

Abstract
Parkin (PRKN) is a ubiquitin E3 ligase that catalyzes the ubiquitination of several proteins. Mutations in the human Parkin gene, PRKN, leads to degeneration of dopaminergic (DA) neurons, resulting in autosomal recessive early-onset parkinsonism and the loss of PRKN function is linked to sporadic Parkinson’s disease (PD). Additionally, several in vitro studies have shown that overexpression of exogenous PRKN protects against the neurotoxic effects induced by a wide range of cellular stressors, emphasizing the need to study the mechanism(s) governing PRKN expression and induction. Here, Prkn was identified as a novel target gene of the aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor and member of the bHLH/PAS (basic helix-loop-helix/Per-Arnt-Sim) superfamily. AhR binds and transactivates the Prkn gene promoter. We also demonstrated that AhR is expressed in DA neurons and that its activation upregulates Prkn mRNA and protein levels in the mouse ventral midbrain. Additionally, the AhR-dependent increase in PRKN levels is associated with a decrease in the protein levels of its target substrate, α-synuclein, in an AhR-dependent manner, because this effect is not observed in Ahr-null mice. These results suggest that treatments designed to induce PRKN expression through the use of nontoxic AhR agonist ligands may be novel strategies to prevent and delay PD.
Keywords: Parkinson’s disease, Aryl hydrocarbon receptor, Parkin, α-Synuclein, TCDD
1. Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease and is clinically characterized by bradykinesia, resting tremor, and postural imbalance induced by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNc). Histopathologically, it is characterized by the presence of intracytoplasmic inclusions of α-synuclein called Lewy bodies [1].
Several reports support the role of Parkin (PRKN) in the development of PD. Parkin is ubiquitously expressed but predominantly in the brain and is an E3 ligase that catalyzes the ubiquitination of several proteins, including Pael-R, α-synuclein, synphilin-1, and Cdc-Rel. Parkin confers protection against several insults to neurons, and impairments in ubiquitination of some of its targets have been linked to the degeneration of dopamine (DA) neurons [2,3]. The neuroprotective effect of Parkin is also mediated by promoting the removal of damaged mitochondria via mitophagy [4] and by regulating the function and stability of excitatory glutamatergic synapses [5].
Mutations in the PRKN in humans are the most common cause of autosomal-recessive early-onset parkinsonism, and reduced Parkin expression has been linked to late-onset PD [6]. Moreover, PRKN haploinsufficiency is associated with an increased risk of developing the sporadic form of PD [7]. In addition to genetic factors, other conditions may alter Parkin function. Several cell stressors, such as hydrogen peroxide, S-nitrosylation, 1-methyl-4-phenylpyridine (MPP+), 6-hydroxydopamine (6-OHDA), paraquat, and rotenone, induce Parkin aggregation [8], while the native structure of PRKN in the brain is modified with age [9]. Therefore, any condition leading to reduced Parkin levels or function compromises neuronal survival.
In agreement with these findings, the overexpression of exogenous Parkin confers protection against the neurotoxic effects induced by a wide range of cellular stressors [10–13], emphasizing the need to study the mechanism(s) governing PRKN gene expression and regulation. However, there is a lack of information regarding the transcription factors involved in Prkn gene regulation.
The aryl hydrocarbon receptor (AhR) mediates the expression of several genes encoding ubiquitin proteasome system proteins [14–16], in particular Ubch7 (also known as UBE2l3 in humans and UbcM4 in mice), an E2 ubiquitin enzyme partner of PRKN. AhR is a ligand-activated transcription factor that mediates the toxicity of environmental pollutants, and its highest-affinity ligand is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Upon binding TCDD, AhR translocates to the nucleus, binds to xenobiotic-responsive elements (XREs), and upregulates the expression of a battery of genes that encode xenobiotic-metabolizing enzymes, including its canonical gene target, Cyp1a1 [17].
On the other hand, it was reported that the promoter of the D. melanogaster homolog of human PRKN has a binding site for AhR [18], suggesting that AhR may be involved in the regulation of Prkn expression. Therefore, we initiated a study to determine whether AhR regulates Prkn expression in the mouse ventral midbrain and its effects on the protein levels of Parkin substrates, in particular α-synuclein (SNCA).
Our data showed that AhR is expressed in mouse DA neurons, and its activation by oral treatment with TCDD induces Parkin expression in the ventral midbrain and decreases the PRKN substrate α-synuclein.
2. Materials and methods
2.1. Materials
SH-SY5Y cells were obtained from the American Type Culture Collection (Manassas, VA, USA). 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) was purchased from AccuStandard (New Haven, CT, USA). Phenylmethanesulfonyl fluoride (PMSF) and dimethyl sulfoxide (DMSO) were acquired from Sigma (St. Louis, MO, USA).
2.2. Animals and treatments
The development of Ahr-null mice was described previously [19]. Wild-type (WT) littermates on a C57BL/6J background were used as controls. The animals were housed in a pathogen-free facility and fed with autoclaved Purina rodent chow (St. Louis, MO, USA) with water available ad libitum. All animal procedures were performed according to the Guide for the Care and Use of Laboratory Animals, as adopted and enforced by the U.S. National Institutes of Health and the Mexican Regulation of Animal Care and Maintenance (NOM-062-ZOO-1999, 2001). The mice were distributed randomly into TCDD and vehicle treatment groups (N = 3, for each of the assay performed), and the treatments were carried out as described previously [20]. TCDD was dissolved in corn oil, and a single dose of TCDD (250 μg/kg) or corn oil alone (vehicle) was administered by gavage to 8- to 9-week-old male C57BL/6J wild-type (WT) and Ahr-null mice. Seven days later, the mice were anesthetized with pentobarbital and euthanized by decapitation. The brains were rapidly removed, and the ventral midbrain was dissected out as follow: the brain was removed, and the dorsal side was placed up. Using a thin blade, a coronal rostral cut was made at the level of the pineal recess, the junction between the caudal part of the interhemispheric scissure and the medial occipital lobe border. Next, a caudal coronal cut in the intercollicular sulcus was made. The midbrain was dissected, and the ventral midbrain section obtained.
Liver sections were also extracted. Brain and liver samples were snap frozen and stored at −70 °C.
2.3. Real-time quantitative polymerase chain reaction (qPCR)
Total RNA was prepared from mouse liver and ventral midbrain using TRIzol reagent according to the manufacturer’s instructions (Invitrogen, Camarillo, CA, USA). RNA was quantified spectrophotometrically at an optical density of 260 nm. RNA integrity was evaluated by electrophoresis on 1% agarose gels. cDNA was prepared for quantitative PCR from 2 μg of total RNA using the SuperScript First-Strand Synthesis kit (Invitrogen, Camarillo, CA, USA) and oligo dTs. PCR was performed on a StepOne Real-Time PCR System (Applied Biosystems, Branchburg, NJ, USA), and the results were analyzed using the comparative threshold cycle (CT) method. mRNAs encoding Ahr, Cyp1a1 Prkn, and 18S ribosomal RNA (rRNA, endogenous), were amplified in a single PCR to allow for the normalization of the mRNA data. The PCR mixture contained 2 μl of cDNA, 1x TaqMan Universal PCR Master Mix (Applied Biosystems, Branchburg, NJ, USA) and 0.9 and 0.25 μM primers and probes, respectively. The primer and probe sequences used for Prkn were 5′-CAAACAAGCAACCCTCACCTT-3′ (forward), 5′-ATCCGGTTTGGAATTAAGACATCGT-3′ (reverse), and CCCA GGGCCCATCTT (probe, FAM). The probes used for Ahr, Cyp1a1 and 18S rRNA were obtained from Applied Biosystems (Branchburg, NJ, USA) with identification numbers Mm01291777_m1, Mm00487218_m1, and Mm00507222_s1, respectively.
2.4. Immunofluorescence
The localization of the AhR protein in the SNc of adult mouse brains was determined by immunofluorescence, and tyrosine hydroxylase (TH) was used to identify DA neurons. Mice were injected with an overdose of pentobarbital (80 mg/kg) and perfused intracardially with saline. Subsequently, the brains were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS), postfixed in the same solution for 48 h and then incubated in 20% sucrose in PBS. Coronal sections (30-μm-thick) were cut with a cryostat and used for immunohistochemical assays. Briefly, free-floating sections were incubated at room temperature for 1 h in 0.25% Triton X-100/PBS buffer and subsequently in 5% BSA and 0.25% Triton X-100/PBS buffer for 3 h. Then, the sections were incubated in 3% BSA and 0.25% Triton X-100/PBS solution containing AhR (1:500) and TH (1:1000) antibodies (Abcam, Cat. ab2770, CA, USA and Cell Signaling Technologies, Cat. 2792, MA, USA, respectively). After 48 h of incubation, the sections were incubated with secondary antibodies (1:400) (Alexa 594 anti-mouse IgG, Cat. A-21203 and Alexa 488 anti-rabbit, Cat. A-11008; Thermo Fisher Scientific, MA, USA) diluted in PBS with 0.25% Triton X-100. The sections were counterstained with DAPI to reveal the nuclei and observed by confocal microscopy. Triple-labeled images were obtained using a confocal laser-scanning microscope (Leica TCS-SPE, Wetzlar, Germany) in the XYZ (Z-stacks) mode using a 40x objective. The Z-stacks (3–4 optical slices) were then converted into three-dimensional projection images using the Leica LAS AF lite software.
