are performed on brain regions that regulate/modulate their effects. Thus, we shall further summarize the role of nNOS on the ANS in the following paragraphs (see below). However, it is important to mention that parasympathetic stimulation is partly regulated by NO which stimulates the release of acetylcholine and controls functions like vascular resistance and myocardial contraction. In vivo inhibition of nNOS in guinea pigs has shown a reduced vagal stimulation leading to tachycardia; this indicates that NO controls antiarrhythmic processes through the vagus nerve (cranial nerve X) originating from the medulla [115,116]. In addition, research also supports the theory that acetylcholine release facilitates the production of NO through nNOS activation via a NO-cGMP pathway within the pacemaker cells present in the sinoatrial and atrioventricular nodes [117]. The role of nNOS on both SANS and PANS will be further described in an integrated way in the following sub-sections.
d). Brain Regions Associated with Cardiovascular Regulation and Functions: Role of Neuronal Nitric Oxide Synthase
The CNS (the brain and the spinal cord) regulates the CVS in two separate processes: First, a feed-forward regulation, commonly known as the “central command” and second, the feedback or reflex regulation such as the baroreflex; both of these general mechanisms integrate in order to contribute to overall cardiovascular homeostasis. Among numerous receptors, neurotransmitters, neuromodulators, and neural mechanisms/pathways that are associated with cardiovascular functions, the NO signaling cascade and nNOS protein are crucial in contributing to their regulatory processes. Both NO and nNOS are present in almost all the neurons within the cardiovascular regulatory centers in the brain, including the nucleus tractus solitarii (NTS), the nucleus ambiguous, the hypothalamus, the dorsal motor nucleus of the vagus, the VLM, and also in vagal afferents [118–120]. After reviewing the literature for the last several decades, we find the VLM as a critical region within the medulla oblongata of the brainstem that is involved in numerous receptor- and neurotransmitter-mediated regulatory processes, integration of afferent and efferent signals from and to the heart and the entire body, and in a vast cascade of mechanisms involving the coordination of the CVS in both healthy and diseased individuals [121–127]. Both anatomically and functionally, the VLM has been separated into the caudal (CVLM) or depressor region and the RVLM or pressor area; this mapping of the VLM was derived from exploratory experiments using several animal species [128–131]. In addition to the VLM, the hypothalamus, particularly the posterior part, plays a critical role in coordinating or fine-tuning all cardiovascular functions, and that both NO molecules and nNOS protein contribute to the overall mechanisms of cardiovascular activity [132–135]. Finally, the NTS region of the brain along with its nNOS associated neuronal network that is located within the dorsal area of the medulla has been implicated in regulating the CVS, and plays the most important role in baroreflex mechanism [136–144]. There are significant interactions and reciprocal connections among the three regions of the brain: the VLM, the NTS and the hypothalamus. For example, the VLM plays a part in the integration of vasopressive responses projecting from the hypothalamus [145]. Therefore, the following paragraphs in this section will separately review the interactions of nNOS with neurotransmission and cardiovascular regulation within the VLM, the NTS, and the hypothalamus.
The RVLM and the CVLM are now established as the two most critical regions in our brains for the maintenance of blood pressure and vasomotor tone, baroreceptor and the exercise pressor reflexes, cardiopulmonary reflex mechanisms, and the overall sympathetic tone [121–127]. Afferent impulses originating from many rostral areas of the brain are also relayed onto these RVLM and/or CVLM neurons where they are integrated and the final responses are either an increase or a decrease in their neuronal activity in order to maintain cardiovascular homeostasis; however, the RVLM being the predominant regulatory center of both the SANS and PANS. Nitric oxide, derived from all NOSs, plays a crucial role in regulating sympathetic nerve activity but the nNOS isoform has a predominant sympathoinhibitory effect under physiological conditions by its action on different regions of the brain, including the hypothalamus, the NTS, and both the RVLM and CVLM [146,147]. On the other hand, nNOS within the RVLM and CVLM is sympathoinhibitory in pathological conditions like congestive cardiac failure, chronic renal failure, hypertension, and diabetes. Both Redox-sensitive inhibition of nNOS-mediated nNOS uncoupling is associated with an overproduction of ROS within the RVLM, and may result in the sympathetic overactivity-induced hypertension that is observed in Metabolic Syndrome [148]. Direct activation of the endocannabinoid G protein-coupled receptor causes hypotension in conscious rats and is mediated via the nNOS isoform [149]. An enhanced orexinergic input along with a potentiated nNOS signaling may modulate the activity of RVLM neurons and result in hypertension in spontaneously hypertensive rats [150].
To add to the plethora of the effects of nNOS, a study demonstrates the existence and increased activation of the nNOS protein within the neurons of the RVLM during static skeletal muscle contraction- or exercise-induced increases in blood pressure and heart rate [151]. All the three eNOS-, nNOS-, and iNOS-cGMP signal transduction mechanisms within the RVLM and CVLM are associated with the regulation of cardiovascular function via alterations in central sympathetic outflow, and that intracerebroventricular (i.c.v.) injection or local administration of specific nNOS antagonists increases the baseline mean arterial blood pressure or MAP [152–154]. In contrast, i.c.v. administration of an NO donor lowers both MAP and heart rate (HR) associated with an increase in the expression of nNOS mRNA within the RVLM [155]. Our laboratory has shown that microdialysis of the drug, 1-(2-trifluoromethylphenyl)-imidazole (TRIM; Tocris Bioscience, Ellisville, MO, USA), a selective nNOS antagonist, at a dose of 1 μM bilaterally into the RVLM for 60 minutes does not alter baseline cardiovascular variables, however, a dose of 100 μM indeed lowers both MAP and HR [13,21]. It should be mentioned that although 1 μM TRIM was microdialyzed into the RVLM, in vitro recovery studies show that the microdialysis membrane only allows about 20±2% of the TRIM to actually diffuse throughout the RVLM area. Thus, the actual amount of TRIM that has been delivered through the mircodialysis probes inserted into the RVLM in our study was in a concentration that is unable to alter baseline blood pressure but was able to modulate the exercise pressor reflex (see below). This is in contrast to other studies where specific nNOS blockers at higher concentrations delivered by either microinjection techniques or by means of i.c.v. administration were able to change the baseline MAP and HR [156]. In addition to alterations of the iNOS isoform and the eNOS protein within the RVLM and CVLM, our laboratory has shown significant changes in the expression of the nNOS enzyme in experiments with a one-sided temporary ischemia using rat models of stroke [21,157]. Expression of the nNOS protein within the ipsilateral RVLM following a transient left-sided focal ischemia or stroke is significantly reduced when compared with the contralateral RVLM, but not in the RVLM of intact rats. In contrast, the expression of the nNOS isoenzyme within the ipsilateral CVLM following an acute episode of stroke is markedly or significantly potentiated relative to that in the contralateral CVLM, suggesting that unilateral ischemic stroke differentially affects ipsilateral nNOS protein abundance within the RVLM and the CVLM [158].
In addition to its increased or decreased expression, there is evidence that the nNOS protein has significant interactions with glutamate, an excitatory amino acid, and gamma aminobutyric acid or GABA, the inhibitory amino acid, in the regulation of the CVS. Our laboratory has demonstrated that alteration in the nNOS activity within the RVLM and CVLM can play differential roles in static muscle contractions-induced increases in cardiovascular responses via an interaction with glutamatergic and GABAergic neurotransmission [159]. Within the RVLM, an nNOS-mediated surge of the release of GABA has been shown to act as a major sympathoinhibitory amino acid neurotransmitter along with decreased superoxide activity [160]. An enhanced expression of nNOS can also increase GABA but attenuate sympathoexcitatory angiotensin II signaling along with glutamate neurotransmission [146]. Swimming exercise has been shown to cause vasodilation mediated by an enhanced NO release, possibly via nNOS and associated with a reduction of glutamate levels within the RVLM in awake rats [161]. We have shown that NO within the RVLM and CVLM differentially modifies cardiovascular responses in addition to altered glutamate and GABA concentrations/release during skeletal muscle contraction or static exercise following stroke [118]. Excitatory glutamatergic and inhibitory GABAergic neurotransmission within the RVLM and CVLM are altered in stroke rats, and that such modulation of neurotransmitter release changes the expression of NOSs and affects cardiovascular functions [158,162]. Blockade of nNOS within the RVLM increases glutamate release and decreases GABA levels, and the result is enhanced pressor and tachycardic responses to exercise, whereas nNOS antagonism within the CVLM causes attenuation of blood pressure and HR increases during exercise mediated via a reduction in neuronal glutamate release and an increase in GABA levels within the CVLM [163]. Thus, we have summarized the interactions among nNOS, cardiovascular functions, and glutamate/GABA interactions within the medulla oblongata. Figure 5 depicts the role of nNOS and neurotransmission in the overall control of cardiac function.
Figure 5. The role of nNOS in autonomic control of cardiac function.
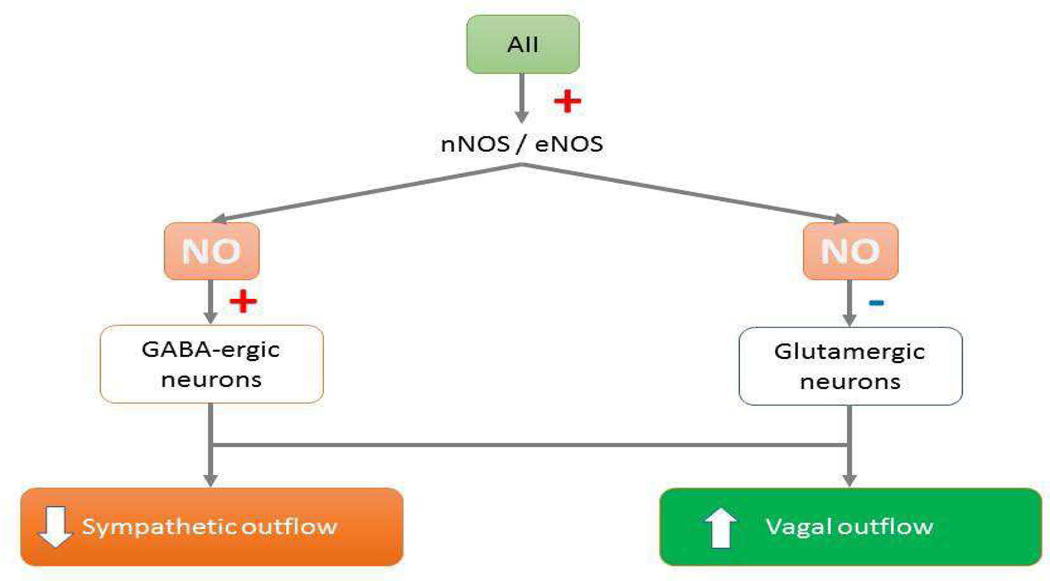
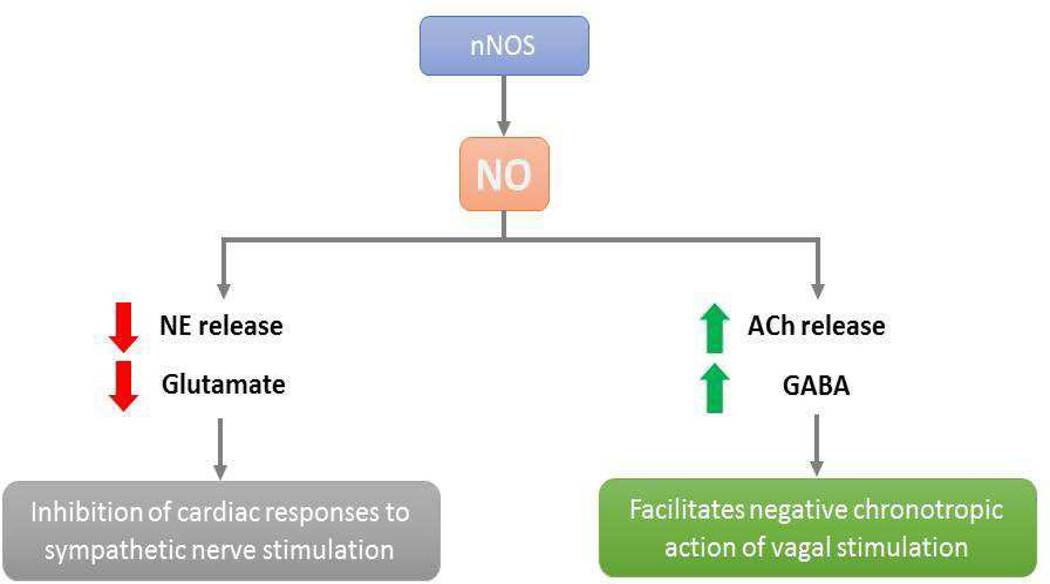
(Top) In the central nervous system, NO (nitric oxide) derived from nNOS (neuronal nitric oxide synthase) and eNOS (endothelial nitric oxide synthase) causes inhibition of sympathetic outflow and stimulation of vagal outflow via augmentation of GABA (γ-amino butyric acid)-mediated neuronal activity and inhibition of glumatergic neurons, respectively. (Bottom) In the periphery, NO derived from nNOS decreases the release of NE (norepinephrine) and possibly glutamate and stimulates the release of ACh (acetylcholine) and GABA that results in the inhibition of cardiac responses to sympathetic nerve stimulation and the facilitation of negative chronotropic action of vagal stimulation, respectively.
In addition to the VLM, studies are also performed to elucidate the role of nNOS within the NTS and the hypothalamus on cardiovascular activity. Inhibition of nNOS within the NTS attenuates the ionotropic activity of the heart in response to an administration of glutamate agonists [142]. Administration of nNOS antagonists within the NTS alters the NO-induced GABA and glutamate release and modulates cardiovascular activity [141]. Because of the fact that the NTS plays the most important role in the baroreflex mechanism, further discussion of the role of NO on baroreflex is described in the following paragraph. On the other hand, the hypothalamus has been implicated in the fine tuning of the overall CVS activities, including the human circadian rhythms and diurnal variations in blood pressure as controlled by the central clock located within the suprachiasmatic nucleus (SCN) of the hypothalamus. Understanding how the hypothalamus is involved in the regulation of the circadian rhythm of blood pressure is critical in the treatment of circadian blood pressure dysfunction and essential hypertension [125]. A recent review summarizes the role of nNOS as well as other neurotransmitters that describes how hypothalamic inflammation elicited by NO over-production can disrupt crucial signaling cascades in order to disturb the central control of blood pressure, and suggests future development of interventional strategies related to the hypothalamic regulation of cardiovascular function, including the inflammatory-sympathetic mechanisms involved in hypertension [126]. Particularly, the paraventricular nucleus (PVN) of the hypothalamus is a very important central fine-tuning site for integration of sympathetic outflow and cardiovascular function, including a role in the exaggerated sympathetic outflow commonly observed in heart failure [64,132]. Within the PVN, the production of NO generated by the nNOS protein plays a critical role in the inhibitory regulation of sympathetic outflow, and that studies in the rat model of experimental heart failure, the nNOS-NO mediated control mechanisms within the PVN is depressed. In summary, we can state that indeed both NO and nNOS within the hypothalamus as well as within the NTS play significant roles in the overall regulation of the CVS.
e). Cardiovascular Reflexes: Role of Neuronal Nitric Oxide Synthase
These following paragraphs will describe the role of nNOS on cardiovascular reflexes such as the baroreflex and the exercise pressor reflex: two important cardiovascular reflexes that occur in our daily lives. It has been recognized by now that nNOS plays a critical function in autonomic neurotransmission [83,157]. The baroreflex or the baroreceptor reflex is a very important homeostatic mechanism involved in the maintenance of blood pressure and HR at a normal physiological range. It is a negative feedback control mechanism where an elevation of blood pressure reflexively lowers HR and vice versa, or a decrease in blood pressure deactivates the baroreflex activation, causing HR to increase: the result is to restore blood pressure at the normal range. However, in situations like exercise, fear, anger, and sexual intercourse the baroreflex is in a reset or shutdown mode where one will observe profound increases in both blood pressure and HR. The baroreceptors, located on the arch of the aorta and at the bifurcation of the common carotid arteries, monitor the fluctuations in arterial blood pressure, and play an important role in dealing with situations like postural or orthostatic hypotension, the tendency for blood pressure to decrease on standing from a supine position [145]. Congruent with current concepts, the medulla oblongata, particularly the NTS and the VLM, controls baroreceptor reflexes and subsequent vascular responses are mediated by alterations in the central sympathetic vasomotor outflow [144,145]. Thus, sympathetic vascular control as well as baroreceptor activity can be specifically attributed to neurons present in the RVLM and the NTS. The glossopharyngeal nerve, the Hering nerve, and the vagus nerve are involved in the control of baroreflex activity. The NO-nNOS system also plays an important role in coordinating this baroreflex mechanism. For example, it has been shown that selective inhibition of nNOS reduces vagally-mediated bradycardia [115,116]. In addition, systemic administration of nNOS inhibitors affects baroreflex activity via modulating peripheral autonomic transmission, postsynaptic responsiveness of target tissues to autonomic stimuli, and autonomic circuits within the CNS [83]. Chronic interference of the nNOS protein dimerization that is responsible for the production of NO within the PVN of the hypothalamus potentiates the elevations in blood pressure via modulating the sympathoexcitation associated with renovascular hypertension [164]. The next paragraph will summarize the other roles of nNOS in the baroreceptor mechanisms.
