

Abstract
The response to systemic infection and injury requires the rapid adaptation of hematopoietic stem cells (HSCs), which proliferate and divert their differentiation toward the myeloid lineage. Significant interest has emerged in understanding the signals that trigger the emergency hematopoietic program. However, the mechanisms that halt this response of HSCs, which is critical to restore homeostasis, remain unknown. Here we reveal that the E3 ubiquitin ligase Speckle-type BTB–POZ protein (SPOP) restrains the inflammatory activation of HSCs. In the absence of Spop, systemic inflammation proceeded in an unresolved manner, and the sustained response in the HSCs resulted in a lethal phenotype reminiscent of hyper-inflammatory syndrome or sepsis. Our proteomic studies decipher that SPOP restricted inflammation by ubiquitinating the innate signal transducer myeloid differentiation primary response protein 88 (MYD88). These findings unearth an HSC-intrinsic post-translational mechanism that is essential for reestablishing homeostasis after emergency hematopoiesis.
Host defense against pathogens and damaged tissue repair requires a well-orchestrated inflammatory response involving a number of blood cell lineages. The continued production of immune and red blood cells is mediated by the bone marrow, in particular the HSCs, which swiftly adapt to stress signals1. After acute infection or injury, HSCs rapidly shift their differentiation toward the myeloid compartment, a process termed ‘emergency hematopoiesis’2. This augmented myeloid output provides rapid protection against the injurious threat3,4. Direct sensing of pathogen-associated molecular patterns (such as Toll-like receptor (TLR) ligands) or inflammatory cytokines5 triggers transcriptional and epigenetic networks that facilitate the switch from steady-state to emergency hematopoiesis6–11. Importantly, the emergency hematopoietic program is transient, because HSCs return to their quiescent state. The dampening or resolution of this response is essential to limit HSCs exhaustion, restore the production of the diverse blood lineages and limit harmful immunopathologies4,12,13.
Uncontrolled inflammation promotes life-threatening diseases such as inflammatory response syndrome, sepsis, and autoimmune and chronic inflammatory diseases. An active coordinated program for resolution, orchestrated by the immune cells themselves, protects the host and ensures tissue repair and homeostasis12. At the site of inflammation, resolution involves a myriad biochemical pathways including the release of lipoxin and other specialized pro-resolving mediators13. Yet, in the bone marrow, the factors that control the resolution of emergency hematopoiesis, a process vital to restoring health, remain elusive. Previously, we have uncovered how protein degradation mediated by ubiquitination regulates HSC functions such as quiescence, self-renewal and differentiation14–16. Here, we reveal a post-translational mechanism that terminates the inflammatory response in the bone marrow, preventing a dysregulated host response. We report that the E3 ubiquitin ligase adapter SPOP controls the stability of the innate adapter MYD88, promoting the switch from emergency to homeostatic programs.
Results
Hematopoietic-specific deletion of Spop promotes lethal neutrophilia.
The ubiquitin–proteasome system (UPS) is fundamental in every key cellular function, including hematopoiesis and response to stress17–19. To identify ubiquitin ligases that potentially control early hematopoiesis, we profiled the expression levels of 602 genes, members of the UPS (consisting of E3s and ubiquitin-accessory proteins), for their expression in mouse hematopoietic stem and early progenitor cells (HSPCs; Supplementary Table 1)20. We found that the Cul3 E3 ubiquitin ligase adapter Spop is one of the highest expressed UPS genes in the long-term HSCs (Lineage-negative (Lin−) Sca-1+Kit+CD135−CD34−; Supplementary Fig. 1a,b), and we raised the possibility that it is important for early stages of blood development. Upon HSC differentiation, SPOP protein abundance was rapidly decreased in myeloid differentiated cells (Supplementary Fig. 1c). To investigate the physiologic role of SPOP in hematopoiesis, we generated a conditionally targeted Spop knockout (KO) allele (SpopF/F) in which exons 4 and 5 were flanked by loxP sites (Supplementary Fig. 1d). Deletion of the floxed exons generates an out-of-frame message leading to a complete loss of protein expression.
Germline deletion of Spop results in embryonic or perinatal lethality21. To conditionally disrupt Spop expression in hematopoietic cells, we first crossed the SpopF/F model to the interferon (IFN)-induced Mx1/Cre-deleted strain (SpopΔMxCre)22. Following a single intraperitoneal polyinosinic:polycytidylic acid (poly(I:C)) injection in 8-week-old mice, we confirmed that SPOP protein was undetectable in bone marrow cells (Supplementary Fig. 1e). We observed rapid weight loss in SpopΔMxCre and complete loss of survival within 60 d after the poly(I:C) injection (Fig. 1a and Supplementary Fig. 1f). Peripheral blood analysis showed high white blood cell counts consistent with a marked expansion of the myeloid CD11b+Gr1+ population and loss of lymphocytes (Fig. 1b–d and Supplementary Fig. 2). Furthermore, the number of red blood cells and hemoglobin concentrations were substantially reduced, and the number of platelets was considerably increased (Fig. 1d). Histopathologic analysis of peripheral blood smears indicated that the expanded myeloid population corresponded to mature neutrophils and left-shifted cells (immature neutrophils), and no blast cells were detected (Fig. 1e). Accordingly, the bone marrow displayed hyper-cellularity and marked granulocytic hyperplasia (Fig. 1e–h). In addition, SpopΔMxCre mice exhibited moderate splenomegaly (data not shown). Immunofluorescence analysis using antibodies to the surface marker Ly6G confirmed increased neutrophil infiltration in the lungs and spleen of mice on day (d)10 after a poly(I:C) treatment that rapidly progressed to a massive lung neutrophilia by d24 (Fig. 1i).
Fig. 1 |. Hematopoietic-specific loss of Spop promotes acute and lethal neutrophilia.
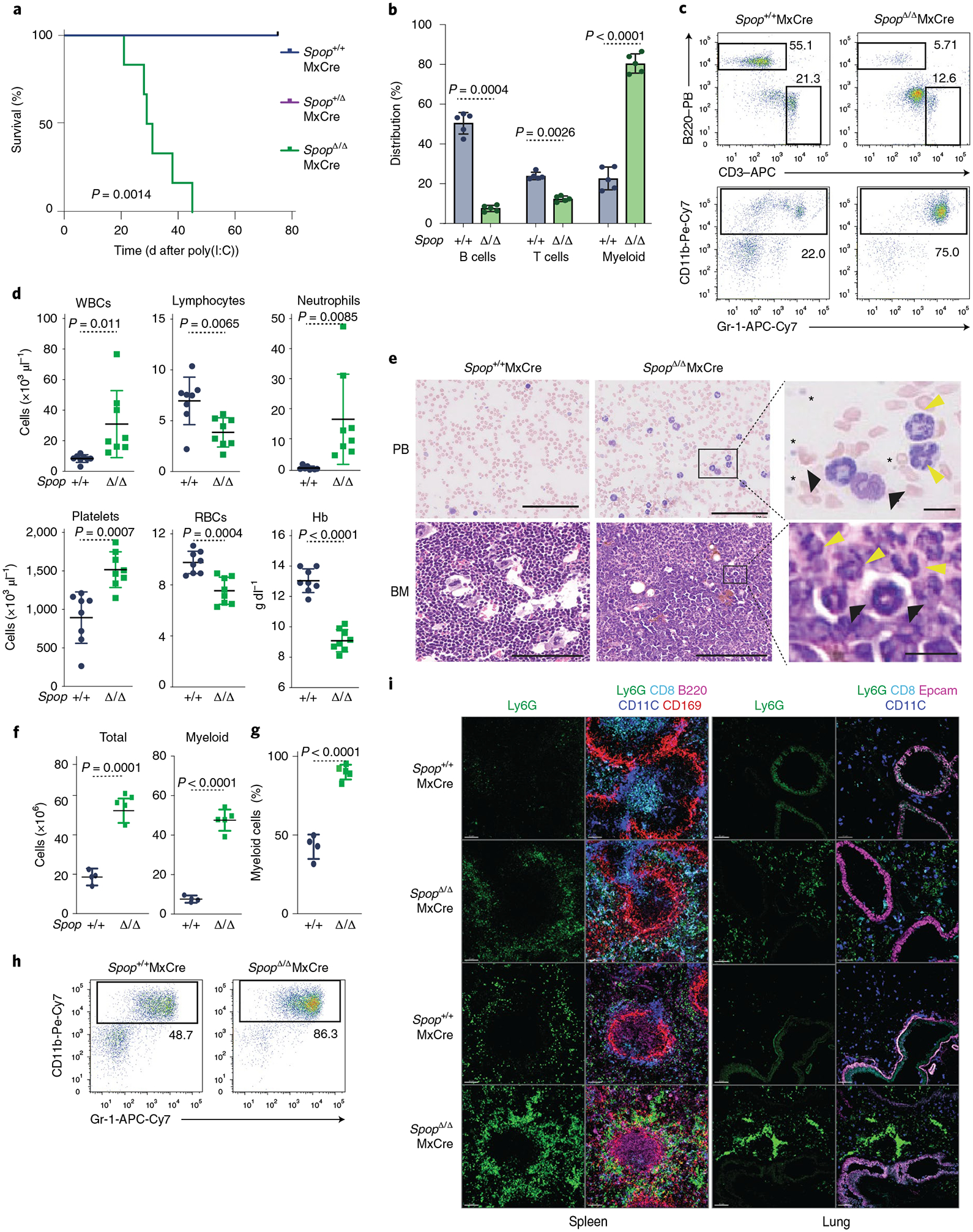
a, Kaplan–Meier analysis of survival of Spop KO and WT control littermates after poly(I:C) injection (n = 6 per genotype; statistical analysis: Mantel–Cox test). b–i, Analysis of Spop KO mice and control littermates on d21 after poly(I:C) treatment. b, Percentage of B (B220+), T (CD3+) and myeloid (CD11b+Gr-1+) cells in peripheral blood (n = 5). c, Representative flow cytometry analysis plots of the proportion of B (B220+), T (CD3+) and myeloid (CD11b+Gr-1+) cells in peripheral blood (n = 5). d, WBC (white blot counts), lymphocytes, neutrophils, platelets, red blot counts and Hb (hemoglobin) levels in peripheral blood (n = 8). e, Peripheral blood smears (left) and hematoxylin and eosin stains of bone marrow sections (right). Yellow arrows indicate mature neutrophils, black arrows indicate immature neutrophils and asterisks indicate platelets. f, Total bone marrow cell number (left) and total myeloid cells (right) collected from one femur and one tibia of the indicated mice (n = 4, Spop+/+; n = 5, SpopΔ/Δ). g, Percentage of myeloid cells (CD11b+Gr-1+) in the bone marrow (n = 4, Spop+/+; n = 5, SpopΔ/Δ). h, Representative flow cytometry analysis plots of the proportion of myeloid (CD11b+Gr-1+) cells in the bone marrow. i, Immunofluorescence showing neutrophil infiltration (Ly6G+) in mouse spleen and lung on d10 and d24 after poly(I:C) treatment. Scale bars, 80 μM. a–i, Data are representative of three independent experiments. Data in b,d and f–h represent mean ± s.d. Statistical analysis: unpaired Student’s t-test (two-tailed) was applied to b,d and f–h. APC, allophycocyanin; BM, bone marrow; PB, Pacific Blue.
Taken together, these data demonstrated that SPOP deficiency in adult animals promotes unrestrained neutrophilia that correlates to rapid lethality. To confirm that the pathology was hematopoietic cell intrinsic, we transplanted lethally irradiated hosts with control (Spop+/+MxCre) or SpopF/FMxCre bone marrow cells. After the donor-derived hematopoiesis was established, a single poly(I:C) injection was sufficient to induce a severe lethal neutrophilia (Supplementary Fig. 1g,h) in the mice reconstituted with SpopΔMxCre bone marrow, confirming that the pathology was exclusively driven by Spop deletion in donor-derived cells.
