

Abstract
Inflammatory bowel disease (IBD) represents a group of chronic relapsing intestinal disorders. Rutaecarpine (RUT), isolated from the Traditional Chinese Medicine (TCM) of Evodia rutaecarpa, was reported to suppress IBD. However, the mechanism by which RUT ameliorates dextran sulfate sodium (DSS)-induced IBD is largely unknown. By use of nuclear factor-erythroid 2–related factor 2 (NRF2) knockout mice, cell-based studies, surface plasmon resonance (SPR), western blotting analysis, and molecular docking studies, the mechanism by which RUT affects DSS-induced colitis was explored. In DSS-treated wild-type mice but not in Nrf2-null mice, RUT significantly improved colitis as revealed by rescued body weight loss, improved histology and inflammation, and induced expression of NRF2 target genes in colon and ileum. Cell-based studies showed that RUT significantly increased the LD50 for hydrogen peroxide (H2O2)-induced cell damage, activated NRF2 nuclear translocation, and suppressed the production of reactive oxygen species in H2O2-treated HCT116 cells, activated NRF2 luciferase reporter activities in HCT116 cells and HepG2 cells, and induced expression of NRF2 target genes in primary intestinal epithelial cells. Molecular docking in silico and SPR assays indicated that RUT interacted with kelch-like ECH-associated protein 1 (KEAP1), and extracellular incubation studies revealed that RUT bound to the KEAP1 kelch domain with a calculated equilibrium dissociation constant Kd of 19.6 μM. In conclusion, these results demonstrate that RUT ameliorates DSS-induced colitis, dependent on NRF2, and could be a potential therapeutic option for IBD patients. Mechanistically, RUT potentiates NRF2 nuclear translocation to upregulate NRF2-mediated antioxidant response by directly inhibiting KEAP1-NRF2 interaction.
Keywords: Dextran sulfate sodium, Inflammatory bowel disease, Kelch-like ECH-Associated protein 1, Nuclear factor-erythroid 2–related factor 2, Rutaecarpine
1. Introduction
Inflammatory bowel diseases (IBD) are chronic relapsing gastrointestinal disorders, commonly exemplified by ulcerative colitis or Crohn’s disease [1]. IBD pathogenesis is closely related to life style, genetic, and environmental etiological factors [2,3], while chronic IBD presents a high risk for colorectal cancer both in mice and humans [4]. Globally, an increasing incidence of IBD has been found over the past decades and IBD has added economic and psychological burden for patients [5]. Although numerous therapeutic advances have emerged in the clinic in recent years [1], no drugs are available to cure this refractory disease, and thus novel therapeutic choices are urgently needed.
Nuclear factor-erythroid 2-related factor 2 (NRF2), a basic leucine zipper redox-sensitive transcriptional factor, binds to antioxidant response element (ARE) upstream of its target genes that serve to neutralize oxidative stress caused by environmental toxicants and diseases [6,7]. NRF2 is located in the cell cytoplasm bound with its repressor kelch-like ECH-associated protein 1 (KEAP1) under normal physiological conditions, while upon exposure to oxidative stress or xenobiotics, the NRF2-binding domain of KEAP1 protein is modified to release NRF2, which can then translocate into the nucleus to trigger cytoprotection by activating its target genes together with small MAF proteins [8]. Although how NRF2 modulates inflammatory disorders differs among previous studies due to different disease models or other experimental conditions used [9,10], accumulating lines of evidence suggest that NRF2 protects against IBD and the associated colorectal cancer. Increased susceptibility to dextran sulfate sodium (DSS)-induced colitis and colitis-associated colorectal cancer was found with Nrf2-null mice compared with wild-type mice [11,12]. It is generally believed that oxidative stress, which is accompanied by intestinal accumulation of hydrogen peroxide (H2O2) and other reactive oxygen species (ROS), plays an important role in IBD pathogenesis and progression [13]. Many agents were found to improve DSS-induced colitis and colitis-associated colorectal cancer via NRF2 activation [14–16]. Therefore, activation of the NRF2-dependent anti-oxidative response is a promising strategy in IBD treatment.
Rutaecarpine (RUT), an alkaloid component isolated from the widely-used Traditional Chinese Medicine (TCM) of Evodia rutaecarpa, has a variety of intriguing biological properties [17], and was demonstrated to protect against oxidative stress-induced HepG2 damage by activating NRF2/ARE signaling [18]. A recent study demonstrated that RUT had a therapeutic potential in treating DSS-induced colitis [19]. However, how RUT influences DSS-induced colitis via NRF2-mediated anti-oxidative signaling is still largely unknown.
To investigate the mechanism by which RUT affects DSS-induced colitis, Nrf2-null (Nrf2−/−) mice and their genetic background-mat ched wild-type (Nrf2+/+) mice were employed to examine whether RUT NRF2-dependently protected against DSS-induced IBD. RUT was found to significantly alleviate DSS-induced IBD and induce mRNA expression of NRF2 target genes only in Nrf2+/+ mice, but not in Nrf2−/− mice, suggesting that RUT ameliorates DSS-induced IBD depending on NRF2 activation. Cell-based in vitro cell culture studies, ligand-docking in silico analysis in combination with surface plasmon resonance (SPR) analysis were performed to explore the potential mechanism. RUT was found to increase the LD50 values for H2O2-treated HCT116 cells, induce NRF2 nuclear translocation and decrease intracellular ROS production in H2O2-treated HCT116 cells, directly bind to the KEAP1 protein kelch domain with an equilibrium dissociation constant Kd of 19.6 μM, activate NRF2 luciferase reporter activity in HCT116 and HepG2 cells, and induce the mRNA expression of NRF2 target genes in primary mouse intestinal epithelial cell cultures. Taken together, RUT was found to NRF2-dependently alleviate DSS-induced IBD in mice. Mechanistically, RUT interfered with KEAP1-NRF2 interaction to induce nuclear NRF2 translocation and then activate the downstream anti-oxidative chemo-protection response. These results support the potential of RUT to be developed as a therapeutic option for clinical IBD patients.
