

Abstract
Gaucher’s disease type 1 (GD1) leads to significant morbidity and mortality through clinical manifestations, such as splenomegaly, hematological complications, and bone disease. Two types of therapies are currently approved for GD1: enzyme replacement therapy (ERT), and substrate reduction therapy (SRT). In this study, we have developed a quantitative systems pharmacology (QSP) model, which recapitulates the effects of eliglustat, the only first‐line SRT approved for GD1, on treatment‐naïve or patients with ERT‐stabilized adult GD1. This multiscale model represents the mechanism of action of eliglustat that leads toward reduction of spleen volume. Model capabilities were illustrated through the application of the model to predict ERT and eliglustat responses in virtual populations of adult patients with GD1, representing patients across a spectrum of disease severity as defined by genotype‐phenotype relationships. In summary, the QSP model provides a mechanistic computational platform for predicting treatment response via different modalities within the heterogeneous GD1 patient population.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Gaucher’s disease type‐1 (GD1) is a monogenetic rare disease with heterogeneous clinical manifestations resulting from the dysregulation of glycosphingolipid metabolism. Currently, two types of therapies are approved for GD1: enzyme replacement therapy (ERT), and substrate reduction therapy (SRT).
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ We developed a multiscale quantitative systems pharmacology (QSP) model that connects the dysregulation of glycosphingolipid metabolism due to GD1 mutations‐induced enzyme deficiency to organ‐level clinical end point. This model was informed by a data‐driven genotype‐phenotype relationship to represent a wide spectrum of patients with mild to severe GD1.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The QSP model is able to recapitulate the range of variability seen in patients with GD1 in the clinic. This provides mechanistic insight into ERT to SRT switch therapy response and prediction of therapeutic response of patients with different disease severities.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUITCS?
☑ This QSP model highlights how real‐world evidence can be incorporated into mechanistic models to predict the therapeutic response of a more diverse patient population compared with that observed from clinical trial data. This model could be used for comparability assessment of drugs with different mechanisms of action, in different GD1 patient subpopulations.
Gaucher’s disease type 1 (GD1) is the non‐neuronopathic form of the autosomal recessive disorder caused by mutations in the gene GBA, which encodes the enzyme acid β‐glucosidase. Acid β‐glucosidase catalyzes the final step in glycosphingolipid (GSL) degradation, the conversion of glucosylceramide (GL‐1) into ceramide. Deficiency in acid β‐glucosidase activity causes progressive accumulation of GL‐1 within cells of multiple organ systems, leading to the heterogeneous clinical manifestations observed in GD1, including splenomegaly, hepatomegaly, anemia, thrombocytopenia, and bone disease. 1 , 2 Accumulation of glucosylsphingosine (lyso‐GL‐1), an additional substrate of acid β‐glucosidase and a cytotoxic disease biomarker, also characterizes the disease. 3 , 4
Two therapeutic approaches are currently approved for the treatment of GD1, both of which help to restore the balance between substrate production and degradation to counteract GL‐1 accumulation. The historic standard of care is enzyme replacement therapy (ERT), which supplements the defective endogenous lysosomal acid β‐glucosidase. ERT for GD1 has been available since 1991, first as human placenta‐derived enzyme (alglucerase, Ceredase) and subsequently, in 1994, as imiglucerase (Cerezyme) a human recombinant form of the enzyme. Alglucerase and imiglucerase have been shown to be therapeutically equivalent in a randomized, two‐arm clinical trial 5 ; and from here on both enzymes are referred to as “Cerezyme”. Cerezyme, administered in biweekly infusions, 6 is efficacious in treating visceral, hematological, and skeletal manifestations of GD1 in both adults and children, in addition to treating growth deficits in children. 7 , 8 , 9 , 10 Substrate reduction therapy slows down the production of GL‐1 by partial inhibition of glucosylceramide synthase. Substrate reduction therapy (SRT) is an oral treatment, and, thus, considered more convenient to ERT’s intravenous mode of administration. The first SRT was miglustat, approved in 2003 as a second‐line treatment for patients unable to tolerate ERT. Eliglustat, a structurally distinct SRT from miglustat, was approved by the US Food and Drug Administration (FDA) in 2014 and by the European Medicines Agency (EMA) in 2015, as a first‐line treatment for adults with GD1 who have poor, intermediate, or extensive cytochrome P450 (CYP) 2D6 metabolizer phenotypes (> 90% of patients 11 ). Eliglustat has demonstrated clinically meaningful improvements in spleen and liver volume, hematological manifestations, and bone mineral density measures in previously untreated adults. 12 , 13 , 14 , 15 , 16 Eliglustat has also shown noninferiority to Cerezyme in the maintenance in adults of clinical parameters previously stabilized on ERT. 17 , 18
Here, we present a quantitative systems pharmacology (QSP) model that describes the pathogenesis of GD1 from subcellular to organ level, to simulate key clinical end points and biochemical markers in treatment naïve patients treated with eliglustat, or patients switching to eliglustat after being previously stabilized on ERT. QSP is a multiscale, mechanistic modeling approach that links the biochemical processes disrupted in the disease state (e.g., deficiency in activity of acid β‐glucosidase) to consequent aberrant behavior at the cellular level (e.g., the formation of lipid‐laden Gaucher cells) and organ level dysfunction (e.g., splenic enlargement). QSP modeling thereby provides an integrative perspective on the disease process and pharmacological effects on biomarkers and clinical end points, and offers mechanistic insight into variability in disease severity and treatment response within the GD1 patient population.
QSP is an emerging discipline gaining considerable traction to support drug development. The utilization of such mechanistic modeling approaches has been endorsed by both the FDA and EMA. QSP models support therapeutic development across a variety of disease areas, including cancer, 19 cardiovascular disease, 20 and diabetes, 21 as well as immunological 22 and neurological conditions. 23 , 24 Although other modeling studies have examined lysosomal storage diseases in general 25 and Gaucher’s disease in particular, 26 these efforts focused on molecular‐level processes or took an empirical modeling approach, respectively. Among other lysosomal storage diseases, QSP has been applied to acid sphingomyelinase deficiency Niemann–Pick disease, supporting the development of the enzyme replacement therapy olipudase alfa. 27 The GD1 QSP model presented here describes key biochemical reactions and organs implicated in this disease, and provides a mechanistic framework for addressing clinically relevant questions related to the substantial heterogeneity observed among patients with GD1.