There have been many studies on the role of NO and nNOS on baroreflex functions. Delivery of an adenovirus-nNOS gene into the RVLM has been shown to improve baroreflex function in rats with congestive heart failure [89,116,165,166]. Another study suggests that nNOS-derived NO facilitates sympathetic baroreflex transmission within the RVLM via a soluble guanylate cyclase-dependent and a superoxide-independent mechanism [167]. The ventral portion of the medial prefrontal cortex (vMPFC) is known to be involved in the modulation of the PANS arm of the baroreflex mechanism. A study suggests that glutamatergic neurotransmission within the vMPFC exerts a tonic facilitatory influence on the PANS component of the baroreflex and mediated via an nNOS-NMDAR interaction within the vMPFC [168]. Studies using nNOS knock-out mice show that the absence of nNOS activity results in an elevated baseline blood pressure and affects the transmission of baroreflex signals [169]. An nNOS-cGMP pathway at the CVLM area also augments the baroreceptor induced reflex bradycardia while that NO, specifically the nNOS isoform, mediates sympathetic cardiac-cardiovascular responses including the baroreflex through its action within the RVLM [170,171]. In addition, protein kinase A-dependent nNOS activation is required for the enhancement of baroreflex activity in response to the action of adrenomedullin, a peptide hormone with several functions including vasodilation, regulation of hormone secretion, promotion of angiogenesis, and antimicrobial activity (https://www.ncbi.nlm.nih.gov/gene/133; Gene ID: 133, updated on 27 January 2018), within the NTS of rats [172]. Moreover, an attenuated expression of nNOS protein within the NTS inhibits sympathetically mediated baroreflex responses in the rat [173]. Even an upregulation of the nNOS protein is seen after activation of central angiotensin II type 2 receptors that improves arterial baroreflex sensitivity in rats with heart failure, and that nNOS attenuates the cardiac sympathetic drive and improves baroreflex control of HR in ovine heart failure [57,174]. Thus, we should recognize the profound influence of nNOS and NO on the baroreflex mechanism in both physiological and pathophysiological states.
The “exercise pressor reflex” was first coined by Dr. Jere H. Mitchell from the Moss Heart Center of the University of Texas Southwestern Medical Center in Dallas, TX, USA. Dr. Mitchell has been a mentor of the first author of this review paper. The exercise pressor reflex can be described as follows: Static exercise or isometric skeletal muscle contraction as observed during push-ups, rowing a boat, weight-lifting and exercising with the dumbbells, sends impulses via the group III and IV muscle afferents within the spinal cord to activate neurons in the medulla oblongata, particularly within the RVLM and the CVLM where they are integrated, and the final results are increases in MAP, HR, myocardial contractility, cardiac output, and sympathetic nerve activity [175–179]. This paragraph will discuss how nNOS within the RVLM and the CVLM modulates cardiovascular responses along with alterations in glutamate and GABA neurotransmission during the exercise pressor reflex. The presence of nNOS within the RVLM plays a key regulatory role in the maintenance of cardiovascular functions in response to changes in mean arterial pressure, pathophysiological conditions and skeletal muscle excitation or relaxation. Our laboratory has several publications on the modulation of cardiovascular functions, alterations of the nNOS, eNOS and iNOS protein expressions, and changes in adrenergic, serotonergic, dopaminergic, glutamatergic and GABAergic neurotransmission in response to static exercise following pharmacological interventions using NO precursor, nNOS, iNOS and eNOS antagonists, Angiotensin II type-2 (AngII type 2) receptor antagonism or blockade of Ang II type-1 receptors, and opioid receptor agonists/antagonists, as well as NMDA and AMPA receptor blockers, among others that date back to 1994 [180–187].
First, expression of nNOS protein within the neurons of the RVLM increases during the exercise pressor reflex [151]. Our data show that nNOS antagonism within the RVLM for greater than two hours results in exaggerated cardiovascular responses (pressor and tachycardic) associated with a decrease in nNOS protein expression within the RVLM [13,159]. Conversely, a two-hour antagonism with a specific nNOS inhibitor administered into the CVLM decreases nNOS expression and attenuates the pressor and tachycardic responses during skeletal muscle contraction. In addition, inhibition of nNOS within the RVLM and CVLM appears to be differentially controlling cardiovascular responses during the exercise pressor reflex via alterations in glutamate and GABA concentrations [13,159]. Pharmacological antagonism of the nNOS protein within the RVLM increases glutamate release and decreases the concentrations of GABA; the result being an augmented or enhanced exercise pressor reflex, while blockade of the nNOS isoform by administration of its antagonist into the CVLM results in attenuating the exercise pressor reflex via reducing neuronal glutamate release and increasing GABA levels [12]. Overall, it appears that the cardiovascular regulatory and reflex mechanisms orchestrated by the VLM in physiological homeostasis are under the influence of an excitatory and an inhibitory coordination among the nNOS protein and its synthesized NO molecules and glutamatergic and GABAergic neurotransmission [12,157]. The role of nNOS on cardiovascular functions in pathophysiological states such as stroke or hypertension will be described in the following sections.
4. Hypertension and Neuronal Nitric Oxide Synthase
Hypertension is one of the leading causes of morbidity and mortality in humans. There are approximately 8 million deaths and 92 million disabilities worldwide every year that are linked to high blood pressure. Hypertension increases the risk of serious cardiovascular diseases, such as coronary heart disease (CHD), congestive heart failure (CHF), ischemic and hemorrhagic stroke, kidney failure, and peripheral arterial/vascular disease. It is also known as the “silent killer” and the chances of debilitating cardiovascular disease associated with hypertension increases if the patient has certain risk-factors such as obesity, diabetes mellitus, and high cholesterol. With proper antihypertensive pharmacological and non-drug therapy, the risks of further cardiovascular and renal complications can be lessened, even though a large segment of hypertensive patients are either untreated or improperly managed. While several classes of antihypertensive drugs are available for the management of hypertension, including resistant hypertension, the exact plethora of factors, etiology, pathogenesis, and pathophysiological mechanisms of almost 90% of the hypertensive population (primary or idiopathic hypertension) is still unclear. However, many mechanisms such as the renin-angiotensin-aldosterone system, SANS, PANS, intravascular volume, and vascular problems associated with hypertension are well-established.
While there have been more than 60,000 scientific publications on NO in the last 20+ years, one interesting definitive conclusion is that people with clinically diagnosed atherosclerosis, diabetes, or hypertension often show impaired NO signaling pathways. Numerous studies also implicate that a high salt dietary intake decreases the production of NO in patients suffering from essential hypertension that affects approximately 90% of the patients. The following paragraphs will discuss past and recent findings about the relationship between hypertension and nNOS-induced NO production. Whether or not the nNOS protein-induced production of NO will be able to prevent or treat any disease in humans is still unknown. However, investigations on nNOS using humans are still ongoing. For example, the importance of nNOS-hydrogen peroxide (H2O2) signaling pathways in the human internal mammary artery and saphenous vein, the commonly used vessels used in coronary artery bypass grafting is still under active research, where the H2O2 produced in the coronary circulation has been shown to increase the susceptibility to accelerated atherosclerosis with subsequent dysfunction of the saphenous vein [188]. Another recent study implies that the nNOS-derived NO plays a very critical role in the physiological regulation of basal systemic vascular resistance and blood pressure in healthy humans [189,190]. Neuronal NOS attenuates the cardiac oxidase enzymes that are shown to ameliorate the sources of oxidative stress-related pathology in diseased hearts [191,192]. Finally, cardiac eNOS protein expression is decreased but the abundance of the nNOS protein along with its activity is significantly increased in hypertension [193]. In the same line, the nNOS isoenzyme abundance is the result of an interplay between angiotensin II (Ang II) type 1 receptor (AT1R) and Ang II type 2 receptor mediated via NADPH oxidase and ROS-dependent nNOS activity in cardiac myocytes. Therefore, the up-stream and/or downstream expression of nNOS in the heart in a case of hypertensive patient may provide further understanding of the underlying mechanisms in hypertensive cardiomyopathy [192].
Vasodilators, angiotensin converting enzymes (ACE) inhibitors, or angiotensin II receptor blockers (ARBs) are commonly used as therapies for the treatment of hypertension and the effect of the drugs is producing vasodilation. In section 3, we have extensively discussed the role of nNOS on blood vessels and therefore we shall not repeat those findings. In addition, another therapy of hypertension is using drugs like clonidine that act on the CNS. The effects of NO and nNOS on the CNS and sympathoinhibition are also described in section 3. Thus, this paragraph will evaluate other past and current findings of the role of nNOS in hypertension. The fact that nNOS produces H2O2 and superoxide which are key mediators in non-neuronal cells signaling, suggests the importance of further exploring the role of nNOS in vascular homeostasis and cardiovascular diseases such as hypertension and atherosclerosis [194]. In addition, nNOS contributes to the control of blood pressure in compensation for a decrease in eNOS expression [195]. Indeed, nNOS-derived H2O2 has been implicated in endothelial dysfunction in hypertension [196]. Finally, the importance of exercise in our daily lives is fully recognized not only for a healthy life but also for the prevention and therapy of diseases like hypertension, stroke or diabetes. We have discussed the role of exercise and oxidative stress in section 4. The levels of NO, nNOS, and oxidative stress within the RVLM system are important determinants of hypertension, and exercise training can restore the high blood pressure to normal blood pressure status by reversing the levels of nNOS expression [197]. In humans, nNOS dysfunction may lead to an impairment of vascular homeostasis in essential hypertension and contributes to stress-induced cardiovascular events [101,198]. Overall, we recognize that nNOS is an important cardiac protector in the diseased heart and plays a major role in arteriosclerosis, the most important cause of hypertension, coronary artery disease and myocardial infarction [192]. In summary, we have discussed the intricate relationship between hypertension and nNOS, particularly about the new findings of nNOS and H2O2 interactions for the development of arteriosclerosis, the most important cause of human cardiovascular diseases. Nevertheless, the achievement of maintaining a normal blood pressure through diet and exercise is possible by every human being, and most importantly, a stable normal blood pressure within the physiological range significantly reduces the risks of death as a result of heart disease and stroke, the development of other debilitating conditions such as kidney failure, and the cost associated with advanced medical care [199,200].
5. Cerebrovascular Accident or Stroke and Neuronal Nitric Oxide Synthase
In the USA, on average, 1 person suffers stroke in every 45 seconds and it continues to escalate due to an increasing population of elderly Americans, obesity, an increase in diabetic population, sedentary living, and lack of physical activity among the general public. Stroke has been identified as the 3rd killer disease in the USA. In 1978, the World Health Organization (WHO) defined stroke as a “neurological deficit of cerebrovascular cause that persists beyond 24 hours or is interrupted by death within 24 hours.” On the other hand, stroke is referred to as a medical situation where there is a reduction or even cessation of blood flow to the brain or a part of the brain that results in ischemia leading to neuronal cell death, necrosis or infarction [201]. Stroke is also termed “cerebrovascular accident” because of its classification into two major categories: “ischemic” and “hemorrhagic”. Ischemic strokes are caused by disruption of blood supply to the brain due to thrombosis or embolism, while hemorrhagic strokes occur due to rupture of a blood vessel, rupture of an aneurysm, or presence of an abnormal blood vessel. Although the major risk factor for an occurrence of stroke is high blood pressure or hypertension, other risk factors such as sedentary lifestyle, cigarette smoking, obesity, high blood cholesterol/triglycerides, diabetes mellitus, previous history of transient ischemic attacks, and atrial/ventricular fibrillation are also implicated as etiologies of strokes [201,202]. Because of the fact that cerebrovascular accidents cause a tremendous impact on the morbidity and mortality of human beings, a lot of money, time, and research have been invested not only for further understanding the pathogenesis and pathophysiology of stroke but also for developing new strategies aimed at prevention of and treatment and rehabilitation of patients with cerebrovascular accidents.
Cerebrovascular accidents or brain ischemia initiate a cascade of events: loss of ATP and subsequent cellular dysfunction; Na+ and Ca2+ influx causing massive Ca2+-induced release of neurotransmitters such as glutamate which results in excitotoxicity and neuronal death mediated via α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) and NMDA receptors; overproduction of uric acid and lactic acid; cell swelling; and finally apoptosis [189,190,203]. Ischemia also activates inflammatory cytokines, causes overexpression of inflammation-related genes, attracts leukocytes into the brain tissue, and promotes cellular adhesion [11,14]. Most importantly, neuronal death following ischemia is attributed to the progressive generation of free radicals and other ROS. Free radicals also directly initiate elements of the programmed cell death cascade by means of redox signaling that damages DNA, proteins and lipids, and compromises the integrity of the blood brain barrier [204–206]. One such free radical is NO, which is generated from neurons, nerve terminals, and/or the endothelium of blood vessels supplying the brain [207–214]. The production of NO following stroke is dependent on the activation of all 3 isoforms of the NOSs enzyme: nNOS, iNOS and eNOS [11,48]. The amount of NO generated by each of these isoforms depends on the elapsed time after a stroke and the expression of each NOSs protein; these factors also determine whether NO is responsible for protecting or destroying neurons within the umbra or penumbra regions [207–214]. The following paragraphs will primarily focus on past and recent discoveries about nNOS and its role in the pathogenesis and management of stroke.
In recent years, the management and therapy for ischemic cerebrovascular accidents in humans have progressed rapidly. The latest protocol of clinical management of an acute episode of stroke in humans has shown the continued development of conceptual ideas and techniques such as mechanical thrombectomy, stenting, and pharmacological interventions with promising results. A recent review with in-depth perspective on the advancement in therapy for ischemic cerebrovascular diseases in humans has shown that indeed, both timely-performed pharmacological and surgical interventions can decrease morbidity and mortality following an ischemic stroke attack [215]. While the role and actions of NO and nNOS on stroke in humans are currently being elucidated, its involvement has been already established in the pathogenesis of ischemic brain damage in experimental animals. The literature suggests that NO has both neuroprotective and neurotoxic effects during the different stages of cerebral ischemia. Which effect occurs first or second depends on the time elapsed since the onset of ischemic injury; the expression of specific NOSs isoforms; cellular sources of NO from the umbra, penumbra, or other regions of the brain; spillover of NO and its metabolites (nitrites and nitrates) into the plasma and cerebrospinal fluid; and the amount of NO ultimately generated by each specific NOSs protein [171,216,217]. For example, one beneficial effect of NO is the inhibition of glutamate-induced Ca2+ influx into neurons, suggesting that NO directly decreases the excitatory glutamatergic neurotransmission [218–220]. Furthermore, at the early stages of cerebral ischemia, a surge in NO release generated by eNOS seems to protect the neurons from death by inducing vasodilatation and inhibiting microvascular aggregation, but in the later stages of stroke, the overproduction of NO by nNOS or iNOS contributes to apoptosis and subsequent neuronal death [170,212,216]. However, nNOS can have beneficial effects on brain infarct size after stroke [7,48,218]. Exactly how much NO is produced by each NOS isoform after cerebral ischemia is unknown, but under inflammatory conditions in the brain, the iNOS isoenzyme can generate thousand times more NO than the nNOS protein [11].
A search of the literature (Last performed on December 20, 2019) reveals very few studies that investigate the relationship of nNOS with ischemic stroke in humans in terms of both pathogenesis and clinical management. Our search includes the International Standard Randomized Controlled Trial Number (ISRCTN), Stroke Trials Registry, Cochrane Stroke Group Trials Register, Embase, ISI Science Citation Indexes, Clinical Trials registry, International Clinical Trials Registry Platform (ICTRP) and MEDLINE. One of the selection criteria has been randomized controlled trials comparing nNOS versus placebo or open control in people within one week of onset following a confirmed stroke. Upon examining all search, there is currently insufficient evidence to recommend the use of the nNOS protein agonists or antagonist in an acute episode of stroke in humans, however, a NO donor, glyceryl trinitrate (GTN) has been assessed in patients with acute stroke, where the drug reduces blood pressure and increases HR, but does not alter the clinical outcome of stroke [221]. Our conclusion has been corroborated by a recent review [221]. We do not have any knowledge if there are other ongoing case-controlled studies or clinical trials. Nevertheless, there are some noteworthy discoveries on the role of nNOS in human disease processes. For example, a case control study performed on the Han Chinese population of North China has shown that there is indeed several gene polymorphism of the nNOS protein that is related to ischemic stroke [222]. Another study assessed whether variants in the nNOS gene were associated with atherosclerosis and stroke susceptibility in the human population, in which it has been shown that the T allele of rs2293050 and A allele of rs2139733 in the nNOS gene may contribute to the increased susceptibility of large-artery atherosclerosis-induced ischemic stroke [223]. However, in humans, the relationship between hypertension and stroke has been well-established. It appears that nNOS plays a significant role in the pathogenesis of hypertension (see “Hypertension and nNOS” section). For example, in humans, nNOS-derived NO has been shown to play a crucial role in the physiological regulation of basal systemic vascular resistance and arterial pressure [189]. Recanalization of atherosclerotic cerebral blood vessel(s) and neuroprotection are the main targeted foci for the specific treatment of acute ischemic stroke in humans. A novel drug, edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), is a potent free radical scavenger that has been used clinically for a long time in order to lessen the neuronal damage following ischemic stroke [224]. Edaravone has a profound effect on NOSs, mediated via increasing the eNOS isoenzyme (neuroprotective NOS after ischemic stroke) and decreasing the nNOS and iNOS proteins or the “so-called neurotoxic NOS” [224]. Furthermore, it has been shown that a thrombolytic drug along with edaravone have the greatest potential to reduce brain edema, decrease the number of stroke-related deaths, and enhance the recovery from neurological deficits in human stroke patients. Nevertheless, more studies are necessary in order to fully establish and justify the use of nNOS agonists or antagonists for the prevention and/or management of stroke using human subjects.