2. Materials and methods
2.1. Chemicals and reagents
Rutaecarpine (RUT) was isolated with a purity > 98% and the structure determined with nuclear magnetic resonance and high-resolution mass spectrometry as described previously [20]. Dextran sulfate sodium salt (DSS, 36,000–50,000 Da) was purchased from MP Biomedicals (Irving, CA). Cell Counting Kit-8 was obtained from Dojindo Molecular Technologies (Rockville, MD). Corn oil, dimethyl sulfoxide (DMSO), Y27632, DL-dithiothreitol, and ethylene diamine tetraacetic acid were purchased from Sigma (St. Louis, MO). Murine recombinant epidermal growth factor and noggin were obtained from Peprotech (Rocky Hill, NJ). Human recombinant R-spondin 1 was purchased from Nuvelo (San Carlos, CA). Fetal bovine serum (FBS), Dulbecco’s modified Eagle’s medium (DMEM) and sodium pyruvate were obtained from Gibco-BRL (Grand Island, NY). Penicillin and streptomycin were obtained from Invitrogen (Carlsbad, CA). TRIzol reagent and lipofectamine 3000 reagent were purchased from Thermo Fisher Scientific (Pittsburgh, PA). Anti-NRF2 (Ab76026), anti-ACTB (Ab8227), and anti-LMNB1 (Ab133741) antibodies were purchased from Abcam (Cambridge, UK). qScriptTM cDNA SuperMix was from Quantabio (Beverly, MA). Sulforaphane (SFN) was obtained from Solarbio (Beijing, China). Recombinant human KEAP1 protein (Cat#, Ag0779) was from Proteintech Group (Chicago, IL).
2.2. Animal studies
Male 6- to 8-week-old C57BL/6J wild-type (Nrf2+/+) and Nrf2-null (Nrf2−/−) mice on the C57BL/6J background were purchased from Jackson Laboratory (Bar Harbor, ME). The mice were maintained in a temperature-controlled environment at (23 ± 1) °C with 60 ± 5% humidity and a 12 h light/12 h dark cycle before starting the experiment and fed standard laboratory chow diet with water ad libitum. The experimental procedures were in compliance with the guidelines for the use of experimental animals of the Peking University Committee on Animal Care and Use (SYXK[Jing]2006–0025) and the animal study protocols were approved by the National Cancer Institute Animal Care and Use Committee.
Nrf2+/+ and Nrf2−/− mice were randomized into three groups: control group, DSS group, and DSS + RUT group. For testing the effect of RUT in protecting against DSS-induced IBD, mice were orally administrated with corn oil for control group and DSS group or 80 mg/kg RUT dissolved in corn oil for DSS + RUT group once a day for three consecutive days prior to DSS treatment. Mice in the DSS group and DSS + RUT group were treated with 2.5% of DSS in their drinking water for 7 days and dosed with corn oil (control group and DSS group) and RUT (DSS + RUT group) by gavage once a day until the end of the experiment. All mice were monitored daily for body weight, stool consistency and rectal bleeding to measure the disease activity index (DAI). The scoring system for the DAI was described in Supplementary Table 1. All animals were killed on day 10. Part of the distal ileum and colon tissues were fixed in 10% buffered formalin for hematoxylin and eosin (H&E) staining, and the left part of ileum and colon tissues were frozen at −80°C for mRNA analysis.
2.3. Histopathology assessment
The mice were killed, ileums and colons removed, and the colon length of each mouse was measured. After washing the tissues with cold phosphate-buffered saline, a part of the ileum and colon tissues of each mouse was cut and immediately fixed in 10% neutral formalin, embedded in paraffin, and then stained with H&E. Histological analysis was performed by microscopic examination. Slide digital images were collected using Pannoramic Viewer software (v. 1.15.2, 3DHISTECH Ltd, Budapest, Hungary). Images shown were representative results of three biological replicates.
2.4. Effect of RUT on hydrogen peroxide (H2O2)-induced cytotoxicity
The HCT116 cell line was obtained from the American Type Culture Collection (Manassas, VA) and cultured in DMEM containing 10% FBS and 1% antibiotics (equal mixture of 0.5% penicillin and 0.5% streptomycin). For the assay, the cells were seeded in 96-well plates overnight at 90% confluence, pretreated with 5 μM of RUT for 18 h, and then treated with 0.031, 0.062, 0.125, 0.25, 0.5, 1.0, 2.0, 4.0, 8.0 or 16 mM of H2O2 for additional 6 h to test the cell viability with the Cell Counting Kit-8 (Apexbio, Houston, TX) using a microplate reader (BioTek Instruments, Winooski, VT).
2.5. Intracellular reactive oxygen species (ROS) production assay
The generation of intracellular ROS induced by H2O2 was determined with the fluorescent probe H2DCF-DA (Wako Pure Chemical Industries, Ltd, Cambridge, MA). Briefly, HCT116 cells were cultured in DMEM containing 10% FBS and 1% antibiotics (equal mixture of 0.5% penicillin and 0.5% streptomycin) with 0, 2.5, 5, or 10 μM RUT for 18 h, H2O2 added to a final concentration of 1 mM for an additional 6 h, and finally incubated with H2DCF-DA in FBS-free DMEM for 20 min at 37°C. To remove H2DCF-DA, the cells were washed with FBS-free DMEM for three times. Fluorescent intensity was measured with an Olympus CKX53 fluorescence microscope equipped with a DP74 camera (Olympus, Tokyo, Japan).