In this study, we present the design, calibration, and qualification of the first GD1 QSP model. We demonstrate that the model can recapitulate patient response trends observed in clinical trials for eliglustat. We also perform a systematic analysis of the genotype‐phenotype relationship in GD1, and apply it to help stratify model‐predicted patient responses across the spectrum of GD1 disease severity.
METHODS
Data sources
The mechanistic detail in QSP models requires integration of data from diverse sources to represent the biology of the disease and the patient phenotypes of interest for pharmacological response evaluation. For the GD1 model, data sources include internal resources (i.e., clinical trial data), published literature, and data collected by the International Collaborative Gaucher Group (ICGG) Gaucher Registry. 28 The ICGG Gaucher Registry (NCT00358943) is a Sanofi Genzyme‐sponsored program, first established in 1991. The ICGG Gaucher Registry is the largest voluntary observational database for Gaucher’s disease, tracking demographics and real‐world clinical outcomes for more than 6,000 patients with Gaucher’s disease from over 60 countries, regardless of treatment history or status. All participants in the eliglustat clinical trials and the ICGG Gaucher Registry provided written informed consent allowing post hoc analysis of de‐identified data. These data were critical in: (i) developing a QSP model that can capture specific clinical attributes of GD1, incorporating markers of GD1 severity, and relevant treatment strategies; (ii) building virtual patient populations that capture the appropriate GD1 phenotypes of interest, and simulating the pharmacological treatment regimens under investigation. Supplemental Table S1 provides a summary of the data sources used in this study, and details at what stage of model development and application different data sources were utilized.
Description of model structure
The GD1 QSP model (Figure 1 ) consists of four submodels representing different scales of biological organization: (i) the pharmacokinetic submodel representing eliglustat; (ii) the molecular‐level submodel describing GSL dynamics within representative cell types of each organ compartment; (iii) the cellular‐level submodel describing the behavior of splenic macrophages; and (iv) the organ‐level submodel describing the spleen volume. A more detailed diagram of the molecular‐level submodel is shown in Supplemental Figure S1 . Further details are included in Supplemental Section S1 .
Figure 1.
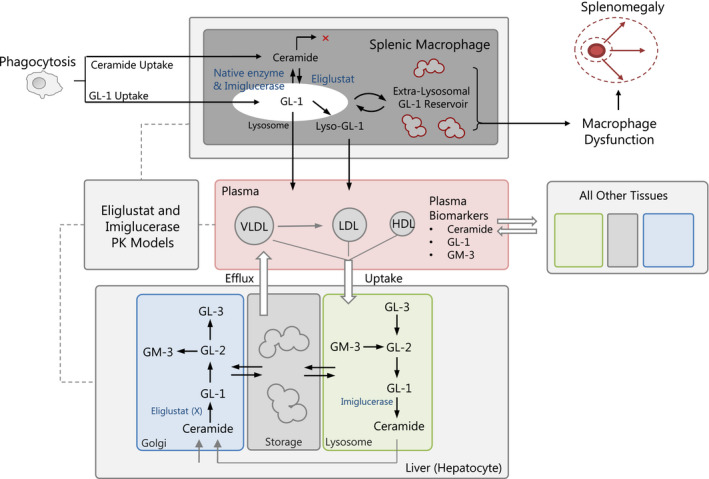
Overview of quantitative systems pharmacology Model Structure. Production and degradation reaction for key glycosphingolipids in their appropriate intracellular organelles are included. Glucosylceramide (GL‐1); glucosylsphingosine (lyso‐GL‐1); lactosylceramide (GL‐2); globotriaosylceramide (GL‐3); monosialodihexosylganglioside (GM‐3). PK, pharmacokinetic.
Parameterization and calibration
The model was calibrated to literature‐derived data and to the clinical end point (spleen volume and multiples of normal (MN)) and four critical biomarkers (plasma GL‐1, lyso‐GL‐1, ceramide, and GM‐3) from the treatment arm of the ENGAGE clinical trial, 12 which evaluated the effects of eliglustat in a population of treatment‐naïve adults with GD1. Unknown model parameters were estimated by applying the Covariance Matrix Adaptation Evolution Strategy. Further details are found in Supplemental Materials Section S2 . Comparison of simulated glycosphingolipid profiles and literature obtained values can be found in Supplemental Figure S2 . Assessment of quality of fits found in Supplemental Figure S3 .
As part of the model calibration strategy, we identified parameters that need to be fitted vs. fixed, based on their role in propagating variability to key biomarkers and end points in the patient population or their potential role in the drug mechanism of action. This is achieved via the use of sensitivity analysis tools, newer approaches like the Linear‐In‐Flux‐Expressions Methodology, 29 and optimization approaches. We considered three categories of parameters, as follows: (1) parameters for which values were extracted directly from the literature—examples of this category include the volumes of different organs, cell types, and organelles, and enzyme kinetic parameters like Michaelis constants; (2) parameters estimated based on literature and other data sources—examples include the rates of phagocytic uptake of ceramide and GL‐1 by splenic macrophages; and (3) parameters calibrated by comparing the model outputs to data. Parameters in this last category were: (i) those for which no measurements or published data could be obtained; (ii) those which represent aggregated rates for complex, multistep biological processes; and/or (iii) those for which notable variability among individuals is expected based on clinical experience or sensitivity analysis. Examples in this category were the patient‐specific residual acid β‐glucosidase activity and rates of disease‐induced spleen growth and repair.