The effects of NO, and particularly that of nNOS, have been studied most extensively by the administration of NO donors, NO precursors, specific NOSs isoform blockers, and glutamate receptor antagonists in both human stroke patients and animal models of cerebral ischemia [22,225]. One study from our laboratory using the standard rat model of stroke shows that a transient left-sided middle cerebral artery occlusion (MCAO)-induced focal ischemia followed by 24 hours of reperfusion significantly attenuates nNOS expression within the left (ipsilateral) RVLM relative to the contralateral RVLM (Figure 6) [158]. Simultaneously, nNOS protein expression within the ipsilateral CVLM following a left-sided MCAO is significantly augmented compared to the contralateral CVLM (Figure 6; Ally et al., 2005). These results suggest that one-sided temporary ischemic stroke affects ipsilateral nNOS protein expression within the VLM, which in turn, may result in the attenuation of cardiovascular responses during static exercise [158,162]. Similar results were obtained with the expression of eNOS protein within the medulla, whereas opposite changes in the expression of the iNOS isoform within the RVLM and CVLM after stroke are observed in our experiments with rats [12,157]. Moreover, the literature has extensive studies regarding the role of nNOS protein during ischemic stroke in animal models and a review of the recent advances in this field is summarized below.
Figure 6:
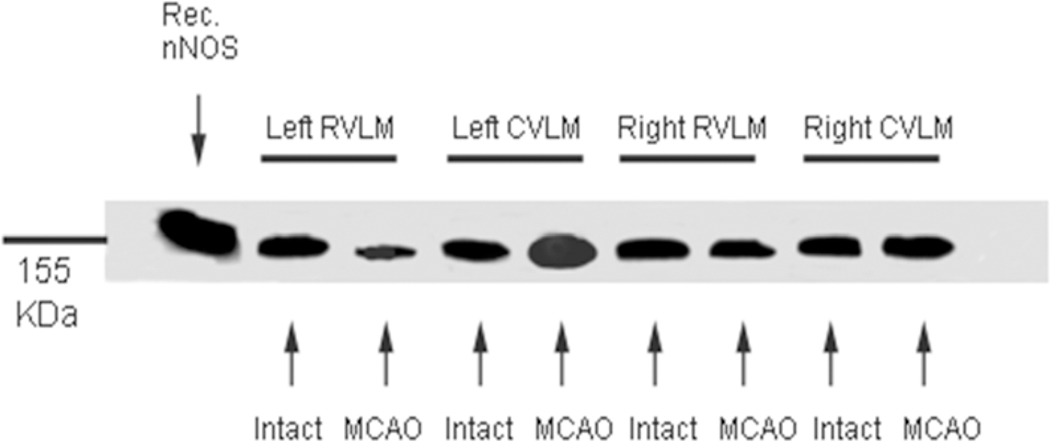
A picture of a gel with Western blots of the 155 KDa recombinant nNOS band and nNOS proteins as derived from each quadrant of the right and left rostral (RVLM) and caudal ventrolateral medulla (CVLM) homogenates of intact or Sham and middle cerebral artery occluded (MCAO) rats. Equal amounts of proteins (20 μg) were loaded in each lane in the same gel. Recombinant nNOS DNA (5μg: Sigma Chemicals, USA) was loaded as control.
In a recent study using mice, it has been shown that the dissociation of nNOS from the postsynaptic density protein 95 (PSD95) is associated with functional recovery after cerebral ischemia that is mediated via decreasing the excessive tonic release of GABA from reactive astrocytes [226]. Note that stroke induces massive releases of both GABA and glutamate from neurons (see above). In the same line, a small aminosulphonate compound called tramiprosate, that is present in various species of red marine algae, provides a dose-dependent neuroprotection both in vitro and in vivo models of ischemic stroke, and that its neuroprotective effect may partially be mediated via disruption of the PSD95 and nNOS interaction, in addition to translocation of the nNOS isoenzyme [148]. Furthermore, atorvastatin, a lipid lowering statin medication that is also involved in phosphorylation of nNOS, has been shown to provide the neuroprotection following ischemic brain injury that is mediated through suppression of the JNK3 signaling pathway, which may emerge as a new concept for the management of stroke in the future [227]. In addition, a study using the rat model of MCAO-induced stroke demonstrates a destructive nNOS/peroxynitrite/AMPK cascade following stroke, and that treatment with S-nitrosoglutathione can attenuate the injurious cycle that in turn, may provide neuroprotection and ameliorate neurological functional activity [228]. Indeed, the nNOS protein has been shown to play a crucial role in the disruption of blood brain barrier following a transient focal cerebral ischemia by MCAO [69]. Overall, the above studies show potential rationales of using nNOS-based therapy in the management of stroke or cerebral ischemia-related pathologies in the future.
The importance of exercise and NO-based therapeutic strategies for preventing and treating stroke cannot be underestimated. According to the American Heart Association, aggressive physical rehabilitation of stroke patients with exercise facilitates recovery of motor functions and also significantly increases peak left ventricular ejection fraction and high-density lipoprotein cholesterol, and decreases resting HR and total serum cholesterol [229,230]. The beneficial effects of exercise intervention, including isometric or static exercise, include improvements in aerobic capacity, functional ability, lipid profile, physical activity, neuroprotection, and a speedy recovery after stroke. It is noteworthy to state that exercise is also recommended to prevent the incidence of stroke [229,230]. In the event of a stroke, both exercise and/or pharmacological/surgical interventions are integral regimens of a treatment protocol. Adjuvant drugs are used to reverse or block the molecular, cellular and biochemical events in the brain occurring as a result of the stroke in order to treat stroke-related signs and symptoms and to reduce brain infarct size. Overall, it has been demonstrated that regular physical exercise, endurance exercise, and even regular swimming improve total antioxidant capacity, prevent neurodegeneration due to redox, and attenuate oxidative damage with increases in antioxidant defenses [231–233].
Exercise is also beneficial and likely exerts its effect, at least in part, through the NO and NOSs system [234–236]. For example, it has been shown that regular exercise training recovers high fat diet-induced nNOS expression within the hippocampus and cerebral cortex of mice [237]. Furthermore, enhanced NO-signaling pathways mediated via the induction of the nNOS protein within the PVN and the dorsomedial hypothalamus (DMH) play a critical role in improving exercise capacity after central administration of an NO precursor, L-Arginine [238]. Regular swimming training by rats can modulate NO-glutamate interaction within the RVLM [235]. Aerobic exercise improves the functioning of the brain mediated via modulation of the nNOS proteins within the brain [239]. In addition, regular exercise restores oxidative stress and NOSs, including nNOS, within the RVLM of hypertensive rats and that exercise induces production of NO molecules [197,236]. Finally, it has been demonstrated that vigorous exercise training protocol indeed improves the reactivity of cerebral arterioles that follows after an acute episode of transient focal ischemia that in turn, reduces brain injury or neuronal damage [240].
A decrease in blood pressure following i.c.v. administration of an NO donor is associated with the overexpression of nNOS mRNA within the RVLM. Following stroke, the surge in glutamate release appears to be toxic, the massive release of GABA being protective, and the amount of NO molecules generated can have either beneficial or deleterious effects on neurons and/or the brain as a whole. Administration of NO donors can be useful within the first few hours after an attack of ischemic stroke in order to induce vasodilation and an increase in blood flow to the affected areas [229,241]. Nitric oxide donors are currently used to treat platelet aggregation and embolus formation in blood vessels, both predisposing factors for stroke, and that human-derived nNOS inhibitors are in the early clinical stages for treating headache and migraine [242]. Thus, studies are focusing on nNOS using both vertebrate animals and humans [7,9,217].
Nonetheless, clinical experience with NO for the treatment of stroke in humans is limited. Many nNOS inhibitors also possess antioxidant properties and can be neuroprotective. Activation of the iNOS protein generates a massive amount of NO that diffuses to other brain regions via the cerebrospinal fluid and therefore, selective iNOS inhibitors could be valuable therapeutic tools for treating human stroke. Therapeutic strategies using NO donors, NO precursors, or selective NOSs inhibitors are rapidly emerging based on their effects on transcription factors at the mRNA level and subsequent expression of the respective protein using a variety of in vitro and in vivo experiments [7,9,217]. It is interesting to speculate whether administration of pharmacological drugs before or after stroke can maintain or recover the normal expression of any or all of the NOSs isoforms and neurochemistry within the VLM, and thus improve cardiovascular functions at rest and also during exercise. In conclusion, this section summarizes the crucial role that the nNOS isoform plays in the pathophysiology and management or treatment strategies of cerebrovascular accidents, and provides further insight into the potential use of the nNOS protein modulators in clinical practice in the future.
6. Nociception and Neuronal Nitric Oxide Synthase
The term “nociception” was first introduced by Dr. Charles Scott Sherrington while describing physiological processes particularly of the sensory nervous activity due to a subjective experience of pain [243–245]. The stimulus or stimuli that may cause pain can be either due to an intense chemical contact such as eating too much chili powder, or as a result of mechanical manipulation like a cut on the skin or a crushing accident on the leg, or can be due to a thermal effect such as too much heat (burn) or exposure to extreme cold [246]. Any of the above-mentioned stimuli activates sensory nerve cells called “nociceptors” and the responding signal travels along a series of nerve fibers to the spinal cord and finally reaches different areas of the brain. The medulla oblongata, the hypothalamus, the PAG, and/or the thalamus within the brain integrate the nociceptive signal(s) in order to evoke a variety of physiological, behavioral and/or defensive responses: responses arising from the subjective experience of pain. The factors and the type of stimulus such as proprioception, thermoception, chemoception and nociception are all integrally connected. The variety of bodily responses caused by pain, for example, how nociception results in autonomic responses before or without reaching unconsciousness e.g., pallor, diaphoresis, tachycardia, hypertension, dizziness, nausea and fainting is well established. One of the responses that are a result of pain is alteration of the cardiovascular function such as blood pressure or HR. The following paragraphs will review the literature and scientific publications that relate to the interaction between the CVS and nociception and how that involvement is associated with NO, particularly with the nNOS isoenzyme.
The various cardiovascular responses due to pain are alterations in blood pressure, HR, sympathetic nerve activity, myocardial contractility, stroke volume, ejection fraction, and changes in EKG properties and they depend on the type of pain, intensity of pain, involvement of specific nociceptor or pathways, and the type(s) of neurotransmitter/receptor(s) involved such as serotonin, opioid, or NMDA/AMPA glutamate receptors. For example, pain generally influences the CVS and that the afferent signals from peripheral pain receptors terminate within different regions in the CNS including the cardiovascular regulatory centers, where the impulses are processed and integrated in order to evoke the above-mentioned reflexes and responses [247–251]. Our laboratory has previously shown that indeed mechanically-induced and heat-mediated thermal nociception increase MAP and HR in an intensity-dependent manner, however, a cold-evoked nociceptive stimulus causes depressor and bradycardic responses [252–255]. Several reviews on pain and cardiovascular activities including recent publications about nociception corroborate our findings. For example, in proportion to the intensity and duration of a pain stimulus, multiple ascending spinal reflexes/impulses activate the SANS in order to increase peripheral vasoconstriction, MAP, HR, and stroke volume in addition to activating the neuroendocrine system, particularly the hypothalamic-pituitary-adrenal system [256,257]. By monitoring EKG changes, blood pressure, respiratory rate and postganglionic sympathetic discharge activity (muscle sympathetic nerve activity or MSNA) in human subjects, scientists reveal a hypothetical possibility that in Fibromyalgia patients, the use of adrenergic antagonists may reduce chronic pain intensity by attenuating the underlying overactive and excessive sympathetic activity [258]. Moreover, a study shows that the African-American populations experience a greater intensity of muscle pain than non-hispanic white population during static or isometric handgrip exercise, however, both group exhibit similar increases in MAP and HR [256]. Finally, high blood pressure is one of the most common co-existing manifestations observed in patients suffering from chronic pain, including the fact that there is a rise in resting MAP due to acute postoperative pain in dental patients after an endodontic surgery [259,260]. In summary, it is well-established that the cardiovascular and nociceptive systems are interrelated and that their interactions lead to several manifestations in bodily homeostasis and defensive/withdrawal reactions.
The multiplicity of mechanisms, vast arrays of neurotransmitter involvement, regions of brain participation, specific receptor interactions, management and treatment, and above all, the relationship between pain and the CVS have been widely studied and are still regarded as active ongoing investigation. A recent chapter in a book describes the medications along with their toxicological side-effects that are used in the treatment of pain in patients with peripheral neuropathy, including their side-effects on cardiovascular function. Our laboratory also investigated the relationship between cardiovascular functions and different receptor-neurotransmitter interactions during several modes of nociceptive stimulus. We have shown that AMPA-receptor blockade within the RVLM attenuates the increases in MAP and HR elicited during mechanically-induced nociception and associated with a reduction in extracellular levels of glutamate, while antagonism of AMPA receptors in the RVLM do not play a role in mediating cardiovascular responses during thermally-evoked nociception [255]. In addition, we show that NMDA receptors within the RVLM play a critical role in altering the enhanced cardiovascular responses during mechanically-induced but not during thermally-evoked nociception by modulating 5-Hydroxytryptamine (5HT), epinephrine (EPI), and dopamine (DA) concentrations within the RVLM [253,254]. Thus, it can be emphasized that indeed nociception involves a wide variety of mechanistic and conceptual information that are yet to be fully delineated in order to understand the overall science of “pain”.
Nitric oxide, the molecule of the year 1992 and the Nobel Prize in 1998, and all three NOSs isoenzymes have been implicated in nociception and cardiovascular responses as a result of pain. For example, BH4 is a crucial cofactor for the production of catecholamine, serotonin and NO, thereby acting as an intrinsic regulator of pain-induced sensitivity. Numerous studies have shown that GTP cyclohydrolase (GCH1), which is a rate-limiting enzyme for the synthesis and production of BH4, is an important modulator of peripheral neuropathy- and inflammation-induced pain [257,261]. However, high concentrations of NO appear to be cytotoxic in situations such as peripheral neuropathy, neurotoxicity and pathological pain [262]. Thus, NO is regarded as an important neurotransmitter involved in the nociceptive processes/pathways. Various reviews and studies in both humans and experimental animals indicate that NO and all three NOSs modulate spinal and sensory neuronal excitability via multi-staged mechanisms that underlie NO’s specific and distinctive roles in pain. Both eNOS and iNOS proteins are involved in neuropathic pain [257] and the following paragraphs will summarize the recent findings about the interactions between nociception and cardiovascular functions in relation to nNOS.
As mentioned earlier, nNOS protein is constitutively expressed in specific neurons of the brain, spinal cord, sympathetic ganglia, adrenal cortex/medulla, peripheral nitroxidergic nerves, epithelial cells and vascular smooth muscles; the nNOS activity on vascular smooth muscle cells being regulated by Ca2+ and CaM mechanism. The overall functions of nNOS are both long-term and short-term regulation of synaptic transmission within the CNS, control and regulation of arterial blood pressure, and smooth muscle relaxation leading to vasodilation [257]. Particularly, nNOS has been shown to play a very important dynamic role in neuronal death due to an episode of cerebrovascular accident such as stroke [48]. However, the role of nNOS on nociception has been the focus of numerous investigations involving both humans and animals. A recent study demonstrates that stimulation of spinal sigma-1 receptors (Sig-1Rs) elicits pain hypersensitivity and is mediated via activating the nNOS enzyme, that leads to an increase in NADP oxidase 2 (NOX2) activity which in turn, may result in a ROS-induced increase in protein kinase C (PKC)-dependent phosphorylation of NMDAR-GluN1 subunit (pGluN1) at the Ser896 site of the spinal cord [263]. In addition, an increase in phosphorylated nNOS isoenzyme has been shown to be associated with neuropathic pain in rats [264]. Overproduction of NO by the nNOS isoform is one of the fundamental cause that underlie many neurodegenerative disorders and neuropathic pain and mediates the upregulation of the expression of the nNOS isoenzyme [58]. Neuronal NOS may also induce neuronal death via peroxynitrite formation, thereby enhancing the phosphorylation of NMDAR and resulting in central sensitization, which in turn, via the production of NO maintains persistent or chronic pain [57]. In addition, the role of nNOS in diabetic neuropathic pain has been studied. In the early stages of diabetes mellitus Type 1 disease, the expression of nNOS within the nitroxidergic neurons is reduced as a result of less production of NO and associated with a defect in the axonal transport of nNOS protein, however, during the late phases of the disease, neurons appear to drastically reduce their nNOS enzyme resulting in irreversible apoptotic degenerative changes [265]. It has been proposed that small molecule inhibitors of the scaffolding protein postsynaptic density 95 kDA (PSD95)-nNOS protein-protein interactions as novel analgesics that act by attenuating pathological pain without producing any unwanted side effects such as motor ataxia [266]. In fact, the discovery of a potent, orally bioavailable and highly selective human nNOS inhibitor, N-(1-(piperidin-4-yl)indolin-5-yl)thiophene-2-carboximidamide has led to the development of pre-clinical trials for the treatment of migraine headache [267]. Other novel compounds that selectively block nNOS-induced NO production has been discovered that are aimed at alleviating neuropathy-associated pain without any cardiovascular side-effects [267,268].
