

SUMMARY
Polycomb repressive complex 2 (PRC2) places H3K27me3 at developmental genes and is causally implicated in keeping bivalent genes silent. It is unclear if that silence requires minimum H3K27me3 levels and how the mark transmits faithfully across mammalian somatic cell generations. Mouse intestinal cells lacking EZH2 methyltransferase reduce H3K27me3 proportionately at all PRC2 target sites, but ~40% uniform residual levels keep target genes inactive. These genes, derepressed in PRC2-null villus cells, remain silent in intestinal stem cells (ISCs). Quantitative chromatin immunoprecipitation and computational modeling indicate that because unmodified histones dilute H3K27me3 by 50% each time DNA replicates, PRC2-deficient ISCs initially retain sufficient H3K27me3 to avoid gene derepression. EZH2 mutant human lymphoma cells also require multiple divisions before H3K27me3 dilution relieves gene silencing. In both cell types, promoters with high basal H3K4me2/3 activate in spite of some residual H3K27me3, compared to less-poised promoters. These findings have implications for PRC2 inhibition in cancer therapy.
In Brief
Loss of PRC2 activity in dividing adult mammalian cells depletes H3K27me3 equally at all marked nucleosomes, owing mainly to replicational dilution by unmarked histones. Individual PRC2 target genes overcome the repressed state in proportion to the number of preceding cell divisions and basal levels of promoter H3K4me2/3.
Graphical Abstract
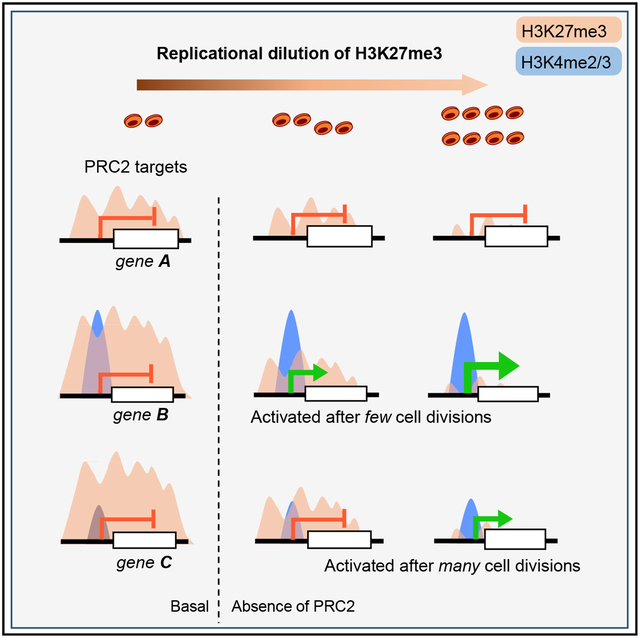
INTRODUCTION
The intervals between cell divisions vary widely among stem cells in different tissues, and transit-amplifying (TA) daughters divide more frequently than stem cells. In symmetric cell divisions, both daughters copy parental histone modifications faithfully (Campos et al., 2014; Reveron-Gomez et al., 2018) and, thus, preserve cell-specific gene activity. This process occurs with fidelity because sister chromatids inherit modified parental nucleosomes almost symmetrically (Petryk et al., 2018) and the ~50% newly recruited, naive nucleosomes later undergo accurate de novo modification (Reveron-Gomez et al., 2018). Thus, histone-modifying enzymes must be recruited to–or stay associated with–active and repressed genomic regions during mitosis. EZH1 or EZH2, two enzymes in Polycomb repressive complex 2 (PRC2) catalyze trimethylation of Lysine 27 on Histone 3 (H3K27me3), a mark associated with silencing of developmental genes (Schuettengruber et al., 2017). Accurate inheritance of H3K27me3 in dividing somatic cells preserves gene repression from one cell generation to the next.
“Off”-state gene memories persist in Drosophila because H3K27me3+ nucleosomes remain associated with marked (e.g., HOX) genes during mitosis and polycomb response elements (PREs) recruit PRC2 to methylate newly inserted nucleosomes (Coleman and Struhl, 2017; Laprell et al., 2017). Deletion of PREs limits de novo trimethylation of H3K27, but HOX genes get derepressed only after PRE-null cells deplete parental H3K27me3 over several cell divisions (Coleman and Struhl, 2017; Laprell et al., 2017). In contrast, mammalian genomes lack consensus PREs (Schuettengruber et al., 2017). Instead, PRC2 binds at sites with pre-existing H3K27me3 or CpG islands low in methylated DNA (Jermann et al., 2014; Mendenhall et al., 2010; Riising et al., 2014), and its activity responds to dense nucleosome compaction (Yuan et al., 2012). Nevertheless, when H3K27me3 is erased in PRC2-null mouse embryonic stem cells (ESCs) and PRC2 activity is later restored, the mark appears accurately at all target regions (Højfeldt et al., 2018), implying that factors other than H3K27me3 hold memory and direct PRC2 to those sites. Moreover, steady-state H3K27me3 levels reflect the equilibrium between methylation, mediated by EZH enzymes; demethylation, catalyzed by KDM6A or KDM6B (Agger et al., 2007; De Santa et al., 2007); and exchange with unmodified histones. Mammalian PRC2 activity has been studied mainly in ESCs; it remains unclear how adult somatic cells implement PRC2-dependent gene silencing at each cell division in vivo and if a critical density of H3K27me3+ nucleosomes is necessary to maintain the repressed state.
PRC2 is often hyperactive or overexpressed in human cancers (Comet et al., 2016). For example, activating EZH2 mutations are found in up to 24% of diffuse large B cell lymphomas (Morin et al., 2010), increase total H3K27me3 levels (Sneeringer et al., 2010; Yap et al., 2011), and promote tumor survival and growth in pre-clinical models (Beguelin et al., 2013). Drugs that target PRC2 function are currently in clinical development; conversely, inactivating EZH2 and other PRC2 mutations are found in other cancers (Comet et al., 2016). The effects of activating or null mutations at individual PRC2 target loci and the genotoxicity of systemic PRC2 inhibition are not well understood.
We studied the mouse intestinal epithelium, where Lgr5+ intestinal stem cells (ISCs) at the bottom of crypt structures sustain high cell turnover (Barker et al., 2007). These ISCs replicate on average every ~3 days (Kozar et al., 2013) to produce TA cells, which occupy most of the crypt (Figure S1A) and replicate every ~6–8 h (Parker et al., 2017). TA cells’ differentiated, post-mitotic progeny migrate upward to line intestinal villi and serve digestive functions. These well-known cell compartments and kinetics (Clevers, 2013) allow rigorous, quantitative investigation of histone marks and gene activity thresholds. Three groups have assessed PRC2 requirements in the intestinal epithelium. PRC2 loss preserved ISC function in one study (Chiacchiera et al., 2016) but impaired Wnt signaling and ISC viability in another (Koppens et al., 2016). Focusing on the consequences in post-mitotic villus cells, we could causally implicate H3K27me3 only in repression of genes that carry bivalent histone marks, i.e., both H3K27me3 and modifications associated with active promoters, H3K4me2 and H3K4me3 (Jadhav et al., 2016). Here, we reconcile the conflicting studies and reveal that in the absence of PRC2, mammalian cells shed H3K27me3 exclusively by replicational dilution of modified nucleosomes. Moreover, our findings indicate that in the absence of PRC2 function, the threshold for target gene activation differs according to the level of basal promoter poising, as reflected in H3K4me2/3 density.
RESULTS
EZH1 Compensation for EZH2 Deficiency Is Partial but Adequate to Maintain Target Gene Silence
PRC2 complexes contain the structural proteins EED and SUZ12 and either EZH1 or EZH2 (Schuettengruber et al., 2017; Shen et al., 2008). Mouse tissues respond to the absence of EZH1 or EZH2 alone, with variable and modest gene derepression (Chen et al., 2009; Ezhkova et al., 2009; Hidalgo et al., 2012; Hirabayashi et al., 2009; Juan et al., 2011), whereas absence of both enzymes (Ezhkova et al., 2011) or the core protein EED (Mirzamohammadi et al., 2016; Xie et al., 2014) leads to severe defects. EZH1 and EZH2 are, therefore, thought to compensate for one another in H3K27 trimethylation. To evaluate this assumption critically in adult mouse intestinal epithelium, we examined the consequences of Villin-CreER-T2-driven (el Marjou et al., 2004) deletion of floxed Eed (Xie et al., 2014) or Ezh2 (Su et al., 2003) alleles. Although EZH2 was lost in both cases (Figure 1A), tissue defects and cell cycle arrest occurred only after EED loss (Figure S1B), as others have reported (Chiacchiera et al., 2016; Koppens et al., 2016). Replication arrest in diverse PRC2-null tissues is attributed to activation of Cdkn2a or Cdkn2b, potent cell cycle inhibitor targets of PRC2 regulation (Chiacchiera et al., 2016; Hidalgo et al., 2012; Xie et al., 2014). Continued cell replication in Ezh2−/− crypts was indeed associated with persistent absence of Cdkn2a mRNA (Figure S1B). Unexpectedly, total H3K27me3 levels were appreciably lower in Ezh2−/− than in wild-type (WT) villi (Figure 1A), and immunoblot quantitation revealed 25% to 62% reduction of all methylated H3K27 forms in Ezh2−/− villus cells (Figures 1B and S1C). Thus, in biochemical terms, EZH1 compensates only partially to methylate H3K27 in the absence of EZH2. This degree of functional redundancy is, however, sufficient to maintain the silence of genes that are derepressed in Eed−/− villus cells (Figure 1C).
Figure 1. Partial Compensation for EZH2 Deficiency Maintains PRC2 Target Gene Silence.
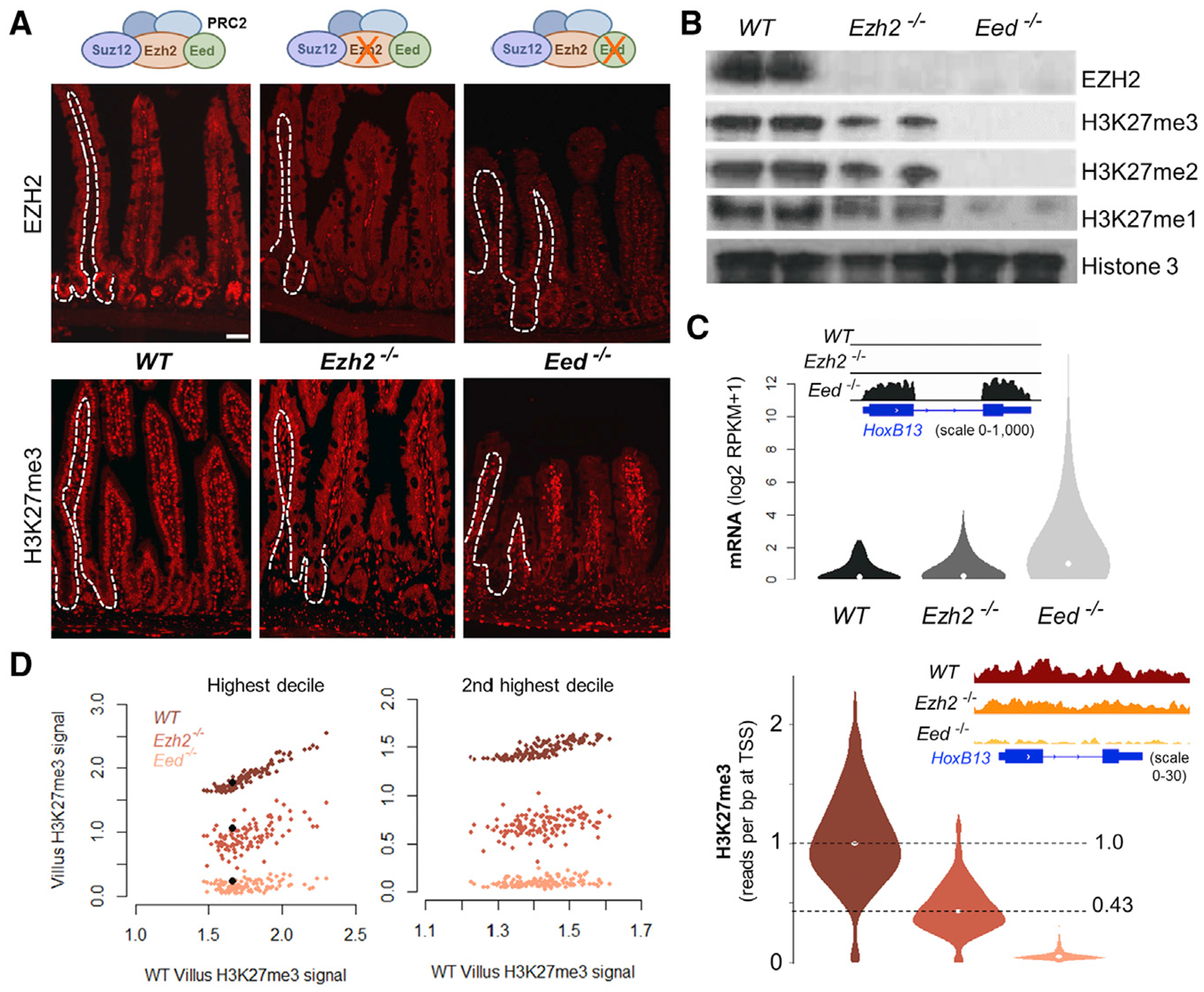
(A) Immunofluorescence in Ezh2−/− and Eed−/− mouse small intestine, showing total loss of EZH2 in the epithelial compartment (single-cell layer outside dotted lines) in both strains. H3K27me3 is reduced in Ezh2−/− and absent in Eed−/− epithelium. Tissues were stained from at least 3 mice of each genotype.