Qualification scenarios
In order to test the predictive capabilities of the model on data not utilized in model calibration, two model qualification scenarios are presented. To this end, a virtual population was constructed as follow: individual model calibrations to plasma lyso‐GL‐1 response from patients in the treatment arm and open‐label period of the ENGAGE trial yielded a set of individual parameter estimates, from which the mean and covariance matrix were used to generate a virtual population of 1,000 subjects. Further details about the generation of the virtual population used in these qualification scenarios can be found in Supplementary Materials Section S4 . In the first qualification scenario, the model was applied to predict the response of ERT‐stabilized patients switched to eliglustat, as evaluated in the ENCORE clinical trial. 17 , 18 Given how treatment‐naïve data were not available for patients enrolled in ENCORE (as initiation on the trial required them to be stabilized on ERT), we assumed similar naïve GSLs and spleen volume dynamics to those observed pretreatment in the ENGAGE trial. We also incorporated a representation of ERT treatment as described in the methods, so that ERT‐treated virtual patients would stabilize to levels comparable to ENCORE baseline data. ERT‐stable patients were generated using the individual treatment‐naïve calibrated parameterizations of the model, and simulated ERT treatment until steady‐state was reached, with only virtual patients reaching spleen volumes < 10 MN (ENCORE’s inclusion criteria) included in the qualification scenario. At this point, ERT to eliglustat switch was simulated. In the second qualification scenario, we generated a virtual population of adults with GD1 replicating the baseline (untreated) spleen volume characteristics of the GD1 population in the ICGG Gaucher Registry and the prevalence of severities (described in Supplemental Section S4 ), and simulated eliglustat treatment outcomes for patients of different disease severity types. Genotype–phenotype analysis is described in Supplemental Materials Section S3 .
Implementation
The model was developed and simulated using MATLAB (The MathWorks, Natick, MA) and using C through the Multiple Interfaces Solver Toolkit. 30 A list of state variables, parameters, equations, ordinary differential equations, and individual calibrations summary can be found in Tables [Link] , [Link] , [Link] , [Link] , [Link] – [Link] , [Link] , [Link] , [Link] , [Link] in Supplemental Material s. MATLAB code can be found in the Supporting Information Section.
RESULTS
Model calibration—molecular level
One of the first steps of model development was to represent the GSL production and degradation pathways of key GSLs, such as GL‐1, GL‐3, and ceramide. Figure 2 illustrates the QSP model behavior and capabilities of describing plasma GSL concentrations, with GD1 state represented with a residual enzyme activity for acid β‐glucosidase of ~ 11%, a value in the range observed for individuals with moderate GD1, as described in more detail below. First, three of the mean molecular‐level model outputs were compared against reported literature measurements 12 , 31 , 32 , 33 for healthy individuals and untreated adults with GD1 (Figure 2a ). These plots indicate that the model structure can effectively capture the differential plasma GSL behavior that arises in the healthy and GD1 state. Next, from the individual calibrations we computed the mean model output for plasma lyso‐GL‐1 baseline and therapeutic response and compared it with the average (across all patients) plasma lyso‐GL‐1 time course observed during the ENGAGE clinical trial (Figure 2b ). This shows that the model is capable of accurately representing the pharmacological effects of eliglustat on this GD1 biomarker over time.
Figure 2.
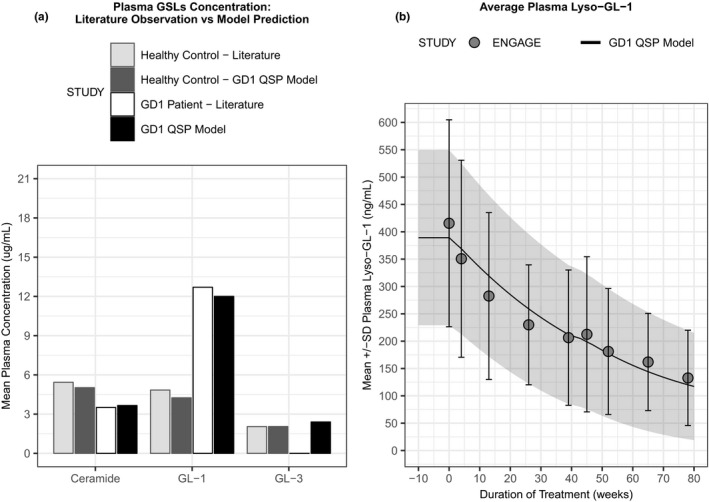
Quantitative systems pharmacology (QSP) model recapitulates plasma glycosphingolipid profiles from healthy and disease state. (a) Simulated steady‐state outputs for plasma ceramide, glucosylceramide (GL‐1), and globotriaosylceramide (GL‐3) levels were compared for healthy (QSP model dark gray vs. literature data light gray bars), (Sanofi internal report 33 , 49 ); and untreated Gaucher’s disease type 1 (GD1; QSP model black vs. literature data white bars). 32 Please note that there was no plasma GL‐3 data available from patients with GD1. (b) Comparison of simulated plasma glucosylsphingosine (lyso‐GL)‐1 in response to eliglustat treatment (mean and SD: solid line with shaded area) overlaid with mean and SD of clinical measurement at each time point (gray circles with error bars) over the course of the ENGAGE clinical trial. 12
Although Figure 2 compared the model output to average GSL values, Figure 3 provides an example of how the model can represent the population variability of baseline and therapeutic response of plasma lyso‐Gl‐1 and spleen volume observed in the ENGAGE clinical trial. Simulation results of baseline (untreated) plasma lyso‐GL‐1 concentrations and the temporal response of this biomarker to treatment with eliglustat are shown in Figure 3a . The mean and SD of the plasma lyso‐GL‐1 response to eliglustat in this virtual population cohort was then compared with clinical observations from the ENGAGE treatment arm, showing model simulations can capture the clinical data.
Figure 3.