Our laboratory has studied the role of NO and all three NOSs enzymes in different regions of the brain that modulate cardiovascular responses and neurotransmission (glutamatergic and GABAergic) during mechanically-induced, heat-induced, and cold-generated nociceptive stimuli. Our first research on nociception began on experiments with modulation of cardiovascular functions via glutamatergic neurotransmission following AMPA-receptor blockade within the RVLM during different modalities of peripheral pain in the year 2001 using rats. Our experimental results show that the AMPA-receptor antagonism within the RVLM attenuates the increases in MAP and HR during mechanical stimulation that is associated with a reduction in extracellular levels of glutamate within the RVLM, however, AMPA receptors blockade within the RVLM do not modulate cardiovascular responses during thermal nociception. Thereafter, we have investigated what role NO plays within the VLM on modulating cardiovascular responses and neurotransmission during mechanical and thermal nociception. Nitric oxide affects cardiovascular responses by altering glutamate concentrations within the RVLM during thermal but not mechanical nociception, suggesting that differential integrative mechanisms regulate the processing of sensory ascending and descending impulses arising from different peripheral nociception [159]. We have also shown that NMDA receptors within the RVLM modulates increases in MAP and HR by changes in the concentrations of 5HT and DA within the RVLM during mechanically-induced but not during thermal nociception [253,254]. In addition, NO within the dorsal dPAG attenuates cardiovascular responses and changes in the concentrations of glutamate during both mechanical and thermal nociceptive stimulation [163]. Bilateral microdialysis of a selective nNOS antagonist, 1-(2-trifluoromethyl phenyl)-imidazole or TRIM into the dPAG has no effect on MAP, HR, glutamate and GABA concentrations as a result of mechanical nociception, however, administration of TRIM into the dPAG significantly enhances the cardiovascular responses and glutamate increases but attenuates concentrations of GABA during a heat-induced thermal nociception, suggesting that nNOS within the dPAG plays a differential role in modulating cardiovascular responses and glutamatergic/GABAergic neurotransmission during different modes of nociception [253]. We have also demonstrated the effects of iNOS and eNOS blockade within the dPAG on cardiovascular responses during mechanically-induced, heat-elicited, and cold-evoked peripheral nociception [119,252].
In summary, uncoupling and over-expression or down-regulation of NOSs isoforms play a crucial role in neuropathic pain mediated via numerous processes or mechanisms, i.e., mediating neuronal excitotoxicity by activating NMDA receptors and numerous transcription pathways (MAPK, JNK, and ERK pathways) that may lead to the death of neurons. All three NOSs appear to generate high concentration of NO, leading to neuronal toxicity by the production of peroxynitrite, a neurotoxic compound. Literature shows that both nNOS and iNOS contribute considerably to the generation of neuropathic pain, however, not much has been investigated about the role of eNOS in neuropathic pain. Nevertheless, pharmacological interventions with NOS isoform agonist and antagonists and their downstream mediators/regulators/signal transduction processes may restart the development of potential therapeutic agents for the treatment of neuropathic and other pain with desirable or mitigated cardiovascular effects.
7. Highlights and Conclusion
In this review, we elaborate past and recent discoveries regarding nNOS and its redox properties along with its diverse role on the CVS in both physiological and pathological states such as stroke and hypertension. In addition, we have narrated the involvement of the nNOS protein in neurotransmission, neuroprotection and neurotoxicity, smooth and skeletal muscle contraction, sexual function/dysfunction, body fluid homeostasis, and atherosclerosis, among others. Consistent with the involvement of nNOS in such various aspects of human biology, we now know that nNOS mRNA and the protein are expressed in numerous tissues. We have shown the genetic organization of the nNOS mRNA and bidomain structure of the nNOS protein isoform. It appears that both its gene structure and expressional regulation are exceedingly complex. The location of nNOS gene on chromosome 12 holds tremendous clinical significance because the same gene encodes NO production and regulation of vascular hypertension. This paradoxical concept plays an important role in the understanding of pathological mechanisms of many cardiovascular diseases.
We describe that the nNOS gene produces multiple mRNA transcripts via a variety of intriguing mechanisms: alternate promoter usage, alternative splicing, cassette insertions/deletions, varied sites for 3’-UTR cleavage and polyadenylation. There are now four different variants of the nNOS protein, i.e., nNOSαa, nNOSαb, nNOSβ, and nNOSγ. Because of the fact that nNOS mediates H2O2-induced arteriosclerosis, it is pertinent to find out which variant is responsible for such an important vasculopathy. If we can determine that, then we can develop and discover specific antagonists of such nNOS variant(s) that in turn, may lead to potential pharmacotherapies of atherosclerosis and atherosclerosis-induced myocardial infarction, stroke, and/or thrombosis. In addition, we describe the specific binding sites and the various receptors associated with nNOS. Taken together, the vast array of properties and structure-related actions of nNOS make the protein as one of the most complex human genes and thus, understanding the intricate genetic regulation of nNOS has been a major focus in continuing further research on nNOS along with its medical, biological, and clinical implications.
While describing the various actions of the nNOS protein on the CVS, we can establish that nNOS-induced NO release is a very important determinant of maintaining basal vascular tone; preventing platelet activation and aggregation; limiting the adhesion of white blood cells to the endothelial wall of arteries; regulating myocyte contractility; and mediating, modulating and integrating the regulation of the overall cardiovascular functions. Neuronal NOS delays the transition to heart failure in response to pressure overload, protects the myocardium from functional deterioration after an episode of myocardial infarction, and decreases mortality after an incident of myocardium infarction or heart attack. In addition, the nNOS protein plays a key role in the pathogenesis of common cardiovascular diseases that includes hypotensive shock, essential hypertension, atherosclerosis, ischemic cardiovascular disease, and heart failure.
The role of nNOS in cardiomyocyte functions and their increasing contractility mediated via the ryanodine receptors and Ca2+ release from the SR has been described, however, the role of nNOS in normal cardiac myocyte function has been shown to be controversial. Neuronal NOS acts as a relay between ryanodine receptors and Ca2+ by inhibiting SR import pump & SERCA, increasing Ca2+ levels in the cytoplasm, and thereby resulting in positive ionotropic and dromotropic effects. On the other hand, inhibition of nNOS activity either by gene disruption or by pharmacological antagonism can potentiate myocardial contractility at physiological temperatures. Such nNOS blockade can also inhibit SERCA over a wide range of HR values and simultaneously augment phospholamban levels in order to cause negative ionotropic and dromotropic effects. Moreover, nNOS-induced production of NO inhibits the L-type Ca2+ currents via the NO-cGMP pathway as a result of attenuating guanylate cyclase-derived increases in peak Ca2+ current. Most probably, in non-damaged myocardium nNOS activity is regulated by beat-to-beat variations in intracellular Ca2+ levels. In pathological conditions of the heart, nNOS can decrease myocardial contractility via inhibition of the L-type Ca2+ current amplitude and the intrinsic Ca2+ transients. The mitochondria and xanthine oxidoreductase enzyme inside a cardiac myocyte are potential targets for nNOS in cardiac disease models. Neuronal NOS, Ca2+-CaMPKII and NOX2 are important mediators in mechanical and chemical transduction processes that occur during cardiac contraction, and can be targeted by new therapeutic approaches for treating stress-induced Ca2+ dysregulation, cardiac arrhythmias, and cardiomyopathy. The role of oxidants, ROS and nNOS on Ca2+ and Na+ movements in healthy and diseased cardiac myocytes in the case of cardiac ischemia-reperfusion injury has been elaborated in our review. Ischemic postconditioning can provide protection of the heart following an ischemia-reperfusion injury to the heart mediated via nNOS located within the SR and mitochondria.
Atrial fibrillation is a growing public health burden and its effective treatment always remains a challenge. Atrial fibrillation leads to electrical remodeling of the atria, which in turn promotes the continued maintenance of atrial fibrillation and resistance to treatment. Although treatment of congestive heart failure and associated atrial fibrillation by ACE inhibitors or ARBs are geared towards cardiac remodeling, however, the main cause of atrial fibrillation still remains poorly understood. In this review, we describe how atrium-specific upregulation of miRNA-32 can be an electrical phenotype in human atrial fibrillation and the key mechanism being the loss of atrial dystrophin and nNOS proteins. Thus, miRNA-31 may represent a potential therapeutic target in cases of atrial fibrillation. Overall, we state that nNOS is a predominant isoform of the NOSs that controls intracellular Ca2+ homeostasis, myocardial contraction, cardiac relaxation, and the signaling pathways, including the balancing of nitroso-redox. Furthermore, we describe that nNOS is induced by the interplay between AngII type 1 receptor and Ang II type 2 receptor and mediated by NADPH oxidase and ROS-dependent nNOS activity in cardiac myocytes. Neuronal NOS can protect the heart from the pathological changes during hypertensive state, and thus can be a basis for future investigation of new drugs targeted towards hypertension-induced diseased myocardium.
While describing the actions of nNOS on vascular tone, we have mentioned that nNOS is also present on the endothelial cells and that it contributes to the modulation of vascular perfusion. In case of a sudden rise in blood pressure, homeostatic mechanisms kick in and trigger the release of NO that will increase the diameter of the vascular lumen and decrease the total peripheral resistance, an important parameter used in the calculation of blood pressure. Both eNOS and nNOS have their separate distinctive roles in the physiological regulation of human microvascular tone in vivo and should be further studied for understanding the pathogenesis of vascular remodeling and vasculopathy in cases of arteriosclerosis and venous thrombosis. We also discuss the role of nNOS on the transcription regulatory systems during angiogenesis, including its role on the dynamics of renal blood flow during vasodilation or vasoconstriction. The nNOS isoform is a critical regulator of renal hemodynamics in the newborn and hence the nNOS-derived NO-cyclic GMP pathway has the potential to be an important target for the treatment of renal circulatory dysfunction such as chronic kidney failure and uremia. Chronic interference of the nNOS protein dimerization that is responsible for the production of NO within the PVN of the hypothalamus potentiates the rises in blood pressure via modulating the sympathoexcitation associated with renovascular hypertension. These effects of nNOS on vascular system can lead to the further development of strategies in the treatment of vascular disorders including ischemic situations. Lastly, but not the least, nNOS has its own distinctive role in the physiological regulation of human coronary vascular tone in vivo. Overall, we have summarized the role of the nNOS protein on the vasculature during both physiological state and pathological situations.
We also discuss the role of nNOS in SANS and PANS including its involvement in several brain regions associated with cardiovascular regulation and functions. We mention the interaction of nNOS with glutamatergic and GABAergic neurotransmission and cardiovascular regulation within the VLM, the NTS, and the hypothalamus. We have discussed that nNOS within the RVLM is sympathoinhibitory in pathological conditions like congestive cardiac failure, chronic renal failure, hypertension, and diabetes, and that there is evidence that the nNOS protein has significant interactions with both glutamate and GABA in the regulation of the overall CVS. Redox-sensitive inhibition of nNOS-mediated nNOS uncoupling is associated with overproduction of ROS within the RVLM, and may result in the sympathetic overactivity-induced hypertension that is observed in Metabolic Syndrome.
We also elaborate that nNOS activity within the RVLM and CVLM plays opposite roles in modifying cardiovascular responses during peripheral stimulus mediated through glutamate and GABA neurotransmission. Blockade of nNOS within the RVLM increases glutamate release and decreases GABA levels, and the result is enhancement of pressor and tachycardic responses as observed during exercise, whereas nNOS antagonism within the CVLM causes attenuation of blood pressure and HR increases in response to exercise by a reduction in neuronal glutamate release and increase in GABA levels within the CVLM. In addition, we discuss that NO-nNOS system plays an important role in baroreflex mechanism in both physiological and pathophysiological states. Selective blockade of nNOS reduces vagally-mediated decreases in HR. Moreover, systemic administration of nNOS inhibitors affects baroreflex activity via modulating peripheral autonomic transmission, postsynaptic responsiveness of target tissues to autonomic stimuli, and the ANS circuitry within the CNS. Finally, any insult during dimerization process of the nNOS protein within the PVN of the hypothalamus may cause renovascular hypertension. The hypothalamus controls human circadian rhythms and diurnal variations in blood pressure and is critical for circadian blood pressure dysfunction and essential hypertension.
We also narrate the role of nNOS in cerebrovascular accident or stroke. The nNOS-induced neuronal release of NO can be considered as a free radical. The production of NO following stroke is dependent on the changes in the abundance of each isoform of NOSs and that nNOS can be either neuroprotective or neurotoxic during the different stages of cerebral ischemia. The pathological changes happening after a stroke depend on the time elapsed after the onset of the stroke and the subsequent expression of specific NOSs isoforms; the amount of NO that was generated by each specific NOSs protein; the effects of NO on the cells within the umbra, penumbra, or other regions of the brain; and on how much NO and its metabolites have entered the plasma or the brain ventricles. At the very early stage of cerebral ischemia, a surge in the release of NO by the eNOS protein appear to protect neuronal death by inducing vasodilatation and inhibiting microvascular aggregation, but later on, too much of NO produced by either nNOS or iNOS may contribute to apoptosis and subsequent neuronal death. Thus, administration of NO donors can be useful within the first few hours after an attack of ischemic stroke and selective iNOS or perhaps nNOS inhibitors could be valuable therapeutic tools as a part of the treatment protocol.
Because nNOS-derived NO plays a crucial role in the physiological regulation of basal systemic vascular resistance, arterial pressure, recanalization of atherosclerotic cerebral blood vessel(s), and neuroprotection, it can be used as a part of the treatment regimen for acute ischemic stroke in humans. Edaravone, a free radical scavenger, has been used clinically for a long time in order to provide neuroprotection following an episode of ischemic stroke. Any thrombolytic drug along with edaravone has the greatest potential to be utilized in human stroke patients. Atorvastatin may also provide additional neuroprotection following an ischemic brain injury. On the other hand, the dissociation of nNOS from the PSD95 is associated with functional recovery after cerebral ischemia. Finally, we can state that regular physical exercise like swimming can increase antioxidant capacity, prevent neurodegeneration due to redox, and attenuate oxidative damages.
In this review, we describe the role of nNOS in hypertension and nociception. It is well known that hypertension is a prevalent risk factor of fatal cardiovascular complications, such as stroke, sudden cardiac death, and heart failure. We discuss past and recent findings about the relationship between hypertension and nNOS-induced NO production. Reduced NO bioavailability in the endothelium is an important precursor of impaired vasodilation and a predisposing factor in the development of hypertension. Neuronal NOS can produce H2O2 and superoxide, key mediators in non-neuronal cells signaling, and they promote arteriosclerosis. Thus, the role of nNOS in vascular homeostasis and cardiovascular diseases such as hypertension and atherosclerosis deserve to be explored. Nevertheless, nNOS has been proposed to play important roles in suppressing arteriosclerotic vascular lesion and that nNOS protects from atherosclerosis [269–272]. We recognize that nNOS is an important cardiac protector in the diseased heart and plays a major role in arteriosclerosis, the most important cause of coronary artery disease and heart attack.
Regarding the nociceptive system, cardiovascular and pain mechanisms lead to various responses and defensive/withdrawal reactions. Nitric oxide and all three NOSs are involved in nociception and cardiovascular functions, and that NO is an important neurotransmitter involved in both CVS and nociceptive processes/pathways. Uncoupling, over-expression, or down-regulation of NOSs isoforms, particularly of nNOS, plays a crucial role in neuropathic pain mediated via numerous processes or mechanisms such as neuronal excitotoxicity by activating NMDAR or via activating MAPK, JNK, and ERK transcription pathways that may lead to the death of neurons. It appears that all three NOSs generate high concentrations of NO leading to neuronal toxicity by the production of peroxynitrite, a neurotoxic compound.
Overall, we have attempted to summarize the numerous roles of the nNOS protein on physiological homeostasis involving its structure-activity relationship, gene variants, the CVS and nociception, and on some important pathological disease states like vascular ischemia, arteriosclerosis, hypertension, congestive cardiac failure, arrhythmias, and stroke. Thus, the importance of nNOS cannot be underestimated and we foresee that both basic and clinical research with the nNOS protein will continue for decades to come and scientists will someday unravel the potential therapeutic value of the nNOS protein, its antagonists, or its variants.
Supplementary Material
Highlights:
- The nitric oxide synthases (NOSs) catalyze the following reaction:
Nitric oxide (NO) modulated by the second messenger cyclic GMP and intracellular calcium is a neuronal mediator for a wide range of physiological, pathophysiological, and pharmacological actions.
Neuronal NOS (nNOS) regulates plasticity of neurons during fetal development and is involved in neuropathic pain.
N-methyl-D-aspartate subtype of the glutamate receptor family regulate NO production, which is altered in hypertension and cerebrovascular accidents.
The RVLM (pressor area) and the CVLM (depressor region) are critical parts in the brain for the maintenance of blood pressure and vasomotor tone, baroreceptor and the exercise pressor reflexes, cardiopulmonary reflexes, and the overall sympathetic tone.
The alteration in the nNOS activity within the RVLM and CVLM provides differential roles in static muscle contractions-induced increases in cardiovascular responses via an interaction with glutamatergic and GABAergic neurotransmission.
The nNOS within the RVLM and CVLM is involved in the sympatho-inhibitory effects in congestive cardiac failure, chronic renal failure, hypertension, and diabetes.
The unilateral ischemic stroke differentially affects ipsilateral nNOS protein abundance within the RVLM and the CVLM.
8. Acknowledgements
The work in our laboratory was supported by the National Institute of Health HL131577 and American Heart Association 19IPLOI34730020.