(B) Immunoblots reveal partial loss of all methylated H3K27 forms in Ezh2−/− and complete loss in Eed−/− villus epithelium. Average signals from duplicate samples are quantified in Figure S1C.
(C) Aggregate quantitative changes in mRNA levels (top) and promoter H3K27me3 ChIP signals (bottom) associated with 1, 322 bivalent PRC2 target genes. RNA-seq data from Eed−/− (Jadhav et al., 2016) and Ezh2−/− (Koppens et al., 2016) villi are from published reports; quantitative H3K27me3 ChIP-seq data with Drosophila spike-in controls are new. Integrative Genome Viewer (IGV; insets) tracks at Hoxb13 show partial H3K27me3 loss, without gene derepression, in Ezh2−/− cells, whereas Eed−/− villus cells show near-total H3K27me3 loss and substantial gene activity.
(D) Uniform reduction in H3K27me3 ChIP signals at all promoters in the two highest deciles of basal H3K27me3 levels. The signal at each promoter is represented in the same vertical line for all villus-cell genotypes. Black dots represent the signal at Hoxb13.
See also Figure S1.
At the 1, 322 bivalent PRC2 target gene promoters in intestinal cells (Jadhav et al., 2016), chromatin immunoprecipitation sequencing (ChIP-seq) with rigorous spike-in controls (see STAR Methods) revealed median H3K27me3 levels in Ezh2−/− and Eed−/− villus cells to be 43.5% and 4.91%, respectively, of the levels in WT cells (Figure 1C). Notably, the loss of H3K27me3 at individual loci in Ezh2−/− and Eed−/− villus cells was proportional to basal (WT) levels, as illustrated at single genes and whole PRC2-regulated domains (Figures 1C and S1D), and residual PRC2 function in Ezh2−/− intestines depleted H3K27me3 uniformly from all marked sites, without locus selectivity (Figures 1C and 1D). H3K27me3 levels were globally and proportionally reduced in Ezh2−/− intestines, and WT patterns of local variation in H3K27me3 density were preserved (Figure S1E). These data reveal quantitative consequences of reduced PRC2 activity and indicate that ~43% residual H3K27me3 is sufficient to repress target genes that are robustly activated when the mark is erased.
Lgr5+ ISCs Are Selectively Spared from Transcriptional Consequences of PRC2 Inactivity
Intestine-specific loss of PRC2 in Villin-CreER-T2 mice activates selected embryonic genes in villus cells (Jadhav et al., 2016), whereas two studies of PRC2 loss driven by naphthoflavone-responsive Ah-Cre found distinct and contradictory effects on ISC function (Chiacchiera et al., 2016; Koppens et al., 2016). To address the discrepancy, we generated Lgr5GFP-Cre(ER-T2); EedFl/Fl mice, noting that Gfp and Cre would be restricted by mosaicism to ISCs in a fraction of intestinal crypts (Barker et al., 2007). We isolated Lgr5+ (GFP+) ISCs 4 days after 5 daily injections of tamoxifen to activate CRE recombinase. Although PRC2 target genes were by then extensively derepressed in post-mitotic villus cells, they stayed silent in ISCs (Figure 2A). To verify that H3K27me3-deficient villus cells with abundant gene deregulation arise from seemingly intact ISCs, we introduced the Rosa26Lox-STOP-Lox-YFP (R26Ryfp) reporter (Srinivas et al., 2001) and traced cells after giving tamoxifen. As a result of mosaicism, PRC2-null (yellow fluorescent protein [YFP]+) and PRC2-proficient (YFP−) crypt-villus units appear side by side, and YFP+ villus cells lacking H3K27me3 were present up to 4 weeks after CRE activation (Figure 2B), indicating persistent ISC activity. TA cell replication had declined >50% within 5 days of CRE induction, but cell division was not perturbed among Lgr5+ ISCs (Figure 2C). Moreover, PRC2 target genes were comparably derepressed in YFP+ crypt and villus cells isolated separately from Lgr5Cre;EedFI/FI;RosaYFP mice, whereas the same genes were silent in Eed−/− ISCs (Figure 2D). In line with these findings, TA cells expressed CDKN2A in the same crypts where Lgr5+ ISCs did not (Figure 2E). Thus, days after PRC2 loss markedly affects TA and villus cell genes and properties, Lgr5+ ISCs are spared from the same consequences. ISCs might either compensate uniquely for H3K27me3 loss or these findings may reflect differential H3K27me3 dilution resulting from vastly different rates of replication (Figure 2F): ~0.3 divisions daily in ISCs (Kozar et al., 2013) and up to 4 divisions daily in TA cells (Parker et al., 2017).
Figure 2. Lack of PRC2 Target Gene Activity in PRC2-Null Lgr5+ ISCs.
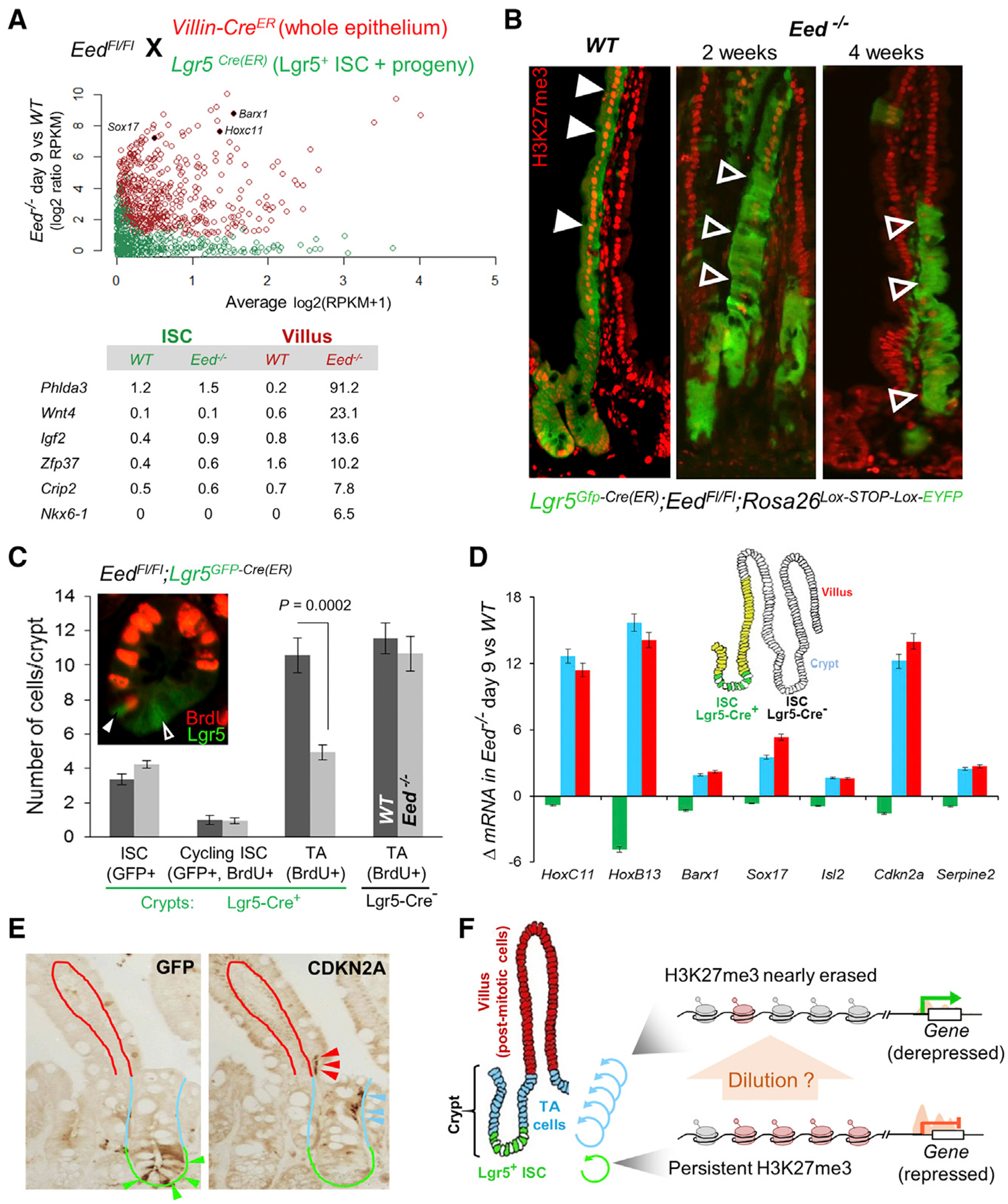
(A) Experimental design to activate CRE and delete Eed in Villin-CreER-T2 (red) or Lgr5Cre(ER-T2) (green) mice. Nine days after the 1st tamoxifen dose, PRC2-null villus epithelium and Lgr5+ ISCs isolated from the respective animals showed markedly different levels of PRC2 target gene expression (RNA-seq). Each gene is represented by a point (villus, red; ISC, green). x axis, average expression in all samples on the given day; y axis, ratio of expression in Eed−/− and WT cells. RPKM values for representative genes are shown below.
(B) Lineage tracing with the R26REYFP reporter coupled with H3K27me3 immunostaining in at least 3 mice at each time point after Eed deletion. Green cells in crypts and villi represent contiguous stripes of Cre-recombined ISC progeny that retain (WT, solid arrows) or lack (Eed−/−, unfilled arrows) H3K27me3 (red nuclear signals). PRC2-null stem cells generate progeny up to 4 weeks after initial CRE activation.
(C) Crypt cell proliferation (bromodeoxyuridine [BrdU+], red) in Eed−/− crypts (EedFl/Fl;Lgr5GFP-Cre(ER)) reveals selective reduction in proliferation of TA cells (BrdU+ only), whereas the numbers of total (GFP+ only) or cycling (GFP+ BrdU+) ISCs remain unchanged. p value was calculated using the t test.