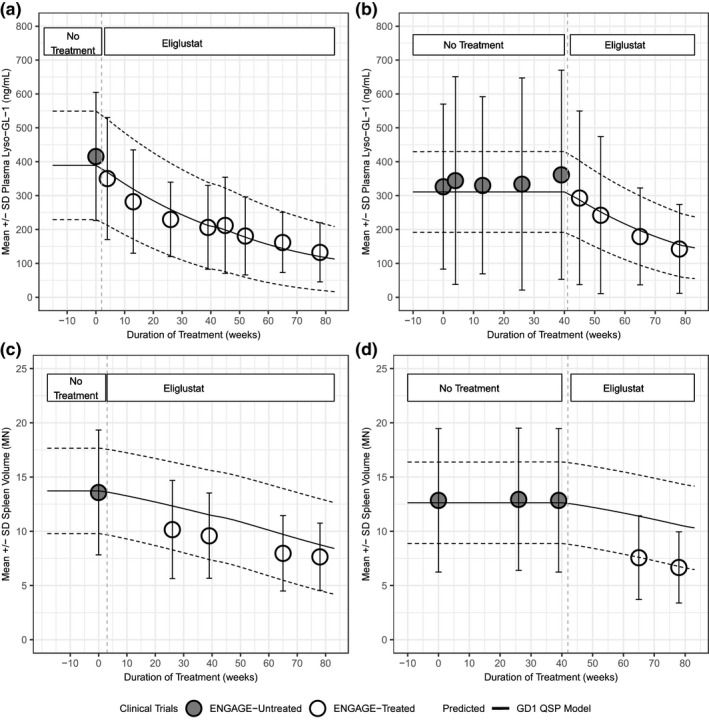
Comparison of simulation results for a population of 1,000 virtual patients to clinical data. Simulated results for plasma glucosylsphingosine (lyso‐GL)‐1 (a) and spleen volume (c) response to eliglustat overlaid with clinical data from active arm from ENGAGE trial. Simulated results for plasma‐lyso‐GL1 (b) and spleen volume (d) response to eliglustat overlaid with clinical data from the placebo arm and open‐label period of the ENGAGE trial. Mean of virtual population (black solid line), and SD (dashed lines). Mean clinical data from patients are shown as points (white circles: treated arm in ENGAGE trial; gray circles: untreated arm in ENGAGE trial) with SD.
As an independent test case, the virtual cohort was used to simulate patients in the ENGAGE placebo arm that began eliglustat treatment only during the open‐label period (after the first 39 weeks of the study). Figure 3b compares data from patients in this cohort with the simulated response from a virtual population selected to match the untreated period. Although larger variability is observed prior to active treatment, the simulation results adequately capture the observed treatment response range.
Model calibration—organ level
Next, the model capabilities for predicting spleen volume in MNs, a key clinical end point in GD1, were explored. As described above for Figure 3a , b , the virtual patient cohort of 1,000 adult GD1 subjects was generated based on the individual calibrations to patients in the treatment arm of the ENGAGE trial. Figure 3c , d shows the corresponding test scenario paralleling shown for plasma lyso‐GL‐1; the same virtual patient cohort was used to simulate spleen volume measurements for the active arm (Figure 3c ) and for the placebo to open‐label period patients (Figure 3d ) of the ENGAGE clinical trial. Again, patient data fall within the model‐predicted range. Overall, Figures 2 , 3 and 2 , 3 demonstrate that the GD1 QSP model can effectively represent patient variability of adult patients with GD1 enrolled in these clinical trials, capturing the baseline and the eliglustat treatment response, in terms of both plasma GSLs and spleen volume changes.
First qualification scenario—ERT to SRT switch
After calibration, the GD1 QSP model was applied to two model qualification scenarios to test the model’s predictive performance compared with data to which the model had not been specifically calibrated. The first scenario examines the ERT‐to‐eliglustat switch in adult patients with GD1 as described in the ENCORE clinical trial. In the ENCORE clinical trial, adults with GD1 who had been stabilized on ERT for 3 or more years were either switched to eliglustat or maintained on imiglucerase. The model was applied to simulate the scenario of eliglustat initiation following ERT stabilization. Figure 4 shows the results of this assessment for plasma GL‐1 (Figure 4a ) and spleen volume (Figure 4b ) in a virtual population of patients with GD1. In Figure 4a , simulation results of plasma GL‐1 concentrations of the virtual GD1 patient population (mean = black solid line and SD = dashed lines) are shown and compared with (i) the baseline data from the ENGAGE trial (gray, closed circles), and (ii) patients with GD1 who switched from ERT to eliglustat, from the ENCORE trial, and extension (white, open circles). 18 Figure 4b shows the analogous comparisons with respect to spleen volume. This analysis demonstrates that the model can adequately represent the response to eliglustat treatment after switching from ERT for plasma GL‐1 and spleen volume, in agreement with the mean results reported in the ENCORE trial. 17 , 18
Figure 4.
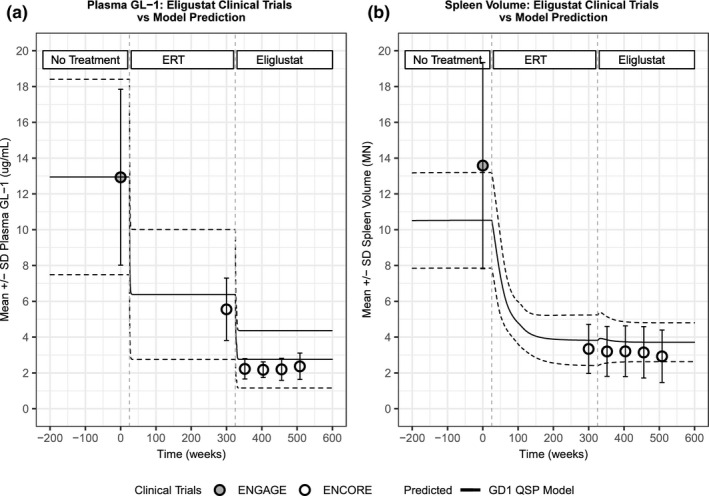
Model recapitulates switch scenario from enzyme replacement therapy (ERT) to eliglustat. Model‐predicted responses (mean: black, solid lines; SD: black, dashed lines) in plasma glucosylceramide (GL)‐1 (a) and spleen volume (b) for previously untreated patients stabilized with Cerezyme and then switched to eliglustat, compared with clinical measurements (mean and SD) from the ENGAGE trial at baseline (gray circles) and the ENCORE trial baseline and after 52 weeks of treatment (white circles). GD, Gaucher’s disease; QSP, quantitative systems pharmacology.