Abbreviations
- VLM
ventrolateral medulla
- PAG
periaqueductal gray matter
- nNOS
neuronal nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- NOS
nitric oxide synthases
- NOSs
nitric oxide synthases
- NO
nitric oxide
- SANS
sympathetic autonomic nervous system
- PANS
parasympathetic autonomic nervous system
- ROS
reactive oxygen species
- CVS
cardiovascular system
- Ca2+
calcium ion
- CaM
calmodulin
- cGMP
cyclic guanosine monophosphate
- BH4
tetrahydrobiopterin
- cAMP
cyclic adenosine monophosphate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
9. References
- [1].Delker SL, Xue F, Li H, Jamal J, Silverman RB, Poulos TL, Role of zinc in isoform-selective inhibitor binding to neuronal nitric oxide synthase, Biochemistry 49 (2010) 10803–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Knowles RG, Moncada S, Nitric oxide synthases in mammals, Biochem J 298 ( Pt 2) (1994) 249–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Morris SM Jr., Enzymes of arginine metabolism, J Nutr 134 (2004) 2743S-2747S; discussion 2765S-2767S. [DOI] [PubMed] [Google Scholar]
- [4].Tapiero H, The 2004 French Medical Academy award to professor Georges Mathe, Biomed Pharmacother 59 (2005) 63. [DOI] [PubMed] [Google Scholar]
- [5].Kishimoto J, Spurr N, Liao M, Lizhi L, Emson P, Xu W, Localization of brain nitric oxide synthase (NOS) to human chromosome 12, Genomics 14 (1992) 802–4. [DOI] [PubMed] [Google Scholar]
- [6].Taylor BS, Kim YM, Wang Q, Shapiro RA, Billiar TR, Geller DA, Nitric oxide down-regulates hepatocyte-inducible nitric oxide synthase gene expression, Arch Surg 132 (1997) 1177–83. [DOI] [PubMed] [Google Scholar]
- [7].Forstermann U, Kleinert H, Gath I, Schwarz P, Closs EI, Dun NJ, Expression and expressional control of nitric oxide synthases in various cell types, Adv Pharmacol 34 (1995) 171–86. [DOI] [PubMed] [Google Scholar]
- [8].Stuehr DJ, Mammalian nitric oxide synthases, Biochim Biophys Acta 1411 (1999) 217–30. [DOI] [PubMed] [Google Scholar]
- [9].Chan JY, Wang SH, Chan SH, Differential roles of iNOS and nNOS at rostral ventrolateral medulla during experimental endotoxemia in the rat, Shock 15 (2001) 65–72. [DOI] [PubMed] [Google Scholar]
- [10].Gusarov I, Starodubtseva M, Wang ZQ, McQuade L, Lippard SJ, Stuehr DJ, Nudler E, Bacterial nitric-oxide synthases operate without a dedicated redox partner, J Biol Chem 283 (2008) 13140–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pannu R, Singh I, Pharmacological strategies for the regulation of inducible nitric oxide synthase: neurodegenerative versus neuroprotective mechanisms, Neurochem Int 49 (2006) 170–82. [DOI] [PubMed] [Google Scholar]
- [12].Ally A, Maher TJ, Transient middle cerebral artery occlusion and reperfusion alters inducible NOS expression within the ventrolateral medulla and modulates cardiovascular function during static exercise, Can J Physiol Pharmacol 89 (2011) 639–46. [DOI] [PubMed] [Google Scholar]
- [13].Ally A, Kabadi S, Phattanarudee S, Patel M, Maher TJ, Neuronal nitric oxide synthase (nNOS) blockade within the ventrolateral medulla differentially modulates cardiovascular responses and nNOS expression during static skeletal muscle contraction, Brain Res 1150 (2007) 21–31. [DOI] [PubMed] [Google Scholar]
- [14].Moro MA, Cardenas A, Hurtado O, Leza JC, Lizasoain I, Role of nitric oxide after brain ischaemia, Cell Calcium 36 (2004) 265–75. [DOI] [PubMed] [Google Scholar]
- [15].Choi DW, Nitric oxide: foe or friend to the injured brain?, Proc Natl Acad Sci U S A 90 (1993) 9741–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Snyder SH, Nitric oxide: first in a new class of neurotransmitters, Science 257 (1992) 494–6. [DOI] [PubMed] [Google Scholar]
- [17].Moncada S, Palmer RM, Higgs EA, The discovery of nitric oxide as the endogenous nitrovasodilator, Hypertension 12 (1988) 365–72. [DOI] [PubMed] [Google Scholar]
- [18].Jiang J, Gan Z, Li Y, Zhao W, Li H, Zheng JP, Ke Y, REM sleep deprivation induces endothelial dysfunction and hypertension in middle-aged rats: Roles of the eNOS/NO/cGMP pathway and supplementation with L-arginine, PLoS One 12 (2017) e0182746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stephan BCM, Harrison SL, Keage HAD, Babateen A, Robinson L, Siervo M, Cardiovascular Disease, the Nitric Oxide Pathway and Risk of Cognitive Impairment and Dementia, Curr Cardiol Rep 19 (2017) 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kanaan GN, Harper ME, Cellular redox dysfunction in the development of cardiovascular diseases, Biochim Biophys Acta Gen Subj 1861 (2017) 2822–2829. [DOI] [PubMed] [Google Scholar]
- [21].Patel D, Bohlke M, Phattanarudee S, Kabadi S, Maher TJ, Ally A, Cardiovascular responses and neurotransmitter changes during blockade of angiotensin II receptors within the ventrolateral medulla, Neurosci Res 60 (2008) 340–8. [DOI] [PubMed] [Google Scholar]
- [22].Schulz R, Rassaf T, Massion PB, Kelm M, Balligand JL, Recent advances in the understanding of the role of nitric oxide in cardiovascular homeostasis, Pharmacol Ther 108 (2005) 225–56. [DOI] [PubMed] [Google Scholar]
- [23].Hewett SJ, Muir JK, Lobner D, Symons A, Choi DW, Potentiation of oxygen-glucose deprivation-induced neuronal death after induction of iNOS, Stroke 27 (1996) 1586–91. [DOI] [PubMed] [Google Scholar]
- [24].Majzunova M, Pakanova Z, Kvasnicka P, Balis P, Cacanyiova S, Dovinova I, Age-dependent redox status in the brain stem of NO-deficient hypertensive rats, J Biomed Sci 24 (2017) 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Falls R, Seman M, Braat S, Sortino J, Allen JD, Neil CJ, Inorganic nitrate as a treatment for acute heart failure: a protocol for a single center, randomized, double-blind, placebo-controlled pilot and feasibility study, J Transl Med 15 (2017) 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bondonno CP, Blekkenhorst LC, Liu AH, Bondonno NP, Ward NC, Croft KD, Hodgson JM, Vegetable-derived bioactive nitrate and cardiovascular health, Mol Aspects Med 61 (2018) 83–91. [DOI] [PubMed] [Google Scholar]
- [27].Rudolph TK, Fuchs A, Klinke A, Schlichting A, Friedrichs K, Hellmich M, Mollenhauer M, Schwedhelm E, Baldus S, Rudolph V, Prasugrel as opposed to clopidogrel improves endothelial nitric oxide bioavailability and reduces platelet-leukocyte interaction in patients with unstable angina pectoris: A randomized controlled trial, Int J Cardiol 248 (2017) 7–13. [DOI] [PubMed] [Google Scholar]
- [28].Mungrue IN, Bredt DS, nNOS at a glance: implications for brain and brawn, J Cell Sci 117 (2004) 2627–9. [DOI] [PubMed] [Google Scholar]
- [29].Bell PD, Franco M, Navar LG, Calcium as a mediator of tubuloglomerular feedback, Annu Rev Physiol 49 (1987) 275–93. [DOI] [PubMed] [Google Scholar]
- [30].Capettini LS, Cortes SF, Silva JF, Alvarez-Leite JI, Lemos VS, Decreased production of neuronal NOS-derived hydrogen peroxide contributes to endothelial dysfunction in atherosclerosis, Br J Pharmacol 164 (2011) 1738–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Guerra DD, Bok R, Lorca RA, Hurt KJ, Protein kinase A facilitates relaxation of mouse ileum via phosphorylation of neuronal nitric oxide synthase, Br J Pharmacol (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Maher A, Abdel Rahman MF, Gad MZ, The Role of Nitric Oxide from Neurological Disease to Cancer, Adv Exp Med Biol 1007 (2017) 71–88. [DOI] [PubMed] [Google Scholar]
- [33].Mount PF, Power DA, Nitric oxide in the kidney: functions and regulation of synthesis, Acta Physiol (Oxf) 187 (2006) 433–46. [DOI] [PubMed] [Google Scholar]
- [34].Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC, Targeted disruption of the neuronal nitric oxide synthase gene, Cell 75 (1993) 1273–86. [DOI] [PubMed] [Google Scholar]
- [35].Sheta EA, McMillan K, Masters BS, Evidence for a bidomain structure of constitutive cerebellar nitric oxide synthase, J Biol Chem 269 (1994) 15147–53. [PubMed] [Google Scholar]
- [36].Alderton WK, Cooper CE, Knowles RG, Nitric oxide synthases: structure, function and inhibition, Biochem J 357 (2001) 593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lowe PN, Smith D, Stammers DK, Riveros-Moreno V, Moncada S, Charles I, Boyhan A, Identification of the domains of neuronal nitric oxide synthase by limited proteolysis, Biochem J 314 ( Pt 1) (1996) 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vandsburger MH, Epstein FH, Emerging MRI methods in translational cardiovascular research, J Cardiovasc Transl Res 4 (2011) 477–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vandsburger MH, French BA, Helm PA, Roy RJ, Kramer CM, Young AA, Epstein FH, Multi-parameter in vivo cardiac magnetic resonance imaging demonstrates normal perfusion reserve despite severely attenuated beta-adrenergic functional response in neuronal nitric oxide synthase knockout mice, Eur Heart J 28 (2007) 2792–8. [DOI] [PubMed] [Google Scholar]
- [40].Kitagawa H, Mori A, Shimada J, Mitsumoto Y, Kikuchi T, Intracerebral adenosine infusion improves neurological outcome after transient focal ischemia in rats, Neurol Res 24 (2002) 317–23. [DOI] [PubMed] [Google Scholar]
- [41].Gorren AC, List BM, Schrammel A, Pitters E, Hemmens B, Werner ER, Schmidt K, Mayer B, Tetrahydrobiopterin-free neuronal nitric oxide synthase: evidence for two identical highly anticooperative pteridine binding sites, Biochemistry 35 (1996) 16735–45. [DOI] [PubMed] [Google Scholar]
- [42].Paige JS, Jaffrey SR, Pharmacologic manipulation of nitric oxide signaling: targeting NOS dimerization and protein-protein interactions, Curr Top Med Chem 7 (2007) 97–114. [DOI] [PubMed] [Google Scholar]
- [43].Venema RC, Ju H, Zou R, Ryan JW, Venema VJ, Subunit interactions of endothelial nitric-oxide synthase. Comparisons to the neuronal and inducible nitric-oxide synthase isoforms, J Biol Chem 272 (1997) 1276–82. [DOI] [PubMed] [Google Scholar]
- [44].Prisco SZ, Priestley JR, Weinberg BD, Prisco AR, Hoffman MJ, Jacob HJ, Flister MJ, Lombard JH, Lazar J, Vascular dysfunction precedes hypertension associated with a blood pressure locus on rat chromosome 12, Am J Physiol Heart Circ Physiol 307 (2014) H1103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Francis BN, Hale A, Channon KM, Wilkins MR, Zhao L, Effects of tetrahydrobiopterin oral treatment in hypoxia-induced pulmonary hypertension in rat, Pulm Circ 4 (2014) 462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Heffernan KS, Fahs CA, Ranadive SM, Patvardhan EA, L-arginine as a nutritional prophylaxis against vascular endothelial dysfunction with aging, J Cardiovasc Pharmacol Ther 15 (2010) 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wilkinson-Berka JL, Deliyanti D, Rana I, Miller AG, Agrotis A, Armani R, Szyndralewiez C, Wingler K, Touyz RM, Cooper ME, Jandeleit-Dahm KA, Schmidt HH, NADPH oxidase, NOX1, mediates vascular injury in ischemic retinopathy, Antioxid Redox Signal 20 (2014) 2726–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Forstermann U, Sessa WC, Nitric oxide synthases: regulation and function, Eur Heart J 33 (2012) 829–37, 837a-837d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nakane M, Schmidt HH, Pollock JS, Forstermann U, Murad F, Cloned human brain nitric oxide synthase is highly expressed in skeletal muscle, FEBS Lett 316 (1993) 175–80. [DOI] [PubMed] [Google Scholar]
- [50].Wang Y, Newton DC, Marsden PA, Neuronal NOS: gene structure, mRNA diversity, and functional relevance, Crit Rev Neurobiol 13 (1999) 21–43. [DOI] [PubMed] [Google Scholar]
- [51].Gachhui R, Abu-Soud HM, Ghosha DK, Presta A, Blazing MA, Mayer B, George SE, Stuehr DJ, Neuronal nitric-oxide synthase interaction with calmodulin-troponin C chimeras, J Biol Chem 273 (1998) 5451–4. [DOI] [PubMed] [Google Scholar]
- [52].Riefler GM, Firestein BL, Binding of neuronal nitric-oxide synthase (nNOS) to carboxyl-terminal-binding protein (CtBP) changes the localization of CtBP from the nucleus to the cytosol: a novel function for targeting by the PDZ domain of nNOS, J Biol Chem 276 (2001) 48262–8. [DOI] [PubMed] [Google Scholar]
- [53].Christopherson KS, Bredt DS, Nitric oxide in excitable tissues: physiological roles and disease, J Clin Invest 100 (1997) 2424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Stricker NL, Christopherson KS, Yi BA, Schatz PJ, Raab RW, Dawes G, Bassett DE Jr., Bredt DS, Li M, PDZ domain of neuronal nitric oxide synthase recognizes novel C-terminal peptide sequences, Nat Biotechnol 15 (1997) 336–42. [DOI] [PubMed] [Google Scholar]
- [55].Balligand JL, Cannon PJ, Nitric oxide synthases and cardiac muscle. Autocrine and paracrine influences, Arterioscler Thromb Vasc Biol 17 (1997) 1846–58. [DOI] [PubMed] [Google Scholar]
- [56].Abu-Soud HM, Yoho LL, Stuehr DJ, Calmodulin controls neuronal nitric-oxide synthase by a dual mechanism. Activation of intra- and interdomain electron transfer, J Biol Chem 269 (1994) 32047–50. [PubMed] [Google Scholar]
- [57].Gao X, Kim HK, Chung JM, Chung K, Reactive oxygen species (ROS) are involved in enhancement of NMDA-receptor phosphorylation in animal models of pain, Pain 131 (2007) 262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mukherjee P, Cinelli MA, Kang S, Silverman RB, Development of nitric oxide synthase inhibitors for neurodegeneration and neuropathic pain, Chem Soc Rev 43 (2014) 6814–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Qureshi I, Chen H, Brown AT, Fitzgerald R, Zhang X, Breckenridge J, Kazi R, Crocker AJ, Stuhlinger MC, Lin K, Cooke JP, Eidt JF, Moursi MM, Homocysteine-induced vascular dysregulation is mediated by the NMDA receptor, Vasc Med 10 (2005) 215–23. [DOI] [PubMed] [Google Scholar]
- [60].Song X, Vaage J, Valen G, The role of neuronal nitric oxide synthase in ischaemia-reperfusion injury of the isolated mouse heart, Acta Physiol Scand 172 (2001) 291–5. [DOI] [PubMed] [Google Scholar]
- [61].Piech A, Dessy C, Havaux X, Feron O, Balligand JL, Differential regulation of nitric oxide synthases and their allosteric regulators in heart and vessels of hypertensive rats, Cardiovasc Res 57 (2003) 456–67. [DOI] [PubMed] [Google Scholar]
- [62].Fridolfsson HN, Patel HH, Caveolin and caveolae in age associated cardiovascular disease, J Geriatr Cardiol 10 (2013) 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Li YF, Wang Y, Channon KM, Schultz HD, Zucker IH, Patel KP, Manipulation of neuronal nitric oxide synthase within the paraventricular nucleus using adenovirus and antisense technology, Methods Mol Med 112 (2005) 59–79. [DOI] [PubMed] [Google Scholar]
- [64].Li FC, Chan JY, Chan SH, Chang AY, In the rostral ventrolateral medulla, the 70-kDa heat shock protein (HSP70), but not HSP90, confers neuroprotection against fatal endotoxemia via augmentation of nitric-oxide synthase I (NOS I)/protein kinase G signaling pathway and inhibition of NOS II/peroxynitrite cascade, Mol Pharmacol 68 (2005) 179–92. [DOI] [PubMed] [Google Scholar]
- [65].Lane P, Gross SS, The autoinhibitory control element and calmodulin conspire to provide physiological modulation of endothelial and neuronal nitric oxide synthase activity, Acta Physiol Scand 168 (2000) 53–63. [DOI] [PubMed] [Google Scholar]
- [66].Kar R, Kellogg DL 3rd, Roman LJ, Oxidative stress induces phosphorylation of neuronal NOS in cardiomyocytes through AMP-activated protein kinase (AMPK), Biochem Biophys Res Commun 459 (2015) 393–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fan JS, Zhang Q, Li M, Tochio H, Yamazaki T, Shimizu M, Zhang M, Protein inhibitor of neuronal nitric-oxide synthase, PIN, binds to a 17-amino acid residue fragment of the enzyme, J Biol Chem 273 (1998) 33472–81. [DOI] [PubMed] [Google Scholar]
- [68].Sharma HS, Wiklund L, Badgaiyan RD, Mohanty S, Alm P, Intracerebral administration of neuronal nitric oxide synthase antiserum attenuates traumatic brain injury-induced blood-brain barrier permeability, brain edema formation, and sensory motor disturbances in the rat, Acta Neurochir Suppl 96 (2006) 288–94. [DOI] [PubMed] [Google Scholar]