Spop deletion impairs the resolution of systemic inflammation.
To explain the underlying causes of the observed neutrophilia, we performed RNA sequencing (RNA-seq) on Ly6G+ sorted neutrophils on d10 after poly(I:C) injection, and we identified 929 deregulated genes in SpopΔMxCre neutrophils compared with wild-type (WT) counterparts (P < 0.05). Gene set enrichment analysis (GSEA) revealed the overexpression of inflammatory response gene signatures (Supplementary Fig. 3a) including the IFN response (Oas2, Ifitm1, Ifitm2, Ifitm3) and TLR–MyD88–NF-κB (nuclear factor κB) signaling pathways (Nfkbia, Map3k8, Tnfsf12, Tnfaip6, Cd14, Bcl3; Supplementary Fig. 3b,c). Ingenuity pathway analysis confirmed the activation of similar pathways (Supplementary Fig. 3d,e). These observations prompted us to hypothesize that the neutrophilia in these animals was due to an acute response to an inflammatory stimulus.
Because poly(I:C) is a TLR3 agonist and mimics a viral infection, peripheral blood myeloid cells23 rapidly expand after the insult and restore normal levels by d5 (Fig. 2a). In contrast, SpopΔMxCre mice showed a persistent increased percentage of neutrophils over time (Fig. 2a), suggesting a sustained inflammatory response. To test whether deletion of Spop in the hematopoietic system promotes spontaneous inflammation or requires immune challenge, we generated a conditional SpopF/FCreERT2 strain, in which tamoxifen injection drives the expression of Cre (Supplementary Fig. 4a). Remarkably, SpopΔCreERT2 mice were viable and developed normally for more than 6 months in the absence of any additional immune challenge (Supplementary Fig. 4b), suggesting that Spop deletion does not promote spontaneous inflammation. However, a single dose of poly(I:C) was sufficient to generate neutrophilia and lethality (Fig. 2b–d). Similar to poly(I:C), bacterial lipopolysaccharide (LPS) triggers innate immune responses through TLR4 activation6,24,25. Notably, SpopΔCreERT2 animals were unable to resolve the inflammatory response triggered by a sublethal injection of LPS, and succumbed to neutrophilia and lethality (Fig. 2e,f and Supplementary Fig. 4c). We next sought to determine whether hematopoietic deleion of Spop resulted in increased mortality from infections. After intranasal influenza inoculation, the percentage of peripheral blood myeloid cells in both WT and SpopΔCreERT2 chimeras increased (Fig. 2g and Supplementary Fig. 4d). Even though 40% of the control mice recovered from the infection, Spop-deleted chimeras showed a continuous loss of body weight, neutrophilia and complete mortality (Fig. 2h,i). Altogether, these data underscore a role for SPOP in the resolution of the inflammation and the reestablishment of homeostasis.
Fig. 2 |. Inflammation triggers dysregulated host response and fatal neutrophilia in Spop-deficient animals.
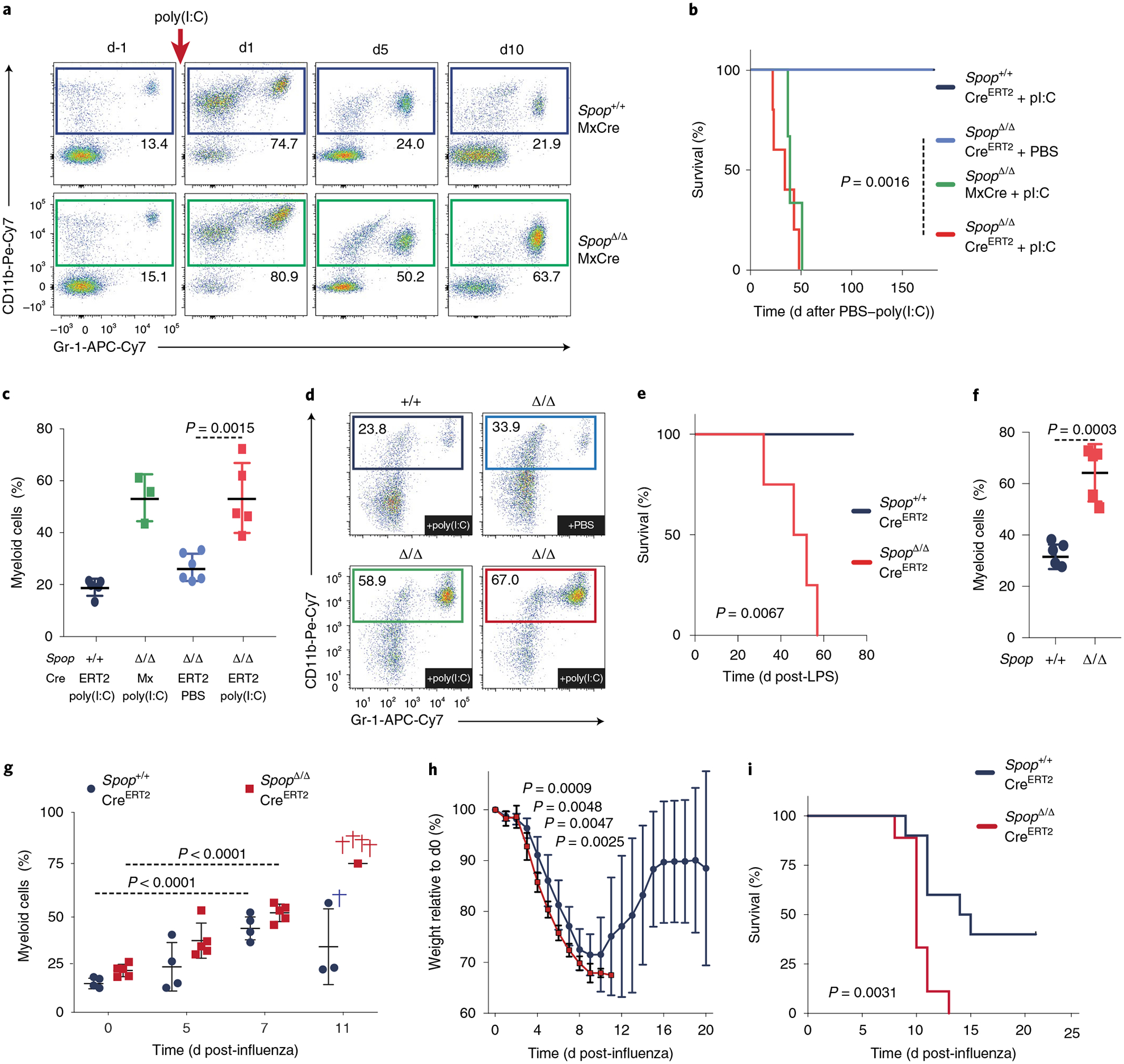
a, Representative flow cytometry analysis plots of the proportion of myeloid (CD11b+Ly6G+) cells in the peripheral blood of Spop KO and WT mice at the indicated times after poly(I:C) injection. b, Kaplan–Meier analysis of survival of the indicated mice after a single poly(I:C) injection (n = 5 per genotype except for SpopΔMxCre, which is n = 3; statistical analysis: Mantel–Cox test). c, Percentage of myeloid (CD11b+Gr-1+) cells in the peripheral blood of Spop KO and control hematopoietic chimeras on d15 after poly(I:C) or PBS injection (n = 5 except for SpopΔ/ΔMxCre, which is n = 3). d, Representative flow cytometry analysis plots of the proportion of myeloid (CD11b+Ly6G+) cells in the peripheral blood of the indicated hematopoietic chimeras on d15 after poly(I:C) or PBS injection. e, Kaplan–Meier analysis of survival of the indicated hematopoietic chimeras after a sublethal LPS injection (n = 4; statistical analysis: Mantel–Cox test). f, Percentage of myeloid (CD11b+Ly6G+) cells in the peripheral blood of the indicated mice on d15 after a sublethal LPS injection (n = 4). g, Percentage of myeloid (CD11b+Gr-1+) cells in peripheral blood of Spop KO CreERT2 (n = 5) and control (n = 4) hematopoietic chimeras at the indicated days after intranasal influenza inoculation. h, Percentage of weight loss of Spop KO CreERT2 (n = 9) and control (n = 10) hematopoietic chimeras after intranasal influenza inoculation. i, Kaplan–Meier analysis of survival of the indicated mice after intranasal influenza inoculation (n = 10). a–i, Data are representative of three independent experiments. Data for c and f–h represent mean ± s.d. Statistical analysis for c and f–h: unpaired Student’s t-test (two-tailed).
We next tested whether the deletion of Spop in neutrophils themselves abrogates the resolution of inflammation. To this purpose, we crossed SpopF/F mice with the granulocytic lineage-specific MRP8–Cre–ires–GFP transgenic mouse, in which Cre recombinase activity is restricted to a fraction of granulocyte monocyte progenitors (GMPs; 10–20%) and virtually all granulocytes (Ly6G+)26. Accordingly, we confirmed normal Spop mRNA abundance in HSPCs and a substantial reduction in granulocytes (Supplementary Fig. 4e). Remarkably, even in the context of inflammation, Spop deletion in the neutrophils was not sufficient to drive pathology (Supplementary Fig. 4f). These results demonstrate that loss of SPOP in granulocytic progenitors or differentiated granulocytes does not impair the resolution of the inflammatory insult, suggesting that SPOP is required for controlling the response within the HSCs and HSPCs.
SPOP deficiency leads to a sustained inflammatory response of HSPCs.
As a result of an inflammatory response, the HSPCs transiently increase in number, and the myeloid-biased and megakaryocyte-biased multipotent progenitors (MPPs) expand substantially4,8,23. We hypothesized that SPOP deficiency results in a defective resolution of the inflammatory response of the HSPCs. In line with our hypothesis, we observed that 10 d after poly(I:C) treatment the control mice recover normal percentages of HSPCs (Lin−Sca-1−Kit+ (LSKs)). In contrast, LSKs remained elevated in SpopΔMxCre mice (Supplementary Fig. 5a). The absolute LSK numbers were notably increased in SpopΔMxCre mice on d21 (Fig. 3a). Within the HSPC population, the megakaryocyte/erythroid-biased (MPP2, LSK–CD135−CD150+CD48+) and myeloid-biased (MPP3, LSK–CD135−CD150−CD48+) MPPs were considerably expanded (Fig. 3b). Conversely, the numbers of HSCs (LSK–CD135−CD150+CD48−) and the lymphoid-biased MPP cells (MPP4: LSK–CD135+) were notably decreased in percentage (Supplementary Fig. 5b,c) and total numbers (Fig. 3b). In addition, we observed a marked expansion of the GMP population (Lin−Sca-1−c-Kit+FcγR+CD34+), which includes the progenitors of monocytes and granulocytes (Fig. 3c and Supplementary Fig. 4b,c). Furthermore, the numbers of the common myeloid progenitors (CMPs; Lin−Sca-1−c-Kit+FcγR−CD34+) and the megakaryocyte and erythrocyte progenitors (MEPs; Lin−Sca-1−c-Kit+FcγR−CD34+) were reduced (Fig. 3c and Supplementary Fig. 4b,c). Annexin V staining showed no differences between WT and SpopΔMxCre (data not shown), suggesting that the loss of HSC, lymphoid and erythroid populations was a consequence of enhanced granulocytic differentiation rather than cell death. Likewise, transcriptomic analysis of HSPCs on d10 after poly(I:C) injection revealed a substantial number of genes (3,007) differentially expressed in SpopΔMxCre LSK cells compared with WT cells (Padj < 0.05; Supplementary Table 2). GSEA analysis revealed a considerable loss of HSC (Meis1, Hlf, Mecom), megakaryocyte–erythrocyte (Gata1, Gata2, Pf4, Mpl) and lymphoid (Ikzf1, Ilr7, Cd79a, Dntt, Rag1, Rag2) gene signatures (Supplementary Fig. 5d,e). In contrast, the MPP3 gene signature and myeloid transcription factors (Cd48, Mpo, Csf1, Gfi, Cebpa, Cebpe, Spi1) were upregulated (Supplementary Fig. 5d,e). Importantly, SpopΔMxCre HSPCs notably upregulated Cebpb encoding C/EBPβ, which is a major transcriptional regulator of emergency granulopoiesis27. These data suggest that SPOP-deficient stem and progenitor cells maintain an emergency granulocytic lineage priming, which is activated during the inflammatory response.