2.5. Western blotting
Ventral midbrain sections were homogenized in buffer containing 20 mM HEPES, 350 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 1% Nonidet P40, 0.5 mM DTT, 0.1% PMSF and Mini cOmplete protease inhibitor cocktail (1 tablet/10 mL, Roche, Mannheim, Germany). The protein suspension was homogenized completely by sonication for 30 s. Protein concentrations were determined using the Bradford assay (Bio-Rad, Hercules, CA, USA). Aliquots (40 μg) were solubilized in sample buffer (60 mM Tris–HCl, pH 6.8, 2% sodium dodecyl sulfate (SDS), 20% glycerol, 2% mercaptoethanol, and 0.001% bromophenol blue) and subjected to 12% SDS polyacrylamide gel electrophoresis. The protein extracts were transferred to a nitrocellulose membrane using a mini trans-blot system (Bio-Rad, Hercules, CA, USA). The transfer was performed at a constant voltage of 80 V for 2 h in transfer buffer (48 mM Tris–HCl, 39 mM glycine, pH 8.3, and 20% methanol). Following the transfer, the membranes were blocked overnight at 4 °C in the presence of 2% nonfat dry milk and 0.5% bovine serum albumin (BSA) in blocking buffer (25 mM Tris–HCl, pH 7.5, and 150 mM NaCl) and subsequently incubated at 4 °C for 3 h with mouse monoclonal anti-Parkin (1:500; Thermo Fisher, Cat. 39–0900, Meridian Rock, IL, USA), rabbit polyclonal anti-Cyp1a1 (1:100, Santa Cruz Biotechnology, Cat. sc-20772, Visalia, CA, USA), and goat anti-β-actin (1:1000; Santa Cruz, Biotechnology, Cat. sc-1616, Visalia, CA, USA), and for 16 h at 4 °C for mouse monoclonal anti-α-synuclein (1:4000; Santa Cruz Biotechnology, Cat. Sc-12767, Visalia, CA, USA). The antibodies were diluted in buffer (25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20, 0.05% nonfat dry milk, and 0.05% BSA). After washing, the membranes were incubated with the relevant horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Thermo Fisher, Cat. 62–6520, Meridian Rock, IL, USA), horse anti-mouse IgG (Vector laboratories, Cat. PI-2000, Burlingame, CA, USA), goat anti-rabbit IgG (Cell Signaling, Cat. 7074, Danvers, USA), and rabbit anti-goat IgG (Thermo Fisher, Cat. 31402, Meridian Rock, IL, USA) secondary antibodies for 2 h at 4 °C. The membranes were washed, and the immunoreactive proteins were detected using an ECL western blotting detection kit (Amersham, Arlington Heights, IL, USA). The integrated optical density of the bands was quantified using scanning densitometry (GS-800 Calibrated Densitometer, Bio-Rad, Hercules, CA, USA).
2.6. In silico analysis
The mouse Prkn promoter was analyzed using the web-based bioinformatics tool EPD (https://epd.epfl.ch//index.php). P < 0.01 was considered significant.
2.7. Chromatin immunoprecipitation (ChIP)
ChIP assays were performed according to the kit protocol (Santa Cruz Biotechnology, Visalia, CA, USA) with an anti-AhR antibody (Thermo Fisher Scientific, Cat. MA1–514, Rockland, IL, USA). The PCR product corresponding to the Prkn proximal gene promoter was generated from an aliquot of immunoprecipitated material. Brain homogenates from WT mice treated with a single oral dose of TCDD or vehicle (corn oil) were washed with PBS buffer and crosslinked with 1% formaldehyde. After chromatin isolation, the DNA was fragmented, and immunoprecipitation was performed. The crosslinking was reversed, the DNA was purified, and PCR amplification was performed as follows: initial denaturation at 94 °C for 3 min and 28 cycles of denaturation at 94 °C for 30 s, annealing at 65 °C for 30 s, and extension at 72 °C for 30 s. A final extension cycle at 72 °C for 10 min was added to the end of the program. The oligonucleotides used for PCR amplification were 5′-AGAAGTGAGCAGGGGGTCGGG-3′ (forward) and 5′-AAGGACCTAC GCGGGCACTG-3′ (reverse).
2.8. Plasmid
The reporter plasmid pGL4/−419-mPrkn, which includes the mouse Prkn gene promoter, was constructed as described. A −419/+1 fragment was generated by PCR amplification using mouse genomic DNA and oligonucleotides (forward: 5′-AGAAGTGAGCAGGGGGTCGGG-3′, reverse: 5′-AAGGACCTACGCGGGCACTG-3′). The fragment was then cloned into the pCR 2.1-TOPO transition vector (Invitrogen, Camarillo, CA, USA). To verify the amplicon orientation as well as its correct sequence, single and double restriction enzyme digestions and sequencing were performed. Then, the fragment was cloned into the KpnI/Xhol site of the pGL4.10 [luc2] reporter vector (Promega, Madison, WI, USA) containing the firefly luciferase gene. This plasmid was designated pGL4/−419_mPrkn.
2.9. Cell culture and transfections
SH-SY5Y cells were obtained from American Type Culture Collection (CRL-2266, Manassas, VA, USA). The cells were grown in 100-mm dishes with high-glucose DMEM (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco, Logan, UT, USA) and 1% antibiotic/antimycotic (Invitrogen, Carlsbad, CA, USA) at 37 °C in a humidified incubator with a 5% CO2 atmosphere. Transfections were performed using Escort IV (Sigma, Saint Louis, MO, USA). Each culture (1 × 106 cells/mL) was transfected with 1, 2.5, 5 or 10 μg of pGL4/−419-mPrkn and 1 μg of pRL-CMV as an internal control. The media was replaced with fresh media containing 10 nM TCDD for 48 h posttransfection. After 48 h, the cells were homogenized by incubation with Passive Lysis Buffer (Promega, Madison, WI, USA) for 15 min at room temperature. A luciferase activity assay was performed using the Dual-Glo Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions with a Modulus luminometer (Turner Biosystems, Sunnyvale, CA, USA). Blanks were obtained by performing the luciferase activity assay on mock-transfected cells. Firefly luciferase activity levels were normalized by comparison to the activity levels of Renilla luciferase.
2.10. Electrophoretic mobility shift assay (EMSA)
Liver and brain tissues from C57BL/6J mouse strain were used to prepare nuclear extracts. Tissues and cells were homogenized in ice-cold buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1.0 mM DTT, and 0.5 mM PMSF) and incubated for 15 min on ice. Finally, the buffer was complemented with 0.5% IGEPAL CA-630 (Sigma-Aldrich), and the mixes were passed through a 22-gauge needle and centrifuged at 2000 × g for 5 min at 4 °C. The supernatant was removed, and the nuclear pellet was homogenized in 20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 1.0 mM PMSF. Nuclear samples were mixed vigorously for 30 min at 4 °C. The nuclear homogenates were centrifuged at 3000 × g for 5 min at 4 °C, and the supernatant was recovered for protein quantification by bicinchoninic acid assay (Sigma-Aldrich).
EMSAs were carried out using a 3′ end-labeled biotin duplex and unlabeled probes. The biotin-labeled reactions were performed according to the manufacturer’s instructions (LightShift Chemiluminescent EMSA kit, Thermo Scientific). The EMSA binding reactions contained 10 mM Tris, pH 8.0, 1 mM MgCl2, 5 mM NaCl, 2.5 mM EDTA, 2.5 mM DTT, 4% glycerol, 1.5 μg nuclear protein extract, 10 mM salmon sperm DNA and 16 fmol of each labeled duplex probe. For competition assays, a 500-fold molar excess of the unlabeled duplex probe was added to the mixture. The samples were separated on pre-run native 5% polyacrylamide gels and transferred to nylon membranes (Amersham Hybond-N+, GE Healthcare). DNA crosslinking to the membranes was achieved by membrane exposure to a UV transilluminator (312 nM bulbs) for 15 min. Finally, the complexes were detected by chemiluminescence according to the manufacturer’s instructions (LightShift Chemiluminescent EMSA kit, Thermo Scientific). The sequences of the primers used for duplex probes were as follows:
XRE1-forward: 5′-TGCGCCCGCCTGGCTCGC-3′, XRE1-reverse: 5′-GCGAGCCAGGCGGGCGCA-3′; XRE consensus-forward: 5′-CTCGAA CTCACGCAACTCCGT-3′, XRE consensus-reverse: 5′-CGGAGTTGCGTG AGTTCGAGC-3′; XRE consensus mut-forward: 5′-CTCGAACTCACTCA ACTCCGT-3′, and XRE consensus mut-reverse: 5′-CGGAGTTGAAGTGA GTTCGAGC-3′.
2.11. Statistical analysis
The results are presented as the mean values ± standard deviation (S.D.). The statistical significance of the data was evaluated using Student’s t-test. In all cases, the differences between the groups were considered to be statistically significant when the p value was less than 0.05.