- [69].Jiang MH, Kaku T, Hada J, Hayashi Y, Different effects of eNOS and nNOS inhibition on transient forebrain ischemia, Brain Res 946 (2002) 139–47. [DOI] [PubMed] [Google Scholar]
- [70].Vaughan CJ, Delanty N, Nitric oxide in acute ischaemic stroke, J Neurol Neurosurg Psychiatry 68 (2000) 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].O’Mahony D, Kendall MJ, Nitric oxide in acute ischaemic stroke: a target for neuroprotection, J Neurol Neurosurg Psychiatry 67 (1999) 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American C Heart Association Statistics, S. Stroke Statistics, Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association, Circulation 135 (2017) e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Dawson TM, Steiner JP, Dawson VL, Dinerman JL, Uhl GR, Snyder SH, Immunosuppressant FK506 enhances phosphorylation of nitric oxide synthase and protects against glutamate neurotoxicity, Proc Natl Acad Sci U S A 90 (1993) 9808–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Vincent JL, Zhang H, Szabo C, Preiser JC, Effects of nitric oxide in septic shock, Am J Respir Crit Care Med 161 (2000) 1781–5. [DOI] [PubMed] [Google Scholar]
- [75].Forstermann U, Xia N, Li H, Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis, Circ Res 120 (2017) 713–735. [DOI] [PubMed] [Google Scholar]
- [76].Jian Z, Han H, Zhang T, Puglisi J, Izu LT, Shaw JA, Onofiok E, Erickson JR, Chen YJ, Horvath B, Shimkunas R, Xiao W, Li Y, Pan T, Chan J, Banyasz T, Tardiff JC, Chiamvimonvat N, Bers DM, Lam KS, Chen-Izu Y, Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling, Sci Signal 7 (2014) ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Prosser BL, Ward CW, Mechano-chemo transduction tunes the heartstrings, Sci Signal 7 (2014) pe7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Harder DR, Ca-induced Ca release: lessons regarding cell models, Am J Physiol Heart Circ Physiol 287 (2004) H1885–6. [DOI] [PubMed] [Google Scholar]
- [79].Zucchi R, Ronca-Testoni S, The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states, Pharmacol Rev 49 (1997) 1–51. [PubMed] [Google Scholar]
- [80].Fabiato A, Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum, Am J Physiol 245 (1983) C1–14. [DOI] [PubMed] [Google Scholar]
- [81].Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, Casadei B, Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling, Circ Res 92 (2003) e52–9. [DOI] [PubMed] [Google Scholar]
- [82].Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B, Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes, Circulation 105 (2002) 3011–6. [DOI] [PubMed] [Google Scholar]
- [83].Sears CE, Ashley EA, Casadei B, Nitric oxide control of cardiac function: is neuronal nitric oxide synthase a key component?, Philos Trans R Soc Lond B Biol Sci 359 (2004) 1021–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Xu KY, Kuppusamy SP, Wang JQ, Li H, Cui H, Dawson TM, Huang PL, Burnett AL, Kuppusamy P, Becker LC, Nitric oxide protects cardiac sarcolemmal membrane enzyme function and ion active transport against ischemia-induced inactivation, J Biol Chem 278 (2003) 41798–803. [DOI] [PubMed] [Google Scholar]
- [85].Gallo MP, Malan D, Bedendi I, Biasin C, Alloatti G, Levi RC, Regulation of cardiac calcium current by NO and cGMP-modulating agents, Pflugers Arch 441 (2001) 621–8. [DOI] [PubMed] [Google Scholar]
- [86].Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O’Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM, Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms, Nature 416 (2002) 337–9. [DOI] [PubMed] [Google Scholar]
- [87].Strasen J, Ritter O, Role of nNOS in cardiac ischemia-reperfusion injury, Trends Cardiovasc Med 21 (2011) 58–63. [DOI] [PubMed] [Google Scholar]
- [88].Hu L, Wang J, Zhu H, Wu X, Zhou L, Song Y, Zhu S, Hao M, Liu C, Fan Y, Wang Y, Li Q, Ischemic postconditioning protects the heart against ischemia-reperfusion injury via neuronal nitric oxide synthase in the sarcoplasmic reticulum and mitochondria, Cell Death Dis 7 (2016) e2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wang Y, Youm JB, Jin CZ, Shin DH, Zhao ZH, Seo EY, Jang JH, Kim SJ, Jin ZH, Zhang YH, Modulation of L-type Ca(2)(+) channel activity by neuronal nitric oxide synthase and myofilament Ca(2)(+) sensitivity in cardiac myocytes from hypertensive rat, Cell Calcium 58 (2015) 264–74. [DOI] [PubMed] [Google Scholar]
- [90].Silva SM, Silva S, Meireles M, Leal S, nNOS is involved in cardiac remodeling induced by chronic ethanol consumption, Toxicology 329 (2015) 98–105. [DOI] [PubMed] [Google Scholar]
- [91].Watts VL, Sepulveda FM, Cingolani OH, Ho AS, Niu X, Kim R, Miller KL, Vandegaer K, Bedja D, Gabrielson KL, Rameau G, O’Rourke B, Kass DA, Barouch LA, Anti-hypertrophic and anti-oxidant effect of beta3-adrenergic stimulation in myocytes requires differential neuronal NOS phosphorylation, J Mol Cell Cardiol 62 (2013) 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Martin SR, Emanuel K, Sears CE, Zhang YH, Casadei B, Are myocardial eNOS and nNOS involved in the beta-adrenergic and muscarinic regulation of inotropy? A systematic investigation, Cardiovasc Res 70 (2006) 97–106. [DOI] [PubMed] [Google Scholar]
- [93].Jones JF, Cardiac defibrillator neurones, J Physiol 587 (2009) 2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Brack KE, Patel VH, Mantravardi R, Coote JH, Ng GA, Direct evidence of nitric oxide release from neuronal nitric oxide synthase activation in the left ventricle as a result of cervical vagus nerve stimulation, J Physiol 587 (2009) 3045–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Reilly S, Liu X, Carnicer R, Rajakumar T, Sayeed R, Krasopoulos G, Verheule S, Fulga T, Schotten U, Casadei B, Evaluation of the role of miR-31-dependent reduction in dystrophin and nNOS on atrial-fibrillation-induced electrical remodelling in man, Lancet 385 Suppl 1 (2015) S82. [DOI] [PubMed] [Google Scholar]
- [96].Boulanger CM, Heymes C, Benessiano J, Geske RS, Levy BI, Vanhoutte PM, Neuronal nitric oxide synthase is expressed in rat vascular smooth muscle cells: activation by angiotensin II in hypertension, Circ Res 83 (1998) 1271–8. [DOI] [PubMed] [Google Scholar]
- [97].Schwarz PM, Kleinert H, Forstermann U, Potential functional significance of brain-type and muscle-type nitric oxide synthase I expressed in adventitia and media of rat aorta, Arterioscler Thromb Vasc Biol 19 (1999) 2584–90. [DOI] [PubMed] [Google Scholar]
- [98].Capettini LS, Cortes SF, Lemos VS, Relative contribution of eNOS and nNOS to endothelium-dependent vasodilation in the mouse aorta, Eur J Pharmacol 643 (2010) 260–6. [DOI] [PubMed] [Google Scholar]
- [99].Capettini LS, Cortes SF, Gomes MA, Silva GA, Pesquero JL, Lopes MJ, Teixeira MM, Lemos VS, Neuronal nitric oxide synthase-derived hydrogen peroxide is a major endothelium-dependent relaxing factor, Am J Physiol Heart Circ Physiol 295 (2008) H2503–11. [DOI] [PubMed] [Google Scholar]
- [100].McCarthy CP, Ibrahim NE, van Kimmenade RRJ, Gaggin HK, Simon ML, Gandhi P, Kelly N, Motiwala SR, Mukai R, Magaret CA, Barnes G, Rhyne RF, Garasic JM, Januzzi JL Jr., A clinical and proteomics approach to predict the presence of obstructive peripheral arterial disease: From the Catheter Sampled Blood Archive in Cardiovascular Diseases (CASABLANCA) Study, Clin Cardiol 41 (2018) 903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Khan M, Dhammu TS, Matsuda F, Singh AK, Singh I, Blocking a vicious cycle nNOS/peroxynitrite/AMPK by S-nitrosoglutathione: implication for stroke therapy, BMC Neurosci 16 (2015) 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].McLeod DS, Baba T, Bhutto IA, Lutty GA, Co-expression of endothelial and neuronal nitric oxide synthases in the developing vasculatures of the human fetal eye, Graefes Arch Clin Exp Ophthalmol 250 (2012) 839–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Rodebaugh J, Sekulic M, Davies W, Montgomery S, Khraibi A, Solhaug MJ, Ratliff BB, Neuronal nitric oxide synthase, nNOS, regulates renal hemodynamics in the postnatal developing piglet, Pediatr Res 71 (2012) 144–9. [DOI] [PubMed] [Google Scholar]
- [104].Melikian N, Seddon MD, Casadei B, Chowienczyk PJ, Shah AM, Neuronal nitric oxide synthase and human vascular regulation, Trends Cardiovasc Med 19 (2009) 256–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Seidel B, Stanarius A, Wolf G, Differential expression of neuronal and endothelial nitric oxide synthase in blood vessels of the rat brain, Neurosci Lett 239 (1997) 109–12. [DOI] [PubMed] [Google Scholar]
- [106].Dieguez G, Garcia-Villalon AL, Dilator and constrictor response of renal vasculature during acute renal hypotension in anesthetized goats. Role of nitric oxide, Vascul Pharmacol 54 (2011) 107–11. [DOI] [PubMed] [Google Scholar]
- [107].Ramachandran R, Ploug KB, Hay-Schmidt A, Olesen J, Jansen-Olesen I, Gupta S, Nitric oxide synthase (NOS) in the trigeminal vascular system and other brain structures related to pain in rats, Neurosci Lett 484 (2010) 192–6. [DOI] [PubMed] [Google Scholar]
- [108].Toda N, Okamura T, Modulation of renal blood flow and vascular tone by neuronal nitric oxide synthase-derived nitric oxide, J Vasc Res 48 (2011) 1–10. [DOI] [PubMed] [Google Scholar]
- [109].Moleda L, Jurzik L, Froh M, Gabele E, Hellerbrand C, Straub RH, Scholmerich J, Wiest R, Role of HSP-90 for increased nNOS-mediated vasodilation in mesenteric arteries in portal hypertension, World J Gastroenterol 16 (2010) 1837–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Seddon M, Melikian N, Dworakowski R, Shabeeh H, Jiang B, Byrne J, Casadei B, Chowienczyk P, Shah AM, Effects of neuronal nitric oxide synthase on human coronary artery diameter and blood flow in vivo, Circulation 119 (2009) 2656–62. [DOI] [PubMed] [Google Scholar]
- [111].Bauser-Heaton HD, Bohlen HG, Cerebral microvascular dilation during hypotension and decreased oxygen tension: a role for nNOS, Am J Physiol Heart Circ Physiol 293 (2007) H2193–201. [DOI] [PubMed] [Google Scholar]
- [112].Zipes DP, Antiarrhythmic therapy in 2014: Contemporary approaches to treating arrhythmias, Nat Rev Cardiol 12 (2015) 68–9. [DOI] [PubMed] [Google Scholar]
- [113].Fernandez SF, Canty JM Jr., Adrenergic and cholinergic plasticity in heart failure, Circ Res 116 (2015) 1639–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].DeMazumder D, Kass DA, O’Rourke B, Tomaselli GF, Cardiac resynchronization therapy restores sympathovagal balance in the failing heart by differential remodeling of cholinergic signaling, Circ Res 116 (2015) 1691–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Markos F, Snow HM, Kidd C, Conlon K, Nitric oxide facilitates vagal control of heart rate via actions in the cardiac parasympathetic ganglia of the anaesthetised dog, Exp Physiol 87 (2002) 49–52. [DOI] [PubMed] [Google Scholar]
- [116].Choate JK, Danson EJ, Morris JF, Paterson DJ, Peripheral vagal control of heart rate is impaired in neuronal NOS knockout mice, Am J Physiol Heart Circ Physiol 281 (2001) H2310–7. [DOI] [PubMed] [Google Scholar]
- [117].Nihei M, Lee JK, Honjo H, Yasui K, Uzzaman M, Kamiya K, Opthof T, Kodama I, Decreased vagal control over heart rate in rats with right-sided congestive heart failure: downregulation of neuronal nitric oxide synthase, Circ J 69 (2005) 493–9. [DOI] [PubMed] [Google Scholar]
- [118].Phattanarudee S, Towiwat P, Maher TJ, Ally A, Effects of medullary administration of a nitric oxide precursor on cardiovascular responses and neurotransmission during static exercise following ischemic stroke, Can J Physiol Pharmacol 91 (2013) 510–20. [DOI] [PubMed] [Google Scholar]
- [119].Chaitoff KA, Toner F, Tedesco A, Maher TJ, Ally A, Effects of inducible nitric oxide synthase blockade within the periaqueductal gray on cardiovascular responses during mechanical, heat, and cold nociception, Neurol Sci 33 (2012) 69–78. [DOI] [PubMed] [Google Scholar]
- [120].Tedesco A, Ally A, Angiotensin II type-2 (AT2) receptor antagonism alters cardiovascular responses to static exercise and simultaneously changes glutamate/GABA levels within the ventrolateral medulla, Neurosci Res 64 (2009) 372–9. [DOI] [PubMed] [Google Scholar]
- [121].Ally A, Ventrolateral medullary control of cardiovascular activity during muscle contraction, Neurosci Biobehav Rev 23 (1998) 65–86. [DOI] [PubMed] [Google Scholar]
- [122].Reis DJ, Golanov EV, Ruggiero DA, Sun MK, Sympatho-excitatory neurons of the rostral ventrolateral medulla are oxygen sensors and essential elements in the tonic and reflex control of the systemic and cerebral circulations, J Hypertens Suppl 12 (1994) S159–80. [PubMed] [Google Scholar]
- [123].Vasquez EC, Lewis SJ, Varner KJ, Brody MJ, Chronic lesion of rostral ventrolateral medulla in spontaneously hypertensive rats, Hypertension 19 (1992) II154–8. [DOI] [PubMed] [Google Scholar]
- [124].Ciriello J, Caverson MM, Polosa C, Function of the ventrolateral medulla in the control of the circulation, Brain Res 396 (1986) 359–91. [DOI] [PubMed] [Google Scholar]
- [125].Douma LG, Gumz ML, Circadian clock-mediated regulation of blood pressure, Free Radic Biol Med 119 (2018) 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Khor S, Cai D, Hypothalamic and inflammatory basis of hypertension, Clin Sci (Lond) 131 (2017) 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Dampney RA, Central mechanisms regulating coordinated cardiovascular and respiratory function during stress and arousal, Am J Physiol Regul Integr Comp Physiol 309 (2015) R429–43. [DOI] [PubMed] [Google Scholar]
- [128].Ross CA, Ruggiero DA, Reis DJ, Projections from the nucleus tractus solitarii to the rostral ventrolateral medulla, J Comp Neurol 242 (1985) 511–34. [DOI] [PubMed] [Google Scholar]
- [129].Ross CA, Ruggiero DA, Joh TH, Park DH, Reis DJ, Rostral ventrolateral medulla: selective projections to the thoracic autonomic cell column from the region containing C1 adrenaline neurons, J Comp Neurol 228 (1984) 168–85. [DOI] [PubMed] [Google Scholar]
- [130].Ross CA, Ruggiero DA, Park DH, Joh TH, Sved AF, Fernandez-Pardal J, Saavedra JM, Reis DJ, Tonic vasomotor control by the rostral ventrolateral medulla: effect of electrical or chemical stimulation of the area containing C1 adrenaline neurons on arterial pressure, heart rate, and plasma catecholamines and vasopressin, J Neurosci 4 (1984) 474–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Ross CA, Armstrong DM, Ruggiero DA, Pickel VM, Joh TH, Reis DJ, Adrenaline neurons in the rostral ventrolateral medulla innervate thoracic spinal cord: a combined immunocytochemical and retrograde transport demonstration, Neurosci Lett 25 (1981) 257–62. [DOI] [PubMed] [Google Scholar]
- [132].Patel KP, Role of paraventricular nucleus in mediating sympathetic outflow in heart failure, Heart Fail Rev 5 (2000) 73–86. [DOI] [PubMed] [Google Scholar]
- [133].Benarroch EE, Paraventricular nucleus, stress response, and cardiovascular disease, Clin Auton Res 15 (2005) 254–63. [DOI] [PubMed] [Google Scholar]
- [134].Coote JH, Homeostasis and stress, Clin Auton Res 15 (2005) 247–8. [DOI] [PubMed] [Google Scholar]
- [135].Coote JH, A role for the paraventricular nucleus of the hypothalamus in the autonomic control of heart and kidney, Exp Physiol 90 (2005) 169–73. [DOI] [PubMed] [Google Scholar]
- [136].Nassar NN, Abdel-Rahman AA, Brain stem adenosine receptors modulate centrally mediated hypotensive responses in conscious rats: A review, J Adv Res 6 (2015) 331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Chen WW, Xiong XQ, Chen Q, Li YH, Kang YM, Zhu GQ, Cardiac sympathetic afferent reflex and its implications for sympathetic activation in chronic heart failure and hypertension, Acta Physiol (Oxf) 213 (2015) 778–94. [DOI] [PubMed] [Google Scholar]
- [138].Paton JF, Wang S, Polson JW, Kasparov S, Signalling across the blood brain barrier by angiotensin II: novel implications for neurogenic hypertension, J Mol Med (Berl) 86 (2008) 705–10. [DOI] [PubMed] [Google Scholar]