Fig. 3 |. Loss of Spop leads to an emergency hematopoietic transcriptional program in HSPCs.
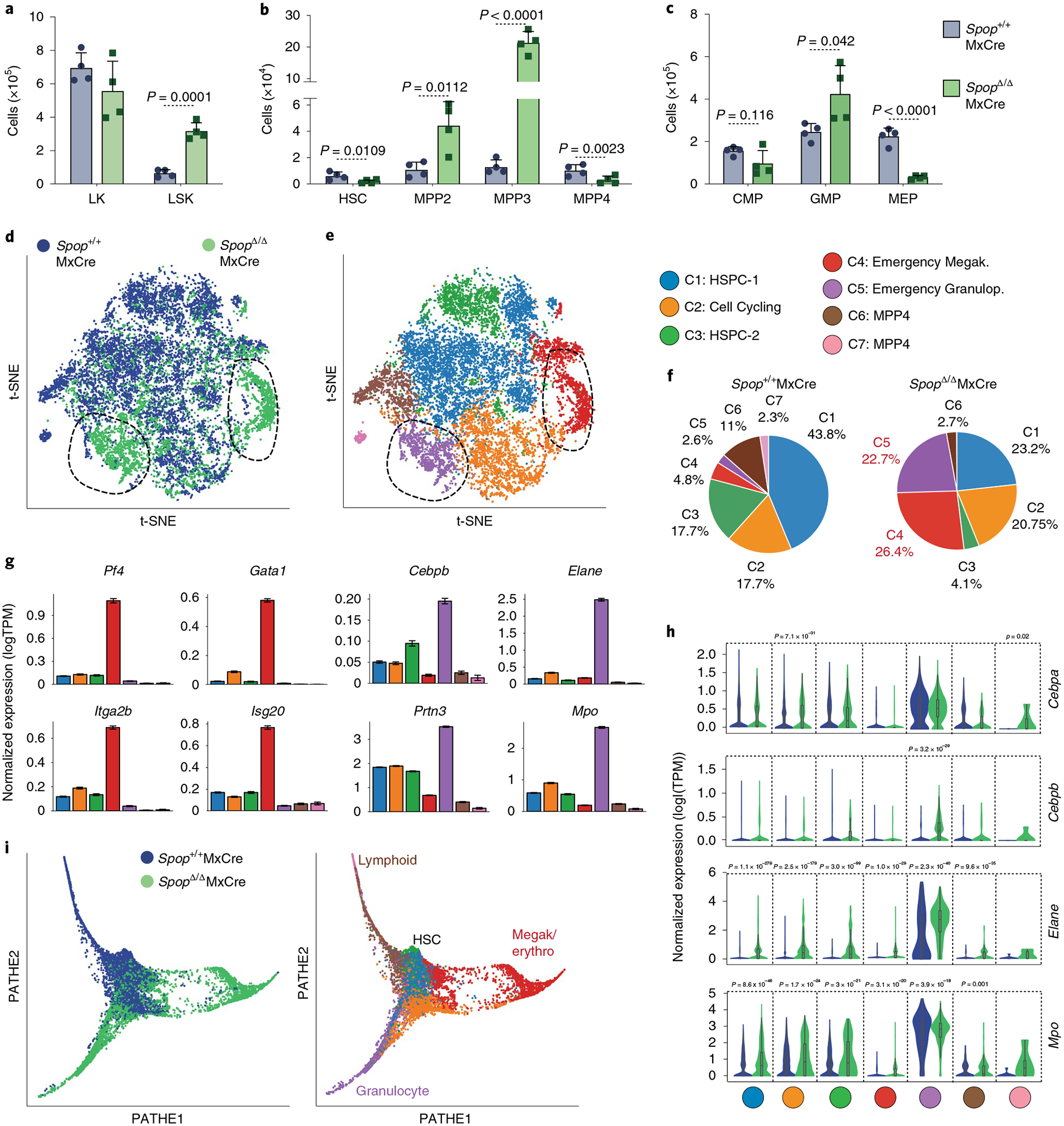
a–c, Total bone marrow cells of the indicated HSPC populations collected from one femur and one tibiae on d21 after poly(I:C) injection: LK (Lin−Sca-1−Kit+), LSK (Lin−Sca-1−Kit+) (a), HSC (LSK–CD135− CD48−CD150+), MPP2 (LSK–CD135−CD48+CD150+), MPP3 (LSK–CD135+CD48+CD150−), MPP4 (LSK–CD135+CD48+CD150−) (b), CMP (LK–CD34+ FcyRlo), GMP (LK–CD34+FcyR+) and MEP (LK–CD34−FcyR−) (c) (n = 4 mice per genotype). Data represent mean ± s.d. Dots represent different mice. Statistical analysis: unpaired Student’s t-test (two-tailed). Data are representative of three independent experiments. d, Spectral t-SNE plot of scRNA-seq data collected from Spop WT (Spop+/+MxCre) and KO (SpopΔ/ΔMxCre) LSK cells analyzed on d10 after poly(I:C) injection (n = 2 mice per group). Dotted line indicates Spop-deleted distinct transcriptional subset. e, Spectral t-SNE plot of color-coded clustering of Spop WT and KO LSK cells based on expression profile. f, Percentage of Spop WT and KO LSK cells per cluster. g, Expression levels of the selected population-specific marker across the clusters (data represent mean ± s.d.). h, Violin plots showing the expression pattern distribution of the indicated genes per cluster and genotype. Violin plots are colored by cell genotype, and width represents the percentage of cells expressing the marker at a given level. Minimum, maximum, median, and 25th and 75th percentiles are shown. Significance was determined using the Wilcoxon rank sum test with Bonferroni multiple comparison correction (C1WT = 3,571, C1KO = 1,143, C2WT = 1,440, C2KO = 1,021, C3WT = 1,146, C3KO = 203, C4WT = 390, C4KO = 1,299, C5WT = 213, C5KO = 1,115, C6WT = 896, C6KO = 134, C7WT = 191, C1KO = 5 cells). i, Cell differentiation trajectory using PHATE visualization of WT and Spop KO cells, genotype coded (left) and cluster coded (right). d–i, Spop+/+MxCre: n = 8,184 cells; SpopΔ/ΔMxCre: n s= 4,920 cells. C1 = 4,714, C2 = 2,461, C3 = 1,649, C4 = 1,689, C5 = 1,328, C6 = 1,030 and C7 = 196 cells. PHATE, potential of heat diffusion for affinity-based trajectory embedding; TPM, transcripts per million.
We then measured the concentrations of an extended panel of proinflammatory cytokines (including IFN-α, IFN-β, IFN-γ, interleukin (IL)-1α, IL-1β, tumor necrosis factor α (TNF-α), IL-6 and granulocyte–macrophage colony-stimulating factor (GM-CSF)) after poly(I:C) challenge. As previously reported8,23, IFN-α rapidly increased in the serum and bone marrow fluids of the stimulated mice (Supplementary Fig. 5f,g), with a somewhat lower increase in the SPOP-deficient mice. In addition, TNF-α concentrations increased in the serum, and IL-1α and IL-1β in the bone marrow at 16 h after the immune challenge. Identical recovery kinetics were observed in both Spop-deleted and control animals (data not shown and Supplementary Fig. 5f,g), suggesting that inflammatory signals are required to initiate the emergency hematopoiesis in Spop-deleted mice, but they are not required to sustain it. These combined studies demonstrate that SPOP is required for the resolution of the inflammatory response of the bone marrow HSPCs.
Single-cell transcriptomic profiling of emergency hematopoietic programs.
Recent studies based on single-cell RNA-seq (scRNA-seq) have redefined a spectrum of transcriptional states that gradually progress into the different hematopoietic lineages28,29,30. Inflammatory signals mostly likely impact on the HSPCs by reshaping their differentiation trajectories. Therefore, to characterize the landscape of the inflammatory transcriptional states sustained in Spop-deleted mice, we conducted scRNA-seq of LSKs of WT (Spop+/+MxCre) and SpopΔMxCre mice at d10 after poly(I:C) administration. After implementing extensive quality-control measures (see Methods), we analyzed 8,147 WT and 4,920 SpopΔMxCre cells using Seurat, an R package for scRNA-seq data31. We visualized all of the WT and SpopΔ transcriptomes together using t-distributed stochastic neighbor embedding (t-SNE; Fig. 3d). An unsupervised graph clustering partitioned the total HSPC population into seven clusters (based on the specific differential expression analysis for every gene in each cluster, relative to the other clusters; Fig. 3e, Supplementary Fig. 6a,b and Supplementary Tables 3 and 4) and confirmed the accumulation of Spop-deleted cells into two main subsets distinct from their WT counterparts (C4 and C5; Fig. 3e,f). The analysis of the specific gene expression patterns related to each cluster allowed us to determine that cells in cluster 4 expressed a gene signature previously described for an emergency megakaryocytic stem-like population32,33, which included hematopoietic markers such as Itga2b (CD41), Pf4 and Gata1, together with IFN response genes such as Isg15 and Isg20 (Fig. 3g and Supplementary Fig. 6c). Accordingly, Spop-deleted marrows showed a substantial expansion of megakaryocyte progenitors (Lin−kit+Sca-1−CD41+CD150+; Supplementary Fig. 6d) at d10 after poly(I:C), which, consistent with previous studies, decreased after 25 d because of their exhaustion33. In addition, cluster 5 included an emergency granulopoietic population highly expressing Cebpb and myeloid-differentiated genes such as Elane, Prtn3 and Ctsg (Fig. 3g and Supplementary Fig. 6e). In contrast, the WT clusters (C1, HSPC-1 and C2, HSPC-2) were notably underrepresented in SPOP-deficient cells, and the lymphoid-primed MPP clusters were almost absent (C6 and C7; Fig. 3f). Furthermore, the emergency granulopoiesis gene signature was also overrepresented in the cells from other clusters (Fig. 3h and Supplementary Fig. 6a), suggesting that this transcriptional state was generalized in the Spop-deleted stem and progenitor cells.
We next applied an enrichment score algorithm to associate each single transcriptome to known signatures for HSC, megakaryocyte/erythrocyte-, myeloid- and lymphoid-biased MPPs34 coupled to a graphical reconstitution of the cell differentiation trajectories (Supplementary Fig. 6f). We observed a transcriptional continuum connecting HSCs and MPPs, which diffused along the different branches for differentiation in the control sample. However, the SpopΔMxCre HSPCs accumulate into two different areas restricted to more differentiated megakaryocyte and granulocyte populations (Fig. 3i and Supplementary Fig. 6f). Cell-cycle gene signature analysis showed that SpopΔMxCre HSPCs contained a higher percentage of proliferative cells than in WT animals (Supplementary Fig. 6g,h), which we verified by in vivo 5-ethynyl-2′-deoxyuridine (EdU) labeling (Supplementary Fig. 6i). Collectively, these findings indicate that Spop deletion in HSPCs promotes an inflammation-induced transcriptional program resulting in the expansion of progenitors committed to the granulocytic and megakaryocytic lineages.