3. Results
Initially, AhR expression in the mouse ventral midbrain and DA neurons was evaluated. AhR mRNA was detected in the mouse ventral midbrain region at one-third of the level in the liver (Fig. 1A). Since the AhR expression is highest in the liver, the observed levels in the ventral midbrain are substantial. We then determined whether AhR protein is expressed in DA neurons. AhR protein was detected by immunofluorescence in the SNc of the adult mouse brain and the signal colocalized with that of tyrosine hydroxylase, a marker of dopaminergic neurons (Fig. 1B).
Fig. 1.
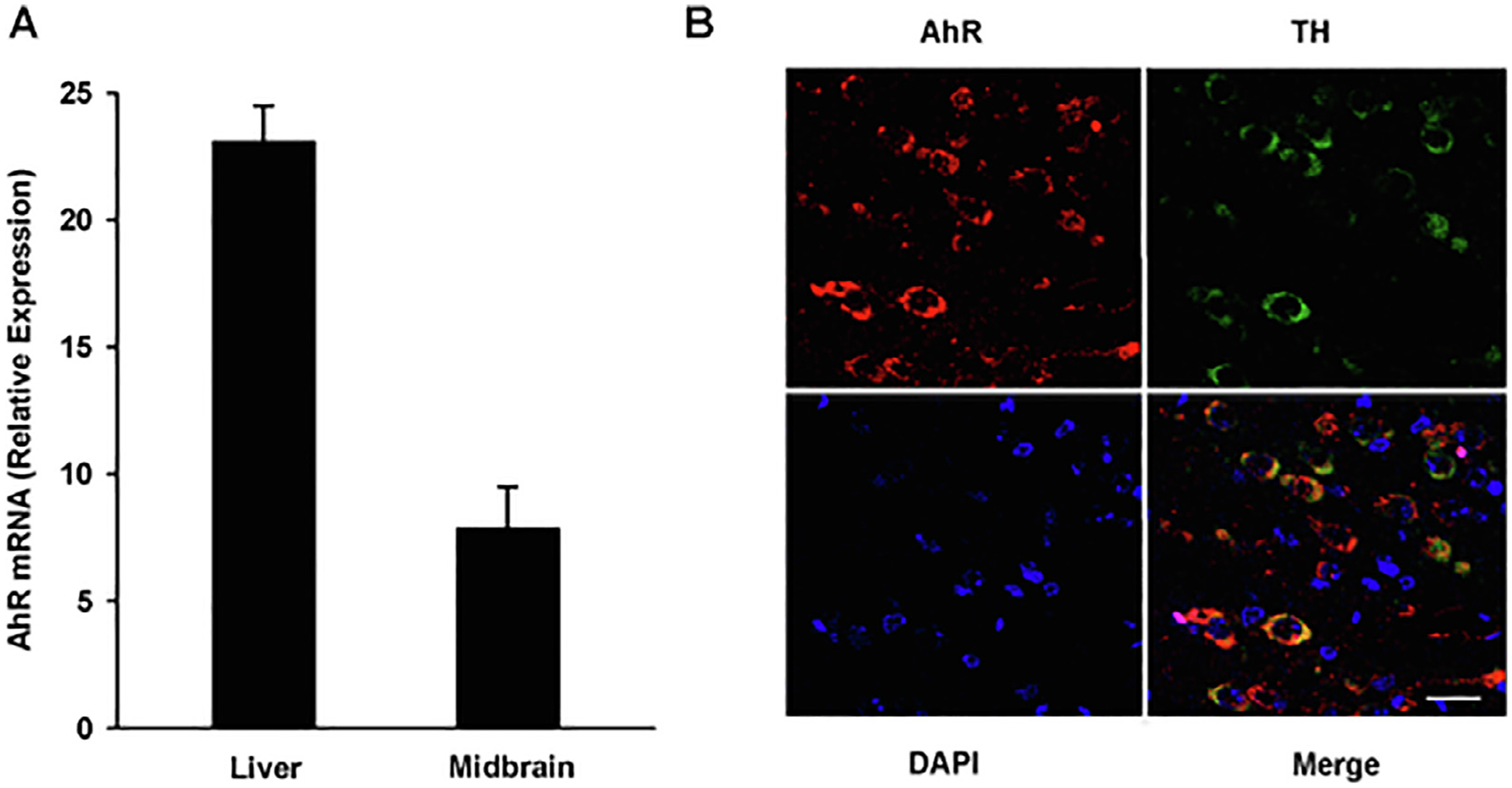
AhR is expressed in the mouse ventral midbrain and DA neurons. A) Total RNA was extracted from the ventral midbrain region and liver of sacrificed mice. AhR mRNA levels were determined by qPCR and normalized to 18S ribosomal RNA. The results are expressed as the mean ± S.D. of samples from three different mice. B) Representative confocal microscopy images of AhR expression in the ventral midbrain region. AhR, TH and DAPI were visualized as red, green, and blue, respectively. Scale bar: 25 μM.
Compared to vehicle treatment, TCDD, an AhR agonist, induced 32- and 3.7-fold increases in the expression of Prkn mRNAs in the mouse liver and ventral midbrain, respectively (Fig. 2A and B). This induction was AhR-dependent, since no induction of Prkn mRNA was observed in TCDD-treated Ahr-null mouse liver and ventral midbrain. As expected, AhR activation induced expression of its canonical target gene, Cyp1a1, in both the liver and ventral midbrain region and the induction was also AhR-dependent (Fig. 2C and D).
Fig. 2.
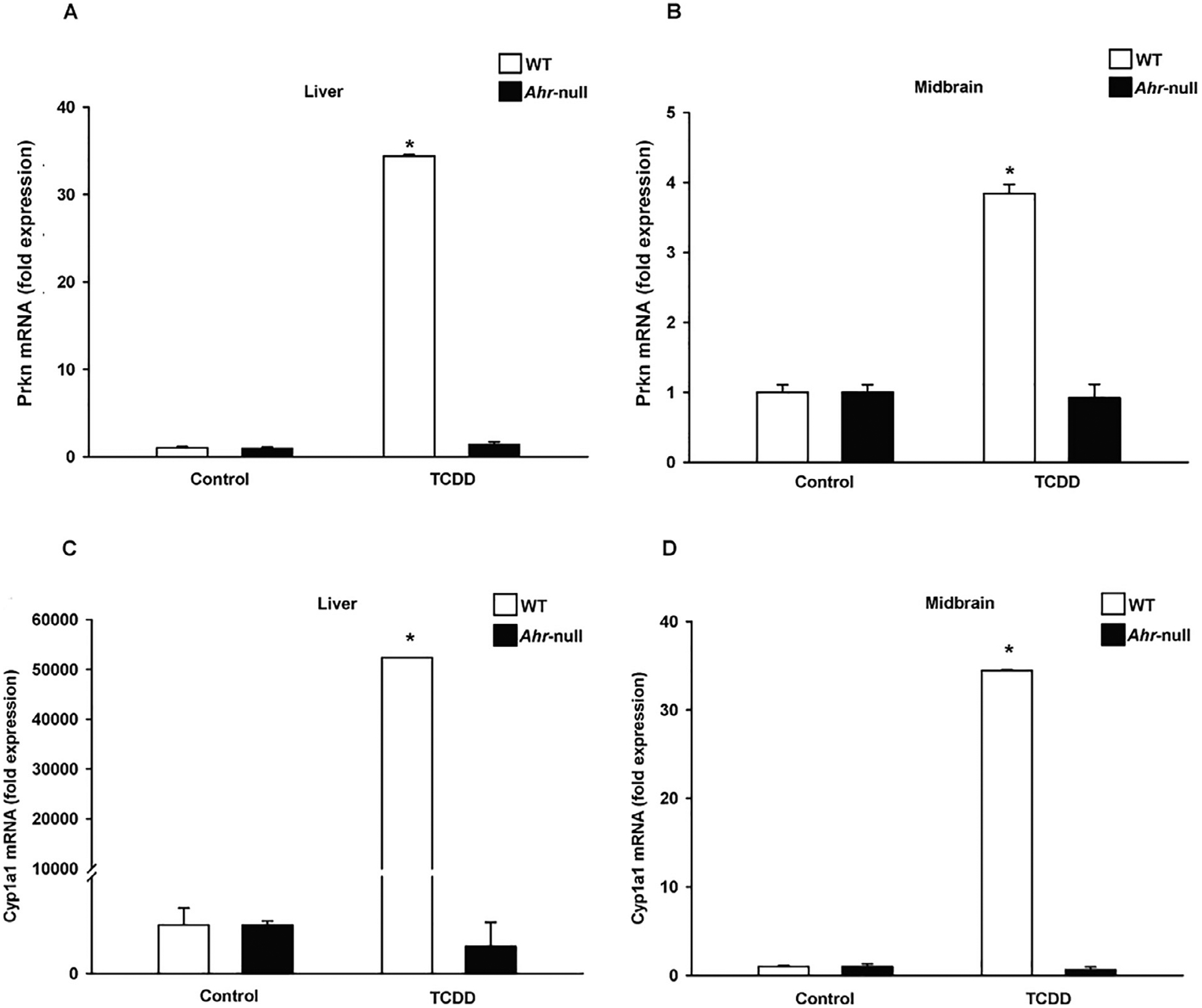
AhR-dependent induction of Cyp1a1 and Prkn mRNA in the ventral mouse liver and ventral midbrain. Wild-type (WT) and Ahr-null mice were treated with a single oral dose of TCDD or corn oil as vehicle. Total RNA was extracted from the liver and ventral midbrain region, and Prkn (A and B) and Cyp1a1 (C and D) mRNA levels were determined by qPCR and normalized to 18S ribosomal RNA. The results are expressed as the mean ± S.D. of samples from three different mice. *p < 0.05, treatment vs. control.