- [139].Paton JF, Waki H, Abdala AP, Dickinson J, Kasparov S, Vascular-brain signaling in hypertension: role of angiotensin II and nitric oxide, Curr Hypertens Rep 9 (2007) 242–7. [DOI] [PubMed] [Google Scholar]
- [140].Hirooka Y, Localized gene transfer and its application for the study of central cardiovascular control, Auton Neurosci 126–127 (2006) 120–9. [DOI] [PubMed] [Google Scholar]
- [141].Zanzinger J, Mechanisms of action of nitric oxide in the brain stem: role of oxidative stress, Auton Neurosci 98 (2002) 24–7. [DOI] [PubMed] [Google Scholar]
- [142].Talman WT, Dragon DN, Ohta H, Lin LH, Nitroxidergic influences on cardiovascular control by NTS: a link with glutamate, Ann N Y Acad Sci 940 (2001) 169–78. [DOI] [PubMed] [Google Scholar]
- [143].Maeda M, Inoue M, Takao S, Nakai M, Central control mechanisms of circulation in the medulla oblongata by nitric oxide, Jpn J Physiol 49 (1999) 467–78. [DOI] [PubMed] [Google Scholar]
- [144].Talman WT, Glutamatergic transmission in the nucleus tractus solitarii: from server to peripherals in the cardiovascular information superhighway, Braz J Med Biol Res 30 (1997) 1–7. [DOI] [PubMed] [Google Scholar]
- [145].Brown DL, Guyenet PG, Electrophysiological study of cardiovascular neurons in the rostral ventrolateral medulla in rats, Circ Res 56 (1985) 359–69. [DOI] [PubMed] [Google Scholar]
- [146].Wang Y, Golledge J, Neuronal nitric oxide synthase and sympathetic nerve activity in neurovascular and metabolic systems, Curr Neurovasc Res 10 (2013) 81–9. [DOI] [PubMed] [Google Scholar]
- [147].Ishide T, Maher TJ, Pearce WJ, Nauli SM, Chaiyakul P, Ally A, Simultaneous glutamate and gamma-aminobutyric acid release within ventrolateral medulla during skeletal muscle contraction in intact and barodenervated rats, Brain Res 923 (2001) 137–46. [DOI] [PubMed] [Google Scholar]
- [148].Wu KL, Chao YM, Tsay SJ, Chen CH, Chan SH, Dovinova I, Chan JY, Role of nitric oxide synthase uncoupling at rostral ventrolateral medulla in redox-sensitive hypertension associated with metabolic syndrome, Hypertension 64 (2014) 815–24. [DOI] [PubMed] [Google Scholar]
- [149].Penumarti A, Abdel-Rahman AA, Neuronal nitric oxide synthase-dependent elevation in adiponectin in the rostral ventrolateral medulla underlies g protein-coupled receptor 18-mediated hypotension in conscious rats, J Pharmacol Exp Ther 351 (2014) 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Lee YH, Tsai MC, Li TL, Dai YW, Huang SC, Hwang LL, Spontaneously hypertensive rats have more orexin neurons in the hypothalamus and enhanced orexinergic input and orexin 2 receptor-associated nitric oxide signalling in the rostral ventrolateral medulla, Exp Physiol 100 (2015) 993–1007. [DOI] [PubMed] [Google Scholar]
- [151].Li J, Nitric oxide synthase (NOS) coexists with activated neurons by skeletal muscle contraction in the brainstem of cats, Life Sci 71 (2002) 2833–43. [DOI] [PubMed] [Google Scholar]
- [152].Patel KP, Li YF, Hirooka Y, Role of nitric oxide in central sympathetic outflow, Exp Biol Med (Maywood) 226 (2001) 814–24. [DOI] [PubMed] [Google Scholar]
- [153].Chan SH, Wang LL, Wang SH, Chan JY, Differential cardiovascular responses to blockade of nNOS or iNOS in rostral ventrolateral medulla of the rat, Br J Pharmacol 133 (2001) 606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Ville Y, Fernandez H, Samuel D, Bismuth H, Frydman R, Pregnancy in liver transplant recipients: course and outcome in 19 cases, Am J Obstet Gynecol 168 (1993) 896–902. [DOI] [PubMed] [Google Scholar]
- [155].Chikada N, Imaki T, Seki T, Harada S, Nakajima K, Yoshimoto T, Naruse M, Demura H, Minami S, Takano K, Distribution of c-fos mRNA in the brain following intracerebroventricular injection of nitric oxide (NO)-releasing compounds: possible role of NO in central cardiovascular regulation, J Neuroendocrinol 12 (2000) 1112–23. [DOI] [PubMed] [Google Scholar]
- [156].Kishi T, Hirooka Y, Sakai K, Shigematsu H, Shimokawa H, Takeshita A, Overexpression of eNOS in the RVLM causes hypotension and bradycardia via GABA release, Hypertension 38 (2001) 896–901. [PubMed] [Google Scholar]
- [157].Ally A, Maher TJ, Endothelial NOS expression within the ventrolateral medulla can affect cardiovascular function during static exercise in stroked rats, Brain Res 1196 (2008) 33–40. [DOI] [PubMed] [Google Scholar]
- [158].Ally A, Nauli SM, Maher TJ, Molecular changes in nNOS protein expression within the ventrolateral medulla following transient focal ischemia affect cardiovascular functions, Brain Res 1055 (2005) 73–82. [DOI] [PubMed] [Google Scholar]
- [159].Ishide T, Maher TJ, Ally A, Role of nitric oxide in the ventrolateral medulla on cardiovascular responses and glutamate neurotransmission during mechanical and thermal stimuli, Pharmacol Res 47 (2003) 59–68. [DOI] [PubMed] [Google Scholar]
- [160].Shinohara K, Hirooka Y, Kishi T, Sunagawa K, Reduction of nitric oxide-mediated gamma-amino butyric acid release in rostral ventrolateral medulla is involved in superoxide-induced sympathoexcitation of hypertensive rats, Circ J 76 (2012) 2814–21. [DOI] [PubMed] [Google Scholar]
- [161].Ogihara CA, Schoorlemmer GH, Lazari Mde F, Giannocco G, Lopes OU, Colombari E, Sato MA, Swimming exercise changes hemodynamic responses evoked by blockade of excitatory amino receptors in the rostral ventrolateral medulla in spontaneously hypertensive rats, Biomed Res Int 2014 (2014) 487129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Ally A, Nauli SM, Maher TJ, Cardiovascular responses and neurotransmission in the ventrolateral medulla during skeletal muscle contraction following transient middle cerebral artery occlusion and reperfusion, Brain Res 952 (2002) 176–87. [DOI] [PubMed] [Google Scholar]
- [163].Ishide T, Preuss CV, Maher TJ, Ally A, Neurochemistry within ventrolateral medulla and cardiovascular effects during static exercise following eNOS antagonism, Neurosci Res 52 (2005) 21–30. [DOI] [PubMed] [Google Scholar]
- [164].Yen DH, Chen LC, Shen YC, Chiu YC, Ho IC, Lou YJ, Chen IC, Yen JC, Protein kinase A-dependent neuronal nitric oxide synthase activation mediates the enhancement of baroreflex response by adrenomedullin in the nucleus tractus solitarii of rats, J Biomed Sci 18 (2011) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Wang X, Shimizu-Sasamata M, Moskowitz MA, Newcomb R, Lo EH, Profiles of glutamate and GABA efflux in core versus peripheral zones of focal cerebral ischemia in mice, Neurosci Lett 313 (2001) 121–4. [DOI] [PubMed] [Google Scholar]
- [166].Wang YX, Pang CC, Functional integrity of the central and sympathetic nervous systems is a prerequisite for pressor and tachycardic effects of diphenyleneiodonium, a novel inhibitor of nitric oxide synthase, J Pharmacol Exp Ther 265 (1993) 263–72. [PubMed] [Google Scholar]
- [167].Wang Y, Patel KP, Cornish KG, Channon KM, Zucker IH, nNOS gene transfer to RVLM improves baroreflex function in rats with chronic heart failure, Am J Physiol Heart Circ Physiol 285 (2003) H1660–7. [DOI] [PubMed] [Google Scholar]
- [168].Mayorov DN, Selective sensitization by nitric oxide of sympathetic baroreflex in rostral ventrolateral medulla of conscious rabbits, Hypertension 45 (2005) 901–6. [DOI] [PubMed] [Google Scholar]
- [169].Resstel LB, Correa FM, Medial prefrontal cortex NMDA receptors and nitric oxide modulate the parasympathetic component of the baroreflex, Eur J Neurosci 23 (2006) 481–8. [DOI] [PubMed] [Google Scholar]
- [170].Fletcher J, Moody WE, Chowdhary S, Coote JH, NO-cGMP pathway at ventrolateral medullary cardiac inhibitory sites enhances the baroreceptor reflex bradycardia in the rat, Brain Res 1123 (2006) 125–34. [DOI] [PubMed] [Google Scholar]
- [171].Carvalho TH, Lopes OU, Tolentino-Silva FR, Baroreflex responses in neuronal nitric oxide synthase knoukout mice (nNOS), Auton Neurosci 126–127 (2006) 163–8. [DOI] [PubMed] [Google Scholar]
- [172].Guo ZL, Tjen ALSC, Fu LW, Longhurst JC, Nitric oxide in rostral ventrolateral medulla regulates cardiac-sympathetic reflexes: role of synthase isoforms, Am J Physiol Heart Circ Physiol 297 (2009) H1478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Rossi NF, Maliszewska-Scislo M, Chen H, Black SM, Sharma S, Ravikov R, Augustyniak RA, Neuronal nitric oxide synthase within paraventricular nucleus: blood pressure and baroreflex in two-kidney, one-clip hypertensive rats, Exp Physiol 95 (2010) 845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Gao J, Zucker IH, Gao L, Activation of central angiotensin type 2 receptors by compound 21 improves arterial baroreflex sensitivity in rats with heart failure, Am J Hypertens 27 (2014) 1248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Ramchandra R, Hood SG, May CN, Central exogenous nitric oxide decreases cardiac sympathetic drive and improves baroreflex control of heart rate in ovine heart failure, Am J Physiol Regul Integr Comp Physiol 307 (2014) R271–80. [DOI] [PubMed] [Google Scholar]
- [176].Ally A, Hand GA, Mitchell JH, Central injection of physostigmine attenuates exercise-induced pressor response in conscious cats, Am J Physiol 273 (1997) R393–9. [DOI] [PubMed] [Google Scholar]
- [177].Ally A, Hand GA, Mitchell JH, Cardiovascular responses to static exercise in conscious cats: effects of intracerebroventricular injection of clonidine, J Physiol 491 ( Pt 2) (1996) 519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Ally A, Wilson LB, Nobrega AC, Mitchell JH, Cardiovascular effects elicited by central administration of physostigmine via M2 muscarinic receptors in conscious cats, Brain Res 677 (1995) 268–76. [DOI] [PubMed] [Google Scholar]
- [179].Mitchell JH, Kaufman MP, Iwamoto GA, The exercise pressor reflex: its cardiovascular effects, afferent mechanisms, and central pathways, Annu Rev Physiol 45 (1983) 229–42. [DOI] [PubMed] [Google Scholar]
- [180].Towiwat P, Phattanarudee S, Maher TJ, Ally A, Modulation of inducible nitric oxide synthase (iNOS) expression and cardiovascular responses during static exercise following iNOS antagonism within the ventrolateral medulla, Mol Cell Biochem 398 (2015) 185–94. [DOI] [PubMed] [Google Scholar]
- [181].Nauli SM, Maher TJ, Pearce WJ, Ally A, Effects of opioid receptor activation on cardiovascular responses and extracellular monoamines within the rostral ventrolateral medulla during static contraction of skeletal muscle, Neurosci Res 41 (2001) 373–83. [DOI] [PubMed] [Google Scholar]
- [182].Nauli SM, Pearce WJ, Amer A, Maher TJ, Ally A, Effects of nitric oxide and GABA interaction within ventrolateral medulla on cardiovascular responses during static muscle contraction, Brain Res 922 (2001) 234–42. [DOI] [PubMed] [Google Scholar]
- [183].Freda BJ, Gaitonde RS, Lillaney R, Ally A, Cardiovascular responses to muscle contraction following microdialysis of nitric oxide precursor into ventrolateral medulla, Brain Res 828 (1999) 60–7. [DOI] [PubMed] [Google Scholar]
- [184].Asmundsson G, Mokler DJ, Ally A, Effects of ventrolateral medullary NMDA-receptor antagonism on biogenic amines and pressor response to muscle contraction, Neurosci Res 32 (1998) 47–56. [DOI] [PubMed] [Google Scholar]
- [185].Caringi D, Maher TJ, Chaiyakul P, Asmundsson G, Ishide T, Ally A, Extracellular glutamate increases in rostral ventrolateral medulla during static muscle contraction, Pflugers Arch 435 (1998) 465–71. [DOI] [PubMed] [Google Scholar]
- [186].Caringi D, Mokler DJ, Koester DM, Ally A, Rostral ventrolateral medullary opioid receptor activation modulates pressor response to muscle contraction, Am J Physiol 274 (1998) H139–46. [DOI] [PubMed] [Google Scholar]
- [187].Asmundsson G, Caringi D, Mokler DJ, Kobayashi T, Ishide T, Ally A, Extracellular serotonin changes in VLM during muscle contraction: effects of 5-HT1A-receptor activation, Am J Physiol 273 (1997) H2899–909. [DOI] [PubMed] [Google Scholar]
- [188].Endlich PW, Aires RD, Goncalves RL, Costa ED, de J Angelo Paula Arantes, Alves LF, da Silva RF, Rezende BA, Cortes SF, Lemos VS, Neuronal nitric oxide synthase-derived hydrogen peroxide effect in grafts used in human coronary bypass surgery, Clin Sci (Lond) 131 (2017) 1015–1026. [DOI] [PubMed] [Google Scholar]
- [189].Shabeeh H, Khan S, Jiang B, Brett S, Melikian N, Casadei B, Chowienczyk PJ, Shah AM, Blood Pressure in Healthy Humans Is Regulated by Neuronal NO Synthase, Hypertension 69 (2017) 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Fadel PJ, Nitric Oxide and Cardiovascular Regulation: Beyond the Endothelium, Hypertension 69 (2017) 778–779. [DOI] [PubMed] [Google Scholar]
- [191].Zhang YH, Nitric oxide signalling and neuronal nitric oxide synthase in the heart under stress, F1000Res 6 (2017) 742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Zhang YH, Jin CZ, Jang JH, Wang Y, Molecular mechanisms of neuronal nitric oxide synthase in cardiac function and pathophysiology, J Physiol 592 (2014) 3189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Zhang YH, Neuronal nitric oxide synthase in hypertension - an update, Clin Hypertens 22 (2016) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Costa ED, Rezende BA, Cortes SF, Lemos VS, Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases, Front Physiol 7 (2016) 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Costa ED, Silva JF, Aires RD, Garcia DC, Kansaon MJ, Wainstein AJ, Rezende BA, Teixeira MM, Silva RF, Cortes SF, Lemos VS, Neuronal nitric oxide synthase contributes to the normalization of blood pressure in medicated hypertensive patients, Nitric Oxide 80 (2018) 98–107. [DOI] [PubMed] [Google Scholar]
- [196].Silva GC, Silva JF, Diniz TF, Lemos VS, Cortes SF, Endothelial dysfunction in DOCA-salt-hypertensive mice: role of neuronal nitric oxide synthase-derived hydrogen peroxide, Clin Sci (Lond) 130 (2016) 895–906. [DOI] [PubMed] [Google Scholar]
- [197].Sousa LE, Magalhaes WG, Bezerra FS, Santos RA, Campagnole-Santos MJ, Isoldi MC, Alzamora AC, Exercise training restores oxidative stress and nitric oxide synthases in the rostral ventrolateral medulla of renovascular hypertensive rats, Free Radic Res 49 (2015) 1335–43. [DOI] [PubMed] [Google Scholar]
- [198].Khan SG, Geer A, Fok HW, Shabeeh H, Brett SE, Shah AM, Chowienczyk PJ, Impaired neuronal nitric oxide synthase-mediated vasodilator responses to mental stress in essential hypertension, Hypertension 65 (2015) 903–9. [DOI] [PubMed] [Google Scholar]
- [199].Overwyk KJ, Dehmer SP, Roy K, Maciosek MV, Hong Y, Baker-Goering MM, Loustalot F, Singleton CM, Ritchey MD, Modeling the Health and Budgetary Impacts of a Team-based Hypertension Care Intervention That Includes Pharmacists, Med Care 57 (2019) 882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Coca A, Economic benefits of treating high-risk hypertension with angiotensin II receptor antagonists (blockers), Clin Drug Investig 28 (2008) 211–20. [DOI] [PubMed] [Google Scholar]
- [201].Donnan GA, Fisher M, Macleod M, Davis SM, Stroke, Lancet 371 (2008) 1612–23. [DOI] [PubMed] [Google Scholar]
- [202].Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA, Connor M, Bennett DA, Moran AE, Sacco RL, Anderson L, Truelsen T, O’Donnell M, Venketasubramanian N, Barker-Collo S, Lawes CM, Wang W, Shinohara Y, Witt E, Ezzati M, Naghavi M, Murray C, Global I Burden of Diseases, S. Risk Factors, G.B.D.S.E.G. the, Global and regional burden of stroke during 1990–2010: findings from the Global Burden of Disease Study 2010, Lancet 383 (2014) 245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Lai TW, Zhang S, Wang YT, Excitotoxicity and stroke: identifying novel targets for neuroprotection, Prog Neurobiol 115 (2014) 157–88. [DOI] [PubMed] [Google Scholar]
- [204].Arundine M, Tymianski M, Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity, Cell Calcium 34 (2003) 325–37. [DOI] [PubMed] [Google Scholar]
- [205].Chan PH, Reactive oxygen radicals in signaling and damage in the ischemic brain, J Cereb Blood Flow Metab 21 (2001) 2–14. [DOI] [PubMed] [Google Scholar]