Spop loss leads to MyD88 stabilization.
Given that SPOP is the substrate-recognizing moiety for an E3 ubiquitin ligase, we postulated that the reported phenotypes are caused by the change of ubiquitylation of one of its substrates. Based on SPOP protein structural details previously characterized35, we generated a dimerization mutant, SPOP(L193P), which lacks catalytic activity and is notably stabilized when expressed in myeloid-committed hematopoietic cells (the K562 myeloid cell line; Supplementary Fig. 7a,b). Analysis of the protein interactome of the SPOP(L193P) mutant identified known interactors (CULLIN3, TRIM24 and CAPRIN1), which served as experimental controls (Fig. 4a and Supplementary Table 5). Importantly, these experiments also identified the MYD88 as an interactor (Fig. 4a). MYD88 is an adapter protein essential for IL-1 receptor (IL-1R) signaling and for all TLR signaling except TLR336. Upon receptor triggering, MYD88 transduces signals to the IL-1R-associated kinase (IRAK) kinases, culminating in the activation of downstream pathways, such as the transcription factor NF-κB and subsequent induction of a number of inflammatory response genes37. Therefore, we sought to validate that MYD88 is a bona fide SPOP substrate. We first tested our proteomic approach using reciprocal co-immunoprecipitation (Fig. 4b,c). By combining deletion mapping with site-directed mutagenesis, we identified the minimal SPOP binding consensus site on the MYD88 protein, which, consistent with previous studies35, corresponded to the conserved 135DSS137 motif (Fig. 4d and Supplementary Fig. 7c). Notably, MyD88D135A/S136A and MyD88S136A/S137A mutants displayed a substantially extended half-life when compared with MYD88WT (Fig. 4e), revealing the SPOP degron as the major regulator of MYD88 half-life. Phosphatase treatment of immunoprecipitated MYD88 did not affect the interaction with SPOP (Supplementary Fig. 7d), and phospho-mimicking mutations of S136 and S137 resulted in ablation of SPOP interaction (Supplementary Fig. 6e), suggesting that MYD88 interaction with SPOP does not require previous phosphorylation.
Fig. 4 |. SPOP directly interacts, ubiquitylates and degrades MYD88.
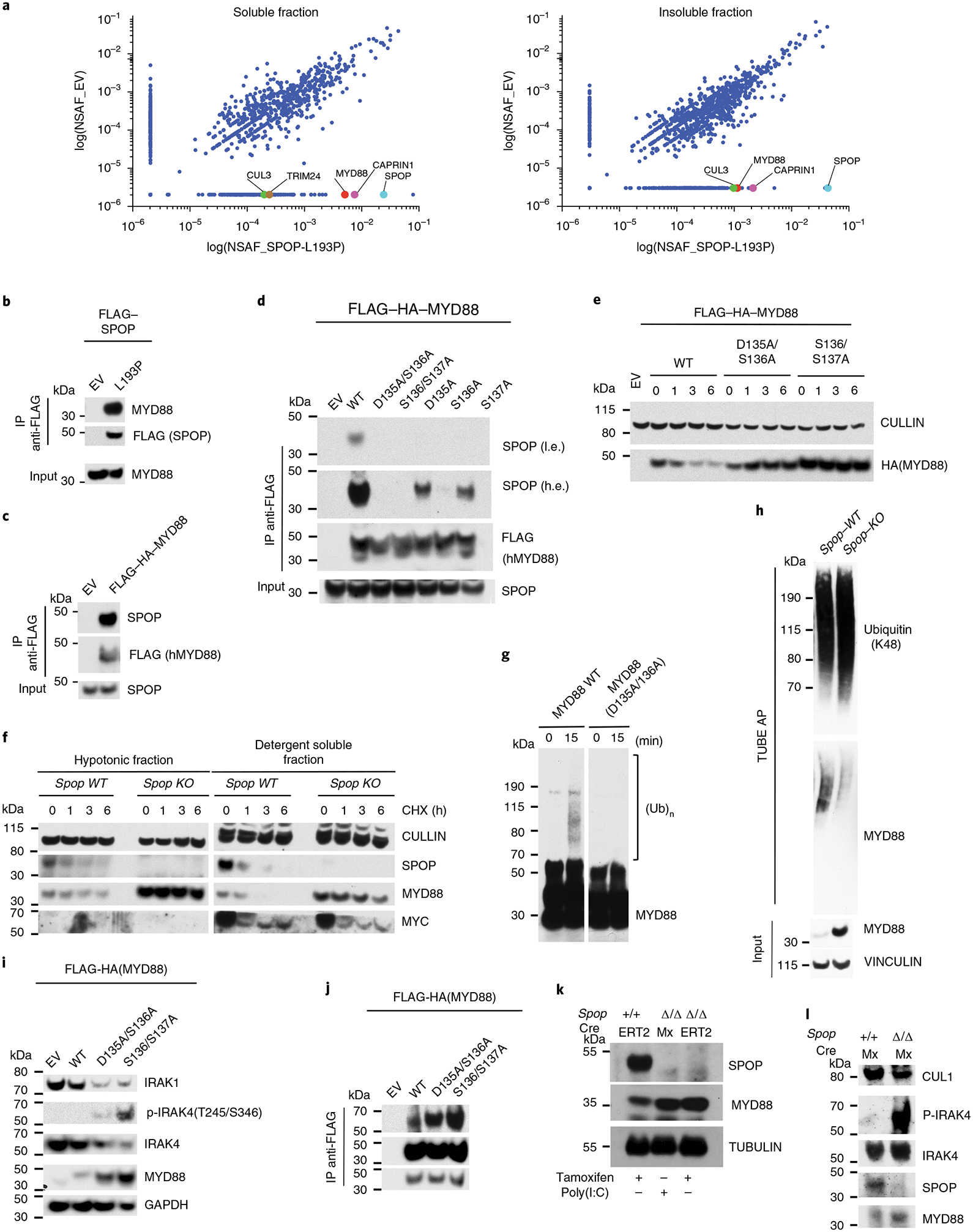
a, Proteomic analysis of FLAG immunoprecipitates from K652 cells stably expressing FLAG–SPOP(L193P). Both NP-40 soluble and insoluble fractions were analyzed. Scatterplot of NSAF values for the EV and SPOP(L193P) conditions are shown. NSAF values are shown for CULLIN3, TRIM24, MYD88, CAPRIN1 and for SPOP itself. For proteins with NSAF = 0, the lowest NSAF value was arbitrarily assigned. b,c, Immunoblot analysis for the indicated proteins of FLAG immunoprecipitates from K562 stably transduced. d, Immunoblot analysis of immunoprecipitated FLAG-tagged WT and mutant (D135A/S136A, S136A/S137A, D135A, S136A, S137A) MYD88 transiently expressed in HEK293T cells. A low and high exposure are shown. e, Immunoblot analysis of whole-cell lysates from K562 cells stably expressing WT or mutant (D135A/S136A, S136A/S137A) HA-tagged MYD88. Cells were treated with CHX for the indicated times. f, Immunoblot analysis of the hypotonic and detergent-soluble fractions of HPC-7 cells Spop WT and Spop KO. Cells were treated with CHX for the indicated times. g, In vitro ubiquitylation reaction of in vitro-translated FLAG-MYD88(WT) and MYD88(D135A/S136A) incubated with purified SPOP-CULLIN3 complex from sf9 cells. h, Lysates from HPC-7 Spop WT and Spop KO were incubated with agarose-coupled TUBE. The affinity precipitates were subjected to immunoblot with the indicated antibodies. i, Immunoblot analysis of whole-cell lysates from K562 cells stably expressing WT or MYD88 mutants (D135A/S136A, S136A/S137A). j, Immunoblot analysis of immunoprecipitated FLAG-tagged MYD88 WT and mutants (D135A/S136A, S136A/S137A) stably expressed in K562 cells. A low and high exposure are shown. k, Immunoblot analysis of HSPCs (c-Kit+ bone marrow cells) of Spop WT, SpopΔ/ΔMxCre and SpopΔ/Δ CreERT mice. l, Immunoblot analysis of whole-cell lysates of HSPCs (Kit+ bone marrow cells) from Spop+/+MxCre and SpopΔ/ΔMxCre mice 20 d after poly(I:C) injection. a–l, Panels show cropped immunoblots. Data are representative of three independent experiments. h.e., high exposure; l.e., low exposure.
Furthermore, endogenous MyD88 protein was stabilized in a mouse HSPC cell line (HPC-7), where Spop was ablated by CRISPR/Cas9-mediated gene editing (Fig. 4f). Next, we incubated the recombinant purified CULLIN3–RBX1–SPOP complex with in vitro-translated MYD88 (Fig. 4g). We confirmed that MYD88 WT was ubiquitylated by the SPOP complex, but the MYD88D135A/S136A mutant was not (Fig. 4g). In addition, the tandem-repeated ubi quitin-binding entity (TUBE) ubiquitination assay demon strated that MyD88 ubiquitination was decreased in Spop-deleted cells (Fig. 4h).
Oligomerization of MYD88 with IRAK kinases complexes and subsequent IRAK4 phosphorylation are essential for the final activation of transcription factors such as NF-κB and AP-1 (ref.38). To understand the downstream mechanism through which MYD88 stabilization drives neutrophilia, we overexpressed increasing amounts of IRAK4 in the ubiquitylation assays and corroborated that MYD88 ubiquitylation was stimulated by co-expression of IRAK4 (Supplementary Fig. 7f,g). Furthermore, stable expression of nondegradable mutants (MYD88D135A/S136A and MYD88S136A/S137A) in K562 cells was sufficient to induce phosphorylation of the MYD88-associated kinase IRAK4 (Fig. 4i). Accordingly, an enhanced association of endogenous phospho-IRAK4 with nondegradable MYD88D135A/S136A and MYD88S136A/S137A mutants was detected (Fig. 4j). Similarly, Irak4 was rapidly phosphorylated in response to LPS in HPC-7 SpopΔ/Δ when compared with Spop+/+ cells (Supplementary Fig. 7h). In line with these findings, we observed MyD88 endogenous stabilization and Irak4 hyper-phosphorylation in Lin−kit+ HSPC bone marrow cells collected from SpopΔMxCre animals (Fig. 4k,l). Collectively, these results suggest that MYD88 protein stability is regulated by SPOP in a ubiquitin-dependent manner. Impaired degradation of MYD88 results in hyper-phosphorylation of the IRAK4 kinase.
Recent studies have shown that the response to inflammation reshapes the chromatin landscape of the HSPCs, promoting their rapid myeloid skewing and an epigenetic reprograming, which promotes a faster immune response of their progeny39,40. In agreement, assay for transposase-accessible chromatin sequencing (ATAC-seq) analysis showed that SpopΔMxCre HSPCs displayed 25,570 gained open chromatin regions relative to WT counterparts (Supplementary Fig. 8a and Supplementary Table 6), which were enriched with genes related to immune response pathways (Ifitm3, Nfkbia, Isg15, Cebpb; Supplementary Fig. 8b,c). In addition, we found increased accessibility at the transcriptional start sites of myeloid transcription factors (Spi1, Cebpa, Cebpe) and granulocytic genes (Elane, Prtn3), and a decreased accessibility of some lymphoid genes such as Rag1 (Supplementary Fig. 8c). Furthermore, transcription factor motif analysis of the regulatory regions gained in SpopΔ HSPCs revealed a notable enrichment for myeloid transcription factors (Pu.1, Cebpa, Cebpe, Cebpg and Cebpb) and the MyD88-IRAK4 downstream transcription factors: NF-κB and AP-1(Jun:Fos), which are important for HSPC activation and myeloid priming upon TLR-induced inflammation10 (Supplementary Fig. 8d). DNA footprinting analysis validated the occupancy of Pu.1, C/EBPβ, AP-1 and NF-κB transcription factors in the regions that gained accessibility in the absence of SPOP (Supplementary Fig. 8e). Taken together, our results demonstrate that an MYd88-dependent inflammatory response is preserved in Spop-deleted HSPCs.