2.6. NRF2 nuclear translocation by immunofluorescence and western blotting
For immunofluorescence assays, HCT116 cells were seeded in glass-bottom petri dishes at a concentration of 2 × 104 per well with DMEM containing 10% FBS and 1% antibiotics (equal mixture of 0.5% penicillin and 0.5% streptomycin) overnight. The cells were then incubated with or without 10 μM RUT for 6 h and fixed with 4% paraformalde-hyde for 10 min. The cells were blocked with 3% bovine serum albumin in TBS (Tris buffered saline) for 40 min, and then incubated with NRF2 antibody (dilution of 1:100) overnight at 4 °C. After washing three times with TBS containing 0.1% Tween 20, the cells were incubated with a fluorescein isothiocyanate (FITC)-conjugated goat-anti-rabbit antibody for 2 h and washed 3 times with TBS containing 0.1% Tween 20. Finally, the cells were stained with 10 μg/mL Hoechst33342 for 15 min and examined with a 3D living cell high speed multicolor laser scanning confocal microscope (Perkin Elmer Life Science, Cambridge, UK) after washed thrice with phosphate-buffered saline.
For western blots, HCT116 cells were seeded into 6-well dishes at a concentration of 2 × 105 per well with DMEM containing 10% FBS and 1% antibiotics. After attachment, cells were incubated with SFN (10 μM) or RUT (5 μM and 10 μM) for 24 h. The Protein Extraction Kit (Beyotime Biotechnology, Shanghai, China) was used to isolate the nuclear and cytosol protein according to the manufacturers protocol. The concentrations of collected protein were determined with the bicinchoninic acid assay and the samples were stored at −80°C until use. ProteinSimple Wes protein analysis (San Jose, CA) was used to detect NRF2 levels according to the user’s guide.
2.7. Surface plasmon resonance (SPR) assay
In the SPR assay, one of the interacting molecules, known as the ligand, was immobilized on a sensor chip surface, while the other interacting molecule, known as the analyte, continuously flowed over the sensor chip surface. When the analyte was binding to the ligand, absorbing molecules caused changes of the local index of refraction and the resonance conditions of the surface plasmon waves. In this experiment, SPR assays were performed on a Biacore T200 instrument (GE Healthcare, Salem, CT) to test if RUT bound to KEAP1. Recombinant human KEAP1 protein with an N-terminal GST was first immobilized on CM5 chips (GE Healthcare, Chicago, IL) via EDC/NHS-mediated crosslinking reaction. Detailed peptide sequence of kelch domain of KEAP1 protein is described in the Supplementary Material. RUT was used at concentrations ranging from 1.56 to 100 μM. The assay was then carried out according to the protocol provided by GE Healthcare. In each analysis, the concentrations were repeated three times at the end of each wash run to confirm the stability of the sensor surface. The parameters of SPR were set as follows: temperature, 25°C; flow rate, 30 μL/min; contact time, 60 s; disassociation time, 150 s. Affinity curve fitting was then performed with a steady-state affinity model to calculate the disassociation constant Kd using the Biacore T200 Evaluation Software (Version 1.0).
2.8. Luciferase reporter assay
For the luciferase assays, AAV-CMV-Nrf2 (mouse) and pCDNA3.1-NRF2 (human) were obtained from Addgene (Cambridge, MA). pGLARE-reporter and pCMV-renilla luciferase vector were provided by Grace L. Guo (Rutgers University, New Brunswick, NJ). HepG2 cells were purchased from the American Type Culture Collection (Manassas, VA). HepG2 cells and HCT116 cells (1 × 104/well) were seeded into 24-well plates for 12 h. Then, the plasmids were transfected using Lipofectamine 3000 reagent. At 24 h following transfection, the cells were treated with 20 μM of SFN, 1.25 μM and 2.5 μM of RUT for 24 h. Dual-Luciferase Reporter Assay System (Promega, Madison, WI) was used to measure the luciferase activity. Renilla luciferase activity was used to normalize the transfection efficiency.
2.9. Molecular docking assay in silico
To investigate the possible binding mode of RUT to KEAP1, the small ligand-binding C-terminal kelch domain of the human KEAP1 (PDB: 4XMB) was selected as described in previous studies [21,22]. Molecular docking analysis of RUT was performed in the kelch pockets of KEAP1 using AutoDock vina.
2.10. Primary intestinal epithelial cell isolation and treatment
Primary intestinal epithelial cells were isolated from C57BL/6N mice as previously reported [23] and seeded on collagen I-coated 12-well plates and cultured with DMEM/F12 containing 10% FBS, 0.1% Y27632, 500 ng/mL of R-spondin 1, 100 ng/mL of noggin, 50 ng/mL of epidermal growth factor, 1% insulin-transferrin-sodium and 1% antibiotics. The intestinal epithelial cells were exposed to 10 μM, 20 μM of RUT or 20 μM of SFN for 24 h, and the cells were washed two times with phosphate-buffered saline and collected for RNA extraction.
2.11. Real-time quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from frozen ileum and colon tissues using TRIzol reagent. cDNA was synthesized from the extracted RNA using qScriptTM cDNA SuperMix. qPCR assays were performed using EvaGreen master mix (Applied Biological Materials Inc, Richmond, Canada) with Applied Biosystems 7500. The primer sequences of primers are listed in Supplementary Table 2. The mRNA expression level for individual gene was calculated and normalized to their corresponding Actb mRNA level.
2.12. Statistical analysis
Statistical analysis was performed on Prism version 7.0 (GraphPad Software, San Diego, CA). Two-tailed Student’s t-test was used to compare the statistic difference between two groups, while One-way ANOVA analysis was used among multiple comparisons. The values are present as mean ± SEM. A value of P < 0.05 was considered as statistically significant.
3. Results
3.1. RUT NRF2-dependently attenuated DSS-induced colitis
To test whether the effects of RUT on DSS-induced colitis is dependent on NRF2, Nrf2−/− and Nrf2+/+ mice were treated by gavage with control corn oil or 80 mg/kg of RUT. Weight loss after DSS treatment was significantly reversed by RUT treatment in Nrf2+/+ mice (Fig. 1A), but not in Nrf2−/− mice (Fig. 1B). In addition, RUT treatment decreased the DAI score in Nrf2+/+ mice (Fig. 1C), but not in Nrf2−/− mice (Fig. 1D). Consistent with the data on body weight change and DAI scores, RUT treatment significantly rescued the DSS-induced decrease of colon length in Nrf2+/+ mice (Fig. 1E), and not in Nrf2−/− mice (Fig. 1F). Similarly, further histopathological analysis showed that the DSS-caused gut damage, including crypt distortion, loss of goblet cells and severe mucosal damage, were markedly alleviated by RUT pretreatment both in colon and ileum only in Nrf2+/+ mice, and not in Nrf2−/− mice (Fig. 2A and B). In addition, the mRNA expression of cytokines including Cox2, Lcn2, Tnfa, Il1b, Il6, and Il10 mRNAs in the ileum and colon were analyzed. RUT pretreatment significantly suppressed the DSS-induced expression of Cox2, Lcn2, Tnfa, and Il6 mRNAs compared with control corn oil treatment both in the ileum (Fig. 3A) and colon (Fig. 3B) of Nrf2+/+ mice, but not in Nrf2−/− mice (Fig. 3C and D).