- [206].Sattler R, Tymianski M, Molecular mechanisms of calcium-dependent excitotoxicity, J Mol Med (Berl) 78 (2000) 3–13. [DOI] [PubMed] [Google Scholar]
- [207].Schwartz-Bloom RD, Sah R, gamma-Aminobutyric acid(A) neurotransmission and cerebral ischemia, J Neurochem 77 (2001) 353–71. [DOI] [PubMed] [Google Scholar]
- [208].Cardenas A, Moro MA, Hurtado O, Leza JC, Lorenzo P, Castrillo A, Bodelon OG, Bosca L, Lizasoain I, Implication of glutamate in the expression of inducible nitric oxide synthase after oxygen and glucose deprivation in rat forebrain slices, J Neurochem 74 (2000) 2041–8. [DOI] [PubMed] [Google Scholar]
- [209].Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME, Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene, J Neurosci 17 (1997) 9157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Iadecola C, Faris PL, Hartman BK, Xu X, Localization of NADPH diaphorase in neurons of the rostral ventral medulla: possible role of nitric oxide in central autonomic regulation and oxygen chemoreception, Brain Res 603 (1993) 173–9. [DOI] [PubMed] [Google Scholar]
- [211].Dawson TM, Bredt DS, Fotuhi M, Hwang PM, Snyder SH, Nitric oxide synthase and neuronal NADPH diaphorase are identical in brain and peripheral tissues, Proc Natl Acad Sci U S A 88 (1991) 7797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH, Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures, Proc Natl Acad Sci U S A 88 (1991) 6368–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Hallenbeck JM, Dutka AJ, Background review and current concepts of reperfusion injury, Arch Neurol 47 (1990) 1245–54. [DOI] [PubMed] [Google Scholar]
- [214].Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA, Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide, Proc Natl Acad Sci U S A 87 (1990) 1620–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Lu J, Wang D, Advances in endovascular therapy for ischemic cerebrovascular diseases, Chronic Dis Transl Med 2 (2016) 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Sadasivan S, Maher TJ, Quang LS, Gamma-Hydroxybutyrate (GHB), gamma-butyrolactone (GBL), and 1,4-butanediol (1,4-BD) reduce the volume of cerebral infarction in rodent transient middle cerebral artery occlusion, Ann N Y Acad Sci 1074 (2006) 537–44. [DOI] [PubMed] [Google Scholar]
- [217].Fletcher BJ, Dunbar SB, Felner JM, Jensen BE, Almon L, Cotsonis G, Fletcher GF, Exercise testing and training in physically disabled men with clinical evidence of coronary artery disease, Am J Cardiol 73 (1994) 170–4. [DOI] [PubMed] [Google Scholar]
- [218].Gordon NF, Gulanick M, Costa F, Fletcher G, Franklin BA, Roth EJ, Shephard T, S.o.E.C.R. American Heart Association Council on Clinical Cardiology, Prevention, N. the Council on Cardiovascular, P.A. the Council on Nutrition, Metabolism, C. the Stroke, Physical activity and exercise recommendations for stroke survivors: an American Heart Association scientific statement from the Council on Clinical Cardiology, Subcommittee on Exercise, Cardiac Rehabilitation, and Prevention; the Council on Cardiovascular Nursing; the Council on Nutrition, Physical Activity, and Metabolism; and the Stroke Council, Circulation 109 (2004) 2031–41. [DOI] [PubMed] [Google Scholar]
- [219].Saleh TM, Cribb AE, Connell BJ, Estrogen-induced recovery of autonomic function after middle cerebral artery occlusion in male rats, Am J Physiol Regul Integr Comp Physiol 281 (2001) R1531–9. [DOI] [PubMed] [Google Scholar]
- [220].Brill PA, Drimmer AM, Morgan LA, Gordon NF, The feasibility of conducting strength and flexibility programs for elderly nursing home residents with dementia, Gerontologist 35 (1995) 263–6. [DOI] [PubMed] [Google Scholar]
- [221].Bath PM, Krishnan K, Appleton JP, Nitric oxide donors (nitrates), L-arginine, or nitric oxide synthase inhibitors for acute stroke, Cochrane Database Syst Rev 4 (2017) CD000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Dai Y, He Z, Sui R, Jiang Z, Ma S, Association of nNOS gene polymorphism with ischemic stroke in Han Chinese of North China, ScientificWorldJournal 2013 (2013) 891581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].He H, Feng M, Qu C, Lu X, Variants in neuronal nitric oxide synthase gene may contribute to increased ischemic stroke susceptibility in a Han Chinese population, Cell Biochem Biophys 70 (2014) 179–87. [DOI] [PubMed] [Google Scholar]
- [224].Yoshida H, Yanai H, Namiki Y, Fukatsu-Sasaki K, Furutani N, Tada N, Neuroprotective effects of edaravone: a novel free radical scavenger in cerebrovascular injury, CNS Drug Rev 12 (2006) 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Saransaari P, Oja SS, Characteristics of GABA release in mouse brain stem slices under normal and ischemic conditions, Neurochem Res 30 (2005) 1549–56. [DOI] [PubMed] [Google Scholar]
- [226].Lin YH, Liang HY, Xu K, Ni HY, Dong J, Xiao H, Chang L, Wu HY, Li F, Zhu DY, Luo CX, Dissociation of nNOS from PSD-95 promotes functional recovery after cerebral ischaemia in mice through reducing excessive tonic GABA release from reactive astrocytes, J Pathol 244 (2018) 176–188. [DOI] [PubMed] [Google Scholar]
- [227].Shao S, Xu M, Zhou J, Ge X, Chen G, Guo L, Luo L, Li K, Zhu Z, Zhang F, Atorvastatin Attenuates Ischemia/Reperfusion-Induced Hippocampal Neurons Injury Via Akt-nNOS-JNK Signaling Pathway, Cell Mol Neurobiol 37 (2017) 753–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Hoque A, Hossain MI, Ameen SS, Ang CS, Williamson N, Ng DC, Chueh AC, Roulston C, Cheng HC, A beacon of hope in stroke therapy-Blockade of pathologically activated cellular events in excitotoxic neuronal death as potential neuroprotective strategies, Pharmacol Ther 160 (2016) 159–79. [DOI] [PubMed] [Google Scholar]
- [229].Shepherd RB, Exercise and training to optimize functional motor performance in stroke: driving neural reorganization?, Neural Plast 8 (2001) 121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Duncan P, Richards L, Wallace D, Stoker-Yates J, Pohl P, Luchies C, Ogle A, Studenski S, A randomized, controlled pilot study of a home-based exercise program for individuals with mild and moderate stroke, Stroke 29 (1998) 2055–60. [DOI] [PubMed] [Google Scholar]
- [231].Franzoni F, Federighi G, Fusi J, Agosta V, Cerri E, Banducci R, Petrocchi A, Bernardi R, Innocenti A, Pruneti C, Daniele S, Pellegrini S, Martini C, Scuri R, Galetta F, Physical Exercise Improves Total Antioxidant Capacity and Gene Expression in Rat Hippocampal Tissue, Arch Ital Biol 155 (2017) 1–10. [DOI] [PubMed] [Google Scholar]
- [232].Joukar S, Vahidi R, Farsinejad A, Asadi-Shekaari M, Shahouzehi B, Ameliorative Effects of Endurance Exercise with Two Different Intensities on Nandrolone Decanoate-Induced Neurodegeneration in Rats: Involving Redox and Apoptotic Systems, Neurotox Res 32 (2017) 41–49. [DOI] [PubMed] [Google Scholar]
- [233].Nonato LF, Rocha-Vieira E, Tossige-Gomes R, Soares AA, Soares BA, Freitas DA, Oliveira MX, Mendonca VA, Lacerda AC, Massensini AR, Leite HR, Swimming training attenuates oxidative damage and increases enzymatic but not non-enzymatic antioxidant defenses in the rat brain, Braz J Med Biol Res 49 (2016) e5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Tomiga Y, Yoshimura S, Ra SG, Takahashi Y, Goto R, Kugimoto I, Uehara Y, Kawanaka K, Higaki Y, Anxiety-like behaviors and hippocampal nNOS in response to diet-induced obesity combined with exercise, J Physiol Sci 69 (2019) 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Raquel Hde A, Masson GS, Barna BF, Zanluqui NG, Pinge-Filho P, Michelini LC, Martins-Pinge MC, Swimming Training Modulates Nitric Oxide-Glutamate Interaction in the Rostral Ventrolateral Medulla in Normotensive Conscious Rats, Front Physiol 7 (2016) 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Nosarev AV, Smagliy LV, Anfinogenova Y, Popov SV, Kapilevich LV, Exercise and NO production: relevance and implications in the cardiopulmonary system, Front Cell Dev Biol 2 (2014) 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Tomiga Y, Yoshimura S, Ito A, Nakashima S, Kawanaka K, Uehara Y, Tanaka H, Higaki Y, Exercise training rescues high fat diet-induced neuronal nitric oxide synthase expression in the hippocampus and cerebral cortex of mice, Nitric Oxide 66 (2017) 71–77. [DOI] [PubMed] [Google Scholar]
- [238].Liu H, Xue J, Involvement of hypothalamic nitric oxide signaling in the modulation of a rat’s exercise capacity, Neuroreport 28 (2017) 408–413. [DOI] [PubMed] [Google Scholar]
- [239].Marton O, Koltai E, Takeda M, Mimura T, Pajk M, Abraham D, Koch LG, Britton SL, Higuchi M, Boldogh I, Radak Z, The rate of training response to aerobic exercise affects brain function of rats, Neurochem Int 99 (2016) 16–23. [DOI] [PubMed] [Google Scholar]
- [240].Arrick DM, Yang S, Li C, Cananzi S, Mayhan WG, Vigorous exercise training improves reactivity of cerebral arterioles and reduces brain injury following transient focal ischemia, Microcirculation 21 (2014) 516–23. [DOI] [PubMed] [Google Scholar]
- [241].Siesjo BK, Katsura K, Mellergard P, Ekholm A, Lundgren J, Smith ML, Acidosis-related brain damage, Prog Brain Res 96 (1993) 23–48. [PubMed] [Google Scholar]
- [242].Olesen J, Ashina M, Emerging migraine treatments and drug targets, Trends Pharmacol Sci 32 (2011) 352–9. [DOI] [PubMed] [Google Scholar]
- [243].Feuerwerker E, Couillard P, Gauthier Y, Herbert Spencer’s influence on the genesis of Sherrington’s concept of the integrative action of the nervous system, Bull Can Hist Med 2 (1985) 205–18. [DOI] [PubMed] [Google Scholar]
- [244].Roaf HE, Sherrington CS, Experiments in examination of the; locked-jaw’ induced by tetanus toxin, J Physiol 34 (1906) 315–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Sherrington CS, Observations on the scratch-reflex in the spinal dog, J Physiol 34 (1906) 1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Hegarty A, Portenoy RK, Pharmacotherapy of neuropathic pain, Semin Neurol 14 (1994) 213–24. [DOI] [PubMed] [Google Scholar]
- [247].Le Bars D, Gozariu M, Cadden SW, Animal models of nociception, Pharmacol Rev 53 (2001) 597–652. [PubMed] [Google Scholar]
- [248].Lovick TA, Midbrain and medullary regulation of defensive cardiovascular functions, Prog Brain Res 107 (1996) 301–13. [DOI] [PubMed] [Google Scholar]
- [249].Bandler R, Keay KA, Columnar organization in the midbrain periaqueductal gray and the integration of emotional expression, Prog Brain Res 107 (1996) 285–300. [DOI] [PubMed] [Google Scholar]
- [250].Lovick TA, Integrated activity of cardiovascular and pain regulatory systems: role in adaptive behavioural responses, Prog Neurobiol 40 (1993) 631–44. [DOI] [PubMed] [Google Scholar]
- [251].Reichling DB, Kwiat GC, Basbaum AI, Anatomy, physiology and pharmacology of the periaqueductal gray contribution to antinociceptive controls, Prog Brain Res 77 (1988) 31–46. [DOI] [PubMed] [Google Scholar]
- [252].Chaitoff KA, Patel D, Ally A, Effects of endothelial NOS antagonism within the periaqueductal gray on cardiovascular responses and neurotransmission during mechanical, heat, and cold nociception, Brain Res 1236 (2008) 93–104. [DOI] [PubMed] [Google Scholar]
- [253].Karlsson GA, Chaitoff KA, Hossain S, Bohlke M, Maher TJ, Ally A, Modulation of cardiovascular responses and neurotransmission during peripheral nociception following nNOS antagonism within the periaqueductal gray, Brain Res 1143 (2007) 150–60. [DOI] [PubMed] [Google Scholar]
- [254].Karlsson GA, Preuss CV, Chaitoff KA, Maher TJ, Ally A, Medullary monoamines and NMDA-receptor regulation of cardiovascular responses during peripheral nociceptive stimuli, Neurosci Res 55 (2006) 316–26. [DOI] [PubMed] [Google Scholar]
- [255].Gray TK, Lewis E 3rd, Maher TJ, Ally A, AMPA-receptor blockade within the RVLM modulates cardiovascular responses via glutamate during peripheral stimuli, Pharmacol Res 43 (2001) 47–54. [DOI] [PubMed] [Google Scholar]
- [256].Umeda M, Williams JP, Marino CA, Hilliard SC, Muscle pain and blood pressure responses during isometric handgrip exercise in healthy African American and non-Hispanic White adults, Physiol Behav 138 (2015) 242–6. [DOI] [PubMed] [Google Scholar]
- [257].Ahlawat A, Rana A, Goyal N, Sharma S, Potential role of nitric oxide synthase isoforms in pathophysiology of neuropathic pain, Inflammopharmacology 22 (2014) 269–78. [DOI] [PubMed] [Google Scholar]
- [258].Zamuner AR, Barbic F, Dipaola F, Bulgheroni M, Diana A, Atzeni F, Marchi A, Sarzi-Puttini P, Porta A, Furlan R, Relationship between sympathetic activity and pain intensity in fibromyalgia, Clin Exp Rheumatol 33 (2015) S53–7. [PubMed] [Google Scholar]
- [259].Biondi DM, Xiang J, Etropolski M, Moskovitz B, Evaluation of blood pressure and heart rate in patients with hypertension who received tapentadol extended release for chronic pain: a post hoc, pooled data analysis, Clin Drug Investig 34 (2014) 565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].King JW, Bair E, Duggan D, Maixner W, Khan AA, The relationship between resting arterial blood pressure and acute postoperative pain in endodontic patients, J Orofac Pain 26 (2012) 321–7. [PMC free article] [PubMed] [Google Scholar]
- [261].Cho HS, Shin YS, Lee YH, Cho WH, Ko YK, Relationship between neuronal nitric oxide synthase and NADPH-diaphorase in the dorsal root ganglia during neuropathic pain, Korean J Anesthesiol 57 (2009) 342–349. [DOI] [PubMed] [Google Scholar]
- [262].Hara MR, Snyder SH, Cell signaling and neuronal death, Annu Rev Pharmacol Toxicol 47 (2007) 117–41. [DOI] [PubMed] [Google Scholar]
- [263].Choi SR, Kwon SG, Choi HS, Han HJ, Beitz AJ, Lee JH, Neuronal NOS Activates Spinal NADPH Oxidase 2 Contributing to Central Sigma-1 Receptor-Induced Pain Hypersensitivity in Mice, Biol Pharm Bull 39 (2016) 1922–1931. [DOI] [PubMed] [Google Scholar]
- [264].Zhou Z, Liang Y, Deng F, Cheng Y, Sun J, Guo L, Xu G, Phosphorylated neuronal nitric oxide synthase in neuropathic pain in rats, Int J Clin Exp Pathol 8 (2015) 12748–56. [PMC free article] [PubMed] [Google Scholar]
- [265].Cellek S, Point of NO return for nitrergic nerves in diabetes: a new insight into diabetic complications, Curr Pharm Des 10 (2004) 3683–95. [DOI] [PubMed] [Google Scholar]
- [266].Lee WH, Xu Z, Ashpole NM, Hudmon A, Kulkarni PM, Thakur GA, Lai YY, Hohmann AG, Small molecule inhibitors of PSD95-nNOS protein-protein interactions as novel analgesics, Neuropharmacology 97 (2015) 464–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Annedi SC, Maddaford SP, Mladenova G, Ramnauth J, Rakhit S, Andrews JS, Lee DK, Zhang D, Porreca F, Bunton D, Christie L, Discovery of N-(3-(1-methyl-1,2,3,6-tetrahydropyridin-4-yl)-1H-indol-6-yl) thiophene-2-carboximidamide as a selective inhibitor of human neuronal nitric oxide synthase (nNOS) for the treatment of pain, J Med Chem 54 (2011) 7408–16. [DOI] [PubMed] [Google Scholar]
- [268].Annedi SC, Maddaford SP, Ramnauth J, Renton P, Speed J, Rakhit S, Andrews JS, Porreca F, 3,5-Disubstituted indole derivatives as selective human neuronal nitric oxide synthase (nNOS) inhibitors, Bioorg Med Chem Lett 22 (2012) 1980–4. [DOI] [PubMed] [Google Scholar]
- [269].Lowenstein CJ, Beneficial effects of neuronal nitric oxide synthase in atherosclerosis, Arterioscler Thromb Vasc Biol 26 (2006) 1417–8. [DOI] [PubMed] [Google Scholar]
- [270].Kuhlencordt PJ, Hotten S, Schodel J, Rutzel S, Hu K, Widder J, Marx A, Huang PL, Ertl G, Atheroprotective effects of neuronal nitric oxide synthase in apolipoprotein e knockout mice, Arterioscler Thromb Vasc Biol 26 (2006) 1539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Schodel J, Padmapriya P, Marx A, Huang PL, Ertl G, Kuhlencordt PJ, Expression of neuronal nitric oxide synthase splice variants in atherosclerotic plaques of apoE knockout mice, Atherosclerosis 206 (2009) 383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Tsutsui M, Neuronal nitric oxide synthase as a novel anti-atherogenic factor, J Atheroscler Thromb 11 (2004) 41–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.