Then, the Prkn and Cyp1a1 protein levels in the mouse ventral midbrain region were evaluated. Compared to vehicle treatment, TCDD treatment increased Prkn protein levels by approximately 3-fold (Fig. 3A and C). Similarly, a 3-fold increase in Cyp1a1 protein was observed (Fig. 3A and B).
Fig. 3.
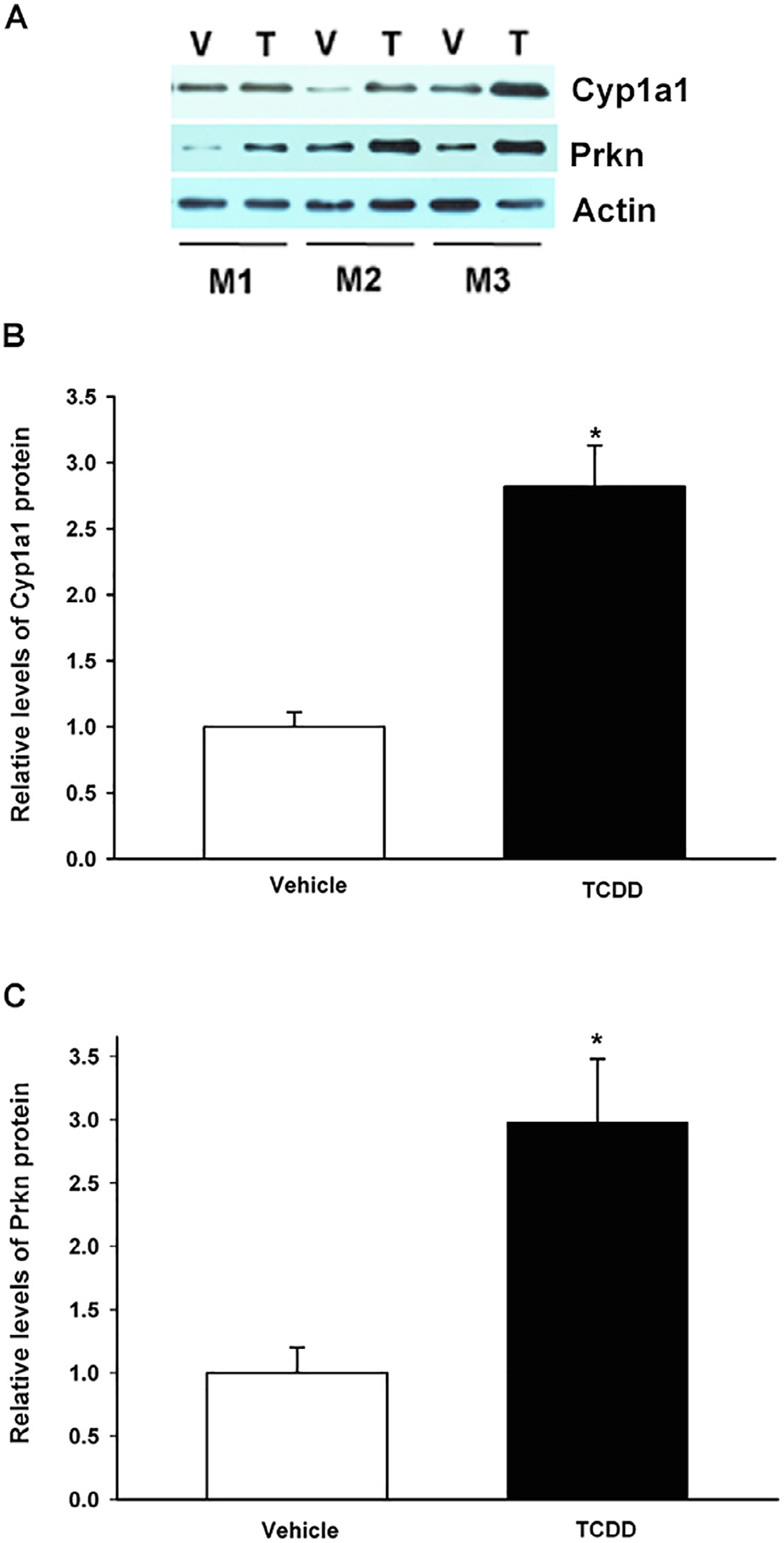
TCDD treatment induced Cyp1a1 and Prkn protein levels in the mouse ventral midbrain. Wild-type mice were treated with a single oral dose of TCDD (T) or corn oil as vehicle (V), and Cyp1a1 and Parkin protein levels were determined by western blotting. Beta actin was used as a loading control (A). Relative Cyp1a1 (B) and Prkn (C) protein levels. The results are expressed as the mean ± S.D. of samples from three different mice (M1, M2, M3). *p < 0.05, treatment vs. control.
The above studies confirm that AhR is expressed in the mouse ventral midbrain, particularly in DA neurons, and indicate that AhR activation upregulates Prkn mRNA and Prkn protein. These results prompted us to investigate and characterize the molecular mechanisms of AhR-dependent Parkin induction. In silico analysis of the mouse Prkn gene promoter was carried out (Fig. 4). Several response elements were identified. Of particular interest for the present study are the three putative concensus AhR binding sites, known as xenobiotic response elements (XREs), located −37, −54 and −349 bp from the Prkn transcription start site. ChIP revealed that AhR indeed binds to the Prkn gene promoter after TCDD treatment (Fig. 5A, lane 4). Moreover, a basal interaction between AhR and the Prkn gene promoter was also observed (Fig. 5A, lane 3), suggesting that this AhR may also be involved in the control of basal Prkn expression possibly through its activation by endogenous ligands.
Fig. 4.
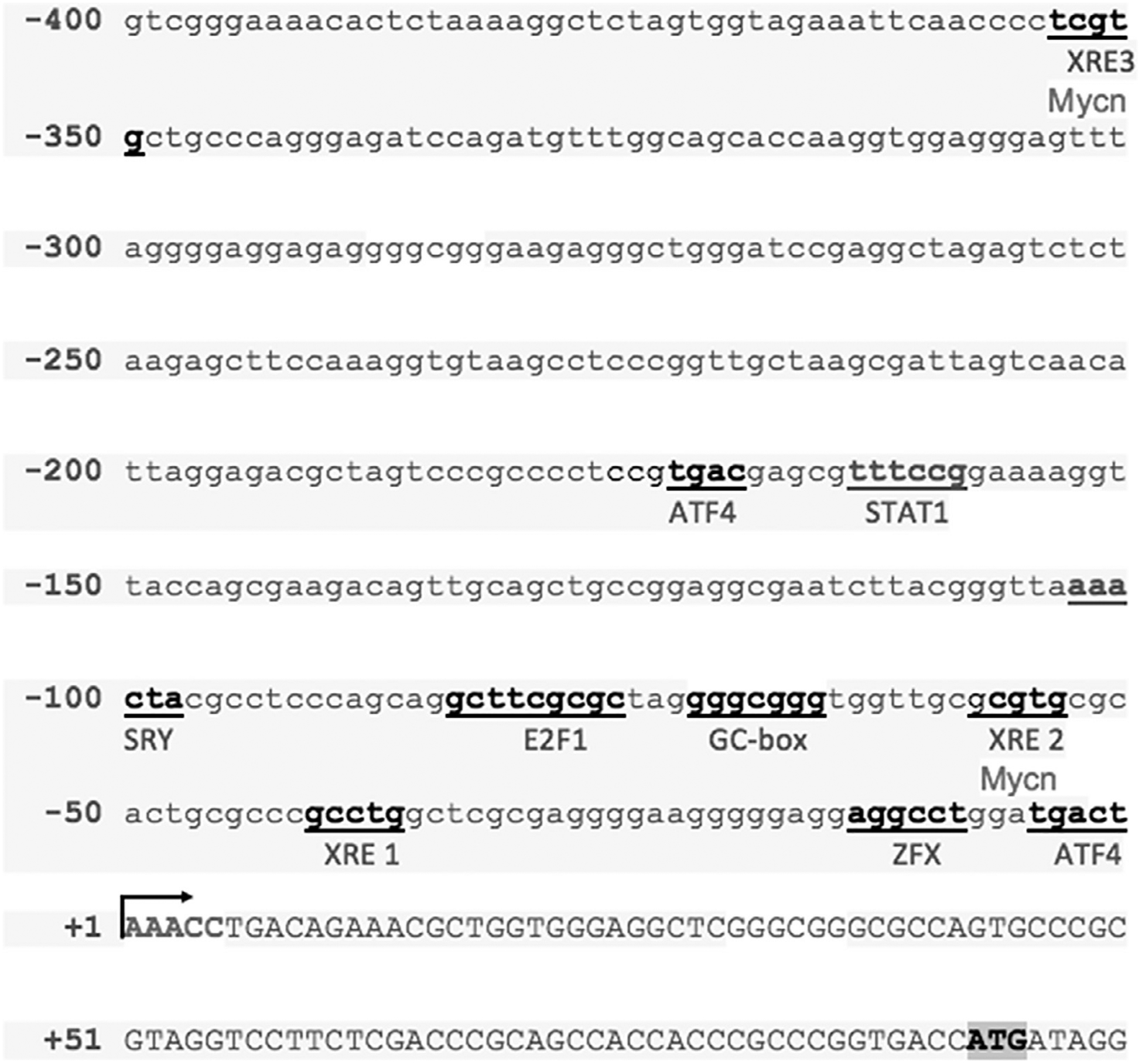
Analysis of the 5′ region of the mouse Prkn gene. The 5′ region of the mouse Prkn gene was analyzed in silico to identify binding sites for known transcription factors (underlined). XRE: xenobiotic response element, Mycn: Mycn proto-oncogene neuroblastoma-derived response element, ATF4: activating transcription factor 4 response element, STAT1: signal transducer and activator of transcription 1 response element, SRY: sex-determining region Y factor response element, E2F: E2F transcription factor response element, GC box, and ZFX: zinc finger protein X-linked response element. The transcription start site is indicated by the arrow and the translation start site codon is indicated by shading.