Fig. 1.

RUT prevented DSS-induced acute colitis depending on NRF2. (A and B), body weight change for Nrf2+/+ mice (A) and Nrf2−/− mice (B). (C and D), DAI score for Nrf2+/+ mice (C) and Nrf2−/− mice (D). (E and F), colon length of Nrf2+/+ mice (E) and Nrf2−/− mice (F). *P < 0.05, **P < 0.01*** and P < 0.001, versus vehicle-treated DSS group. Significance was determined by using one-way ANOVA. Data are presented as the mean ± SEM, N = 6 per group.
Fig. 2.

H&E staining analyses of distal colon and ileum sections. (A), H&E staining analyses of colon sections for both Nrf2+/+ and Nrf2−/− mice. (B), H& E staining analyses of ileum sections for both Nrf2+/+ and Nrf2−/− mice. Scale bar size = 100 μm.
Fig. 3.
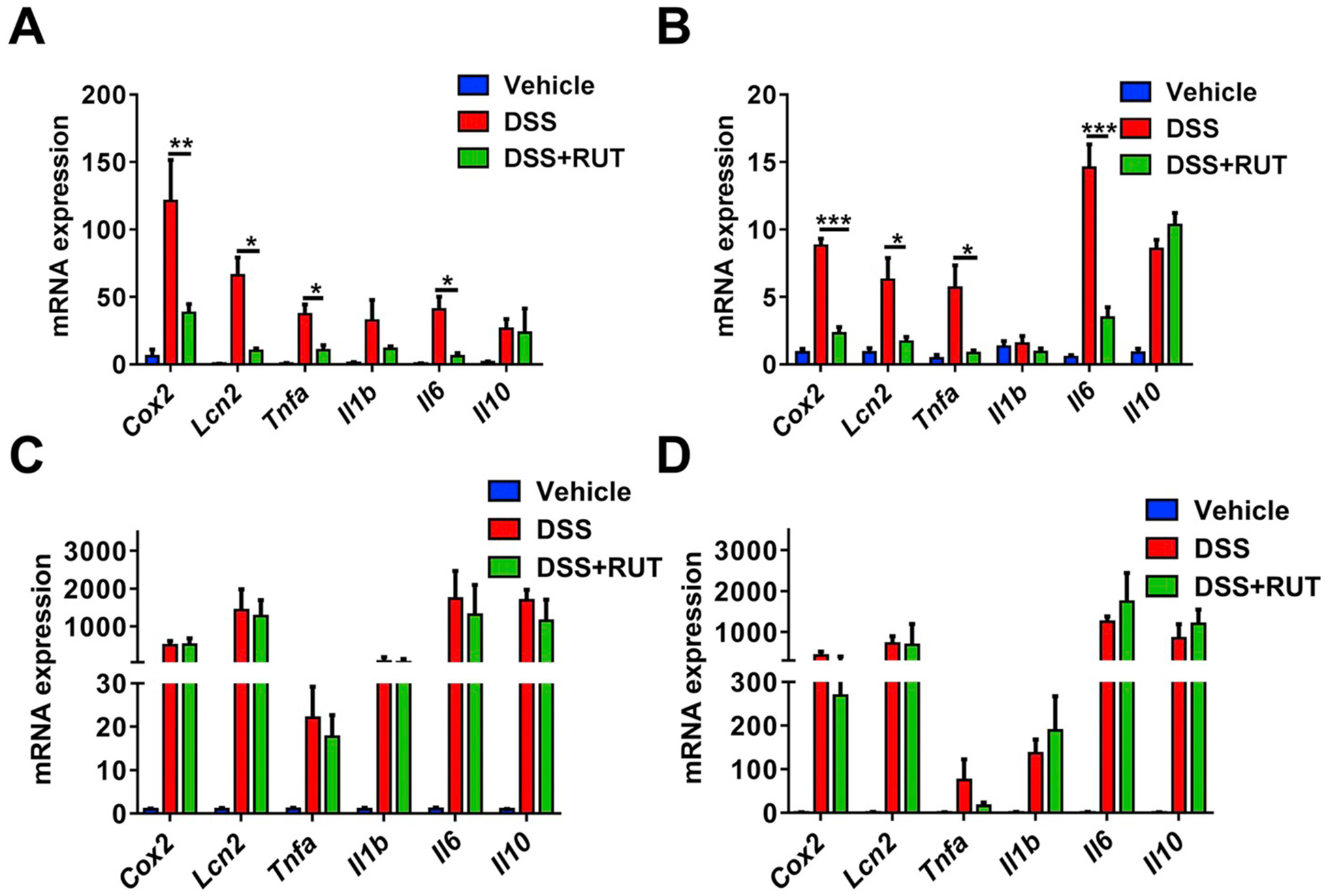
Analysis of proinflammatory gene mRNAs in colon and ileum. (A), Expression of proinflammatory gene mRNAs in colons of Vehicle-, DSS-and DSS+RUT-treated Nrf2+/+ mice; (B), Expression of proinflammatory gene mRNAs in ileum of Vehicle-, DSS- and DSS+RUT-treated Nrf2+/+ mice. (C), Expression of proinflammatory gene mRNAs in colon of Vehicle-, DSS-and DSS+RUT-treated Nrf2−/− mice. (D), Expression of proinflammatory gene mRNAs in ileum of Vehicle-, DSS- and DSS+RUT-treated Nrf2−/− mice. *P < 0.05, **P < 0.01, and ***P < 0.001, versus DSS group. Statistical significance was determined by one-way ANOVA. Data are presented as the mean ± SEM, N = 6 for each group.