Second qualification scenario—phenotypic expansion of virtual patients
The second qualification scenario tests the capability of the QSP model to describe the heterogeneous‐nature of the GD1 patient population, which includes a more diverse presentation of genotypes and disease severities than typically represented in clinical trials. There is a recognized association between patient genotype and clinical characteristics, such as spleen volume and age of diagnosis, 34 although there is significant disease severity variability among patients of the same genotype. We examined clinical trial data from previously untreated patients in the ENGAGE and phase II trials and observed that when disease severity was classified by genotype alone, the average spleen volume for patients with mild, moderate, or severe disease genotype followed the expected trend of larger spleens for patients with more severe genotypes (Supplemental Table S2 and Table S3 ).
This assessment of clinical trial data was supplemented by analysis of spleen volume data from GD1 adults in the ICGG Gaucher Registry. Spleen volumes reported in the ICGG Gaucher Registry were measured volumetrically by magnetic resonance imaging, computed tomography, or ultrasonography, and listed as MN spleen size predicted for body weight, 35 with a standard formula used to convert ultrasound measurements to volume equivalents. 36 , 37 To analyze the ICGG Gaucher Registry data, we identified two confounding factors that affect the range of spleen volumes reported. The first was the method of measurement; we excluded ultrasound measurements from the analysis because these estimates of spleen volume are recognized to be less accurate compared with measurements taken by magnetic resonance imaging or computed tomography scan. 38 The second factor was the availability of treatment at the time of the measurement. We noted that when considering all adult untreated patients in the ICGG Gaucher Registry, the spleen volumes in each disease severity category were smaller on average compared with when the same analysis was performed on a restricted set of adult untreated patients from the years prior to the approval of ERT (Supplemental Table S4 ). A reasonable explanation for this observation is that once ERT was available as a standard of care, Cerezyme treatment was initiated in the patients with more severe symptomatic GD1 before they reached adulthood. Hence, they would have not been included in an analysis of treatment‐naïve adult patients. 39 For this reason, to analyze spleen volume in a treatment‐naïve population with different severities, we chose to exclude data obtained after the year of Cerezyme approval, to ensure that our summary of the ICGG Gaucher Registry was an accurate representation of the range of spleen volumes possible for untreated patients with adult GD1. When these restrictions were applied, however, no ICGG Gaucher Registry patients with a severe genotype were left. Thus, in the analysis of the prevalence of patients in each genotype category, we included all patients in the ICGG Gaucher Registry, regardless of their year of enrollment or of their treatment status, to obtain a more complete picture of the adult GD1 population.
To guide the development of virtual populations for each severity category, we used both the ICGG Gaucher Registry and clinical trial data (additional details can be found in Supplemental Section S4 ). The clinical data summary (Supplemental Table S2 ) is affected by the restrictions on spleen volume in the trial inclusion criteria, but can supplement the ICGG Gaucher Registry data (Supplemental Table S4 ). Using both the ICGG Gaucher Registry data, after the above‐mentioned criteria were imposed, and data from phase II eliglustat and ENGAGE clinical trials, we generated a final summary of spleen volume statistics for patients of each genotype severity category, which is shown in Supplemental Table S5 and Figure 5 (observed bars). Although there is significant overlap in the range of spleen volumes for each genotype‐based severity category, we note that the average spleen volume and overall spleen volume distributions of each category follow the expected trend of larger spleens in patients with more severe genotypes. Even when the criteria were imposed, the majority of patients included were mild or moderate in severity (mild = 44 patients, moderate = 65 patients, and severe = 6 patients). This is due to the larger prevalence of the N370S homozygous genotype in the ICGG Gaucher Registry cohort, which is usually associated with less severe presentation of the disease. All other genotypes were categorized as moderate unless they were specifically identified as severe mutations. Supplementary Table S5 also shows the prevalence used to construct different cohorts of the virtual population, based on the prevalence of genotype severities across the ICGG Gaucher Registry population.
Figure 5.
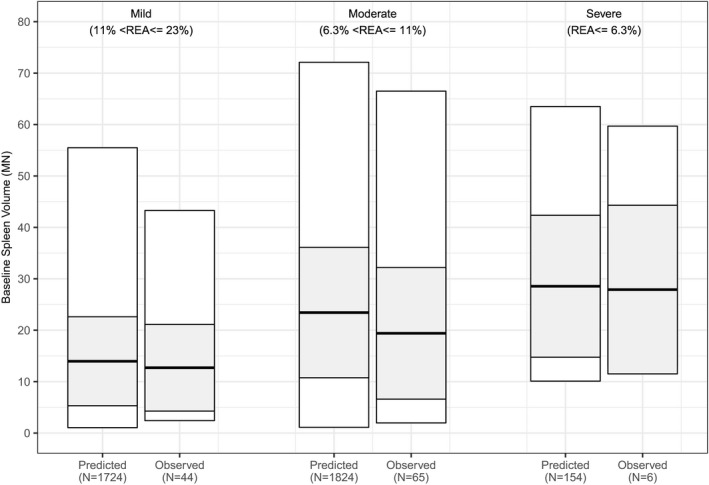
Quantitative systems pharmacology (QSP) model describes the diversity of genotype‐based severities observed. Comparison of predicted ranges of baseline spleen volumes mapped to different genotype‐based severities. Observed spleen volume ranges reflect those shown in Supplemental Table S5 (minimum to maximum: white rectangle; mean and SD: thick black line and shaded rectangle). Predicted spleen volume ranges reflect virtual populations simulated using the Gaucher’s disease type 1 QSP model with the range spleen volumes identified in the genotype‐phenotype analysis. Observed patients included those in the ENGAGE and phase II eliglustat trial as well as treatment‐naïve patients with magnetic resonance imaging or computed tomography spleen volume measurements in the International Collaborative Gaucher Group Gaucher Registry prior to 1992.
Next, we generated virtual populations corresponding to these genotype‐based severity categories by assigning each severity category to a corresponding range of spleen volumes observed per severity category and prevalence (Figure 5 ). The resulting virtual populations in each severity category contained predicted residual enzyme activity values that encompass the range of residual enzyme activity measured in patients with GD1, 12 , 40 , 41 , 42 , 43 suggesting that the model predicted residual enzyme activity: spleen volume relationship captured by the model is valid. This result highlights how the model can effectively capture the ranges of residual enzyme activity in each severity category, despite that these ranges were not imposed during model calibration.