Fig. 5.
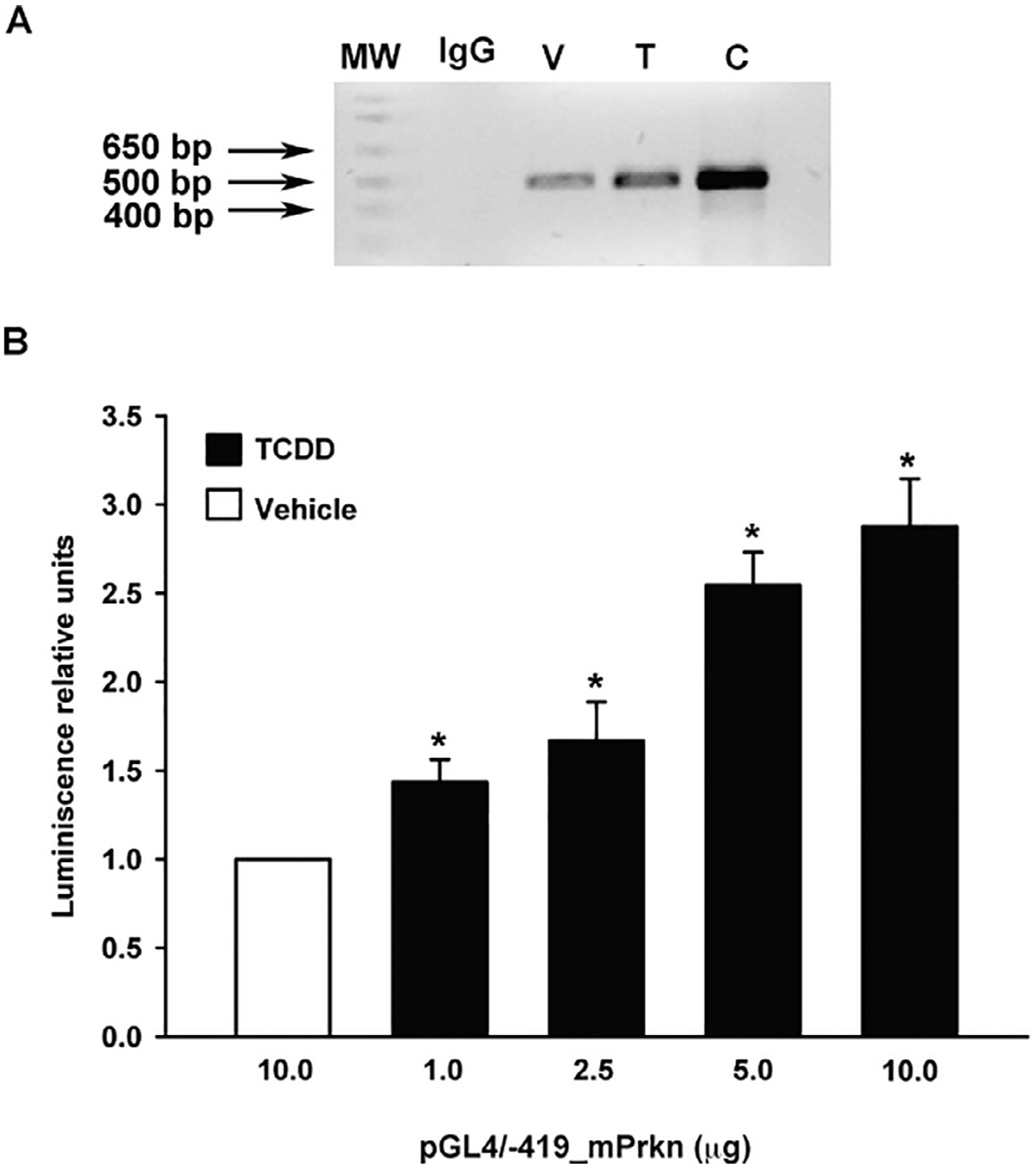
AhR binds and transactivates the Prkn promoter gene. A) ChIP analysis of AhR binding to the mouse Prkn gene promoter region. Material for immunoprecipitation was obtained from the brain tissues of mice treated with corn oil as vehicle (V, lane 3) or a single oral dose of TCDD (T, lane 4). PCR products corresponding to the −407/+74 bp region of the Prkn gene promoter were generated. Genomic DNA was used as the positive control (C, lane 5) and IgG antibody was used as the negative control (IgG, lane 2). MW (lane 1) represents molecular weight markers. A representative gel of two independent experiments is shown. B) The luciferase activity of SH-SY5Y cells (1 × 106) transfected with 1, 2.5, 5 or 10 μg of the pGL4/−419_mPrkn vector containing the firefly luciferase open reading frame under the control of the mouse Prkn gene promoter and treated with 10 nM of TCDD or DMSO (vehicle) for 48 h was determined. The luciferase activity was normalized to Renilla luciferase activity and is expressed as the mean ± S.D. of three independent experiments. * p < 0.05, control vs. treatment.
To determine whether AhR binding to the Prkn promoter results in its transactivation, a luciferase reporter plasmid (pGL4/−419-mPrkn), driven by the mouse Prkn promoter, was constructed and transfected into SH-SY5Y human neuroblastoma cells. A dose response of increased luciferase expression from 1.0 to 10 μg of transfected plasmid was found after 10 nM TCDD treatment, up to a 3-fold increase with 10 μg of the reporter construct, relative to vehicle-treated cells (Fig. 5B).
According to the in silico study, among the three XREs identified, XRE1 exhibited the highest score. Therefore, the interaction between AhR and XRE1 was investigated by EMSAs. The addition of nuclear extracts from mouse livers and ventral midbrains treated with TCDD to the XRE1 motif resulted in the formation of a DNA-protein complex as represented by the shifted band (Fig. 6A–B, lane 2). This binding was specific since an excess of unlabeled XRE1 or consensus XRE probes blocked formation of this complex (Fig. 6A–B lanes 3 and 4), whereas an unlabeled mutated consensus XRE probe did not compete with the labeled XRE1 probe (Fig. 6A–B lane 5). Together, these results indicate that treatment with TCDD promotes AhR binding to XRE1, transactivates the Prkn gene promoter and induces Prkn mRNA and Prkn protein expression.
Fig. 6.