Amelioration of the inflammatory response by blocking the expression of proinflammatory cytokines could result from NRF2 activation [24]. To further confirm whether RUT exerts its protective effects by activating the NRF2 signaling pathway, expression of NRF2 target genes in the ileum and colon were analyzed in Nrf2+/+ and Nrf2−/− mice. Compared with control corn oil treatment, the antioxidant target gene mRNAs of NRF2, including Nqo1, Gsta1, Hmox1 and Sod1 were increased by RUT treatment only in Nrf2+/+ mice, but not in Nrf2−/− mice (Fig. 4A and B). These data demonstrate that RUT, at dose of 80 mg/kg, markedly prevents DSS-induced colitis dependent on the presence of NRF2.
Fig. 4.

Analysis of NRF2 target gene mRNAs in colon and ileum or primary intestinal epithelial cells. (A and B), Expression of NRF2 target gene mRNAs in colon (A) and ileum (B) from Nrf2+/+ and Nrf2−/− mice treated with DSS +/− RUT. (C), Expression of NRF2 target gene mRNAs in RUT or SFN-treated primary intestinal epithelial cells. *P < 0.05 and **P < 0.01 versus the Nrf2+/+ DSS-treated group for Panels A and B. *P < 0.05, *P < 0.01 and ***P < 0.001 versus the Vehicle-treated group for Panel C, Data are presented as the mean ± SEM. Statistical significance was determined by two-tailed Student’s t-test or one-way ANOVA. N = 6 for each group.
3.2. RUT induced the expression of NRF2 target genes in primary intestinal epithelial cells
To characterize the direct effect of RUT on the NRF2 pathway in intestinal cells, primary mouse intestinal epithelial cells were isolated and analyzed. In primary mouse intestinal epithelial cells, RUT at 10 μM and 20 μM, significantly induced expression of NRF2 target gene mRNAs including Nqo1, Gsta1 and Gclc, with SFN at 20 μM used as the positive control (Fig. 4C). These data confirm the direct effect of RUT in activating NRF2 in primary intestinal epithelial cells in vitro.
3.3. RUT suppressed H2O2-induced cytotoxicity and intracellular ROS accumulation
Increased oxidative stress is accompanied by the accumulation of H 2O2. ROS was frequently found in the intestine of preclinical IBD mouse models and clinical IBD patients, and is highly correlated with IBD pathogenesis and progression [13]. Thus, H2O2 was used to induce oxidative stress in cultured cells to test the effect and mechanism of RUT treatment on oxidative stress. In the HCT116 cell line, 5 μM of RUT increased the LC50 for H2O2-induced cell damage from 2.11 mM to 3.61 mM (Fig. 5A and B). To further determine whether RUT also suppressed H2O2-induced ROS generation, intracellular level of ROS was quantified using the fluorescent dye H2DCF-DA. In H2O2-treated HCT116 cells, compared with control vehicle treatment, RUT pretreatment significantly decreased the fluorescent signal in a concentration-dependent manner (Fig. 5C). These data suggest that RUT pretreatment suppresses H2O2-induced cell cytotoxicity and intracellular generation of ROS.
Fig. 5.

Suppression of RUT to H2O2-induced cytotoxicity and ROS accumulation. (A and B), LC50 of H 2O2-treated HCT116 cells without (A) or with (B) 5 μM of RUT pretreatment, N = 3 for each group; (C), Representative images of H2O2-induced ROS accumulation in HCT116 cells after control vehicle or RUT pretreatment at the dose of 2.5 μM, 5.0 μM or 10.0 μM (N = 3 for each group).
3.4. RUT increased NRF2 nuclear translocation
NRF2 nuclear translocation is a prerequisite to activate its downstream anti-oxidative response target genes. To investigate whether RUT induced NRF2 translocation, an immunofluorescence assay was performed to detect the intracellular distribution of NRF2 in HCT116 cells treated with control vehicle or 10 μM of RUT. Double immunofluorescent staining for nuclei (Hoechst) and NRF2 protein (FITC) in HCT116 cells were performed, revealing that NRF2 (green staining) was evenly localized in the cytoplasm of control cells, while the intensity of green staining was markedly increased in nuclei or concentrated on the edge of nuclei of cells treated with RUT (Fig. 6A). These data suggest that RUT significantly increases nuclear translocation of NRF2 protein.
Fig. 6.

Immunofluorescent assay for NRF2 nuclear translocation after RUT treatment. (A), Double immunofluorescent staining for nucleus (Hoechst, blue color) and NRF2 protein (FITC, green color) by control vehicle or 10 μM of RUT in HCT116 cells, magnification time, 400×. N = 3 for each group. (B), Representative western blot analysis of cytosol NRF2 and nuclear NRF2 levels in RUT or SFN-treated HCT116 cells. NRF2, when phosphorylated to be activated, was detected at 116 kDa. β-Actin (ACTB) and Lamin B (LMNB1) were used a cytosolic and nuclear loading controls. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The effect of RUT on the cellular distribution of NRF2 in HCT116 cells was further investigated using western blotting with SFN as a positive control. When treated with RUT (5 μM and 10 μM) and SFN (10 μM) for 24 h, no obvious changes in the expression of cytosolic NRF2 protein was found (Fig. 6B), while translocation of the nuclear NRF2 protein was significantly increased in the RUT and SFN-treated groups (Fig. 6C).
3.5. RUT interfered with the interaction between KEAP1 and NRF2 by binding to KEAP1 protein
Binding to the KEAP1 kelch domain to achieve the inhibition of KEAP1-NRF2 interaction is a well-documented mechanism for NRF2 activation [25], and the SPR assay was developed to examine the kinetics of KEAP1-NRF2 interaction [25,26]. To determine whether RUT could interfere with KEAP1-NRF2 protein interaction, an SPR assay was performed to examine the interaction between RUT and the KEAP1 Kelch domain. The equilibrium binding curve fits were developed and the equilibrium dissociation constant (Kd, calculated with Biacore T200 Evaluation Software) for the extracellular binding affinity of RUT with KEAP1 was 19.6 μM (Fig. 7A). SPR further demonstrated that RUT, at 12.5 μM and 37.5 μM, was able to disrupt NRF2-KEAP1 interaction (Fig. 7B). These results indicate that RUT could directly bind to KEAP1 and disrupt the KEAP1–NRF2 interaction. To further investigate the possible binding mode between RUT and KEAP1 protein, molecular ligand docking in silico was performed between RUT and the KEAP1 kelch domain using AutoDock vina. RUT had a fully rigid structure, with five rings in the same plane, and the conformation occupied a small space (Fig. 7C). Docking analysis showed that RUT entered a large hydrophobic cavity and formed a hydrophobic interaction with Gly364, Tyr334 and Ala556. Oxygen atoms on the amide group formed a hydrogen bond interaction with Arg415 (Fig. 7C). These results suggest that RUT could directly bind with KEAP1 protein and thereby has the potential for inducing NRF2 release and activation.
Fig. 7.

RUT activates NRF2 by interfering with the interaction between KEAP1 and NRF2. (A), SPR assay for interaction of RUT with kelch domain KEAP1 protein; (B), SPR assay for NRF2-KEAP1 interaction treated with 0, 12.5, 37.5 μM of RUT; (C) Ligand docking of RUT in the kelch domain of human KEAP1 protein; (D and E) Luciferase assays for NRF2 activation in HCT116 (D) and HepG2 (E). Data are presented as the mean ± SEM, n = 3 per group. **P < 0.01, ***P < 0.001, versus control group, was determined by one-way ANOVA test.
3.6. RUT activated NRF2 in luciferase reporter assays
To further confirm whether RUT could activate NRF2, luciferase reporter gene assays were performed with ARE-driven luciferase reporter and human NRF2 overexpression plasmids. With SFN as a positive control, RUT dose-dependently activated NRF2/ARE luciferase activity both in HCT116 and HepG2 cell lines (Fig. 7D and E). These data support the view that RUT is a potent NRF2 activator.
4. Discussion
The effects of RUT on IBD was presumed to be through suppression of inflammation, as suggested in an earlier study where it reduced DSS-induced inflammation [19]; however, the mechanism was not revealed in this study. A previous publication showed that RUT could induce NRF2 luciferase activity and decrease tert-butyl H2O2-induced hepatotoxicity [18]. Several mechanisms are known to activate the NRF2/ARE cascade by inhibiting KEAP1-NRF2 interaction to induce NRF2 nuclear translocation, or by directly enhancing NRF2 expression levels post-transcriptionally and/or posttranslationally [6–8]. RUT was shown to activate NRF2 via enhancing the phosphorylation of protein kinase B and Ca2+/calmodulin-dependent protein kinase II [18]. However, whether and how RUT directly interferes with KEAP1-NRF2 interaction is still unknown. Thus, in the current study, experiments were conducted to determine whether RUT decreased IBD in a NRF2-dependent manner by using Nrf2−/− mice in vivo, and whether RUT modulated KEAP1-NRF2 interaction to activate NRF2/ARE signaling in vitro.
In mice, RUT was found to elicit its pharmacological effect on DSS-induced IBD dependent on the presence of NRF2. The dose of RUT used in the current study was 80 mg/kg, which was shown to produce no toxicity in mice [20]. In primary intestinal epithelial cells, RUT significantly induced expression of the NRF2 target gene mRNAs Nqo1, Gsta1 and Gclc, that could contribute to attenuation of IBD by increasing levels of glutathione, which further supports the view that RUT could directly activate intestinal NRF2 signaling to decrease DSS-induced colitis. The effects of RUT on the KEAP1-NRF2 pathway were then analyzed both by SPR assays, cell-based studies in vitro and ligand-docking studies in silico. RUT inhibited the KEAP1-NRF2 protein-protein interaction by directly binding to KEAP1, which released NRF2 allowing its nuclear translocation and activation of NRF2 target genes and the downstream anti-oxidative response (Fig. 8).
Fig. 8.

Proposed mechanism and pathway of RUT in suppressing DSS-induced IBD in mice. In cytoplasm, KEAP1 protein is usually bound to NRF2 to stabilize NRF2 in the cytoplasm. Upon oxidative stress treatment, RUT directly binds to KEAP1 resulting in NRF2 release and translocation to the nucleus to bind to AREs and initiate the anti-oxidative response by upregulating the expression of downstream target genes including Nqo1, Hmox1, and Sod1, which then decreases intracellular ROS production and alleviates oxidative stress-induced cytotoxicity. Pharmacologically, RUT improved DSS-induced colitis and alleviated intestinal inflammation dependent on NRF2 activation.
H2O2 is among the major causes of oxidative stress in mouse IBD models and in IBD patients [13], and NRF2 nuclear translocation to release NRF2 from KEAP1 binding is an essential upstream step to initiate its downstream antioxidative response [8]. RUT pretreatment was found to protect against the H2O2-induced damage and decrease H2O2-induced ROS generation in HCT116 cells. NRF2 was translocated to the nucleus after RUT treatment in H2O2-treated HCT116 cells, supporting the view that NRF2 nuclear translocation mediates the cytoprotection by RUT. Luciferase reporter gene assays confirmed the activating effect of RUT in NRF2 signaling in both HepG2 and HCT116 cell lines. In line with these data, a previous report found a similar effect of RUT on tert-butyl H2O2-induced cell damage and activation of NRF2 luciferase activity in HepG2 cells [18]. The current study not only used intestinal or colon cancer cell lines to further extend the effect of RUT in activating intestinal NRF2 activity and NRF2-mediated chemoprotection, but also employed primary intestine cells to demonstrate that RUT could efficiently activate NRF2 activity in normal tissue-derived cells.
KEAP1-NRF2 protein-protein interaction is a major mechanism for maintaining a lower level of NRF2 in cell cytoplasm. KEAP1 protein contains three conserved domains (BTB, linker, C-terminal kelch), among which the C-terminal kelch domain could bind to the Neh2 domain of NRF2 [27]. Under homeostatic conditions of cellular function, NRF2 is negatively regulated by KEAP1 via ubiquitin conjugation and degradation to remain at low cellular concentrations. Thus, disrupting the KEAP1–NRF2 protein-protein interaction represents an attractive strategy to activate NRF2. The SPR-based solution competition assay was developed for selective screening for NRF2 activators that directly inhibit KEAP1-NRF2 interaction [25]. By using the SPR assay, RUT was identified as a novel inhibitor that could disrupt the KEAP1–NRF2 interaction. Our molecular docking analysis in silico also supports the direct binding ability of RUT to KEAP1 protein kelch domain by hydrophobic and hydrogen bond interactions. Similar with the present findings, an earlier report also showed that an inhibitor of KEAP1-NRF2 protein-protein interaction protected colon cells and alleviated experimental colitis [14]. Although it is still possible that RUT could activate NRF2 signaling via other signaling pathways, the current study demonstrates a novel mechanism by which RUT activates NRF2 at least in part due to direct binding of RUT with the KEAP1 kelch domain to induce NRF2 nuclear translocation and activation.
Pharmacological targeting the NRF2 and KEAP1 is efficient in treating various types of chronic diseases in experimental animal models. An NRF2 activator, dimethyl fumarate received clinical approval and several other NRF2 modulators are in various stages of clinical development for treating multiple sclerosis, psoriasis and various other diseases [28,29]. However, these NRF2 activators also show adverse effects and some NRF2 activators, such as SFN, are unstable at room temperature [28]. The fumaric acid ester group of the NRF2 activator could lead to mild to moderate abdominal pain, flushing, diarrhea, nausea and even the serious symptom of leukopenia due to its target-off effects [28,30]. Given that the disadvantages of safety, stability and off-target effects of existing NRF2 activators and the fact that still no NRF2 activators are available in clinical development beyond preclinical IBD models for treating IBD, developing novel NRF2 modulators as anti-IBD therapeutics is still warranted. In this study, RUT was demonstrated to activate NRF2 as one novel member of NRF2-KEAP1 interaction inhibitors, which are believed to have improved target selectivity [28]. RUT, as a natural compound, has a long history as a component of traditional herbs, and is generally believed to be safe after long-term intake [20]. RUT as well as other NRF2-KEAP1 inhibitors represent a recent focus of NRF2 modulator discovery and could be novel candidates for anti-IBD drug discoveries.
In summary, the present study demonstrated that the traditional herb-derived RUT is an inhibitor of the KEAP1-NRF2 interaction leading to activation of NRF2. By using DSS-induced IBD as a pathological disease model in combination with the Nrf2-null mice, RUT was shown to elicit its pharmacological effect on DSS-induced colitis at least in part dependent on NRF2. RUT could be a therapeutic option in treating IBD.
Supplementary Material
Acknowledgments
This project was supported by the National Key R&D Program of China (2018YFC1704500, 2018YFC1704506), the National Natural Science Foundation of China (81773865), and the National Cancer Institute Intramural Research Program.
Abbreviations:
- ARE
antioxidant response element
- DAI
disease activity index
- DSS
dextran sulfate sodium
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
dimethyl sulfoxide
- H2O2
hydrogen peroxide
- TCM
Traditional Chinese Medicine
- IBD
inflammatory bowel disease
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- KEAP1
kelch-like ECH-associated protein 1
- LD50
50% of lethal dose
- NRF2
nuclear factor-erythroid 2–related factor 2
- RUT
rutaecarpine
- ROS
reactive oxygen species
- SPR
surface plasmon resonance assay
- SFN
Sulforaphane
- TBS
Tris buffered saline
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.freeradbiomed.2019.12.012.
References
- [1].Neurath M, Current and emerging therapeutic targets for IBD, Nat. Rev. Gastroenterol. Hepatol 14 (2017) 688. [DOI] [PubMed] [Google Scholar]
- [2].Ng SC, Bernstein CN, Vatn MH, Lakatos PL, Loftus EV Jr., C. Tysk, C. O’Morain, B. Moum, J.F. Colombel, Epidemiology, D. Natural history task force of the international organization of inflammatory bowel, geographical variability and environmental risk factors in inflammatory bowel disease, Gut 62 (2013) 630–649. [DOI] [PubMed] [Google Scholar]
- [3].Ng SC, Tang W, Leong RW, Chen M, Ko Y, Studd C, Niewiadomski O, Bell S, Kamm MA, de Silva HJ, Kasturiratne A, Senanayake YU, Ooi CJ, Ling KL, Ong D, Goh KL, Hilmi I, Ouyang Q, Wang YF, Hu P, Zhu Z, Zeng Z, Wu K, Wang X, Xia B, Li J, Pisespongsa P, Manatsathit S, Aniwan S, Simadibrata M, Abdullah M, Tsang SW, Wong TC, Hui AJ, Chow CM, Yu HH, Li MF, Ng KK, Ching J, Wu JC, Chan FK, SungC.s JJ. Asia-Pacific AG Colitis epidemiology study, Environmental risk factors in inflammatory bowel disease: a population-based case-control study in Asia-Pacific, Gut 64 (2015) 1063–1071. [DOI] [PubMed] [Google Scholar]
- [4].Rhodes JM, Campbell BJ, Inflammation and colorectal cancer: IBD-associated and sporadic cancer compared, Trends Mol. Med 8 (2002) 10–16. [DOI] [PubMed] [Google Scholar]
- [5].Bernstein CN, Review article: changes in the epidemiology of inflammatory bowel disease-clues for aetiology, Aliment. Pharmacol. Ther 46 (2017) 911–919. [DOI] [PubMed] [Google Scholar]
- [6].Hayes JD, Dinkova-Kostova AT, The Nrf2 regulatory network provides an interface between redox and intermediary metabolism, Trends Biochem. Sci 39 (2014) 199–218. [DOI] [PubMed] [Google Scholar]
- [7].Nguyen T, Yang CS, Pickett CB, The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress, Free Radic. Biol. Med 37 (2004) 433–441. [DOI] [PubMed] [Google Scholar]
- [8].Suzuki T, Yamamoto M, Molecular basis of the Keap1-Nrf2 system, Free Radic. Biol. Med 88 (2015) 93–100. [DOI] [PubMed] [Google Scholar]
- [9].Gerstgrasser A, Melhem H, Leonardi I, Atrott K, Schafer M, Werner S, Rogler G, Frey-Wagner I, Cell-specific activation of the Nrf2 antioxidant pathway increases mucosal inflammation in acute but not in chronic colitis, J. Crohns. Colitis 11 (4) (2017) 485–499. [DOI] [PubMed] [Google Scholar]
- [10].Kim J, Cha YN, Surh YJ, A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders, Mutat. Res 690 (2010) 12–23. [DOI] [PubMed] [Google Scholar]
- [11].Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN, Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis, Cancer Res. 66 (2006) 11580–11584. [DOI] [PubMed] [Google Scholar]
- [12].Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, Cheung WK, Chan JY, Reddy BS, Yang CS, Kong AN, Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer, Cancer Prev. Res 1 (2008) 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE, Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases, Physiol. Rev 94 (2014) 329–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lu MC, Ji JA, Jiang YL, Chen ZY, Yuan ZW, You QD, Jiang ZY, An inhibitor of the Keap1-Nrf2 protein-protein interaction protects NCM460 colonic cells and alleviates experimental colitis, Sci. Rep 6 (2016) 26585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang Y, Wang H, Qian C, Tang J, Zhou W, Liu X, You Q, Hu R, 3-(2-Oxo-2-phenylethylidene)-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(1 1bH)-one (compound 1), a novel potent Nrf2/ARE inducer, protects against DSS-induced colitis via inhibiting NLRP3 inflammasome, Biochem. Pharmacol 101 (2016) 71–86. [DOI] [PubMed] [Google Scholar]
- [16].Yang Y, Cai X, Yang J, Sun X, Hu C, Yan Z, Xu X, Lu W, Wang X, Cao P, Chemoprevention of dietary digitoflavone on colitis-associated colon tumorigenesis through inducing Nrf2 signaling pathway and inhibition of inflammation, Mol. Cancer 13 (2014) 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee SH, Son JK, Jeong BS, Jeong TC, Chang HW, Lee ES, Jahng Y, Progress in the studies on rutaecarpine, Molecules 13 (2008) 272–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jin SW, Hwang YP, Choi CY, Kim HG, Kim SJ, Kim Y, Chung YC, Lee KJ, Jeong TC, Jeong HG, Protective effect of rutaecarpine against t-BHP-induced hepatotoxicity by upregulating antioxidant enzymes via the CaMKII-Akt and Nrf2/ARE pathways, Food Chem. Toxicol 100 (2017) 138–148. [DOI] [PubMed] [Google Scholar]
- [19].luo D, Li F, Zou Y, Therapeutic effects of rutaecarpine on dextran sodium sulfate-induced experimental colitis in mice, Natl. Med. J. China (Peking) 98 (7) (2018) 533–538. [DOI] [PubMed] [Google Scholar]
- [20].Zhang Y, Yan T, Sun D, Xie C, Zheng Y, Zhang L, Yagai T, Krausz KW, Bisson WH, Yang X, Gonzalez FJ, Structure-activity relationships of the main bioactive constituents of Euodia rutaecarpa on aryl hydrocarbon receptor activation and associated bile acid homeostasis, Drug Metab. Dispos 46 (2018) 1030–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Georgakopoulos ND, Gatliff J, Wells G, Development of Keap1-interactive small molecules that regulate Nrf2 transcriptional activity, Curr. Opin. Toxicol 1 (2016) 1–8. [Google Scholar]
- [22].Jain AD, Potteti H, Richardson BG, Kingsley L, Luciano JP, Ryuzoji AF, Lee H, Krunic A, Mesecar AD, Reddy SP, Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators, Eur. J. Med. Chem 103 (2015) 252–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gracz AD, Puthoff BJ, Magness ST, Identification, isolation, and culture of intestinal epithelial stem cells from murine intestine, Methods Mol. Biol 879 (2012) 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, Yamamoto M, Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription, Nat. Commun 7 (2016) 11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Abed DA, Goldstein M, Albanyan H, Jin H, Hu L, Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents, Acta Pharm. Sin. B 5 (2015) 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen Y, Inoyama D, Kong AN, Beamer LJ, Hu L, Kinetic analyses of Keap1-Nrf2 interaction and determination of the minimal Nrf2 peptide sequence required for Keap1 binding using surface plasmon resonance, Chem. Biol. Drug Des 78 (2011) 1014–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li X, Zhang D, Hannink M, Beamer LJ, Crystal structure of the Kelch domain of human Keap1, J. Biol. Chem 279 (2004) 54750–54758. [DOI] [PubMed] [Google Scholar]
- [28].Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, Attucks OC, Franklin S, Levonen AL, Kensler TW, Dinkova-Kostova AT, Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases, Nat. Rev. Drug Discov 18 (4) (2019) 295–317. [DOI] [PubMed] [Google Scholar]
- [29].Mrowietz U, Christophers E, Altmeyer P, Treatment of psoriasis with fumaric acid esters: results of a prospective multicentre study. German Multicentre Study, Br. J. Dermatol 138 (3) (1998) 456–460. [DOI] [PubMed] [Google Scholar]
- [30].Chen H, Assmann JC, Krenz A, Rahman M, Grimm M, Karsten CM, Koeh J, Offermanns S, Wettschureck N, Schwaninger M, Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate’s protective effect in EAE, J. Clin. Investig 124 (5) (2014) 2188–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.