Myd88 deletion protects from lethal neutrophilia.
To functionally assess whether accumulation of MyD88 contributes to the neutrophilic phenotype of SPOP-deficient animals, we crossed SpopF/F MxCre mice to MyD88-deficient animals. Sixteen hours postinjection, the LSK compartments of Myd88−/− and SpopΔMxCre Myd88−/− hematopoietic chimeras were comparably expanded to WT and SpopΔMxCre (Supplementary Fig. 9a). We collected Lin−c-Kit+ progenitor cells 1 week postinjection and confirmed that Spop protein expression was identically disrupted in all animal strains (Supplementary Fig. 9b,c). As expected, SpopΔMxCre chimeras developed rapid neutrophilia and lethality. In contrast, SpopΔMxCre Myd88−/− animals recovered from the inflammatory insult and did not develop any neutrophilia (Fig. 5a–c). Furthermore, heterozyzgous deletion of Myd88 was sufficient to rescue or considerably delay neutrophilia in Spop-deleted mice (Fig. 5a–c), suggesting that decreasing MyD88 protein levels can reverse the immunopathologies occurring in SPOP-deficient animals.
Fig. 5 |. Loss of MyD88 protein rescues Spop-induced lethal neutrophilia.
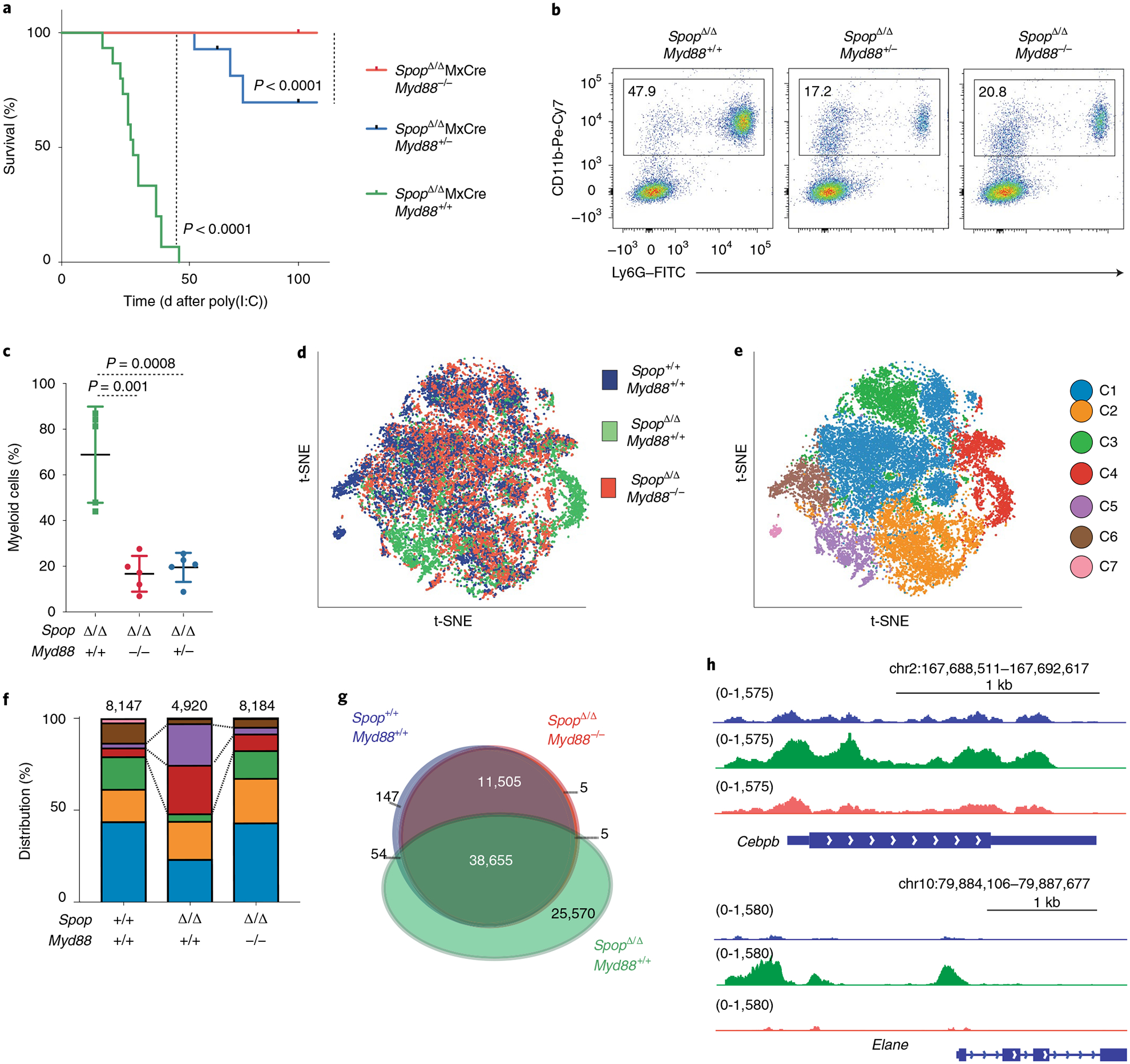
a, Kaplan–Meier analysis of survival of Spop KO (SpopΔ/ΔMxCre, n = 15 mice), Spop–Myd88 dKO (SpopΔ/ΔMxCre MyD88−/−, n = 7 mice) and SpopΔ/ΔMxCre MyD88+/− (n = 14 mice) hematopoietic chimeric mice after a single poly(I:C) injection. Statistical analysis: Mantel–Cox test. b, Representative flow cytometry analysis plots of the proportion of myeloid (CD11b+Ly6G+) cells in the peripheral blood of the indicated hematopoietic chimeric mice on d14 after poly(I:C) injection. Data are representative of three independent experiments. c, Percentage of myeloid (CD11b+Ly6G+) cells in the peripheral blood of the indicated hematopoietic chimeric mice after poly(I:C) injection (n = 5 mice per genotype). Data represent mean ± s.d. Dots represent different mice. Statistical analysis: unpaired Student’s t-test (two-tailed). Data are representative of three independent experiments. d, Spectral t-SNE plot of scRNA-seq data collected from Spop WT (n = 8,184), Spop KO (n = 4,920) and dKO (n = 8,184) LSK cells analyzed on d10 after poly(I:C) injection (cells were sorted from two mice per group). e, Spectral t-SNE plot of color-coded clustering of Spop WT (n = 8,184), Spop KO (n = 4,920) and dKO (n = 8,184) LSK cells based on expression profile. f, Distribution of WT, Spop KO and dKO LSK cells per cluster. g, Venn diagram showing the ATAC-seq signals common and unique open regions for the indicated LSKs (Lin−c-Kit+Sca-1+ bone marrow cells) collected at d10 after poly(I:C). h, Genome Browser plots showing the normalized ATAC-seq profiles at the promoter and distal elements of the indicated genes for WT (Spop+/+MxCre, blue), KO (SpopΔ/ΔMxCre, green) and dKO (SpopΔ/ΔMxCre MyD88−/−, orange) LSKs.
To study the molecular mechanism of this phenotypic rescue, we performed single-cell RNA-seq on LSKs purified from Spop+/+MxCre (control), SpopΔMxCre and SpopΔMxCre Myd88−/− (double-knockout (dKO)) littermates 10 d after a single poly(I:C) injection. Notably, the dKO progenitors were distributed, pre-dominantly through the HSPC-1, HSPC-2 and proliferative clusters (C1, C2 and C3), in an almost identical fashion as their WT counterparts, supporting the reported phenotypic rescue (Fig. 5d–f, Supplementary Fig. 9d and Supplementary Table 7). Likewise, dKO HSPCs showed similar proliferation rates than controls (Supplementary Fig. 9e). Moreover, ATAC sequencing confirmed a similar distribution of open chromatin regions between Spop WT and dKO cells (Fig. 5g,h). Collectively, these results demonstrate that MyD88 ablation is sufficient to rescue the SpopΔMxCre emergency hematopoietic transcriptional program, restoring the differentiation through all of the hematopoietic lineages.
Targeting of IL-1R:MYD88 signaling protects from lethal neutrophilia.
Poly(I:C) directly activates the only TLR that is independent of MYD88 signaling. However, it induces a transient increase in the amounts of IL-1α and IL-1β in the bone marrow. Furthermore, previous studies have shown that in vivo IL-1β stimulation functions as an emergency signal that directly instructs HSCs through activation of the AP-1 and NF-κB pathways and through the engagement of a PU.1-dependent myeloid gene program10. Therefore, we hypothesized that IL-1 signaling triggered neutrophilia in poly(I:C)-stimulated Spop-deleted mice. To test this hypothesis, we injected SpopΔMxCre mice with IL-1R blocking antibodies41. Remarkably, IL-1R blocking entirely halted the neutrophilia in SPOP-deficient mice (Fig. 6a–c), suggesting that IL-1R signaling is essential for the development of the poly(I:C)-dependent phenotypes reported in the SpopΔMxCre animals. In contrast, blocking of TNF, which does not signal through MYD88, had no impact in Spop-deleted driven neutrophilia (Supplementary Fig. 9f,g). Taken together, our results support a model in which IL-1R signaling initiates an emergency hematopoietic program in HSPCs and, subsequently, SPOP-mediated regulation of its downstream effector MYD88 is necessary for the resolution of this response.
Fig. 6 |. IL-1-Myd88 signaling promotes neutrophilia in Spop-deficient animals.