The effect of eliglustat treatment on spleen volume change for treatment‐naïve virtual patients with diverse severity categories was then simulated. This set of virtual patients represents a more heterogeneous population of patients with GD1 than those enrolled in clinical trials used in the model calibration. The simulation results were compared with those observed in the phase II trial, a clinical data set not used during model calibration and found that this heterogeneous virtual population is predicted to respond as well to eliglustat treatment as those patients included in the clinical data set (Figure 6 ).
Figure 6.
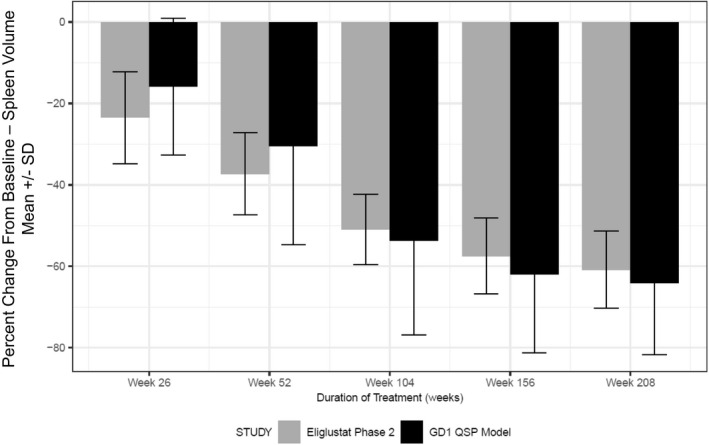
Quantitative systems pharmacology (QSP) model‐predicted responses in spleen volume changes. Simulation results of virtual population constructed in Figure 5 was treated with eliglustat (mean and SD: dark, closed bars) compared with clinical measurements (mean and SD: gray, closed bars) from the phase II eliglustat clinical trial. GD1, Gaucher’s disease type 1.
DISCUSSION
We present the first QSP model to describe GD1 and the response of adult patients to both ERT and eliglustat. The model uses a multiscale, mechanistic framework to describe the GSL biochemical pathway that is perturbed by the causal genetic mutation in GD1, the effect of this perturbation on plasma biomarkers, including lyso‐GL‐1, and the clinically important disease end point of splenomegaly. The QSP model provides a biologically grounded basis for explaining disease and drug response variability and for supporting future therapeutic development.
Two qualification scenarios were performed to illustrate how the mechanistic QSP strategy enables the model to address clinically relevant questions. First, the model was applied to assess the ERT to eliglustat switch in adult patients with GD1. Biomarker and clinical end point predictions for this scenario corresponded well to clinical observations from the ENCORE trial. Overall, the first qualification scenario showed that the GD1 QSP model can predict both plasma GL‐1 and spleen volume responses in a clinically meaningful scenario with two different therapeutic modalities. Although the model was calibrated with data from the ENGAGE trial, which included adults with GD1 who were previously untreated with ERT or had been off‐treatment for > 9 months, it is also capable of accurately describing the switch to eliglustat in adults with GD1 who were previously stabilized on ERT. Second, the model was applied to predict eliglustat response in the more diverse GD1 population represented by the ICGG Gaucher Registry. Genotype‐based stratification of splenomegaly severity provided an example of how the model could provide mechanistic insight into observed patient variability. This qualification exercise showcases how real‐world data, analyzed with the appropriate assumptions, can be implemented to inform a QSP approach to provide a more accurate representation of the variability in disease presentation found in GD1.
Although the genotype‐based categorization has been helpful in differentiating patients who are expected to need adjustments to their therapy to reach the therapeutic goal for spleen volume, it is recognized that there is substantial variability in disease even among patients with the same genotype, 44 which is reflected in the wide range of spleen volumes reported in Supplementary Table S2 . In the future, there could be more insight gained into potential patient low‐responders to treatment by considering additional stratification factors, such as the severity of bone disease or other disease biomarkers.
A significant challenge noticed during the genotype‐phenotype analysis in the second qualification scenario was properly identifying the most representative ICGG Gaucher Registry subpopulation to consider in the severity analysis. The goal was to obtain an unbiased view of naive spleen volume ranges for each genotype‐based disease severity category. By identifying confounding factors, we eliminated some bias introduced by (i) less accurate methods of measurement; and (ii) the lack of spleen volume data for severe treatment‐naïve patients once ERT became available. However, it is likely that there are other factors contributing to the lack of data on the severe GD1 subgroup. Prior to availability of Cerezyme treatment, the Gaucher population was reduced in size due to the increased early mortality of patients with GD1, 45 which would be expected to be pronounced for more severe patients, resulting in a survivor bias for the mild/moderate patients that reach adulthood. Moreover, there was less recognition of the disease prior to disease awareness campaigns initialized by Genzyme at the time of Cerezyme approval, so fewer patients overall were diagnosed with GD1. 39 These types of challenges in obtaining data are particularly relevant for the rare diseases, and can impact data availability and model development.
As more information becomes available on the natural history 46 and the phenotypic variability 47 of GD1, this data can be integrated into the model on an ongoing basis to describe other relevant clinical manifestations of GD1, such as skeletal and hematological disease, or to determine how the model can be adapted to represent other populations. On a broader level, the GD1 QSP model and the QSP model for acid sphingomyelinase deficiency both contribute to the design of an integrated lysosomal storage disease modeling platform. 48
In summary, the GD1 QSP model provides a mechanistic basis for capturing and quantifying the systemic disease, treatment response, and patient variability within the GD1 population. The calibration results demonstrate that the model can replicate clinically observed trends in biomarkers and spleen volume. The qualification scenarios illustrate how the model can be applied to gain insight into clinically meaningful questions, thereby supporting decision making and adding value to the therapeutic development process. Expansion and refinement of the QSP model will continue as additional data become available, improving its capabilities to support both research and development in GD1.
Funding
This study was funded by Sanofi.
Conflict of Interest
R.A., M.T., R.J.L., J.L., B.N., M.J.P., and S.N.Z. are current employees of Sanofi. C.D.K., Z.V.R., J.S.B., and K.A. were employees of Sanofi while this study was conducted. P.J. and J.T. are employees of RES and were contracted by Sanofi while this research was conducted. G.S., F.R., and L.M. were contracted by Sanofi while this research was conducted.