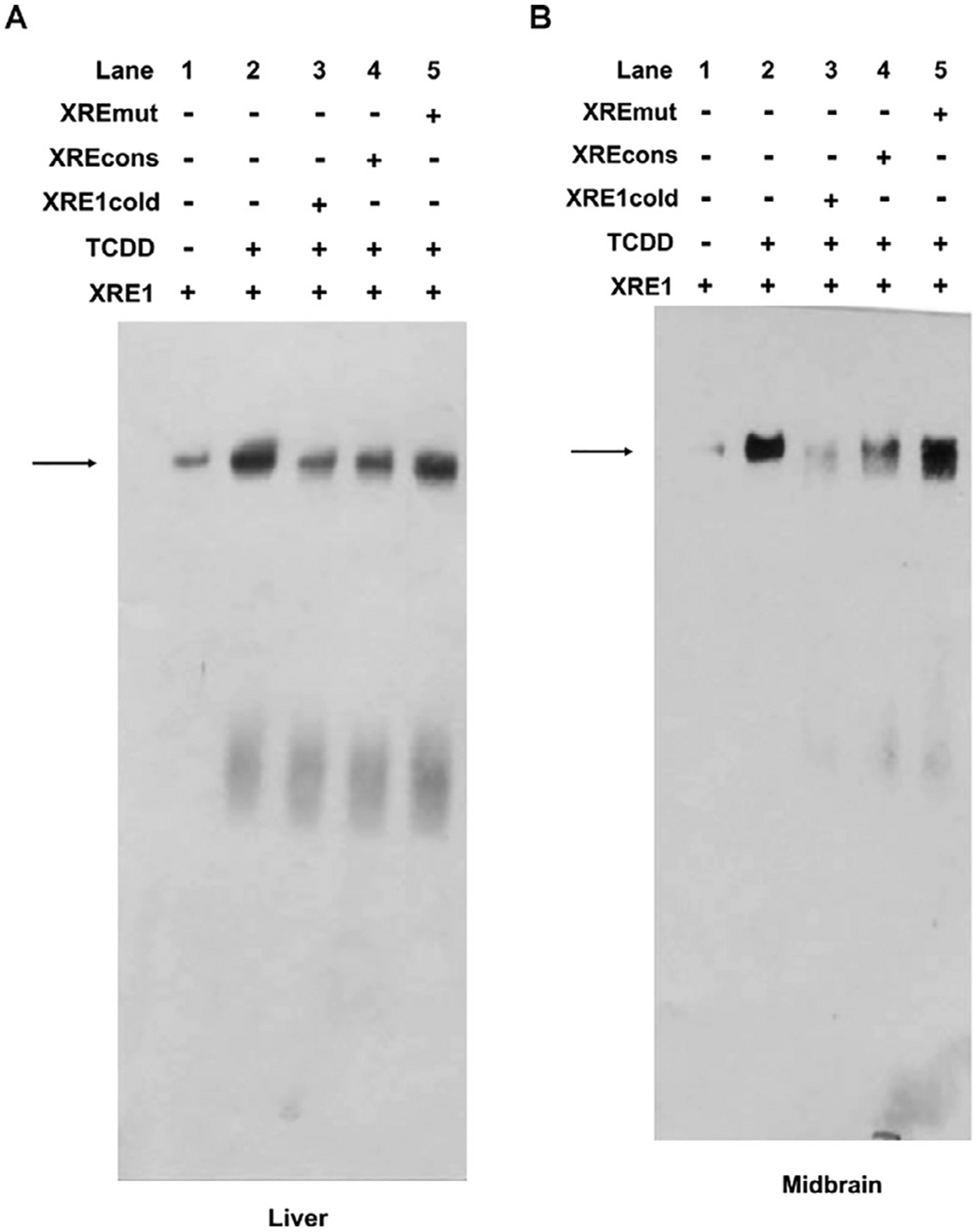
EMSA analysis of protein binding to the putative XRE1 identified in the Prkn gene promoter. Nuclear extracts from the livers (A) or ventral midbrains (B) of mice treated with corn oil (lane 1) or 250 μg/kg TCDD (lanes 2–5) were incubated with a labeled putative XRE1 probe only (lane 2), with a labeled putative XRE1 probe plus a 500-fold molar excess of an unlabeled putative XRE1 probe (lane 3), with the putative XRE1 labeled probe plus an unlabeled consensus XRE probe characterized from the mouse Cyp1a1 gene (lane 4), or with a labeled putative XRE1 probe plus an unlabeled mutated consensus XRE probe characterized from the mouse Cyp1a1 gene (lane 5). The arrow indicates the shifted bands. The image is representative of 2 independent experiments. The nuclear extract was obtained from the ventral midbrain from 3 mice.
Next, we sought to determine whether the AhR-dependent increase in Parkin levels is associated with a decrease in the protein levels of their target substrates. For this purpose, WT and Ahr-null mice were treated with TCDD, and Snca protein levels in the ventral midbrain region were determined by western blot. After TCDD treatment, WT mice exhibited an approximately 10-fold decrease in α-synuclein protein levels compared to vehicle treatment (Fig. 7), and this change was significant. In contrast, this effect was not observed in Ahr-null samples.
Fig. 7.
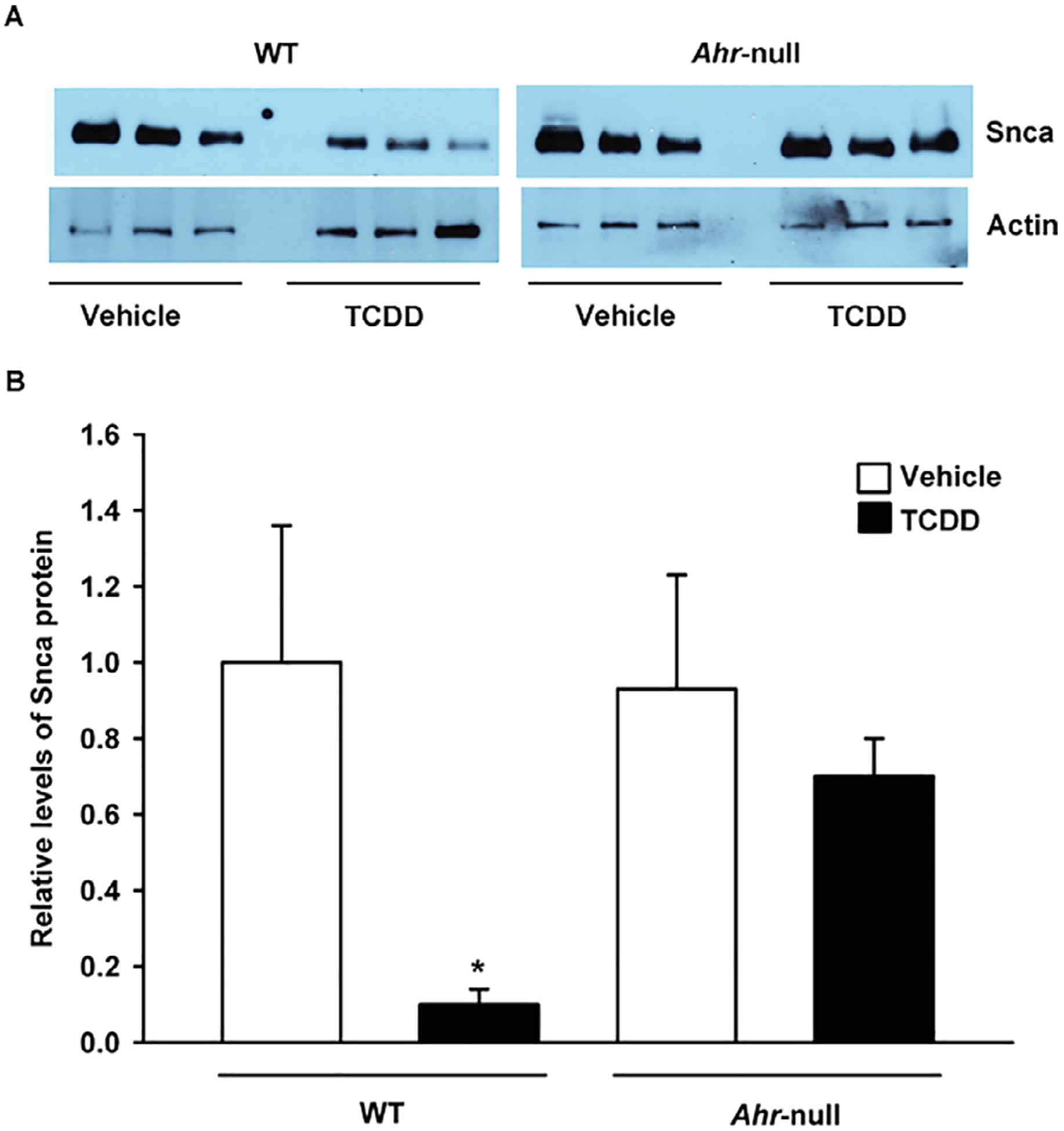
AhR activation by TCDD results in a decrease in Snca protein levels in the mouse ventral midbrain. Wild-type (WT) and Ahr-null mice were treated with a single oral dose of TCDD or corn oil as vehicle, and Snca protein levels were determined by western blotting. Beta actin was used as a loading control (A). Relative Scna protein levels (B). The results are expressed as the mean ± S.D. of samples from three different mice. *p < 0.05, treatment vs control.
4. Discussion
A deficit in Parkin levels and/or function have been associated with the etiology of several neurodegenerative disorders, including PD. Mutations in the human PRKN gene result in autosomal-recessive early-onset parkinsonism, and reduced Parkin expression has been linked to late-onset PD. Conversely, the overexpression of Parkin in vitro and in vivo protects DA neurons from a variety of cellular stressors that produce oxidative stress, excitotoxicity, mitochondrial dysfunction, endoplasmic reticular stress, apoptosis, and proteotoxicity [21,22]. Thus, gaining insight into the molecular mechanism that direct Parkin expression is crucial for the design of novel treatments based on the induction of Parkin levels.
Here, we show that Parkin is transcriptionally upregulated by TCDD via AhR activation. First, we demonstrated that this transcription factor is expressed in the mouse ventral midbrain, particularly in DA neurons. Considering that the liver is among the organs and tissues with the highest levels of AhR, the levels of its transcript in the mouse ventral midbrain were substantial; the levels in the ventral midbrain were one third of those observed in liver, suggesting that AhR has important functions in this brain region.
The induction of Cyp1a1 mRNA in the ventral midbrain after TCDD treatment indicates that TCDD crosses the blood-brain barrier and reaches brain tissues to activate AhR. Similar to Cyp1a1 but to a lesser extent, TCDD promotes the overexpression of Parkin. In both cases, the effect is AhR-dependent since no induction is observed in Ahr-null mice. At the protein level, compared to vehicle treatment, TCDD treatment results in a 3-fold induction in Prkn expression. These results clearly demonstrate that the activation of AhR upregulates Prkn gene expression.
The in silico analysis of the Prkn gene promoter identified 3 putative AhR binding sites, namely, XRE1, XRE2, and XRE3, which are located −37, −54 and −349 bp, respectively, from the transcription start site. The Prkn gene promoter in Drosophila melanogaster contains a binding site for AhR [18] and the human PRKN promoter contains 3 AhR binding sites, suggesting that the gene encoding Parkin may be under the control of AhR in several diverse species and thus this Ahr-driven signaling pathway must be of developmental and/or physiological consequence.
ChIP, EMSA, and transactivation studies confirmed that TCDD promotes AhR recruitment to the Prkn gene promoter, inducing Parkin expression. ChIP also showed an interaction between AhR and the promoter under control conditions, that is, without TCDD treatment. The above results suggest the possible presence of an endogenous ligand and that AhR may also control basal Parkin expression.
Prkn is transcriptionally regulated by the restriction of trophic factors and nutrients [23], p53 [24], activating transcription factor 4 (ATF4) [25], and N-myc (Mycn) [26]. Consistent with this, the in silico study identified the presence of ATF4 and Mycn binding sites. No p53 responsive elements were recognized because they are located in intron 1 and −3332 bp from the ATG site. Of particular interest is that Mycn and AhR share similar responsive elements within the Prkn promoter gene. In contrast to AhR, Mycn, a critical transcription factor involved in neural development [27], downregulates Parkin expression. The latter occurs in the context of differentiation when Parkin expression is observed in the latest stages of embryonic development and when Mycn expression is the lowest [26]. These data suggest that Mycn may negatively control Parkin expression by blocking AhR binding to Mycn/AhR responsive elements.
Additionally, a sex-determining region Y factor (SRY) response element located −98 bp from the transcription start site was identified. Immunohistochemical studies showed that SRY is expressed in DA neurons and that 6-OHDA induces its expression, while its knockdown exacerbates cell death produced by ROS [28]. It would not be unexpected for the reported SRY protective response to be mediated, at least in part, by the induction of Parkin expression. Similarly, the induction of ATF4 plays an important neuroprotection role against rotenone-induced DA neuronal death, presumably through the induction of Parkin levels [29].
TCDD treatment results in a significant decrease in the protein levels of α-synuclein (Snca) in the mouse ventral midbrain in an AhR-dependent manner. Snca function is not well understood, but it may play a role in synaptic functioning and neuroplasticity by regulating neuro-transmitter release. It is present in several brain regions, including the SNc [30], and its progressive accumulation in cells leads to the degeneration of nigrostriatal circuits, resulting in several neurodegenerative diseases, such as PD. Cell stress, mutations, and posttranslational modifications disrupt the native Snca conformation exposing the central hydrophobic motif leading to its misfolding and aggregation. Particularly, phosphorylation at serine 129 (S129) has been associated with Snca fibrillation and the formation of cytoplasmic inclusions [31].
Snca clearance is carried out by the ubiquitin-proteosome system (UPS). However, there is no evidence indicating that S129-Snca is a parkin substrate. In contrast, in association with UbcH7, parkin ubiquitinate and promotes the proteasome degradation of O-linked glycosylated Snca [32]. Therefore, the decrease in Snca protein levels observed in the present study may be, in part, the result of the degradation of the O-linked glycosylated forms.
We previously reported that activation of AhR also results in UbcH7 induction together with a decrease in synphilin-1 (Sncaip) protein levels in the mouse ventral midbrain [16]. These data, together with those reported here, suggest that AhR-mediated increased Parkin and UbcH7 levels may be responsible for the decreased of both the O-linked glycosylated Snca and Sncaip protein levels. Snca degradation is also carried on by the autophagy-lysosomal pathway. We have reported recently that TCDD treatment, in an AhR-dependent way, induce the lysosome degradation of NFκB [33]. Thus, it is possible that the observed decrease in Snca is also due to lysosomal degradation.
The above may explain the parkinsonian effects derived from dieldrin exposure [34], a pesticide that has AhR antagonist properties [35]. On the other hand, AhR agonists such as beta-naphthoflavone and chemical mixtures containing AhR agonists such as cigarette smoke protect against MPP+-induced dopamine depletion [36]. In fact, several epidemiological studies have reported a negative association between PD and cigarette smoking [37,38].
In addition to TCDD and beta-naphthoflavone, a highly toxic compounds, several endogenous or natural products such as kynurenine, FICZ (6-formylindolo (3,2b) carbazole), ITE (2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester) and I3C (indole-3-carbinol) act as AhR agonists. In particular, tangeritin, a citrus flavonoid with AhR agonistic activity [39], protects against parkinsonian effects induced by 6-OHDA in a rat model [40]. Similarly, carnosic acid, a diterpene with AhR agonist activity that is found in rosemary extract [41], protects SH-SY5Y cells from 6-OHDA-induced apoptosis [42].
Several investigations have shown that Parkin, in addition to playing a role in neuronal function, may act as a tumor suppressor gene. Mutations in the PRKN gene have been reported in colorectal and gastric cancer, lung carcinoma and breast cancer [43,44], while its overexpression inhibits the growth of colon [45], hepatocarcinoma [46], breast [47] and glioblastoma cancer cells [48]. Therefore, the induction of Parkin levels may have an effect on the development of carcinogenic processes.
Taken together, our findings provide in vivo and in vitro evidence suggesting that Parkin is transcriptionally upregulated by AhR, which is associated with reduced protein levels of Snca in the mouse ventral midbrain. The already established protective role of Parkin in DA neuron survival together with the present data strongly suggest that treatments designed to induce Parkin expression through the use of nontoxic AhR agonist ligands may prevent and delay the onset and progression of neurodegenerative disorders such as PD.
Acknowledgement
This work was supported by Consejo Nacional de Ciencia y Tecnología, México, grants 153377 and 280897.
References
- [1].Kish SJ, Shannak K, Hornykiewicz O, Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications, N. Engl. J. Med 318 (14) (1988) 876–880. [DOI] [PubMed] [Google Scholar]
- [2].Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR, Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons, Neuron 36 (6) (2002) 1007–1019. [DOI] [PubMed] [Google Scholar]
- [3].Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B, Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila, Neuron 37 (6) (2003) 911–924. [DOI] [PubMed] [Google Scholar]
- [4].Winklhofer KF, Parkin and mitochondrial quality control: toward assembling the puzzle, Trends Cell Biol. 24 (6) (2014) 332–341. [DOI] [PubMed] [Google Scholar]
- [5].Helton TD, Otsuka T, Lee MC, Mu Y, Ehlers MD, Pruning and loss of excitatory synapses by the parkin ubiquitin ligase, Proc. Natl. Acad. Sci. U.S.A 105 (49) (2008) 19492–19497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].West AB, Maraganore D, Crook J, Lesnick T, Lockhart PJ, Wilkes KM, Kapatos G, Hardy JA, Farrer MJ, Functional association of the parkin gene promoter with idiopathic Parkinson’s disease, Hum. Mol. Genet 11 (22) (2002) 2787–2792. [DOI] [PubMed] [Google Scholar]
- [7].Pankratz N, Kissell DK, Pauciulo MW, Halter CA, Rudolph A, Pfeiffer RF, Marder KS, Foroud T, Nichols WC, Parkin dosage mutations have greater pathogenicity in familial PD than simple sequence mutations, Neurology 73 (4) (2009) 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL, Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function, Hum. Mol. Genet 14 (24) (2005) 3885–3897. [DOI] [PubMed] [Google Scholar]
- [9].Pawlyk AC, Giasson BI, Sampathu DM, Perez FA, Lim KL, Dawson VL, Dawson TM, Palmiter RD, Trojanowski JQ, Lee VM, Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age, J. Biol. Chem 278 (48) (2003) 48120–48128. [DOI] [PubMed] [Google Scholar]
- [10].Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, Aebischer P, Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease, Proc. Natl. Acad. Sci. U.S.A 101 (50) (2004) 17510–17515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jiang H, Ren Y, Zhao J, Feng J, Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis, Hum. Mol. Genet 13 (16) (2004) 1745–1754. [DOI] [PubMed] [Google Scholar]
- [12].Ren Y, Jiang H, Yang F, Nakaso K, Feng J, Parkin protects dopaminergic neurons against microtubule-depolymerizing toxins by attenuating microtubule-associated protein kinase activation, J. Biol. Chem 284 (6) (2009) 4009–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liu B, Traini R, Killinger B, Schneider B, Moszczynska A, Overexpression of parkin in the rat nigrostriatal dopamine system protects against methamphetamine neurotoxicity, Exp. Neurol 247 (2013) 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Reyes-Hernandez OD, Mejia-Garcia A, Sanchez-Ocampo EM, Cabanas-Cortes MA, Ramirez P, Chavez-Gonzalez L, Gonzalez FJ, Elizondo G, Ube2l3 gene expression is modulated by activation of the aryl hydrocarbon receptor: implications for p53 ubiquitination, Biochem. Pharmacol 80 (6) (2010) 932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mejia-Garcia A, Gonzalez-Barbosa E, Martinez-Guzman C, Torres-Ramos MA, Rodriguez MS, Guzman-Leon S, Elizondo G, Activation of AHR mediates the ubiquitination and proteasome degradation of c-Fos through the induction of Ubcm4 gene expression, Toxicology 337 (2015) 47–57. [DOI] [PubMed] [Google Scholar]
- [16].Gonzalez-Barbosa E, Mejia-Garcia A, Bautista E, Gonzalez FJ, Segovia J, Elizondo G, TCDD induces UbcH7 expression and synphilin-1 protein degradation in the mouse ventral midbrain, J. Biochem. Mol. Toxicol 31 (10) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rowlands JC, Gustafsson JA, Aryl hydrocarbon receptor-mediated signal transduction, Crit. Rev. Toxicol 27 (2) (1997) 109–134. [DOI] [PubMed] [Google Scholar]
- [18].Bae YJ, Park KS, Kang SJ, Genomic organization and expression of parkin in Drosophila melanogaster, Exp. Mol. Med 35 (5) (2003) 393–402. [DOI] [PubMed] [Google Scholar]
- [19].Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ, Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor, Science 268 (5211) (1995) 722–726. [DOI] [PubMed] [Google Scholar]
- [20].Huang P, Ceccatelli S, Hoegberg P, Sten Shi TJ, Hakansson H, Rannug A, TCDD-induced expression of Ah receptor responsive genes in the pituitary and brain of cellular retinol-binding protein (CRBP-I) knockout mice, Toxicol. Appl. Pharmacol 192 (3) (2003) 262–274. [DOI] [PubMed] [Google Scholar]
- [21].Feany MB, Pallanck LJ, Parkin: a multipurpose neuroprotective agent? Neuron 38 (1) (2003) 13–16. [DOI] [PubMed] [Google Scholar]
- [22].Wahabi K, Perwez A, Rizvi MA, Parkin in Parkinson’s disease and cancer: a double-edged sword, Mol. Neurobiol 55 (8) (2018) 6788–6800. [DOI] [PubMed] [Google Scholar]
- [23].Klinkenberg M, Gispert S, Dominguez-Bautista JA, Braun I, Auburger G, Jendrach M, Restriction of trophic factors and nutrients induces PARKIN expression, Neurogenetics 13 (1) (2012) 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z, Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect, Proc. Natl. Acad. Sci. U.S.A 108 (39) (2011) 16259–16264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D, Gasser T, Augustin R, Trumbach D, Irrcher I, Park DS, Wurst W, Kilberg MS, Tatzelt J, Winklhofer KF, Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress, Cell Death Differ. 18 (5) (2011) 769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].West AB, Kapatos G, O’Farrell C, Gonzalez-de-Chavez F, Chiu K, Farrer MJ, Maidment NT, N-myc regulates parkin expression, J. Biol. Chem 279 (28) (2004) 28896–28902. [DOI] [PubMed] [Google Scholar]
- [27].Knoepfler PS, Cheng PF, Eisenman RN, N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation, Genes Dev. 16 (20) (2002) 2699–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Czech DP, Lee J, Correia J, Loke H, Moller EK, Harley VR, Transient neuroprotection by SRY upregulation in dopamine cells following injury in males, Endocrinology 155 (7) (2014) 2602–2612. [DOI] [PubMed] [Google Scholar]
- [29].Wu L, Luo N, Zhao HR, Gao Q, Lu J, Pan Y, Shi JP, Tian YY, Zhang YD, Salubrinal protects against rotenone-induced SH-SY5Y cell death via ATF4-parkin pathway, Brain Res. 1549 (2014) 52–62. [DOI] [PubMed] [Google Scholar]
- [30].Lashuel HA, Overk CR, Oueslati A, Masliah E, The many faces of alpha-synuclein: from structure and toxicity to therapeutic target, Nat. Rev. Neurosci 14 (1) (2013) 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Smith WW, Margolis RL, Li X, Troncoso JC, Lee MK, Dawson VL, Dawson TM, Iwatsubo T, Ross CA, Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells, J. Neurosci 25 (23) (2005) 5544–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ, Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease, Science 293 (5528) (2001) 263–269. [DOI] [PubMed] [Google Scholar]
- [33].Dominguez-Acosta O, Vega L, Estrada-Muniz E, Rodriguez MS, Gonzalez FJ, Elizondo G, Activation of aryl hydrocarbon receptor regulates the LPS/IFNgamma-induced inflammatory response by inducing ubiquitin-proteosomal and lysosomal degradation of RelA/p65, Biochem. Pharmacol 155 (2018) 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baltazar MT, Dinis-Oliveira RJ, de Lourdes Bastos M, Tsatsakis AM, Duarte JA, Carvalho F, Pesticides exposure as etiological factors of Parkinson’s disease and other neurodegenerative diseases–a mechanistic approach, Toxicol. Lett 230 (2) (2014) 85–103. [DOI] [PubMed] [Google Scholar]
- [35].Long M, Laier P, Vinggaard AM, Andersen HR, Lynggaard J, Bonefeld-Jorgensen EC, Effects of currently used pesticides in the AhR-CALUX assay: comparison between the human TV101L and the rat H4IIE cell line, Toxicology 194 (1–2) (2003) 77–93. [DOI] [PubMed] [Google Scholar]
- [36].Shahi GS, Das NP, Moochhala SM, 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity: partial protection against striato-nigral dopamine depletion in C57BL/6J mice by cigarette smoke exposure and by beta-naphthoflavone-pretreatment, Neurosci. Lett 127 (2) (1991) 247–250. [DOI] [PubMed] [Google Scholar]
- [37].Godwin-Austen RB, Lee PN, Marmot MG, Stern GM, Smoking and Parkinson’s disease, J. Neurol. Neurosurg. Psychiatry 45 (7) (1982) 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gallo V, Vineis P, Cancellieri M, Chiodini P, Barker RA, Brayne C, Pearce N, Vermeulen R, Panico S, Bueno-de-Mesquita B, Vanacore N, Forsgren L, Ramat S, Ardanaz E, Arriola L, Peterson J, Hansson O, Gavrila D, Sacerdote C, Sieri S, Kuhn T, Katzke VA, van der Schouw YT, Kyrozis A, Masala G, Mattiello A, Perneczky R, Middleton L, Saracci R, Riboli E, Exploring causality of the association between smoking and Parkinson’s disease, Int. J. Epidemiol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Busbee PB, Rouse M, Nagarkatti M, Nagarkatti PS, Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders, Nutr. Rev 71 (6) (2013) 353–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Datla KP, Christidou M, Widmer WW, Rooprai HK, Dexter DT, Tissue distribution and neuroprotective effects of citrus flavonoid tangeretin in a rat model of Parkinson’s disease, NeuroReport 12 (17) (2001) 3871–3875. [DOI] [PubMed] [Google Scholar]
- [41].Amakura Y, Yoshimura M, Takaoka M, Toda H, Tsutsumi T, Matsuda R, Teshima R, Nakamura M, Handa H, Yoshida T, Characterization of natural aryl hydrocarbon receptor agonists from cassia seed and rosemary, Molecules 19 (4) (2014) 4956–4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fu RH, Huang LC, Lin CY, Tsai CW, Modulation of ARTS and XIAP by Parkin is associated with carnosic acid protects SH-SY5Y cells against 6-hydroxydopamine-induced apoptosis, Mol. Neurobiol 55 (2) (2018) 1786–1794. [DOI] [PubMed] [Google Scholar]
- [43].Liu J, Zhang C, Zhao Y, Yue X, Wu H, Huang S, Chen J, Tomsky K, Xie H, Khella CA, Gatza ML, Xia D, Gao J, White E, Haffty BG, Hu W, Feng Z, Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression, Nat. Commun 8 (1) (2017) 1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N, The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data, Cancer Discov. 2 (5) (2012) 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo F, Cantley LC, Wyllie AH, Adams DJ, Arends MJ, PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice, Proc. Natl. Acad. Sci. U.S.A 107 (34) (2010) 15145–15150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang F, Denison S, Lai JP, Philips LA, Montoya D, Kock N, Schule B, Klein C, Shridhar V, Roberts LR, Smith DI, Parkin gene alterations in hepatocellular carcinoma, Genes Chromosom. Cancer 40 (2) (2004) 85–96. [DOI] [PubMed] [Google Scholar]
- [47].Tay SP, Yeo CW, Chai C, Chua PJ, Tan HM, Ang AX, Yip DL, Sung JX, Tan PH, Bay BH, Wong SH, Tang C, Tan JM, Lim KL, Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells, J. Biol. Chem 285 (38) (2010) 29231–29238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, Holland EC, Paty PB, Mischel PS, Liau L, Cloughesy TF, Mellinghoff IK, Solit DB, Chan TA, Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies, Nat. Genet 42 (1) (2010) 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]