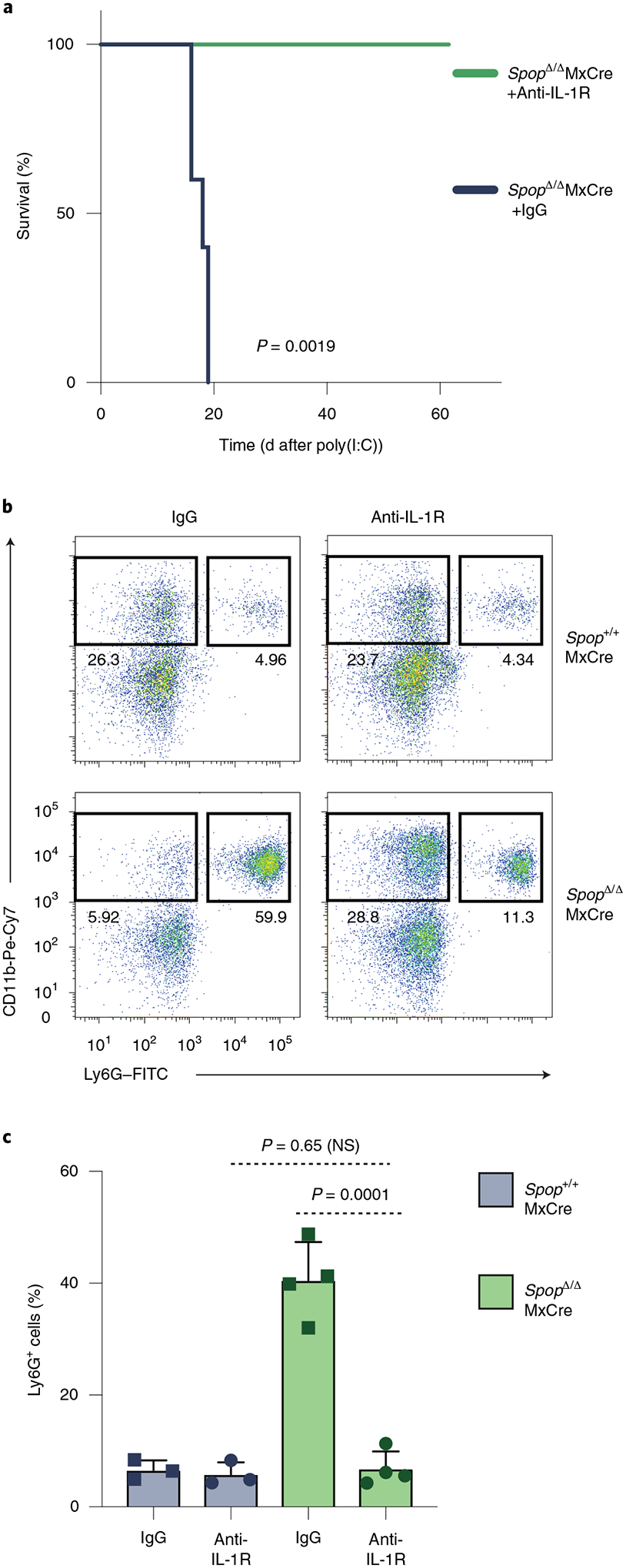
Spop KO mice were treated with anti-IL-1R or IgG for 7 consecutive days starting at day −1 of poly(I:C) challenge. a, Kaplan–Meier analysis of survival of Spop KO treated with IgG or anti-IL-1R (n = 5 per condition; statistical analysis: Mantel–Cox test). b, Representative flow cytometry analysis plots of the granulocyte (CD11b+Ly6G+) distribution in the peripheral blood of Spop KO and control mice treated with IgG or anti-IL-1R on d14 after poly(I:C) injection. c, Percentage of granulocyte (CD11b+Ly6G+) cells in the peripheral blood of Spop KO and control mice treated with IgG or anti-IL-1R at d10 after a poly(I:C) injection. Data represent mean ± s.d. Spop+/+: n = 3 mice per condition; SpopΔ/Δ: n = 4 mice per condition. Statistical analysis: unpaired t-test (two-tailed). a–c, Data are representative of two independent experiments. NS, not significant.
Discussion
Genetic, epigenetic and environmental factors are determinants on the heterogeneity of the host response to infections42,43. Until now, most studies have extensively evaluated the mechanism of inflammation resolution initiated at local sites and how a dysregulated inflammatory response of the immune cells leads to pathologies such as sepsis, hyper-inflammation and autoimmune diseases. In contrast, the means by which HSPCs switch off inflammatory response is less clear. The importance of this event is major, because failure of inflammation resolution could lead to stem cell exhaustion and failure of blood cell production. Also, it is possible that uncontrolled inflammatory responses within populations that are characterized by self-renewal properties could lead to transformation and induction of hematologic neoplasms. This notion has been reinforced by the finding that a substantial portion of elderly individuals are characterized by clonal hematopoiesis44. Because the presence of clonal hematopoiesis mutations has been associated with an increased propensity of progression to leukemia, it is conceivable that dysregulated inflammatory responses could cooperate with acquired mutations to lead to transformation. Moreover, a number of recent reports describe interactions between a (normal and inflamed) bone marrow microenvironment and leukemia cells45,46.
Our results identify a role for SPOP in restraining inflammation-induced emergency hematopoiesis and promoting inflammation resolution in the bone marrow. Mechanistically, we identify MYD88 as an SPOP substrate. Interestingly, Myd88 was first described as a gene upregulated during IL-6-induced myeloid differentiation47. Since then, it has emerged as a central and highly conserved node in inflammatory responses by transducing signals from IL-1Rs and TLRs in complexes that also include the IRAK kinases33,36. Our results demonstrate that SPOP-mediated MYD88 ubiquitylation restrains emergency hematopoiesis by directly controlling the HSPC differentiation fate. We propose that MyD88 stabilization, caused by Spop deletion, synergizes with proinflammatory signals (poly(I:C), LPS, flu virus) to trigger a program of emergency hematopoiesis. This program is characterized by the activation of a number of transcription factors that control myeloid differentiation (PU.1, C/EBPβ, C/EBPα) and inflammatory response (NF-κB and AP-1). It will be intriguing to see whether MYD88 protein stability is controlled by SPOP also in terminally differentiated innate immune subsets that are important for the resolution of inflammation outside of the marrow.
Our studies suggest that SPOP is a key regulator of emergency hematopoiesis and inflammation sensing in the bone marrow. Interestingly, a number of E3 ubiquitin ligases and deubiquitinases control a complex network that fine-tunes the inflammatory response, including the LUBAC (linear ubiquitin chain assembly complex) complex, TRAF6, A20 and CYLD48. More recently, the deubiquitinase OTULIN was shown to interact with the LUBAC, controlling its auto-ubiquitination and TNF-mediated inflammatory response49. Importantly, SPOP functions upstream of these signaling pathways, specifically targeting MyD88 through K48-mediated ubiquitination switching off the inflammatory response.
Finally, we were able to show that impaired degradation of MYD88, via SPOP deficiency or expression of MyD88 mutants incapable of binding to SPOP, induces constitutive IRAK4 phosphorylation or stabilization of phospho-IRAK4. Oligomerization of MYD88 and IRAK kinases in a complex termed Myddosome38 constitutes a key event during TLR activation. Given the fact that IRAK4 facilitates SPOP-mediated MYD88 ubiquitination, we speculate that SPOP terminates a MYD88 protein pool at the level of the Myddosome. Recent studies have highlighted the benefits of pro-resolving agents as a therapy for uncontrolled inflammation13. Although future experimentation is required, it is tempting to hypothesize that likewise some immunopathologies can be controlled using small molecules that specifically modify SPOP or IRAK4 activity50.
Methods
Animal models.
The Spop-targeted mice used for this project were created from the embryonic stem cell clone (Spop_D01) generated by the Wellcome Trust Sanger Institute and made into mice by the KOMP Repository (https://www.komp.org/) and the Mouse Biology Program (https://mbp.mousebiology.org/) at the University of California, Davis. Sperm from this strain was used for in vitro fertilization according to general protocols. The generated Spop Frt mice were initially crossed to a germline Flp deleter (Rosa26:FLPe knock-in) to eliminate the LacZ and neomycin cassettes, and subsequently to the Mx1–Cre and to the Rosa26–CreERT2 to generate conditional allele SpopF/F. Mx–Cre mice received one injection of poly(I:C) (10 μg g−1). Alternatively, Cre–ERT2 mice received four injections (every other day) of tamoxifen (T5648, 75 μg g−1; Sigma). Males and females from 8–12 weeks old were used for the in vivo experiments. All mice were housed at New York University School of Medicine under pathogen-free conditions. All procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees at New York University School of Medicine.
Transplantation.
Total nucleated bone marrow cells were collected by flushing the femur and tibias of CD45.2+ mice with PBS using a 22.5-gauge needle. The cell suspension was treated with red cell lysis buffer and resuspended in PBS. Donor cells (0.5 × 106 per genotype per mouse) were transplanted via retro-orbital injection into lethally irradiated (2 × 550 rad) sex-matched congenic CD45.1+ recipients (B6.SJL–Ptprca). Reconstitution was monitored 4 weeks after transplantation.
Immune challenge.
One week after the last tamoxifen injection, SpopΔ/ΔCreERT2 and control littermate mice received one intraperitoneal injection of poly(I:C) (10 μg g−1) or LPS (Escherichia coli serotype, O111-B4; 5 μg g−1; Sigma). For influenza infection, mice were anesthetized with ketamine–xylazine and infected intranasally (30 μl) with 300 50% embryo infective dose of the mouse-adapted influenza A H1N1 strain Puerto Rico/34/8 from Charles Rivers (material number 10100374 and batch number 4XP160412).
Plasmids.
Human Spop cDNA cloned into pcDNA3.1–hemagglutinin (HA) was kindly provided by K. Wellen and subsequently subcloned into pcDNA3.1–FLAG–streptavidin. Human MYD88 open reading frame was amplified from BJAB cells cDNA and subcloned in pcDNA3.1–FLAG–HA. Human IRAK4 was kindly provided by D. Brady and subcloned in pcDNA3.1–HA. N-terminal truncation mutants were generated by standard PCR methods. C-terminal truncation mutants and point mutants were generated by using the QuickChange Site-directed Mutagenesis kit (Stratagene). For the production of retrovirus, cDNAs encoding FLAG/HA-tagged SPOP(L193P) mutant were subcloned into the retroviral vector pMSCV-Puro. cDNAs encoding FLAG/HA-tagged and untagged MyD88 were subcloned in the retroviral vectors pBABE puro and MIGR1, respectively. MSCV_ Cas9_puro (Addgene plasmid no. 65655) and LGR lentiviral plasmids (Addgene plasmid no. 65656) were obtained from Addgene. Spop and Myd88 guide RNA sequences were obtained from published studies51 and cloned into LGR according to previously established protocols52.
Cell culture.
HEK293T cells were maintained in Dulbecco’s Modified Eagle’s medium containing 10% fetal bovine serum (FBS). K562 cells were maintained in RPMI 1640 medium containing 10% FBS. HPC-7 cells53 in Iscove’s Modified Dulbecco’s Medium contained 10% FBS supplemented with 0.15 mM NaHCO3, 6.5 μl l−1 monothioglycerol and 0.1 μg ml−1 mouse stem cell factor (SCF). The following drugs were used: cycloheximide (CHX; 50 μg ml−1; Sigma-Aldrich) and LPSs from E. coli O111:B4 (10 μg ml−1; Sigma-Aldrich). Stable cell lines were established after selection with puromycin (0.5 μg ml−1; Sigma-Aldrich). HEK293T cells were transfected using polyethylenimine. GP-293 packaging cells (Clontech) were used for retrovirus production. The virus-containing medium was collected after 72 h post-transfection, and the cells were spin infected in the presence of 10 μg ml−1 polybrene (Sigma) at 1,800–2,300 r.p.m. for 30–90 min. To generate an HPC-7–Spop KO cell line, we transduced HPC-7 cells with retroviral particles carrying Cas9, and 24 h later they were treated with puromycin (1 μg ml−1). Stable expression of Cas9 was determined by immunoblot. A Cas9–HPC-7 stable clone was transduced with lentiviral particles carrying the guide RNA against Spop gene or Rosa26 control. GFP+ cells were single cell sorted into 96-well plates 72 h later. After their expansion, three different clones in which Spop deletion was confirmed by immunoblot were selected for the proteomic experiments.
Ubiquitinome transcriptional profiling of the HSPC.
A list of the E3 ubiquitin ligases and accessory proteins was obtained from published databases54. The RNA-seq expression data for those genes were acquired from Lara-Astiaso et al.20 (GeoRef: GSE60101_1256271; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60101).
Flow cytometry.
Bone marrow cells were isolated by flushing the long bones in PBS with 3% FBS. The cells were dissociated to a single-cell suspension by filtering through a 70-μm nylon mesh. Red blood cells were lysed with ammonium–chloride–potassium buffer, and the remaining cells were resuspended in PBS with 3% FBS. Nonspecific antibody binding was blocked by incubation with anti-CD16/CD32 clone 24G2 (BD Pharmingen). Cells were incubated with primary antibodies for 60 min and streptavidin conjugates, when applicable, for 15 min on ice. All antibodies were purchased from BD Pharmingen or eBioscience (see Reporting Summary). Bone marrow–lineage antibody cocktail included CD11b, Gr-1, NK1.1, TER-119, CD4, CD8 and B220.
Immunohistochemistry.
Tissues were dissected from mice for fixation overnight in 10% formalin (Fisher). Fixed tissues were dehydrated and embedded in paraffin for sectioning. Paraffin sections (5 μm) were prepared and stained with hematoxylin and eosin (H&E; Leica Autostainer XL).
Tissue preparation for immunofluorescence and confocal microscopy.
Tissue were fixed in paraformaldehyde, lysine and sodium periodate buffer (0.05 M phosphate buffer, 0.1 M l-lysine, pH 7.4, 2 mg ml−1 NaIO4, and 10 mg ml−1 paraformaldehyde) overnight at 4 °C followed by 30% sucrose overnight at 4 °C and subsequent embedding in OCT media. Twenty-micrometer frozen tissue sections were sectioned using Leica CM3050S cryostat. FcR blocked with anti-CD16/CD32 Fc block (BioLegend) was diluted in PBS containing 2% goat serum and 2% FBS for 1 h at 25 °C. Sections were stained with the indicated antibodies that were diluted in PBS containing 2% goat serum, 2% FBS and 0.05% Fc block for 1 h at 25 °C. For the intracellular staining, all of the antibodies including the Fc block were diluted in PBS containing 2% goat serum, 2% FBS and 0.1% Triton-X100. Images were acquired using a Zeiss LSM 880 confocal microscope (Carl Zeiss) with the Zen Black software. The imaging data were processed and analyzed using Imaris software v.9.0.1 (Bitplane; Oxford Instruments).
Cytokine quantification.
For collecting bone marrow fluids, the four long bones (two femurs and two tibiae) of each mouse were flushed with the same 200 μl of PBS. Cells were spun down by centrifugation at 1,000 r.p.m. for 5 min. Next, supernatants were clarified by a second centrifugation at 12,000 r.p.m. for 5 min. IL-1α and IL-1β were measured by ELISA (mouse IL-1β and mouse IL-1α ELISA kit; R&D). IFN-α, IL-1β and other cytokines (data not shown) were measured using the Legendplex Anti-virus response panel (BioLegend).
RNA extraction and population-based RNA-seq.
Total RNA was extracted from bone marrow sorted samples (Ly6G+ cells for neutrophils and Lin−, CD117+, SCA-1+ for LSK) using the RNeasy Plus Micro Kit (Life Technologies). Real-time PCR was carried out with SYBR Green Master Mix (Roche) and a LightCycler 480 II (Roche). RNA-seq libraries were generated with SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing and Low Input Library Prep Kit v.2 (634899; Takara Bio), and sequenced on an Illumina HiSeq 4000 sequencer. Results were demultiplexed and converted to FASTQ format using Illumina bcl2fastq software. The sequencing reads were aligned to the mouse genome (mm10/GRCm38) using the splice-aware STAR aligner. The feature Counts program55 was used to generate counts for each gene based on how many aligned reads overlap its exons. These counts were then normalized and used to test for differential expression using negative binomial generalized linear models implemented by the DESeq2 R package.
Nuclear extraction.
Cells were lysed in hypotonic buffer (50 mM Tris, pH 8.0, 1 mM EDTA, 0.02% Nonidet-P40 (NP-40), 10% glycerol) containing protease inhibitors. The hypotonic fraction was collected after centrifugation at 2,400 r.p.m. for 3 min at 4 °C. The pellets (intact nuclei) were lysed with radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% Na deoxycholate, 1% NP-40) containing protease inhibitors. After 5 min of lysis, the samples were spun at 15,000 r.p.m. for 5 min at 4 °C. The supernatants (detergent-soluble fraction) were collected.
Immunoprecipitation.
Cells expressing FLAG-tagged proteins were lysed in NP-40 buffer (15 mM Tris, pH 7.4, 1 mM EDTA, 150 mM NaCl, 1 mM MgCl2, 10% glycerol, 0.1% NP-40) containing protease inhibitors. After 5 min of lysis, the samples were spun at 15,000 r.p.m. for 5 min at 4 °C. The supernatants were incubated with anti-FLAG/M2 affinity gel agarose beads (15 μl of slurry per sample; A2220; Sigma) for 2 h at 4 °C. After five washes with lysis buffer, the immunoprecipitates were eluted with 2× Laemmli buffer (240 mM Tris, pH 6.8, 8% SDS, 0.04% bromophenol blue, 5% β-mercaptoethanol, 40% glycerol) followed by boiling.
λ-Phosphatase treatment.
Following the washes of the immunoprecipitation protocol, the beads were divided equally per condition and were added to the reaction mix (25 mM Tris, pH 7.4, 50 mM NaCl, 0.1 mM MnCl2, 1× NEB buffer PMP). λ-Phosphatase (P0753, 400 U; New England Biolabs) was added to the mix for 30 min at 30 °C. Proteins were eluted with 2× Laemmli buffer.
Immunoblotting.
Whole-cell lysates were generated by lysis with RIPA buffer or with a urea lysis buffer (48% urea (Bio-Rad), 15 mM Tris–HCl, pH 7.5, 8.7% glycerin, 1% SDS, 0.004% bromophenol blue, 143 mM β-mercaptoethanol) and quantified using BCA protein assay (Pierce). Equal amounts of proteins were applied to a 6% polyacrylamide gel for electrophoresis followed by transfer on a polyvinylidene difluoride membrane (Immobilon-P; Millipore) for 2 h at 80 V. Membranes were incubated with the primary antibody for 2 h or overnight followed by incubation with the secondary antibody. Proteins were detected by incubating the membranes with a chemiluminescent substrate and subsequent exposure to autoradiography films. All antibodies were used at a dilution of 1:1,000 unless otherwise specified (see Reporting Summary).
Ubiquitylation assays.
HEK293T cells were lysed in 1% SDS. After sonication, the lysates were diluted to 0.1% SDS in NP-40 buffer and immunoprecipitated with anti-FLAG/M2 affinity gel agarose beads. After elution, the samples were subjected to SDS–polyacrylamide gel electrophoresis and analyzed by immunoblotting.
FLAG-tagged SPOP and MYC-tagged CULLIN3 were co-expressed in Sf9 insect cells. The ubiquitylation of in vitro-translated FLAG-tagged MyD88(WT) and FLAG-tagged MyD88(135/136) was carried out as described previously56. The ubiquitin reaction mix contains E1 (20 ng μl−1), UbcH5c (14 ng μl−1), ubiquitin (2.5 μg μl−1) and ATP (2 mM). The reaction was incubated at 30 °C.
TUBE.
HPC-7 Spop+/+ and Spop−/− cells were lysed in NP-40 buffer (15 mM Tris, pH 7.4, 1 mM EDTA, 150 mM NaCl, 1 mM MgCl2, 10% glycerol, 0.1% NP-40) containing protease inhibitors and N-ethylmaleimide. After 5 min of lysis, the samples were spun at 15,000 r.p.m. for 5 min at 4 °C. The supernatants were incubated with agarose-coupled TUBE (UM401; LifeSensor) affinity gel agarose beads for 2 h at 4 °C. After five washes with lysis buffer, the immunoprecipitates were eluted with 2× Laemmli buffer followed by boiling.
Purification and analysis of SPOP(L193P) interactors.
Approximately 5 × 108 K562 cells stably expressing an empty vector (EV) or FLAG-tagged SPOP(L193P) were harvested and subjected to nuclear extraction. The intact nuclei were lysed in NP-40 buffer. The resulting supernatants (soluble fraction) were collected. The resulting pellets (insoluble fraction) were resuspended in RIPA buffer and subjected to five cycles of sonication. Both fractions were incubated with an anti-FLAG agarose resin (Sigma) and after washing, proteins were eluted with FLAG peptides (Sigma). The eluates were separated by SDS–polyacrylamide gel electrophoresis, and proteins were stained by Silver Staining kit (Life Technology). The final eluate was then precipitated with trichloroacetic acid.
MudPIT analysis.
Trichloroacetic acid-precipitated proteins were urea denatured, reduced, alkylated and digested with endoproteinase Lys-C (Roche), followed by modified trypsin (Roche), as described previously57,58. Peptide mixtures were loaded onto 100-μm fused silica microcapillary columns packed with 5-μm C18 reverse phase (Aqua; Phenomenex), strong cation exchange particles (Luna; Phenomenex) and reverse phase59. Loaded microcapillary columns were placed in-line with a Quaternary Agilent 1100 series HPLC pump and an LTQ linear ion trap mass spectrometer equipped with a nano-LC electrospray ionization source (Thermo Scientific). Fully automated ten-step MudPIT runs were carried out on the electrosprayed peptides, as described previously57. Tandem mass spectra were interpreted using SEQUEST60 against a database of 61,318 sequences, consisting of 30,449 nonredundant human proteins (downloaded from National Center for Biotechnology Information on 27 August 2012), 160 usual contaminants (such as human keratins, IgGs and proteolytic enzymes) and, to estimate false discovery rates, 30,659 randomized amino acid sequences derived from each nonredundant protein entry. Peptide–spectrum matches were sorted and selected using DTASelect with the following criteria set: spectra–peptide matches were retained only if they had a DeltCn of at least 0.08 and a minimum XCorr of 1.8 for singly-, 2.0 for doubly- and 3.0 for triply-charged spectra. In addition, peptides had to be fully tryptic and at least seven amino acids long. Combining all runs, proteins had to be detected by at least two such peptides, or one peptide with two independent spectra. Under these criteria, the final FDRs at the protein and spectral levels were 2.1 ± 0.3% and 0.94 ± 0.03%, respectively. Peptide hits from multiple runs were compared using CONTRAST61. To estimate relative protein abundance, we calculated normalized spectral abundance factors (NSAFs) for each detected protein, as described previously57,62,63.
Single-cell library preparation and sequencing.
Ten days after poly(I:C) intraperitoneal injection, we sorted Lin−, CD117+, SCA-1+ cells from two WT, two Spop KO and two SpopΔ/Δ Myd88−/− (dKO). After sorting, we pooled samples by genotype. Single-cell RNA-seq libraries were prepared using the following Single Cell 3′ Reagent Kits v.2: Chromium Single Cell 3′ Library & Gel Bead Kit v.2, PN-120237; Single Cell 3′ Chip Kit v.2 PN-120236 and i7 Multiplex Kit PN-120262 (10x Genomics)64 and following the Single Cell 3′ Reagent Kits v.2 User Guide (Manual Part no. CG00052 Rev A). Libraries were run on an Illumina HiSeq 4000 as 150-bp paired-end reads, one full lane per sample.
scRNA-seq processing.
Sequencing results were demultiplexed and converted to FASTQ format using Illumina bcl2fastq software. The Cell Ranger Single-Cell Software Suite (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger) was used to perform sample demultiplexing, barcode processing and single-cell 3′ gene counting. The cDNA insert was aligned to the mm10/GRCm38 reference genome. Only confidently mapped, non-PCR duplicates with valid barcodes and UMIs were used to generate the gene–barcode matrix. Further analysis including the identification of highly variable genes, dimensionality reduction, standard unsupervised clustering algorithms and the discovery of differentially expressed genes was performed using the Seurat R package65. Extreme outliers in terms of library complexity in the top and bottom 2% quantiles of the distribution of genes detected by cell and cells with more than 10% of mitochondria transcripts were excluded. Data were normalized by the total expression, then multiplied by a scale factor of 10,000, and the result was log transformed.
To account for technical batch differences between the three libraries, we used the Seurat alignment method for data integration31. After filtering, the mean and median numbers of detected genes per cell were 3,255.9 and 3,157, respectively. Aligned canonical correlation analysis was used as a basis for partitioning the combined dataset into clusters using a smart local moving community detection algorithm. To find markers that define individual clusters, we performed differential expression analysis using Wilcoxon rank sum test for each cluster compared with all other cells for genes detected in at least 20% of the cluster cells. We excluded small clusters of contaminating cells (n = 144 in WT, n = 112 in dKO cells) expressing very high levels of lineage-differentiated hematopoietic markers (Ly6g, S100a8, Elane, Cd74, Ctss, Tox, Rora, Ly6d, Ilr7, Id2).
For visualizing the data, we further reduced the dimensionality of the entire 21,251-cell dataset to project the cells in two-dimensional space using t-SNE based on the aligned canonical correlation analysis, and we divided the cells into seven clusters. We first examined the transcriptional profile of each cluster and sample comparing the WT (n = 8,147) with the Spop KO cells (n = 4,920) and excluding the SpopΔ/Δ Myd88−/− (dKO) cells (n = 8,184) (Fig. 3d). In a subsequent analysis, we examined all three populations (Fig. 5f). We used the PHATE (Potential of Heat-diffusion for Affinity-based Transition Embedding) method for visualizing high-dimensional data that aims to preserve a range of patterns such as continual progression, branching and clusters (Fig. 5i and Supplementary Fig. 5f).
We calculated the average expression levels of all genes across all cells of each cluster and used the gene set variation analysis package66 to determine cluster-level gene set enrichment scores of the relevant gene signatures. The scores were computed with the single-sample GSEA method67. The scores were normalized by the absolute difference between the minimum and the maximum, and plotted in a heatmap.
ATAC-seq preparation processing and footprinting analysis.
Chromatin accessibility was accessed using the ATAC-seq protocol68. ATAC-seq libraries were generated from 5 × 104 sorted LSKs. Libraries were sequenced on an Illumina HiSeq 4000 using 50-bp paired-end reads. Reads were trimmed for adapter sequences and low-quality score bases, and were mapped to the mouse reference genome. Only reads that were properly paired and had a unique alignment (mapping quality > 10) were retained. Duplicate reads were removed using sambamba markdup. Peaks were determined algorithmically using Model-based Analysis of ChIP-Seq 2 algorithm with q > 0.05. Differential peaks were called using the DiffBind package. Differential peaks were subject to ontology analysis using Genomic Regions Enrichment of Annotation Tool with whole mouse genome (GRCm38/mm10) as the background. Motif analysis of these regions was performed with the MEME-ChIP tool using JASPAR Vertebrates and UniPROBE Mouse as the set of known motifs. Transcriptional factor footprints were calculated for KO sequencing data using wellington_footprints.py function from pyDNase tool and including–A parameter, which corrects for differences between DNaseI and Tn5 activities. For further analysis, only those footprints that overlapped with differential peaks were considered. For each transcriptional factor considered, individual occurrences across the genome were calculated using the MEME FIMO command line tool using Motifs in meme format downloaded from JASPAR. To assess which footprint corresponded to each transcriptional factor, we overlapped ATAC-seq footprints in differential peaks with each transcriptional factor individual occurrence.
Statistical analysis.
Except for RNA-seq, scRNA-seq and ATAC-seq data analysis (see Methods), GraphPad Prism software was used to determine the statistical significance. Group means were compared with two-tailed unpaired Student’s t-test when only two experimental groups were involved. For other analyses, one- or two-way ANOVA with multiple-comparison correction was used. All P values were two-sided, and statistical significance was assessed at or less than 0.05. Data represent results from samples collected from at least three different mice (as indicated in each figure legend), and they are representative of at least two independent experiments. For RNA-seq, differential expression analysis was performed using DESeq2 using Padj < 0.05 as significant. For scRNA-seq, differential expression analysis was determined by using the Wilcoxon rank sum test. For ATAC-seq, differential peaks were determined by DiffBind package with q > 0.05.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. In addition, raw data generated in this study (bulk RNA-seq, scRNA-seq, ATAC-seq) are available at the GEO database under the accession number GSE112542. Raw data for the proteomics Ms/Ms analysis are available at https://www.ebi.ac.uk/pride/archive/projects/PXD009469.
Supplementary Material
Acknowledgements
We would like to thank all members of the Aifantis laboratory for discussions throughout the duration of this project, specifically E. Wang and B. Aranda for expert advice in CRISPR/Cas9 technology and K. Hockemeyer for expert advice in preparing the manuscript; A. Heguy and the NYU Genome Technology Center (supported in part by National Institutes of Health, National Cancer Institute grant P30CA016087-30) for expertise with sequencing experiments; the NYU Histology Core (5P30CA16087-31) for assistance; C. Loomis and L. Chiriboga for immunohistochemistry experiments; and S. Naik for her intellectual input. This work used computing resources at the High-Performance Computing Facility at the NYU Medical Center. I.A. is supported by the National Institutes of Health (grant nos. R01CA133379, 5R01CA173636, RO1CA216421, RO1CA133379), the Leukemia and Lymphoma Society (TRP Program) and the NYSTEM program of the New York State Health Department (NYSTEM-N11G-255). L.B. is supported in part by grants (nos. R00-CA166181-04 and R01-CA207513-01) from the National Cancer Institute and Gilead Sciences Research Scholars Program in Hematology/Oncology. I.A. dedicates this work to the memory of his mentor H. von Boehmer.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability and associated accession codes are available at https://doi.org/10.1038/s41590-019-0454-6.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41590-019-0454-6.
Peer review information: Laurie Dempsey was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
References
- 1.King KY & Goodell MA Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol 11, 685–692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boettcher S & Manz MG Regulation of inflammation- and infection-driven hematopoiesis. Trends Immunol. 38, 345–357 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R Origin and physiological roles of inflammation. Nature 454, 428–435 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Pietras EM Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood 130, 1693–1698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirantes C, Passegué E & Pietras EM Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp. Cell Res 329, 248–254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagai Y et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24, 801–812 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Luca K et al. The TLR1/2 agonist PAM(3)CSK(4) instructs commitment of human hematopoietic stem cells to a myeloid cell fate. Leukemia 23, 2063–2074 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Essers MA et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458, 904–908 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Schuettpelz LG et al. G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leukemia 28, 1851–1860 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pietras EM et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol 18, 607–618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baldridge MT, King KY, Boles NC, Weksberg DC & Goodell MA Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ortega-Gomez A, Perretti M & Soehnlein O Resolution of inflammation: an integrated view. EMBO Mol. Med 5, 661–674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serhan CN & Levy BD Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J. Clin. Invest 128, 2657–2669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson BJ et al. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J. Exp. Med 205, 1395–1408 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King B et al. The ubiquitin ligase Huwe1 regulates the maintenance and lymphoid commitment of hematopoietic stem cells. Nat. Immunol 17, 1312–1321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao J et al. The CUL4-DDB1 ubiquitin ligase complex controls adult and embryonic stem cell differentiation and homeostasis. eLife 4, e07539 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strikoudis A, Guillamot M & Aifantis I Regulation of stem cell function by protein ubiquitylation. EMBO Rep. 15, 365–382 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rape M Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol 19, 59–70 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Bhoj VG & Chen ZJ Ubiquitylation in innate and adaptive immunity. Nature 458, 430–437 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Lara-Astiaso D et al. Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai H & Liu A Spop promotes skeletal development and homeostasis by positively regulating Ihh signaling. Proc. Natl Acad. Sci. USA 113, 14751–14756 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuhn R, Schwenk F, Aguet M & Rajewsky K Inducible gene targeting in mice. Science 269, 1427–1429 (1995). [DOI] [PubMed] [Google Scholar]
- 23.Pietras EM et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J. Exp. Med 211, 245–262 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takizawa H et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell 21, 225–240.e225 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Delano MJ et al. MyD88−dependent expansion of an immature GR-1(+) CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med 204, 1463–1474 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Passegue E, Wagner EF & Weissman IL JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 119, 431–443 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Hirai H et al. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat. Immunol 7, 732–739 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Olsson A et al. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 537, 698–702 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nestorowa S et al. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–e31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giladi A et al. Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat. Cell Biol 20, 836–846 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haas S et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell 17, 422–434 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Medzhitov R et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 2, 253–258 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Tikhonova AN et al. The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhuang M et al. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol. Cell 36, 39–50 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen P TheTLR and IL-1 signalling network at a glance. J. Cell Sci 127, 2383–2390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akira S & Takeda K Toll-like receptor signalling. Nat. Rev. Immunol 4, 499–511 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Gay NJ, Gangloff M & O’Neill LA What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 32, 104–109 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Kaufmann E et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 172, 176–190.e119 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Netea MG et al. Trained immunity: a program of innate immune memory in health and disease. Science 352, aaf1098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naik S et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550, 475–480 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y et al. A functional genomics approach to understand variation in cytokine production in humans. Cell 167, 1099–1110.e1014 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Ter Horst R et al. Host and environmental factors influencing individual human cytokine responses. Cell 167, 1111–1124.e1113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowman RL, Busque L & Levine RL Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell 22, 157–170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medyouf H The microenvironment in human myeloid malignancies: emerging concepts and therapeutic implications. Blood 129, 1617–1626 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Barreyro L, Chlon TM & Starczynowski DT Chronic immune response dysregulation in MDS pathogenesis. Blood 132, 1553–1560 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lord KA, Hoffman-Liebermann B & Liebermann DA Nucleotide sequence and expression of a cDNA encoding MyD88, a novel myeloid differentiation primary response gene induced by IL6. Oncogene 5, 1095–1097 (1990). [PubMed] [Google Scholar]
- 48.Hu H & Sun SC Ubiquitin signaling in immune responses. Cell Res. 26, 457–483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heger K et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 559, 120–124 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Wang Z, Wesche H, Stevens T, Walker N & Yeh WC IRAK-4 inhibitors for inflammation. Curr. Top. Med. Chem 9, 724–737 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi J et al. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol 33, 661–667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pinto do OP, Kolterud A & Carlsson L Expression of the LIM-homeobox gene LH2 generates immortalized steel factor-dependent multipotent hematopoietic precursors. EMBO J. 17, 5744–5756 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Medvar B, Raghuram V, Pisitkun T, Sarkar A & Knepper MA Comprehensive database of human E3 ubiquitin ligases: application to aquaporin-2 regulation. Physiol. Genom 48, 502–512 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Busino L et al. Fbxw7α- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat. Cell Biol 14, 375–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Florens L & Washburn MP Proteomic analysis by multidimensional protein identification technology. Methods Mol. Biol 328, 159–175 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Washburn MP, Wolters D & Yates JR 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol 19, 242–247 (2001). [DOI] [PubMed] [Google Scholar]
- 59.McDonald WH, Ohi R, Miyamoto DT, Mitchison TJ & Yates JR Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. Int. J. Mass Spectrom 219, 245–251 (2002). [Google Scholar]
- 60.Eng JK, McCormack AL & Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989 (1994). [DOI] [PubMed] [Google Scholar]
- 61.Tabb DL, McDonald WH & Yates JR 3rd DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res 1, 21–26 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paoletti AC et al. Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc. Natl Acad. Sci. USA 103, 18928–18933 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zybailov B et al. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res 5, 2339–2347 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Zheng GX et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Macosko EZ et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hanzelmann S, Castelo R & Guinney J GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barbie DA et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 2921–2929 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. In addition, raw data generated in this study (bulk RNA-seq, scRNA-seq, ATAC-seq) are available at the GEO database under the accession number GSE112542. Raw data for the proteomics Ms/Ms analysis are available at https://www.ebi.ac.uk/pride/archive/projects/PXD009469.