Author Contributions
R.A., C.D.K., M.T., R.J.L., L.M., K.A., and S.N.Z. wrote the manuscript. R.A., C.D.K., J.S.B., L.M., M.J.P., K.A., and S.N.Z designed the research. R.A., C.D.K., M.T., R.J.L., G.S., F.R., B.N., P.J., and J.T. performed the research. R.A., C.D.K., M.T., R.J.L., G.S., F.R., P.J., J.T., B.N., J.L., L.M., M.J.P., K.A., and S.N.Z. analyzed the data. C.D.K. and Z.V.R. contributed new reagents/analytical tools.
Supporting information
Acknowledgments
The authors thank Greg Fagan, Julie Batista, and Eva Stamenovic (ICGG Gaucher Registry) for providing data access; Sebastiaan Gaemers, Davorka Sekulic, Tim Olson, Lisa Underhill, and Laura Croal (Sanofi‐Genzyme) for critical review and support in preparing this manuscript.
References
- 1. Mistry, P.K. , Weinreb, N.J. , Kaplan, P. , Cole, J.A. , Gwosdow, A.R. & Hangartner, T. Osteopenia in Gaucher disease develops early in life: response to imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells Mol. Dis. 46, 66–72 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grabowski, G.A. , Petsko, G.A. & Kolodny, E.H. Gaucher disease In The Online Metabolic and Molecular Bases of Inherited Disease [Internet] (eds. Beaudet A.L., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A. & Gibson K.M., et al.). (The McGraw‐Hill Companies, Inc., New York, NY, 2014. [cited 2018 May 23]). <ommbid.mhmedical.com/content.aspx?aid=1102895727>. [Google Scholar]
- 3. Murugesan, V. et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 91, 1082–1089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walkley, S.U. & Vanier, M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta 1793, 726–736 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grabowski, G.A. et al. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose‐terminated glucocerebrosidase from natural and recombinant sources. Ann. Intern. Med. 122, 33–39 (1995). [DOI] [PubMed] [Google Scholar]
- 6. Charrow, J. & Scott, C.R. Long‐term treatment outcomes in Gaucher disease. Am. J. Hematol. 90 (suppl. 1), S19–S24 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Weinreb, N.J. et al. Long‐term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J. Inherit. Metab. Dis. 36, 543–553 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weinreb, N. , Taylor, J. , Cox, T. , Yee, J. & vom Dahl, S. A benchmark analysis of the achievement of therapeutic goals for type 1 Gaucher disease patients treated with imiglucerase. Am. J. Hematol. 83, 890–895 (2008). [DOI] [PubMed] [Google Scholar]
- 9. Andersson, H. , Kaplan, P. , Kacena, K. & Yee, J. Eight‐year clinical outcomes of long‐term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics 122, 1182–1190 (2008). [DOI] [PubMed] [Google Scholar]
- 10. Weinreb, N.J. et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am. J. Med. 113, 112–119 (2002). [DOI] [PubMed] [Google Scholar]
- 11. Peterschmitt, M.J. et al. A pooled analysis of adverse events in 393 adults with Gaucher disease type 1 from four clinical trials of oral eliglustat: evaluation of frequency, timing, and duration. Blood Cells Mol. Dis. 68, 185–191 (2018). [DOI] [PubMed] [Google Scholar]
- 12. Mistry, P.K. et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA 313, 695–706 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lukina, E. et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz‐112638) treatment: 2‐year results of a phase 2 study. Blood 116, 4095–4098 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lukina, E. et al. A phase 2 study of eliglustat tartrate (Genz‐112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood 116, 893–899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mistry, P.K. et al. Outcomes after 18 months of eliglustat therapy in treatment‐naïve adults with Gaucher disease type 1: the phase 3 ENGAGE trial. Am. J. Hematol. 92, 1170–1176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lukina, E et al. Outcomes after 8 years of eliglustat therapy for Gaucher disease type 1: final results from the phase 2 trial. Am. J. Hematol. 94, 29–38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cox, T.M. et al. Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open‐label, non‐inferiority trial. Lancet Lond. Engl. 385, 2355–2362 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Cox, T.M. et al. Eliglustat maintains long‐term clinical stability in patients with Gaucher disease type 1 stabilized on enzyme therapy. Blood 129, 2375–2383 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Byrne‐Hoffman, C. & David, I.I. A quantitative systems pharmacology perspective on cancer immunology. Processes 3, 235–256 (2015). [Google Scholar]
- 20. Ming, J.E. et al. A quantitative systems pharmacology platform to investigate the impact of Alirocumab and cholesterol‐lowering therapies on lipid profiles and plaque characteristics. Gene Regul. Syst. Biol. 11, 1177625017710941 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dalla Man, C. , Rizza, R.A. & Cobelli, C. Meal simulation model of the glucose‐insulin system. IEEE Trans. Biomed. Eng. 54, 1740–1749 (2007). [DOI] [PubMed] [Google Scholar]
- 22. Hosseini, I. & Gabhann, F.M. Mechanistic models predict efficacy of CCR5‐deficient stem cell transplants in HIV patient populations. CPT Pharmacomet. Syst. Pharmacol. 5, 82–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wronowska, W. , Charzyńska, A. , Nienałtowski, K. & Gambin, A. Computational modeling of sphingolipid metabolism. BMC Syst. Biol. 9, 47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clausznitzer, D. et al. Quantitative systems pharmacology model for Alzheimer disease indicates targeting sphingolipid dysregulation as potential treatment option. CPT Pharmacometrics Syst. Pharmacol. 7, 759–770 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Conzelmann, E. & Sandhoff, K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev. Neurosci. 6, 58–71 (1983). [DOI] [PubMed] [Google Scholar]