(D) qRT-PCR analysis of representative PRC2 target genes in Lgr5Gfp-Cre(ER);EedFl/Fl;R26REYFP intestinal compartments. YFP+ Eed−/− cell populations were purified by flow cytometry: ISC (GFP+ YFP+), crypt epithelium (depleted of villi), and villus epithelium (depleted of crypts). Nine days after the 1st dose of tamoxifen, target genes were comparably derepressed in villus and crypt (dominated by TA) cells but remained silent in Lgr5+ ISCs. Data represent biological triplicates.
(E) Immunohistochemical analysis of GFP (left) and CDKN2A (right) in serial tissue sections from 2 independent EedFl/Fl;Lgr5GFP-Cre mouse intestines. Crypt base ISCs express GFP (green arrows) but lack CDKN2A, which is present in their presumptive progeny in higher crypt tiers (blue arrows) and the villus base (red arrows).
(F) Hypothesis that these findings may reflect replicational dilution of marked histones.
Replication-Based H3K27me3 Erasure and Target Gene Reactivation in PRC2-Deficient ISCs
To distinguish between these possibilities, we omitted the R26Ryfp reporter, which would be expressed both in ISCs, and in their progeny, to allow capture of PRC2-null Lgr5Gfp-Cre ISCs over an extended period. Knowing that PRC2-null crypt cells are at a proliferative disadvantage compared to those that escape bi-allelic Eed deletion (Jadhav et al., 2016), we maintained selective pressure with chronic tamoxifen exposure to eliminate Eed-proficient cells (Figure 3A). The fraction of PRC2-null GFP+ crypts (Figure 3A) and GFP+ cells (Figure 3B) declined progressively in treated Lgr5Cre;EedFl/Fl mice, indicating that ISCs may delay rather than escape consequences of PRC2 loss. Moreover, although the H3K27-specific demethylase KDM6A (Agger et al., 2007) was present at thousands of active promoters in WT intestinal cells, it was conspicuously absent at PRC2 target genes (Figure S2A; antibodies against KDM6B gave no ChIP signals), suggesting that demethylation is not a significant factor in locus H3K27me3 dynamics. To measure H3K27me3 levels in Eed−/− ISCs, we purified Lgr5+ ISCs by GFP flow cytometry at different days after the first dose of tamoxifen (Figure 3A). The number of ISCs collected at any time was sufficient for RNA sequencing (RNA-seq), and cells collected through 21 days were adequate for rigorously quantitative H3K27me3 ChIP-seq. The mark was erased by day 9 in crypt TA cells or post-mitotic villus cells (Figures 1C and S2B), whereas substantial H3K27me3 remained at that time at PRC2 target sites in Eed−/− ISCs but declined gradually over subsequent days (Figure 3C; data quantified in Figure S2C).
Figure 3. Replicational Dilution of H3K27me3 at Target Genes in PRC2-Deficient ISCs.
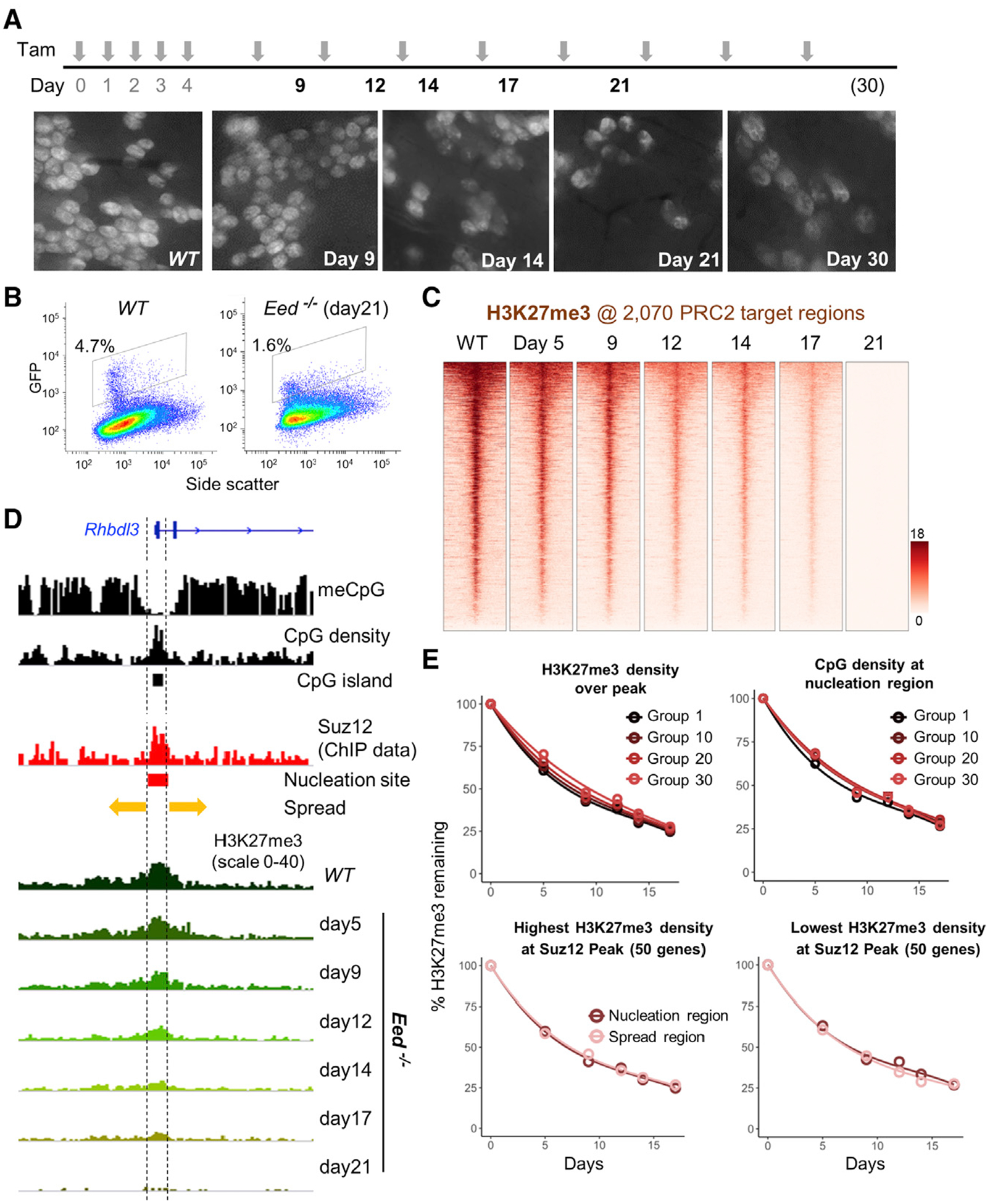
(A) Experimental scheme showing induction of PRC2 loss, induced by tamoxifen (TAM), and ISC collection times to quantify H3K27me3 and mRNA levels. Whole-mount fluorescence micrographs show progressive depletion of crypts containing GFP+ ISCs in EedFl/Fl;Lgr5GFP-Cre(ER) mouse duodenum. Images represent intestines from 2 mice at 30 days and 3 mice at all other times.
(B) Quantitation of Lgr5+ ISCs by GFP flow cytometry before and 21 days after PRC2 loss.
(C) Progressive loss of H3K27me3 at 2, 070 target promoters (TSS ± 10 kb) in the absence of PRC2 function. Genes in the heatmaps are ordered by decreasing H3K27me3 signals in WT ISCs.
(D) IGV tracks at a representative promoter, Rhbdl3, showing the variables known to influence H3K27me3 distribution, including methylated DNA, CpG density (CpG islands), and Suz12 binding (nucleation region). Kinetics of H3K27me3 decay in Eed−/− ISCs are not influenced by these variables, as indicted by the uniform proportional loss across the locus.
(E) In aggregate, the progressive loss of H3K27me3 in Eed−/− ISCs occurs with similar kinetics at sites with high or low starting H3K27me3, CpG dinucleotide density over the Suz12 peak, and in the nucleation or spread regions. Promoters were placed in 50-gene groups from highest to lowest. Graphs are shown for groups 1 (top 50), 10 (genes 501–550), 20 (genes 1, 001–1, 050) and onward. For analysis of nucleation versus spread regions (bottom), groups with the highest (left) and lowest (right) H3K27me3 density at the Suz12 peak are shown.
See also Figure S2.
Sequential H3K27me3 ChIP from purified PRC2-null cells of a single kind provided powerful data to examine the kinetics of H3K27me3 clearance in relation to parameters such as DNA methylation and starting H3K27me3 density. In mammalian cells, which lack PREs, H3K27me3 marks concentrate at unmethylated CpG islands, in inverse proportion to the degree of DNA methylation over the region (Xie et al., 2013). Here, PRC2 binds at central “nucleation” sites and then spreads its enzymatic activity laterally in either direction (Oksuz et al., 2018), as exemplified in intestinal cells at the Rhbdl3 locus (Figure 3D). In this light, we placed all H3K27me3-marked regions in bins of 50 each, from those with the highest to the lowest ChIP signal, either in aggregate or at nucleation sites (PRC2/SUZ12 ChIP peaks; Jadhav et al., 2019). Separately, we also considered ChIP signals in nucleation or spread regions and 50-region bins from highest to lowest density of methylated CpG dinucleotides in the nucleation regions. Throughout the period of observation, the rates of H3K27me3 decay after PRC2 depletion were essentially indistinguishable across these bins and at nucleation or spread regions (Figures 3D and 3E). Thus, H3K27me3 is cleared uniformly across all regions, in proportion to the starting levels, much as the mark is proportionally reduced at all regions in Ezh2−/− intestinal cells (Figure 1D). These findings implicate replicational dilution as the principal, and perhaps only, agent in clearing H3K27me3.
If clearance of H3K27me3 by replicational dilution is the limiting factor in PRC2 target gene derepression, then ISCs should activate those genes upon continued H3K27me3 clearance after day 9. Indeed, GFP+ ISCs isolated at increasing time after Cre induction gradually activated the same genes that were derepressed considerably earlier in villus cells, and the pace of gene derepression matched that of H3K27me3 depletion (Figure 4). A total of >99% of intestinal PRC2 targets were silent on the 9th day and 17% were active by day 14, rising to 76% with significant expression by day 30 (Figure 4A). Cdkn2a and Hoxb13 mRNA levels, for example, increased >150-and >1, 500-fold, respectively (Figure 4B; additional representative data in Figure S2D), by day 30, reflecting a long delay after Eed deletion. Thus, the many TA cell divisions, and fewer ISC divisions in the same period, explain the different kinetics of PRC2 target gene derepression in vivo. Gradual Cdkn2a activation particularly explains why PRC2-proficient, Cdkn2a− crypts replace Eed-null ISCs over time.
Figure 4. Gradual Reactivation of PRC2 Target Genes in Lgr5+ ISCs.
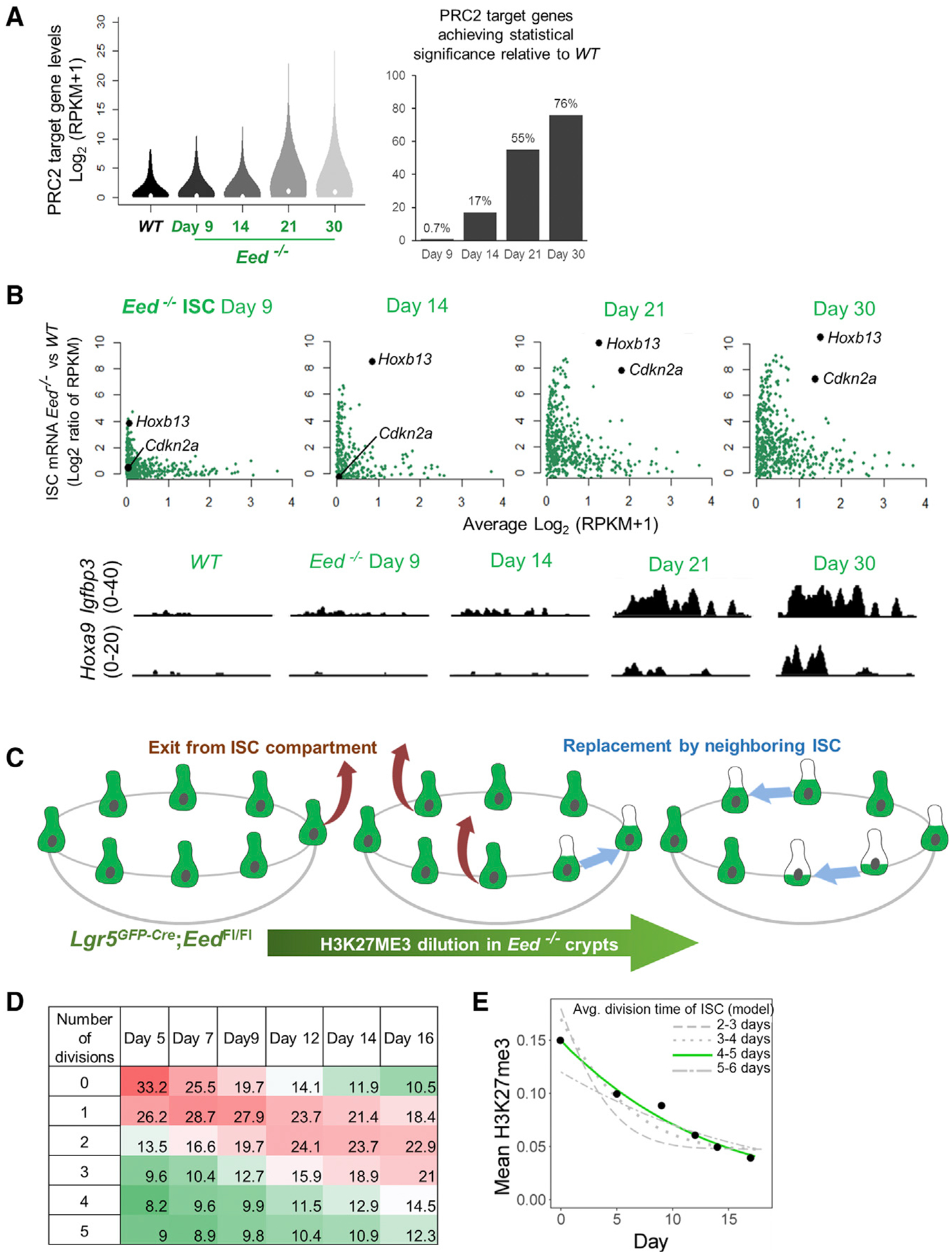
(A) Aggregate analyses in Eed−/− ISCs (RNA-seq data) reveal cumulative activation of 76% of PRC2 target genes over an extended period of PRC2 absence. The analysis is restricted to bivalent genes activated in Eed−/− villi (red dots in Figure 2A), showing mRNA levels of all these genes in the violin plot and those that achieved significance (false discovery rate, q < 0.05) in the bar graph.
(B) Discrete analysis of PRC2 targets that were activated in villus cells (red dots in Figure 2A), showing delayed and progressive activation in Eed−/− ISCs. Representative IGV tracks show low levels of Igfbp3 appearing by day 9, whereas Hoxa9 is measurably expressed after 14 days.
(C) H3K27me3 status in the Lgr5+ ISC compartment after PRC2 loss and replicational dilution influenced by the stochastic nature of ISC divisions.
(D) Predicted fractions of ISCs that have completed the indicated number of cell divisions by the indicated days, considering 16 starting ISCs per crypt and known features of neutral drift.
(E) Computational model of H3K27me3 decay by dilution in ISCs. Each gray curve represents the rate of decay, averaged across all PRC2 target promoters, as the model predicts for different intervals (2–3 days, 3–4 days, etc.) between ISC divisions. Experimental data (black dots, mean H3K27me3 at target promoters at different times after CRE activation in vivo) best fit the green curve (~4-day interval between ISC divisions).
See also Figure S2.
Target Gene Activation Is a Function of Both H3K27me3 Depletion and Basal Promoter Poising
Although the opposing trends in H3K27me3 depletion (Figure 3C) and PRC2 target activation (Figure 4A) are clear, the measurements are challenging to interpret in bulk Lgr5+ ISCs. Because these cells divide stochastically (Lopez-Garcia et al., 2010; Snippert et al., 2010), the population at any time is a mix of cells that divided repeatedly, hence depleting H3K27me3, and others that seldom divided, thus retaining marked nucleosomes. To study H3K27me3 dynamics and target gene activation in this light, we derived a computational model for stochastic ISC replication and exit from the crypt base, with nearby parental or sibling ISCs randomly filling vacated spots (Figure 4C). From this model, where the nodes of a graph describe single cells and its edges keep track of the nearest ISC neighbors (Chakrabarti et al., 2018), we estimated the fraction of ISCs that had replicated a specified number of times by given days after PRC2 depletion (Figure 4D). To simulate H3K27me3 decay in the ISC population over time, we halved H3K27me3 levels at every division, choosing at random which cells divide and which cells leave the compartment. As expected, the rate of H3K27me3 decay for different cell division frequencies (gray curves) was slower than the interval between cell divisions (Figure 4E) because only some ISCs will have divided, so the population loses <50% of H3K27me3 at each locus. The theoretical curve that best fits the experimental ChIP-seq data for H3K27me3 decay (Figures 3C and S2C) corresponds to a mean cell division interval of ~4 days (Figure 4E, green curve), a value that was insensitive to the number of Lgr5+ cells in the crypt base and whether we considered H3K27me3 levels over 500 bp or 1 kb at target promoters (Figure S2E). Considering the expected lag between tamoxifen exposure and loss of PRC2 activity, this value mirrors the reported average time of 3 to 3.5 days between ISC divisions (Kozar et al., 2013). Because our model considers only replicational H3K27me3 dilution, this agreement with the reported ISC division rate–or even a little slower–suggests that other factors, such as enzymatic demethylation, have a minor or no role.
Even by 9 days, the pool of Eed−/− ISCs should contain some cells that have divided ≥3 times (Figure 4D), likely accounting for small but discernible target gene activity (Figure 4B, e.g., Hoxb13), but factors other than absolute residual H3K27me3 levels might also contribute. We, therefore, examined mRNAs expressed in Lgr5+/GFP+ ISCs at a single-cell (sc) resolution on different days after mice received a single, 2-mg dose of tamoxifen (STAR Methods). In aggregate, 11, 123 cells were informative, and by uniform manifold approximation and projection (UMAP analysis; McInnes et al., 2018), cells harvested on days 7 and 12 clustered together, whereas those isolated on day 16 were distinctive (Figure 5A). Some target transcripts were, however, present in single cells on day 12 and even on day 7, whereas others demonstrated comparable activity only in ISCs isolated 16 days after Eed deletion (Figures 5A and 5B). Igf2 and Prss16, for example, were activated early, whereas Cdkn2a and Hoxc9 were mostly activated later (Figures 5A and S3B).
Figure 5. Progressive Derepression of PRC2 Targets Is Associated with Promoter Poising.
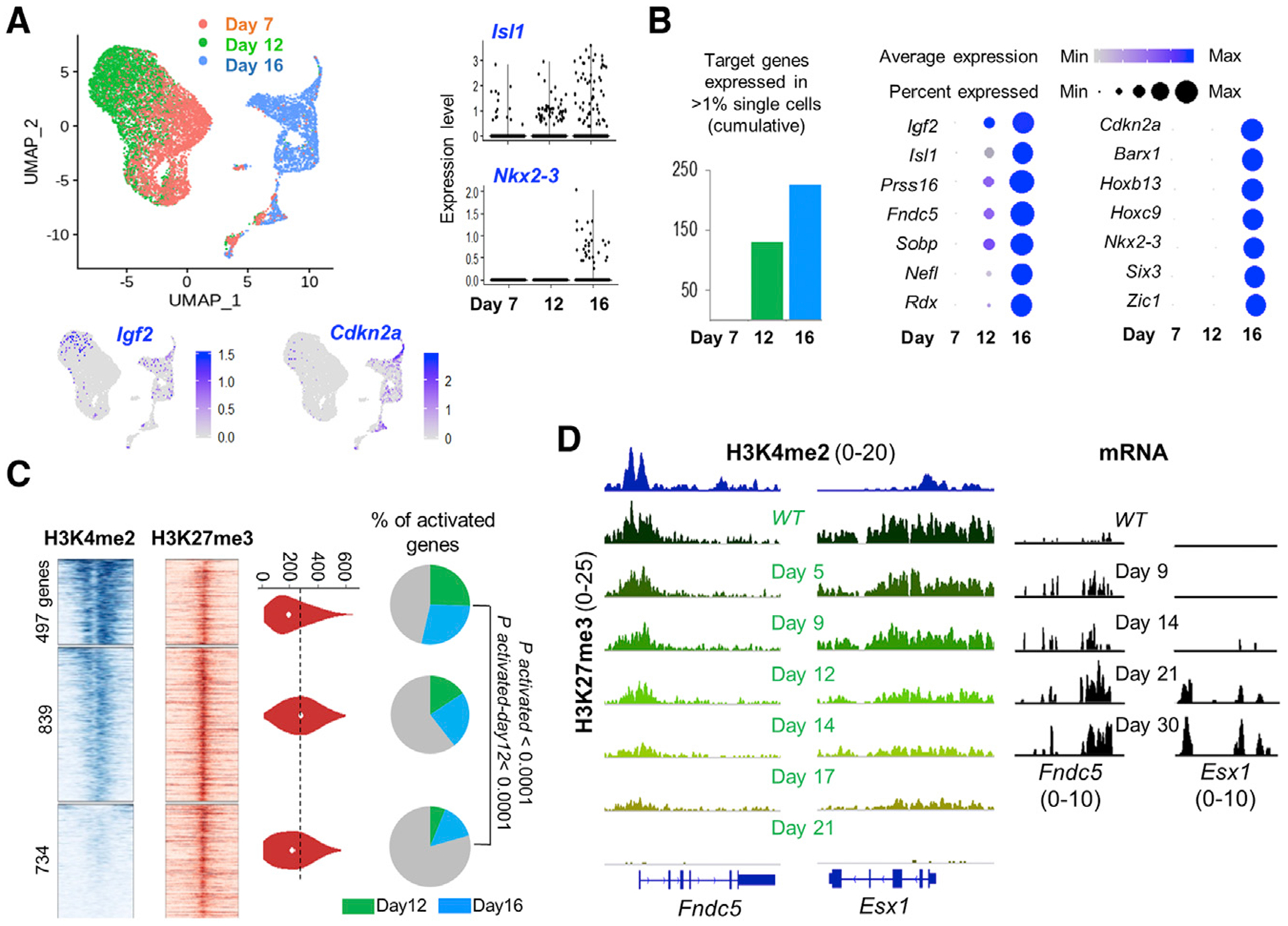
(A) Uniform manifold approximation and projection (UMAP) plots from RNA analysis of single Eed−/− ISCs harvested on different days after PRC2 inactivation. Representative target genes Igf2 and Cdkn2a (projected on the UMAP template) and Isl1 and Nkx2–3 (violin plots, right) show early (day 12) or delayed (day 16) activation.
(B) Left, increasing numbers of PRC2 target transcripts were detected in >1% of cells on days 12 and 16, compared with their absence on day 7. Right, progressive gains in RNA levels (color scale) and the fraction of cells with measurable RNA (dot diameter) of PRC2 target genes, starting with their silence as late as 7 days after Eed deletion. Some targets activate earlier than others.
(C) Promoter H3K4me2 (TSS ± 1.5 kb) and H3K27me3 (TSS ± 10 kb) signals in ISCs at 2, 070 PRC2 target genes, grouped in 3 k-mean clusters based on H3K4me2 signal density. H3K27me3 signals in the 3 clusters are similar (quantified in violin plots). Pie charts show that genes with high promoter H3K4me2 have higher (q < 0.0001, “N-1” chi-square test) activation, both overall and specifically on day 12 versus day 16.
(D) Representative IGV tracks at Fndc5 and Esx1, genes with different degrees of promoter poising (H3K4me2). Upon PRC2 loss, H3H27me3 is cleared proportionally, whereas Fndc5 (high promoter H3K4me2) activates sooner than Esx1 (low H3K4me2).
See also Figure S3.
As H3K27me3 decays proportionally at all target genes (Figures 3C–3E), this difference across hundreds of targets must have a basis other than absolute H3K27me3 levels. We, therefore, revisited the role of promoter bivalency (Jadhav et al., 2016) and clustered PRC2 target genes according to promoter H3K4me2 levels in WT ISCs; clusters with high, low, or virtually no promoter H3K4me2 showed minimal differences in basal H3K27me3 (Figure 5C). The information content in sc RNA libraries is sparse because many cells lack specific transcripts owing simply to inefficient capture (Yuan et al., 2017). Nevertheless, among all target genes detected in >1% of ISCs at any time, more genes with high basal H3K4me2 appeared at all and were present earlier than those with low H3K4me2 (Figure 5C). Thus, replicational H3K27me3 dilution is a dominant but not the only factor in PRC2 target gene activation. Given that H3K27me3 clears uniformly at all sites, these data indicate that well-poised promoters activate in the face of higher residual H3K27me3 levels than less-poised promoters, which activate only upon further H3K27me3 erasure. Representative loci illustrate this contrast and that earlier gene activation does not reflect more rapid clearance of H3K27me3 (Figure 5D).
Cell-Replication-Dependent H3K27me3 Dilution Is Generally Responsible for Gene Derepression
Replication-dependent dilution of H3K27me3 has been demonstrated in Drosophila larvae (Coleman and Struhl, 2017; Laprell et al., 2017) and at various phases of the cell cycle in cultured human HeLa cells (Reveron-Gomez et al., 2018). Nevertheless, the observed PRC2 target gene kinetics in ISCs could in principle reflect time-dependent sensitivity to EED loss for some reason other than the frequency of cell division. To exclude this possibility, we stained the EZH2 mutant lymphoma cell line SU-DHL-10 (Drexler, 2001) with CellTrace. This permeable fluorescent dye binds cytoplasmic proteins stoichiometrically and dilutes 50% at each mitosis, hence allowing isolation of cells by flow cytometry after precisely 1, 2, 3, and further cell divisions (Filby et al., 2015). Fluorescence in SU-DHL-10 cells was reduced by 50% on consecutive days, reflecting near-synchronous cell divisions within the known ~24-h doubling time (Drexler, 2001). To examine the consequences of PRC2 loss, we treated cells with the EZH inhibitor UNC1999 or its inactive analog UNC2400 (Xu et al., 2015), and to test the need for cell replication in PRC2 effects, we applied the cell cycle inhibitor palbociclib, which arrested the serial dilution of fluorescent signals (Figure 6A). Most cells treated with 5 μM UNC1999 and 2 μM palbociclib, alone or together, survived.
Figure 6. Cell-Replication-Dependent H3K27me3 Dilution Underlies PRC2 Target Gene Derepression in Lymphoma Cells.
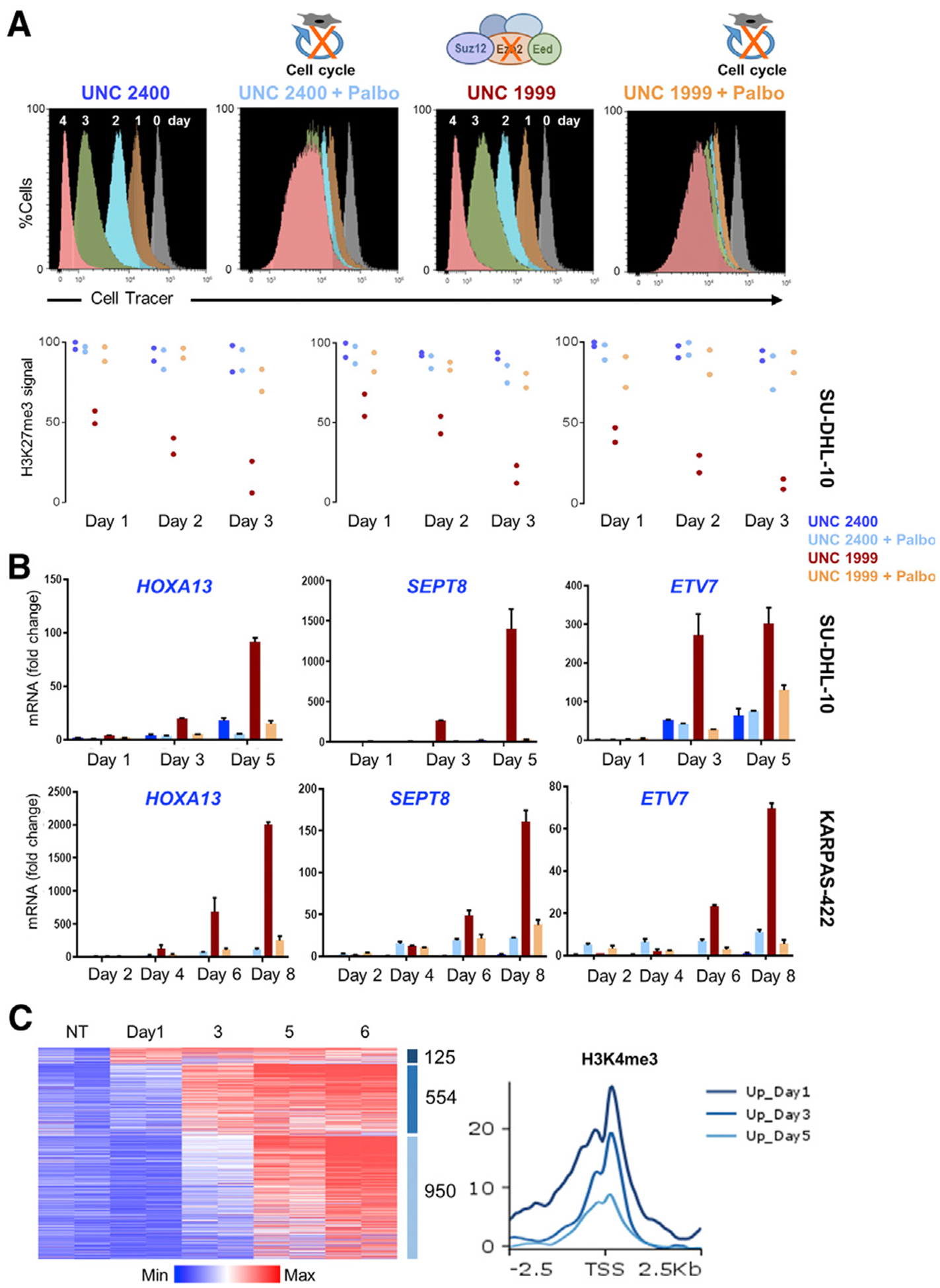
(A) Histograms from flow cytometry of CellTrace-labeled SU-DHL-10 cells, showing ~50% reduced fluorescence each day, reflecting replicational dilution of the cytoplasmic dye. Cell cycle inhibitor palbociclib (Palbo) arrests this dilution, and inhibition of PRC2 by the small molecule UNC1999 or an inactive analog (UNC2400) does not materially deter cell division. qRT-PCR analysis (representing 2 or 3 biological replicates) shows gradually increasing expression of representative PRC2 target genes in UNC1999-treated SU-DHL-10 cells and attenuation of this increase in cells co-treated with Palbo. ChIP-qPCR for H3K27me3 at the corresponding promoters reveals persistent signals in control cells, ~50% reduction in UNC1999-treated cells after each division, and attenuated reduction of H3K27me3 in cells co-treated with Palbo.
(B) Likewise, PRC2 target genes increase expression only after repeated cell division in UNC1999-treated KARPAS-422 cells and Palbo attenuates this increase. Data are from 2 or 3 biological replicates.
(C) Progressive activation of PRC2 target genes, measured by RNA-seq analysis of DMSO-treated cells (controls) and on different days after UNC1999 exposure of SU-DHL-10 cells. Data (log2 RPKM+1) are shown for duplicate samples from each condition. Aggregate quantitation of H3K4me3 around promoters (TSS ± 2.5 kb) of genes expressed 1, 3, or 5 days (and after). Average H3K4me3 signals are higher at promoters that activate early.
See also Figure S4.
To identify PRC2 target genes in SU-DHL-10, we performed ChIP-seq for H3K27me3 and H3K4me3. In line with EZH2 hyperactivity, H3K27me3 extended broadly at about one-half of all loci, including expressed genes, but the marking at ~3, 000 transcription start sites (TSSs) ended abruptly near dense H3K4me3 deposits (Figure S4A). By ranking all H3K27me3-marked TSSs according to H3K4me3 levels, 5, 288 promoters showed low H3K27me3, abundant H3K4me3, and RNA expression in spite of nearby H3K27me3 (Figure S4B). We, hence, regard this group as PRC2-insensitive and focus on the 8, 211 unexpressed (<1 reads per kb of transcript per million mapped reads [RPKM]) H3K27me3+ genes (Figure S4C) that may represent bona fide targets, analogous to non-lymphoma cells. In SU-DHL-10 cells collected serially after drug treatment, ChIP-qPCR at representative promoters (STAR Methods) showed a geometric decline of H3K27me3 after successive cell divisions, and cell cycle arrest attenuated this loss (Figures 6A and S4C). Having, thus, validated the experimental model, we first assessed selected target genes by quantitative RT-PCR. These targets were marginally derepressed by the third day after PRC2 inhibition and massively increased by day 5, and gene activation was negated upon co-treatment with palbociclib (Figures 6B and S4D). Another EZH2 mutant lymphoma cell line, KARPAS-422, which cycles ~2.5 times slower than SU-DHL-10, confirmed this cell-replication-dependent activation of representative PRC2 target genes (Figures 6B and S4D).
To extend these findings across all targets, we treated SU-DHL-10 cells with UNC1999, followed by RNA-seq analysis of biological replicates collected by CellTrace flow cytometry after specified numbers of cell divisions. Considering all target genes detected at any time, we observed a clear global trend of increasing activation after successive cell divisions (Figure 6C). As with Eed−/− ISCs (Figure 5), again some target genes activated sooner than others, and the differences in the onset of gene activity correlated with basal levels of promoter H3K4me3 (Figure 6C). To exclude non-specific effects of UNC1999, we treated SU-DHL-10 cells separately with the EED-targeted compound MAK-683 (Huang et al., 2017). Again, most PRC2 target genes were activated after 4 cell divisions, with a minority activated after fewer divisions (Figure S4E). Thus, when PRC2 function is absent, both mouse intestinal and human B lymphoma cells erase H3K27me3 by diluting parental nucleosomes at each cell division, and bivalent PRC2 target genes become active when the local H3K27me3 density falls below some critical level. The correlation between target gene activation and basal H3K4me3 levels implies that activation thresholds are lower for well-poised promoters, i.e., they activate with more residual H3K27me3, than less-poised promoters, which require further removal of H3K27me3 for gene activity. H3K4me3 could be a crucial modification promoting gene activity or a proxy for some other feature that promotes gene expression.
DISCUSSION
Across adult somatic cell generations, two mechanisms are possible for the loss of covalent histone marks: enzymatic erasure or dilution of parental nucleosomes over daughter chromatids. Our study of distinct cell compartments in the mammalian gut epithelium reveals that replicational dilution is the dominant mode for the loss of H3K27me3, which occurs in proportion to the frequency of cell divisions. Because ISCs replicate 5–10 times less frequently than TA cells (Kozar et al., 2013; Parker et al., 2017), they retain H3K27me3 for extended periods after PRC2 inactivation, thus keeping PRC2 target genes repressed, whereas H3K27me3 levels in TA cells and their post-mitotic progeny rapidly drop below the levels necessary to maintain gene silence. Our observations leave open the possibility that KDM6-mediated demethylation contributes toward H3K27me3 erasure during transitions from ISCs to TA and post-mitotic villus cells; in PRC2-null ISCs, however, replicational dilution fully explains H3K27me3 kinetics, independent of basal H3K27me3 or CpG dinucleotide density and without the need to invoke active demethylation. These findings extend to the in vivo setting in mice the elegant prior work on related questions in Drosophila embryos (Coleman and Struhl, 2017; Laprell et al., 2017) and in cultured HeLa cells at different phases of the cell cycle (Reveron-Gomez et al., 2018). The consequences of PRC2 inactivity in mouse intestines (Jadhav et al., 2016) suggest that EZH and EED inhibitors will cause significant intestinal toxicity. Our findings explain why tissue defects vary according to the number of divisions by PRC2-null ISCs (Chiacchiera et al., 2016; Koppens et al., 2016) and suggest that clinical toxicity could be mitigated by drug regimens that preserve sufficient H3K27me3 in the ISC compartment.
Although the correlations we report are robust and internally coherent, our conclusions rest largely on indirect evidence. In the future, ChIP-seq analysis at a sc resolution (scChIP) might definitively demonstrate the relations between gene activation thresholds and serial dilution of H3K27me3 marks. Chromatin access can now be determined at sc resolution (Buenrostro et al., 2018), but scChIP remains challenging (Yuan et al., 2017), especially when histone marks such as H3K27me3 are spread over large genomic distances. To examine H3K27me3 dynamics in PRC2-null ISCs, we developed a method to model the stochasticity of neutral drift among Lgr5+ ISCs. Our computational approach replicated the process correctly, in that crypts became monoclonal in the expected time (Lopez-Garcia et al., 2010; Snippert et al., 2010), and accurately predicted our experimental data on H3K27me3 decay and locus-to-locus variance, based on the established value of 3 to 3.5 days for the interval between ISC divisions (Kozar et al., 2013). These computational simulations, thus, support the idea that PRC2-null cells lose H3K27me3 when unmodified histones dilute marked parental nucleosomes during cell division.
Lone Ezh2 deletion has discernible, albeit mild, effects in some mouse tissues (Chen et al., 2009; Ezhkova et al., 2009; Hidalgo et al., 2012; Hirabayashi et al., 2009; Juan et al., 2011) but not the intestine. Our rigorous analysis of H3K27me3 in Ezh2- and Eed-null intestinal cells is instructive in this light. Unexpectedly, Ezh2−/− cells retain only ~40% of basal H3K27me3, a level that is sufficient to maintain the silence of all PRC2 target genes. In contrast, Eed−/− ISCs and PRC2-null lymphoma cells require 3 or more cell divisions to derepress many genes, implying that the threshold for their gene activity is ≤12.5% of native H3K27me3 levels. An important conclusion of this study is that the critical threshold is not the same for all PRC2 targets. Genes with high basal promoter poising, reflected in high H3K4me2/3 levels, overcome silencing with more residual H3K27me3 than less-poised genes, which require additional clearance of H3K27me3 before they become active. Of course, H3K27me3 levels likely differ depending on cell cycle times, PRC2 concentration (Stafford et al., 2018), and the cell lineage, and different promoters are better or less poised in different tissues (Jadhav et al., 2016). Thus, fractions of H3K27me3 loss sufficient for gene derepression–activation “thresholds”–will vary not only by tissue differences in promoter poising but also by differences in basal H3K27me3 levels at different genes.
The quantitative loss of H3K27me3 in Ezh2−/− intestines is nearly identical across PRC2 target loci, and even subtle variation among nucleosomes is accurately preserved in Ezh2−/− cells. Moreover, in the absence of PRC2 function, regions with high basal H3K27me3, high CpG density, and PRC2-nucleated or spread sites all lose H3K27me3 marking at essentially the same rate. Our findings suggest that tissue differences in compensation for the absence of EZH2 largely reflect the amount of available EZH1 to keep H3K27me3 levels above the activation thresholds and not qualitative differences in methyltransferase activity at different genes. This point is pertinent to drug toxicity because both EZH2 and EED inhibitors are currently in clinical development.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests should be directed to the Lead Contact, Ramesh Shivdasani (ramesh_shivdasani@dfci.harvard.edu), and will be fulfilled after execution of a suitable Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Lgr5EGFP-IRES-CreERT (Barker et al., 2007) and ROSA26lsl-yfp mice were purchased from The Jackson Laboratories, and Ezh2+/Fl mice (Su et al., 2003) from the Mutant Mouse Regional Resource Centers. VillinCreER-T2 (el Marjou et al., 2004) and Eedfl/fl mice (Xie et al., 2014) were generous gifts from S. Robine (Institut Pasteur, France) and S. Orkin (Boston Children’s Hospital), respectively. Mice were housed in a colony maintained at 23 ± 1°C, 55 ± 15% humidity, and 12 hr light/dark cycles, and handled according to protocols approved by the Animal Care and Use Committee of the Dana-Farber Cancer Institute. Mice were genotyped by PCR before weaning and again at the time of experiments.
METHOD DETAILS
Mouse treatments
Animals 8 weeks or older were injected with 2 mg intraperitoneal (i.p.) tamoxifen on 5 consecutive days, followed in some experiments by 1 mg tamoxifen every 3rd day. For detection of proliferating cells, 1 mg BrdU in PBS was administered i.p. 1 h before euthanasia and intestinal harvest.
Isolation of intestinal villus, crypt and stem cells
RNA and chromatin studies and histologic analyses were done on tissue or cells from the proximal 1/3 small intestine. To isolate villi, fresh tissue was washed with cold phosphate-buffered saline (PBS) and rotated for 40 min in 5 mM EDTA in PBS (pH 8) at 4°C, with manual shaking every 10 min. To recover villus epithelium, the resulting suspension was passed over 70-μm filters. To purify Lgr5+ ISC from Lgr5EGFP-IRES-CreERT2 mouse intestines, villi were scraped away with glass slides and intestines were washed in PBS, followed by rotation for 40 min in 5 mM EDTA in PBS (pH 8) at 4°C with manual shaking every 10 min and a change of EDTA solution after 30 minutes. Villus fragments were removed using 70-μm filters and crypts, collected from the filtrate by centrifugation, were rotated in 4% TrypLE solution (Invitrogen) at 37°C for 30 min to generate single-cell suspensions. Viable ISC (GFPhi DAPI−) were then isolated by flow cytometry in a BD FACSAria II SORP instrument. To collect YFP-labeled cells from Lgr5Cre;EedFl/Fl;RosaYFP mice, purified crypts or villi were digested with 4% TrypLE solution for 30–50 minutes at 37°C, followed by FACS isolation using the YFP channel.
Detection of cells and proteins
Intestines were fixed overnight in 4% paraformaldehyde at 4°C, followed by dehydration in an ethanol series and embedding in paraffin. To examine tissue morphology, 5-μm sections were stained with hematoxylin and eosin. Alternatively, sections were treated with 10 mM sodium citrate (pH 6) for antigen retrieval and incubated overnight at 4°C with Ab against H3K27me3 (Millipore 07–449, 1:1000), EZH2 (Cell Signaling D2C9, 1:2500), or KI67 (Vector VP-K452, 1:500) diluted in PBS. Sections were washed with PBS and incubated with anti-rabbit or anti-mouse IgG conjugated to Cy3 or biotin (Jackson Laboratories, 1:1000). Signals were detected by fluorescence or after staining with Vectastain Elite ABC Kit (Vector) and 3, 3′ diaminobenzidine tetrahydrochloride (Sigma P8375). Consecutive tissue sections were used to identify basal Lgr5Gfp cells (GFP Ab, Santa Cruz SC9996, 1:200) and CDKN2A-expressing cells (CDKN2A Ab, Santa CruzSC32748, 1:200) in the same crypts. To detect lineage tracing from Lgr5Gfp ISC, fixed mouse intestines were flash-frozen in OCT compound (Tissue-Tek, 4583) and 5-μm sections were visualized by fluorescence microscopy. To visualize proliferating cells, sections were stained with BrdU Ab (AbD Serotec OBT0030CX) followed by Alexa Fluor 488-conjugated anti-rat IgG. Tissues were analyzed from at least 4 mice of each genotype.
Culture, treatment, and molecular analysis of lymphoma cells
SU-DHL-10 and KARPAS-422 cells were cultured in RPMI medium (GIBCO 11875–093) supplemented with 20% fetal bovine serum (FBS) and antibiotics. Cells were seeded at 3 × 105 cells/mL and treated with 5 μM (for SU-DHL-10) or 2.5 μM (for KARPAS-422) UNC1999 or the same concentrations of UNC2400 (R&D Systems), alone or in combination with 2 μM or 5 μM palbociclib (Sigma-Aldrich PD0332991), respectively, or with 10 μM MAK-683 (B1972–5, BioVision), as specified. Cells were collected on the indicated days after drug treatment and suspended in Trizol reagent. Total RNA was isolated with RNeasy Mini Kit (QIAGEN), followed by on-column DNase I digestion. cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), followed by qRT-PCR using PowerSYBR Green (Applied Biosystems) and primers listed in Suppl. Table S1. Values for each transcript were normalized to those for Gapdh mRNA. Treated SU-DHL-10 were stained with 5 μM CellTrace Violet Cell Proliferation Kit (Life Technologies C34557) and sorted using a Sony SH800z flow cytometer on the specified days. Live cells were identified using LIVE/DEAD Fixable Far-Red Dead Cell Stain Kit (Thermo Fisher Scientific L-34973). RNA-seq methods are described below. To determine effective concentrations of PRC2 inhibitors, treated SU-DHL-10 cells were lysed in Triton Extraction Buffer (TEB) containing 0.5% Triton X-100 and 2 mM phenylmethylsulfonylfluorde (PMSF) in PBS for 10 min, followed by overnight extraction of histones in 0.2 N HCl at 4°Cand immunoblotting with H3K27me3 (Millipore 07–449, 1:1000) and Histone3 (Cell Signaling 9715S, 1:1000) Ab.
RNA- and ChIP-seq
Intestinal villus, crypt, or SU-DHL-10 cells were pelleted and resuspended in Trizol reagent (Life Technologies); FACS-isolated Lgr5+ ISC and YFP+ crypt or villus cells were collected directly in Trizol. Total RNA was purified using the manufacturer’s instructions and used to prepare libraries with TruSeq RNA Sample Preparation Kit V2 (Illumina RS-122–2001), followed by sequencing on a NextSeq 500 instrument (Illumina) to obtain 75-bp single-end reads. For scRNA-seq, 104 FACS-purified ISC were loaded onto Chromium Chip B using the 3′ GEM Library & Gel Bead Kit v3 (10X Genomics), followed by reverse transcription, cDNA amplification, and library preparation according to the manufacturer’s instructions. Libraries were sequenced on a HiSeq4000 instrument (Illumina).
For ChIP-seq, cells were fixed by rotation in 1% formaldehyde solution for 25 min at room temperature. 1×106 to 5×106 fixed cells were lysed by mixing in a buffer containing 30 mM Tris-HCl (pH 8), 1% SDS, 10 mM EDTA, and protease inhibitors (Roche), followed by sonication in a Covaris E210 sonicator for 5- to 10-min cycles at 4°C. Sheared chromatin was centrifuged to remove debris and incubated overnight at 4°C with antibody (Ab) against H3K27me3 (Millipore 07–449), H3K4me3 (Diagenode, C15410003), or KDM6A (Cell Signaling, D3Q1I), together with spike-in chromatin (10 ng Drosophila Line 2, Active Motif 53083) and spike-in Ab (2 μg, Drosophila-specific histone variant H2Av, Active Motif 61686). Ab-chromatin complexes were isolated using magnetic beads (Dynal) and sequentially washed twice in low-salt (20 mM Tris-HCl pH 8.1, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100), high-salt (20 mM Tris-HCl pH 8.1, 500 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100), and lithium chloride (10 mM Tris pH 8.1, 0.25 M LiCl, 1 mM EDTA, 1% NP-40, 1% deoxycholate) buffers. Cross-links were reversed by incubation in 1% SDS and 0.1 M NaHCO3 for 6 h at 65°C, followed by DNA purification using columns (QIAGEN). ChIP-seq libraries were prepared using the ThruPLEX kit (Rubicon, R400427) and 500 pg of library DNA was analyzed using high sensitivity DNA Chip with Bioanalyzer 2100 (Agilent Genomics) to determine DNA size distribution. Libraries were sequenced to generate 75-bp single-end (NextSeq 500 instrument, Illumina) or 150-bp paired end (HiSeq X instrument, Novogene) reads.
ChIP-qPCR
SU-DHL-10 cell were pelleted, followed by fixation in 1% formaldehyde, rotating for 10 min at ambient temperature. ChIP assays were performed as described above, using 5×106 fixed cells and H3K27me3 Ab. Primers (listed in Suppl. Table S1) were designed within 1 kb of PRC2 target TSSs. Equal amounts of ChIP (from various days after UNC-1999 treatment) and input DNA (sonicated chromatin collected before ChIP) were amplified with PowerSYBR Green. Each target region was assessed in biological duplicates and results were normalized to the input DNA.
QUANTIFICATION AND STATISTICAL ANALYSIS
Immunohistochemistry (Figure S1B) and immunofluorescence (Figures 1A, 2B, and 2C) were conducted using tissue from at least 3 independent animals (N = 3). In cell proliferation assays using BrdU co-localization with Lgr5Gfp ISC or TA cells (Figure 2C), at least 50 crypts with Lgr5Gfp ISC were analyzed. To quantify precisely and compare H3K27me3 signals across various conditions (Wild-Type, Ezh2−/−, Eed−/−) at various times, we used Drosophila chromatin spike-in with an antibody specific to Drosophila histone variant H2Av during ChIP-seq experiments; details are mentioned in the section on Computational Analyses. qRT-PCR and ChIP-qPCR on lymphoma cells (Figures 6A–6C, S4B, and S4C) were run on biological duplicates.
Computational analyses
Raw RNA-seq reads were aligned to the mouse genome (Mm9, NCBI build 37) using STAR aligner v2.5.3a (Dobin et al., 2013) followed by RSeQC v2.6.2 assessment (Wang et al., 2012) to determine per-base sequence quality, per-read GC content (~50%), comparable read alignments to ± strands, exon versus intron read distributions, and 3′ bias. Transcript levels were expressed as read counts using HTSeq v0.6.1 (Anders et al., 2015) and normalized across libraries using DESeq2 (Love et al., 2014), followed by conversion into reads per kb of transcript length per 1M mapped reads (RPKM). We determined differential expression between samples using DESeq2, with false-discovery rate (FDR) < 0.05.
Raw ChIP-seq reads were aligned to the mouse genome (Mm9, NCBI build 37) using Bowtie2 v2.3.4.3 (Langmead and Salzberg, 2012); reads aligned to multiple locations and PCR duplicates were removed from further analysis. For 150-bp paired-end sequence data, only the Read1 (forward) files were trimmed to 75 bp and used for further processing. For quantitative comparison of H3K27me3 signal across various samples, libraries were down-sampled to similar read depth. For libraries with Drosophila chromatin spike-in, reads were aligned to the Drosophila genome (dm6) and a normalization factor (NF = Drosophila reads in library with the lowest count / Drosophila reads in individual library) was derived for each, followed by down-sampling of read counts in proportion to this NF. Reads were converted into signal files (bigWig) using DeepTools v2.1.0 (Ramírez et al., 2016) and visualized using the Integrated Genome Viewer (IGV) (Robinson et al., 2011). H3K27m3-marked regions were detected using SICER v0.0.1 (Xu et al., 2014), and normalized H3K27me3 read numbers per bp were determined using BedTools v2.26.0 (Quinlan and Hall, 2010) (Figures 1C, 1D, S2B, and S2C) for peaks associated with TSSs in WT cells or total read numbers for genomic loci (Figures S1D). Scatter (e.g., Figures 2A and 4B) and violin plots (e.g., Figures 1C, S2B, S2C, 4A, and 5C) for gene expression and H3K27me3 ChIP-seq signal estimates were generated in base R (version 3) or using ggplot2 package v2.2.1(Wickham, 2009). ChIP-seq heatmaps for KDM6A and aggregate density profiles of read distribution were generated using DeepTools.
To determine candidate PRC2 target genes in lymphoma cells, we plotted H3K27me3 ChIP signals from SU-DHL-10 cells along 27,135 RefSeq loci (Figure S4A, genes divided in 5 equal-sized bins and 2-kb regions upstream of TSSs [1st column] and downstream of transcription end sites [last column]). Genes were clustered by k-means (k = 5) based on ChIP signal strength, and 13,439 genes (top 3 clusters, Figure S4A) were analyzed for RNA expression and presence of H3K4me3 at the TSS. 5,228 genes with high promoter H3K4me3, active expression (RPKM ≥ 1), and lacking H3K27me3 over gene bodies were designated as PRC2-insensitive; the remaining 8,211 genes lacking expression (RPKM < 1) were considered putative PRC2 targets.
Single-cell (sc) RNA-seq
We used Cell Ranger v3.0.2 (10X Genomics) to align scRNA-seq libraries with 95,947,874 (Day 7), 118,804,298 (Day 12), 98,335,287 (Day 16) raw reads to the GRCm37/mm9 mouse genome and to estimate unique molecular identifiers (UMIs). Raw aligned features were processed using the Seurat v3.0.2 package (Butler et al., 2018) in R version 3.6.1 (R Core Team, 2013). We then excluded cells with < 3,000 UMIs, < 1,200 unique genes, > 20% mitochondrial gene contribution, and expression novelty (log10 Genes per UMI) < 0.8 (Figure S3A). The remaining 11,123 cells (3,749 cells from day 7, 4,641 cells from day 12, and 2,733 cells from day 16) were used for differential mRNA analysis over the different time points.
Normalization, reduction of dimensionality, and clustering
We used Seurat to normalize the data and to detect variable genes, applying Variance Stabilization transformation, followed by scaling and principal component (PC) analysis. Cell clusters were defined using the top 10 PCs to construct a Shared Nearest Neighbor graph. Dimensionality was reduced using the Uniform Manifold Approximation and Projection (UMAP) technique (McInnes et al., 2018). For all 2,070 PRC2 target genes, we determined the fraction of ‘expressing’ cells, i.e., those with transcripts detected in ≥ 1% of cells on a given day. Given the limited representation of transcripts in scRNA-seq data, we did not observe any target gene expression on Day 7 (Figure 5B). PRC2 targets were classified as early- or late-activated depending on detectable expression on Day 12 or Day 16 (Figures 5B and 5C).
Mathematical modeling
We based the model on previous evidence for neutral ISC drift (Lopez-Garcia et al., 2010; Snippert et al., 2010). To implement a model where ISC are arranged geometrically in a ring, we followed our earlier work on tracking stochastic cell divisions and lineages using graphs (Chakrabarti et al., 2018). A ring comprising 14–16 cells was constructed using the R package iGraph (Csardi and Nepus, 2006), such that each cell represents a node on the graph and undirected edges of the graph connect the nearest neighbors in the ring. Each node is ascribed attributes that keep track of cellular birth time, division time, lineage, H3K27me3 levels and number of prior divisions.
ChIP-seq data (reads per bp over the TSS ± 1 kb) from all genes and replicates were used to derive the mean and variance in H3K27me3 at days 0, 5, 9, 12, 14 and 17 (filled black circles in Figure 4E). For a given trajectory, H3K27me3 levels in the 16 ISC were chosen from a normal distribution N(μi = μ, σ), where i denotes the trajectory number and σ represents the variability in H3K27me3 levels among ISC in a single crypt. σ was chosen as 0.01, but the results were insensitive to its precise value. μ was treated as a free parameter and we varied it over a large range centered around the experimental value at day 0. The other free parameters in the model were average ISC division time and the maximum number of divisions a cell could undergo. For each parameter combination, we simulated ~2,000 trajectories of H3K27me3 decay. In each trajectory, an exponentially distributed random number (rate l) was generated to define the time when an ISC exits the compartment and is replaced by a daughter, sibling or other ISC. This rate λ in turn sets the average ISC division time. Exiting cells were chosen randomly from the 16 extant cells, the next cell to divide was chosen randomly from 1 of its 2 neighbors, and daughter cells were allocated 1/2 the H3K27me3 value of their mother. This process was executed as long as a cell chosen for division had not passed the maximum limit of divisions; in this case, the exiting cell was not replaced and left a vacant spot in the one-dimensional ring, consistent with the reduction in Lgr5+ crypts observed over time in the experimental data (Figure 3A). This 2-step process of cell exit coupled with neighboring cell division and H3K27me3 dilution (Figure 4C) was repeated until 17 days, thus completing one trajectory of the simulation. The trajectory was stored for analysis only if average cell division times fell within the ranges < 1 day, 1–2 days, 2–3 days, 3–4 days, 4–5 days, or 5–6 days.
Methylation decay kinetics were computed by averaging H3K27me3 values at any time over the ~2,000 accepted trajectories for every range of average cell division intervals. At specified times (days 5, 7, 9, 12, 14 and 16) the number of times each ISC had divided was also recorded in each trajectory, giving estimates of the fraction that had undergone a certain number of divisions. Histograms from each of the ~2,000 accepted trajectories were averaged to generate robust estimates (Figure 4D). Distance of the simulation results to the experimental data and the best-fitting theoretical curve were computed using weighted least-squares (χ2), where the squared deviation of the simulation results from the experimental ChIP data was weighted by the standard deviation in H3K27me3 levels at each time point. The three free parameters in the model - average ISC division time, the value μ of mean methylation at time 0, and the maximum number of allowable divisions, were systematically varied over a discrete grid and χ2 values were generated for each parameter combination. Reducing the ISC pool to 14 cells or computing H3K27me3 levels with ChIP-seq data over TSS ± 500 bp and TSS ± 1 kb did not affect the main results of the model (Figure S2E).
DATA AND CODE AVAILABILITY
The accession number for the data reported is GEO Database: GSE128671.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ezh2 | Cell Signaling Technology | Cat# 5246; RRID: AB_10694683 |
| Trimethyl-Histone H3 (Lys27) Antibody | EMD Millipore | Cat# 07-449; RRID: AB_310624 |
| GFP | Santa Cruz | Cat#SC9996; RRID: AB_627695 |
| CDKN2A | Santa Cruz | Cat#SC32748; RRID: AB_628071 |
| BrdU | Bio-Rad | Cat#OBT0030; RRID: AB_609568 |
| Drosophila-specific histone variant H2Av | Active Motif | Cat# 61686; RRID: AB_2737370 |
| Ki-67 | Santa Cruz Biotechnology | Cat# VP-K452; RRID: AB_2314697 |
| KDM6A | Cell Signaling Technology | Cat# 33510; RRID: AB_2721244 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma | SKU# T5648 |
| TrypLE Select Enzyme (10X) | ThermoFisher | A1217702 |
| Drosophila Line 2 spike-in chromatin | Active Motif | 53083 |
| OCT compound | Tissue-Tek | 4583 |
| RPMI | GIBCO | 11875-093 |
| UNC1999 | R&D Systems | 4904/10 |
| UNC2400 | R&D Systems | 4905/10 |
| MAK-683 | BioVision | B1972-5 |
| Palbociclib | Sigma | PD0332991 |
| Trizol reagent | ThermoFisher | 15596026 |
| Critical Commercial Assays | ||
| TruSeq RNA Library Prep Kit v2 | Illumina | RS-122-2001 |
| ThruPLEX DNA-seq 48S Kit | Rubicon Genomics | R400427 |
| 3,3′ diaminobenzidine tetrahydrochloride | Sigma | P8375 |
| Agencourt AMPure XP | Beckman Coulter | A63881 |
| Qubit dsDNA HS Assay Kit | ThermoFisher | Q32854 |
| QIAquick PCR Purification Kit | QIAGEN | 28106 |
| MinElute PCR Purification Kit | QIAGEN | 28004 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| RNase-Free DNase Set | QIAGEN | 79254 |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 |
| Vectastain Elite ABC Kit | Vector | PK-6100 |
| Power SYBR Green PCR Master Mix | ThermoFisher | 4367659 |
| High-Capacity cDNA Reverse Transcription Kit | ThermoFisher | 4368814 |
| cOmplete, mini protease inhibitor cocktail | Sigma | 11836153001 |
| Chromium Single Cell B Chip | 10X Genomics | 1000073 |
| Chromium Next GEM Single Cell 3′ GEM, Library & Gel Bead Kit | 10X Genomics | 1000128 |
| Deposited Data | ||
| Raw and analyzed DNA and RNA sequence data | This paper | GEO Database: GSE128671 |
| Source files for immunoblots, histology, immunofluorescence | This paper | Mendeley Data: https://doi.org/10.17632/3ph6mx54gr.2 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Villin-CreER(T2) | el Marjou et al., 2004 | N/A |
| Mouse: Lgr5EGFP-IRES-CreERT | The Jackson Laboratory | N/A |
| Mouse: Eedfl/fl | Xie et al., 2014 | N/A |
| Mouse: Ezh2fl/fl | Su et al., 2003 | N/A |
| Oligonucleotides | ||
| All primer sequences are listed in Table S1 | N/A | |
| Software and Algorithms | ||
| Bedtools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| deepTools v2.5 | Ramírez et al., 2016 | https://github.com/deeptools/deepTools |
| STAR aligner v2.5.3a | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| DeSeq2 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ggplot2 | Wickham, 2009 | https://ggplot2.tidyverse.org/ |
| GraphPad Prism 7 | N/A | https://www.graphpad.com/scientific-software/prism/ |
| HTSeq v0.6.1 | Anders et al., 2015 | https://www-huber.embl.de/HTSeq/doc/overview.html |
| IGV | Robinson et al., 2011 | http://software.broadinstitute.org/software/igv/ |
| SICER v0.0.1 | Xu et al., 2014 | https://github.com/dariober/SICERpy |
| R | R Core Team, 2013 | https://www.R-project.org/ |
| RSeQC v2.6.2 | Wang et al., 2012 | http://rseqc.sourceforge.net/ |
| Cell Ranger v3.0.2 | 10X Genomics | https://github.com/10XGenomics/cellranger |
| Seurat v3.0.2 | Butler et al., 2018 | https://github.com/satijalab/seurat |
Highlights.
EZH2 loss reduces all H3K27me3 equally; ~40% retained marks keep target genes silent
Replicational dilution is the major path for mammalian cells to clear H3K27me3
H3K27me3 levels halve at each cell division irrespective of nucleosomal location
The balance of promoter H3K4me2/3 and H3K27me3 levels determines gene activity
ACKNOWLEDGMENTS
This work was supported by NIH awards U01DK103152 (a stem cell consortium funded by NIDDK and NIAID), R01DK081113 (to R.A.S.), K01DK113067 (to U.J.), and P50CA127003; the DFCI-Novartis Drug Discovery Program; gifts from the Lind family; and microscopy facilities in the Harvard Digestive Disease Center (P30DK034854). We thank S. Robine and S.H. Orkin for gifts of Villin-CreER T2 and EedFI/FI mice, respectively; the Mutant Mouse Regional Resource Centers for providing Ezh2+/Fl mice; and J. Pyrdol and A. Luoma for technical assistance with scRNA libraries.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.01.017.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, and Helin K (2007). UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449, 731–734. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, and Clevers H (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. (2013). EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY, and Greenleaf WJ (2018). Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell 173, 1535–1548.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos EI, Stafford JM, and Reinberg D (2014). Epigenetic inheritance: histone bookmarks across generations. Trends Cell Biol. 24, 664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Paek AL, Reyes J, Lasick KA, Lahav G, and Michor F (2018). Hidden heterogeneity and circadian-controlled cell fate inferred from single cell lineages. Nat. Commun 9, 5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, and Kim SK (2009). Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 23, 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiacchiera F, Rossi A, Jammula S, Zanotti M, and Pasini D (2016). PRC2 preserves intestinal progenitors and restricts secretory lineage commitment. EMBO J. 35, 2301–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2013). The intestinal crypt, a prototype stem cell compartment. Cell 154, 274–284. [DOI] [PubMed] [Google Scholar]
- Coleman RT, and Struhl G (2017). Causal role for inheritance of H3K27me3 in maintaining the OFF state of a Drosophila HOX gene. Science 356, eaai8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comet I, Riising EM, Leblanc B, and Helin K (2016). Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 16, 803–810. [DOI] [PubMed] [Google Scholar]
- Csardi G, and Nepus T (2006). The igraph software package for complex network research. Int. J. Complex Sys 1695, 1–9. [Google Scholar]
- De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, and Natoli G (2007). The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130, 1083–1094. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler HG (2001). SU-DHL-10 The Leukemia-Lymphoma Cell Line FactsBook (Academic Press; ), p. 270. [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, and Robine S (2004). Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193. [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, and Fuchs E (2009). Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 136, 1122–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezhkova E, Lien WH, Stokes N, Pasolli HA, Silva JM, and Fuchs E (2011). EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 25, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filby A, Begum J, Jalal M, and Day W (2015). Appraising the suitability of succinimidyl and lipophilic fluorescent dyes to track proliferation in non-quiescent cells by dye dilution. Methods 82, 29–37. [DOI] [PubMed] [Google Scholar]
- Hidalgo I, Herrera-Merchan A, Ligos JM, Carramolino L, Nuñez J, Martinez F, Dominguez O, Torres M, and Gonzalez S (2012). Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell 11, 649–662. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M, and Gotoh Y (2009). Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63, 600–613. [DOI] [PubMed] [Google Scholar]
- Højfeldt JW, Laugesen A, Willumsen BM, Damhofer H, Hedehus L, Tvardovskiy A, Mohammad F, Jensen ON, and Helin K (2018). Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol 25, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhang J, Yu Z, Zhang H, Wang Y, Lingel A, Qi W, Gu J, Zhao K, Shultz MD, et al. (2017). Discovery of first-in-class, potent, and orally bioavailable Embryonic Ectoderm Development (EED) inhibitor with robust anticancer efficacy. J. Med. Chem 60, 2215–2226. [DOI] [PubMed] [Google Scholar]
- Jadhav U, Nalapareddy K, Saxena M, O’Neill NK, Pinello L, Yuan GC, Orkin SH, and Shivdasani RA (2016). Acquired tissue-specific promoter bivalency is a basis for PRC2 necessity in adult cells. Cell 165, 1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav U, Cavazza A, Banerjee KK, Xie H, O’Neill NK, Saenz-Vash V, Herbert Z, Madha S, Orkin SH, Zhai H, et al. (2019). Extensive recovery of embryonic enhancer and gene memory stored in hypomethylated enhancer DNA. Mol. Cell 74, 542–554.e545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermann P, Hoerner L, Burger L, and Schübeler D (2014). Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc. Natl. Acad. Sci. USA 111, E3415–E3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan AH, Derfoul A, Feng X, Ryall JG, Dell’Orso S, Pasut A, Zare H, Simone JM, Rudnicki MA, and Sartorelli V (2011). Polycomb EZH2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev. 25, 789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppens MA, Bounova G, Gargiulo G, Tanger E, Janssen H, Cornelissen-Steijger P, Blom M, Song JY, Wessels LF, and van Lohuizen M (2016). Deletion of Polycomb Repressive Complex 2 from mouse intestine causes loss of stem cells. Gastroenterology 151, 684–697.e612. [DOI] [PubMed] [Google Scholar]
- Kozar S, Morrissey E, Nicholson AM, van der Heijden M, Zecchini HI, Kemp R, Tavaré S, Vermeulen L, and Winton DJ (2013). Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell 13, 626–633. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprell F, Finkl K, and Müller J (2017). Propagation of Polycomb-repressed chromatin requires sequence-specific recruitment to DNA. Science 356, 85–88. [DOI] [PubMed] [Google Scholar]
- Lopez-Garcia C, Klein AM, Simons BD, and Winton DJ (2010). Intestinal stem cell replacement follows a pattern of neutral drift. Science 330, 822–825. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv, arXiv: 1802.03426 https://arxiv.org/abs/1802.03426. [Google Scholar]
- Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, and Bernstein BE (2010). GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 6, e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzamohammadi F, Papaioannou G, Inloes JB, Rankin EB, Xie H, Schipani E, Orkin SH, and Kobayashi T (2016). Polycomb repressive complex 2 regulates skeletal growth by suppressing Wnt and TGF-β signalling. Nat. Commun 7, 12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. (2010). Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet 42, 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksuz O, Narendra V, Lee CH, Descostes N, LeRoy G, Raviram R, Blumenberg L, Karch K, Rocha PP, Garcia BA, et al. (2018). Capturing the onset of PRC2-mediated repressive domain formation. Mol. Cell 70, 1149–1162.e1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker A, Maclaren OJ, Fletcher AG, Muraro D, Kreuzaler PA, Byrne HM, Maini PK, Watson AJ, and Pin C (2017). Cell proliferation within small intestinal crypts is the principal driving force for cell migration on villi. FASEB J. 31, 636–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryk N, Dalby M, Wenger A, Stromme CB, Strandsby A, Andersson R, and Groth A (2018). MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 361, 1389–1392. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reveron-Gomez N, Gonzalez-Aguilera C, Stewart-Morgan KR, Petryk N, Flury V, Graziano S, Johansen JV, Jakobsen JS, Alabert C, and Groth A (2018). Accurate recycling of parental histones reproduces the histone modification landscape during DNA replication. Mol. Cell 72, 239–249.e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, and Helin K (2014). Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell 55, 347–360. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdoittir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Bourbon HM, Di Croce L, and Cavalli G (2017). Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171, 34–57. [DOI] [PubMed] [Google Scholar]
- Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, and Orkin SH (2008). EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 32, 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, and Copeland RA (2010). Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 707, 20980–20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, and Clevers H (2010). Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 743, 134–144. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, and Costantini F (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol 7, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Lee CH, Voigt P, Descostes N, Saldana-Meyer R, Yu JR, Leroy G, Oksuz O, Chapman JR, Suarez F, et al. (2018). Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv 4, eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, and Tarakhovsky A (2003). Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat. Immunol 4, 124–131. [DOI] [PubMed] [Google Scholar]
- R Core Team (2013). R: A language and environment for statistical computing. https://www.R-project.org/.
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2: Elegant graphics for data analysis (SpringerVerlag).
- Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, et al. (2013). Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 753, 1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, and Orkin SH (2014). Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 74, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Grullon S, Ge K, and Peng W (2014). Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods Mol. Biol 7750, 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, Allison DF, Cai L, Rockowitz S, Liu S, et al. (2015). Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 725, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. (2011). Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 777, 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, Li X, Xu M, Zhang Z, Niu T, et al. (2012). Dense chromatin activates Polycomb Repressive Complex 2 to regulate H3 Lysine 27 methylation. Science 337, 971–975. [DOI] [PubMed] [Google Scholar]
- Yuan GC, Cai L, Elowitz M, Enver T, Fan G, Guo G, Irizarry R, Kharchenko P, Kim J, Orkin S, et al. (2017). Challenges and emerging directions in single-cell analysis. Genome Biol. 78, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the data reported is GEO Database: GSE128671.