- 26. Vigan, M. et al. Modeling changes in biomarkers in Gaucher disease patients receiving enzyme replacement therapy using a pathophysiological model. Orphanet J. Rare Dis. 9, 95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaddi, C. et al. Quantitative systems pharmacology model of acid sphingomyelinase deficiency and the enzyme replacement therapy olipudase alfa is an innovative tool for linking pathophysiology and pharmacology. CPT Pharmacometrics Syst. Pharmacol. 7, 442–452 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weinreb, N.J. & Kaplan, P. The history and accomplishments of the ICGG Gaucher registry. Am. J. Hematol. 90 (suppl. 1), S2–S5 (2015). [DOI] [PubMed] [Google Scholar]
- 29. McQuade, S.T. , Abrams, R.E. , Barrett, J.S. , Piccoli, B. & Azer, K. Linear‐in‐flux‐expressions methodology: toward a robust mathematical framework for quantitative systems pharmacology simulators. Gene Regul. Syst. Bio. 11, 1177625017711414 (2017). 10.1177/1177625017711414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Rijn, Z. , Kaddi, C. & Azer, K. Multiple‐Interface Solver Toolkit (MIST): efficient and modular quantitative systems pharmacology modeling framework. 14th Annual Conference on Frontiers in Applied and Computational Mathematics, Newark, New Jersey, June 24–25, 2017. [Google Scholar]
- 31. Ferraz, M.J. et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 590, 716–725 (2016). [DOI] [PubMed] [Google Scholar]
- 32. Ghauharali‐van der Vlugt, K. et al. Prominent increase in plasma ganglioside GM3 is associated with clinical manifestations of type I Gaucher disease. Clin. Chim. Acta. 389, 109–113 (2008). [DOI] [PubMed] [Google Scholar]
- 33. Dawson, G. , Kruski, A.W. & Scanu, A.M. Distribution of glycosphingolipids in the serum lipoproteins of normal human subjects and patients with hypo‐ and hyperlipidemias. J. Lipid Res. 17, 125–131 (1976). [PubMed] [Google Scholar]
- 34. Sibille, A. , Eng, C.M. , Kim, S.J. , Pastores, G. & Grabowski, G.A. Phenotype/genotype correlations in Gaucher disease type I: clinical and therapeutic implications. Am. J. Hum. Genet. 52, 1094–1101 (1993). [PMC free article] [PubMed] [Google Scholar]
- 35. Barton, N.W. et al. Replacement therapy for inherited enzyme deficiency — macrophage‐targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 324, 1464–1470 (1991). [DOI] [PubMed] [Google Scholar]
- 36. Elstein, D. , Hadas‐Halpern, I. , Azuri, Y. , Abrahamov, A. , Bar‐Ziv, Y. & Zimran, A. Accuracy of ultrasonography in assessing spleen and liver size in patients with Gaucher disease: comparison to computed tomographic measurements. J. Ultrasound Med. 16, 209–211 (1997). [DOI] [PubMed] [Google Scholar]
- 37. El‐Beshlawy, A. et al. Long‐term hematological, visceral, and growth outcomes in children with Gaucher disease type 3 treated with imiglucerase in the International Collaborative Gaucher Group Gaucher Registry. Mol. Genet. Metab. 120, 47–56 (2017). [DOI] [PubMed] [Google Scholar]
- 38. Heymsfield, S. , Ross, R. , Wang, Z. & Frager, D. Imaging techniques of body composition: advantages of measurement and new uses Chapter 5. In Emerging Technologies for Nutrition Research: Potential for Assessing Military Performance Capability [Internet] (eds. Carlson‐Newberry SJ. & Costello RB.) 127–150 (National Academies Press, Washington, DC, 1997). [PubMed] [Google Scholar]
- 39. Mistry, P.K. et al. Transformation in pretreatment manifestations of Gaucher disease type 1 during two decades of alglucerase/imiglucerase enzyme replacement therapy in the International Collaborative Gaucher Group (ICGG) Gaucher Registry. Am. J. Hematol. 92, 929–939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sawkar, A.R. et al. Gaucher disease‐associated glucocerebrosidases show mutation‐dependent chemical chaperoning profiles. Chem. Biol. 12, 1235–1244 (2005). [DOI] [PubMed] [Google Scholar]
- 41. Montfort, M. , Chabás, A. , Vilageliu, L. & Grinberg, D. Functional analysis of 13 GBA mutant alleles identified in Gaucher disease patients: pathogenic changes and “modifier” polymorphisms. Hum. Mutat. 23, 567–575 (2004). [DOI] [PubMed] [Google Scholar]
- 42. Grace, M.E. , Newman, K.M. , Scheinker, V. , Berg‐Fussman, A. & Grabowski, G.A. Analysis of human acid beta‐glucosidase by site‐directed mutagenesis and heterologous expression. J. Biol. Chem. 269, 2283–2291 (1994). [PubMed] [Google Scholar]
- 43. Turner, B.M. & Hirschhorn, K. Properties of beta‐glucosidase in cultured skin fibroblasts from controls and patients with Gaucher disease. Am. J. Hum. Genet. 30, 346–358 (1978). [PMC free article] [PubMed] [Google Scholar]
- 44. Germain, D.P. , Puech, J.P. , Caillaud, C. , Kahn, A. & Poenaru, L. Exhaustive screening of the acid beta‐glucosidase gene, by fluorescence‐assisted mismatch analysis using universal primers: mutation profile and genotype/phenotype correlations in Gaucher disease. Am. J. Hum. Genet. 63, 415–427 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weinreb, N.J. et al. Life expectancy in Gaucher disease type 1. Am. J. Hematol. 83, 896–900 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang, A.C. et al. Early manifestations of type 1 Gaucher disease in presymptomatic children diagnosed after parental carrier screening. Genet. Med. 19, 652–658 (2017). [DOI] [PubMed] [Google Scholar]
- 47. Hassan, S. , Sidransky, E. & Tayebi, N. The role of epigenetics in lysosomal storage disorders: uncharted territory. Mol. Genet. Metab. 122, 10–18 (2017). [DOI] [PubMed] [Google Scholar]
- 48. Kaddi, C. et al. Integrated quantitative systems pharmacology (QSP) model of lysosomal diseases provides an innovative computational platform to support research and therapeutic development for the sphingolipidoses. Mol. Genet. Metab. 123, S73–S74 (2018). [Google Scholar]
- 49. Ferraz, M.J. et al. Lyso‐glycosphingolipid abnormalities in different murine models of lysosomal storage disorders. Mol. Genet. Metab. 117, 186–193 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials