

Abstract
There has been considerable progress in our understanding of cardiac cell metabolism in health and disease, yet important gaps remain in basic knowledge and its translation to clinical care. AMP-activated protein kinase (AMPK) functions either to conserve ATP or to promote alternative methods of ATP generation. Since the discovery of AMPK more than three decades ago and demonstration of its expression in the heart, interest has grown exponentially in this major fuel gauge as a modulator of the cellular response to ischemia. Such pathway may potentially explain the strong association between metabolic syndrome and ischemic heart disease. Still missing from our most recent cardiology textbooks, this article aims to summarize our understanding so far of the role of AMPK in coordinating the cellular response to ischemic stress and reperfusion injury in the heart. We aim to provide a focused update on the pharmacological agents activating AMPK for treatment of diabetes that show potential cardioprotective effects. Our hope is to stimulate future researchers to the potential benefits of harnessing the AMPK signaling pathway, or better one of its novel downstream targets for the treatment of myocardial ischemia.
Keywords: Ischemia-reperfusion injury, Myocardial infarction, AMPK, Myocardial ischemia, Diabetes, Agonists, Glucose homeostasis
1. Introduction
Heart disease remains the leading cause of death worldwide in spite of the dramatic improvements in the management of acute myocardial infarction. While the underlying pathophysiology of the atherosclerotic process is not significantly different in diabetic subjects, the prothrombotic and procoa-gulant state with which diabetes is associated is thought to contribute to the higher incidence of and worse prognosis after myocardial infarction1. Following reperfusion with percutaneous coronary intervention or thrombolytics, infarct size propagation seemed inevitable2. That was when attention has turned to cellular adaptive mechanisms in the hope to improve mortality and morbidity of myocardial infarction. AMPK has stood out over the past decade as a master conductor orchestrating the intracellular metabolic process, both at the cellular level and in experimental animal models3–6. It functions as a sensor during low energy states, such as ischemia, to change substrate utilization and thereby increase ATP synthesis. AMPK also seemed to put the cell in ‘survival mode’ conserving energy that would otherwise be consumed in non-urgent anabolic processes7–9. Since then AMPK has emerged as a very promising target for cytoprotective therapy10–15. Our perception of the AMPK pathway continues to evolve as we better understand the intricate relationships between AMPK and its upstream and downstream kinases. The aim of the current review is to discuss the benefits and potential harms of AMPK activation during myocardial infarction, especially in the diabetic population and shed light on the pros and cons of currently available AMPK activators.
2. Structure of AMPK
AMPK is a widely distributed, highly conserved heterotrimeric enzyme composed of a catalytic α (63 kDa) subunit and the non-catalytic β and γ subunits5 (Fig. 1). There are two genes encoding isoforms of both the α and β subunits (α1, α2, β1 and β2). The α2 isoform is the subunit of AMPK found predominantly within cardiac muscles16. AMPK is activated by AMPK kinase (AMPKK) by phosphorylation of threonine-172 at the N-terminal end of the α subunits17,18. The close proximity of AMPK to cellular glycogen stores allows it to rapidly affect changes in glycogen metabolism in response to changes in the metabolic demands19. Binding sites for AMP are found on the regulatory γ subunits which also bind ATP16,20. Of clinical significance is the observation that mutations in the γ2 subunit, the dominant isoform in the heart (gene symbol PRKAG2), are associated with abnormal glycogen accumulation in cardiomyoctes manifesting as Wolff–Parkinson–White syndrome and familial hypertrophic cardiomyopathy21. The binding of AMP and ATP to AMPK occurs in a mutually exclusive manner17. An increased AMP to ATP ratio leads to a conformational change in AMPK that makes it a poorer substrate for degrading protein phosphatases and a better substrate for AMPKK5,17. Because of its dual effect, small rises in AMP levels can induce a dramatic rise in the activity of AMPK22. Adenylate kinase maintains its reaction (2ADP↔ATP+AMP) close to equilibrium, therefore the AMP:ATP ratio varies as the square of the ADP:ATP ratio, and this makes the AMP:ATP ratio a very sensitive indicator of cellular energy charge23. Similar positive allosterism is also observed with an increased creatinine:phosphocreatine ratio20. Activation of AMPK leads to phosphorylation of many target proteins important for ATP synthesis and utilization as well as the inhibition of ATP-consuming pathways such as fatty acid synthesis11,24. Once the energy stores are replenished, the binding of ATP maintains the activity of AMPK low25.
Figure 1.
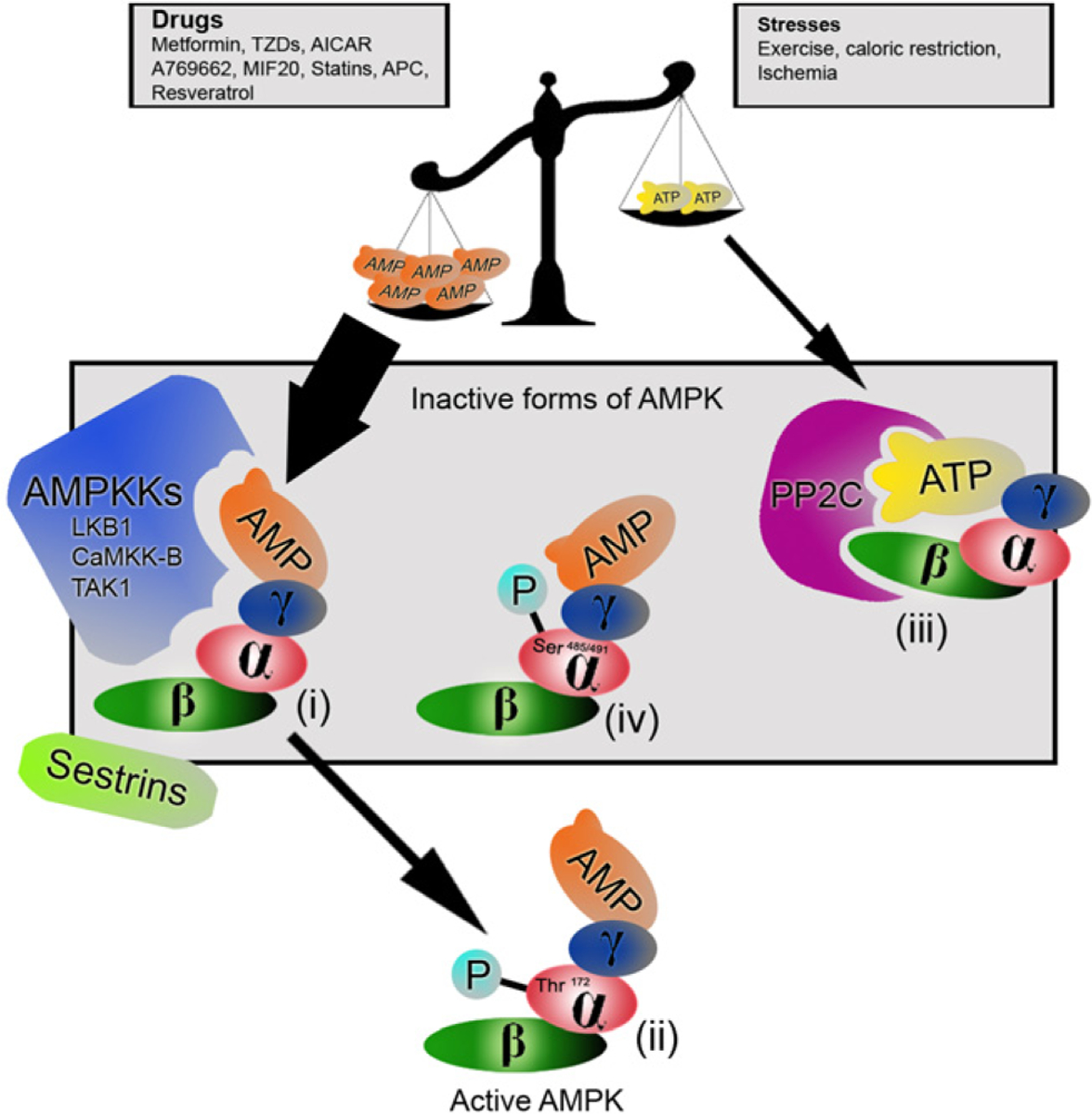
Schematic representation of the different forms of AMPK. Increases in AMP relative to ATP, sensed as a low energy state, allows AMPKK to phosphorylate Thr172 and activate AMPK at the N-terminal of the α subunit (i). This occurs in situations of stress and may be simulated by different pharmacological agents. The active form of AMPK (ii) is now available to lead the myriad of changes in cellular metabolism. Abundance of ATP, as in high energy states, allows ATP to bind to AMPK causing a conformational change that renders it vulnerable to degrading phosphatases (iii). Phosphorylation of AMPK by Akt at Ser485/491 in response to insulin causes another conformational change that prevents AMPKKs from binding and activating AMPK (iv).
3. Upstream regulators of AMPK
There are at least three different upstream AMPK kinases (AMPKKs), the most widely expressed being LKB1 (with two accessory subunits, STRADα/β and MO25α/β) which is critical for the gluconeogenic flux and consequently glucose homeostasis26,27. AMPK phosphorylation during myocardial ischemia however seems to be related, in addition, to AMPKKs other than LKB1 as observed by Altarejos et al.28 AMPK activity was noted to be increased in mild ischemia before the classic shift in the AMP:ATP and creatine:phosphocreatinine ratios17,29. One other activator of AMPK is Ca2+-calmodulin–protein kinase kinase β (CaMKK-β) which phosphorylates and activates AMPK in response to increased Ca2+ fluxes providing a mechanism for cells to anticipate the increased demand for ATP30,31. An interesting phenomenon described by Anderson et al.32,33 is the autophosphorylation of CaMKK-β after sustained exposure to Ca2+. This might contribute to sustained myocardial stunning and ischemia reperfusion injury (RI) following myocardial infarction. The third AMPK phosphorylating kinase is transforming growth factor-β -activated kinase 1 (TAK1)34. TAK1, a member of the mitogen-activated protein kinase kinase kinase (MAPKKK) family, is a key mediator of proinflammatory and stress signals29,35. TAK1 was initially found to activate the AMPK analogue in yeast, Snf1 before being linked to mammalian AMPK34. A large body of promising research is being invested in each upstream kinase to determine which one is predominantly expressed in the heart and is the first domino in the cardiomyocyte response to ischemic stress cascade and post-MI remodeling. Detailed discussion of each of these kinases is beyond the scope of this article and is readily available for the avid reader. Most likely the cellular response to ischemia initiating AMPK activation is interplay of all three kinases and others. Adiponectin, a plasma protein with cardioprotective, antidiabetic and anti-inflammatory properties also exerts its action through AMPK activation36,37. Of note is that adiponectin levels were found to correlate inversely with insulin resistance, obesity and coronary artery disease possibly explaining why patients with diabetes and ischemic heart disease have worse prognoses38. Furthermore sestrins, a family of conserved proteins that accumulate in cells exposed to stress, and lack kinase activity have been recently shown to potentiate the activation of AMPK independent of AMP:ATP39. Little is known about which phosphatase regulates AMPK. Both type 2A and type 2C protein phosphatases (PP2A and PP2C, respectively) are good candidates since they can dephosphorylate and deactivate AMPK in vitro40.
4. Downstream targets of AMPK in the heart
Activation of AMPK in the heart leads to a myriad of changes affecting glucose and lipid metabolism, protein synthesis and gene expression3,41. AMPK activation induces the translocation of glucose transporter 4 (GLUT4) to the plasma membrane by a mechanism distinct to that of insulin and not clearly understood42,43. It augments the binding of transcription factor myocyte enhancing factor-2 (MEF-2) to promoters in the GLUT4 gene increasing its expression44. Under hypoxic conditions in the heart, AMPK activates phosphofructokinase-2 (PFK-2), the product of which is fructose 2, 6-biphosphate (Fru2, 6-P2)45,46. This molecule allosterically regulates phosphofructokinase-1 and in the liver fructose 1, 6-biphosphatase, favoring glycolysis over gluconeogenesis. This phenomenon, recognized as the Pasteur Effect is responsible for increased ATP production through anaerobic glycolysis during ischemia45. AMPK also inhibits both isoforms of Acetyl-CoA carboxylase (ACC1 and ACC2) through phosphorylation47. This eventually leads to decreased inhibition of carnitine palmitoyltransferase I (CPTI) in the mitochondria and increased β-oxidation of fatty acids4,6. Increased fatty acid oxidation, like increased glycolysis will lead to valuable increases in ATP production in stress conditions. One important concept in energy production is the Randle cycle where the utilization of one nutrient inhibits the use of the other directly and without hormonal mediation48. The glucose-fatty acid cycle is thus a biochemical mechanism that controls fuel selection and adapts substrate supply and demand in normal tissues in coordination with hormones controlling substrate concentrations in the circulation49.
In addition AMPK phosphorylates and inactivates HMG-CoA reductase, hormone-sensitive lipase, as well as transcription factors hepatic nuclear factor 4α (HNF4α) and sterol regulatory element binding proteins 1c and −2 (SREBP-1c and −2)50. This reduces energy consumption at the cellular level and in the long term atherogenic dyslipidemia. AMPK further conserves ATP expenditure by inhibiting liver glycogen synthase and creatine kinase51.
With regards to protein metabolism, activation of AMPK phosphorylates and activates endothelial nitric oxide synthase (eNOS) in cardiac endothelium causing NO-mediated vasodilatation and improved perfusion4,42. NO activation in platelets leads to a decrease in thrombin-induced aggregation, thereby, limiting the pro-coagulant effects of platelet activation52. AMPK activation regulates protein synthesis through inhibition of elongation factor 2 (eEF2) and indirectly through inhibition of mammalian target of rapamycin (mTOR)53. Of clinical significance is the use of mTOR inhibitors in drug-eluting stents to inhibit protein synthesis and restenosis following reestablishing blood flow. Another promising transcription factor and downstream target of AMPK recently linked to improved cardiac oxidative stress is Forkhead box protein (FoxO)54. Mice with deletion of FoxO1 and FoxO3 were found to have increased scar formation, induction of stress-responsive signaling, and apoptotic cell death in response to ischemia55. Peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) is a multi-functional transcriptional coactivator involved in the regulation of cardiac mitochondrial biogenesis and oxidative phosphorylation56. It is activated by AMPK and sirtuin 1 (SIRT1) through phosphorylation and deacetylation, respectively57. Phosphorylated AMPK also indirectly activates PGC-1α through SIRT1 by promoting an intracellular increase in NAD+ levels, the rate-limiting substrate for the deacetylase activity of SIRT158. Once turned on, PGC-1α boosts mitochondrial activity to adapt to the increased energy requirements.
5. AMPK regulates autophagy
AMPK is also involved in triggering autophagy in the heart during ischemia and reperfusion59. Autophagy is a lysosomal pathway involved in the turnover of cell’s own macromolecules and organelles9. This has been identified as an adaptive cell response allowing the cell to survive otherwise lethal challenges by removing damaged organelles and misfolded proteins. The principle is to conserve energy that would otherwise be used in non-essential pathways. In addition free fatty acids and amino acids produced by the degradation of these organelles are then recycled to maintain mitochondrial ATP production and ribosomal protein synthesis60. Recent studies have shown that AMPK is involved in triggering autophagy in the heart in response to ischemia59. Activated AMPK inhibits mammalian target of rapamycin complex 1 (mTORC1/mTOR) activity via a pathway involving tuberous sclerosis complex 1 and 2 (TSC1/2) thus stimulating autophagy53. Recent studies unveiled that ULK1 is a mediator for AMPK modulating autophagy9,61,62.
6. AMPK and myocardial ischemia
Utilizing the AMPK pathway seems to have a beneficial effect during myocardial ischemia11,12,14,31,63,64. There is a fairly stereotypical sequence of physiologic changes that develop during an episode of spontaneous transmural ischemia3,20. Under physiological conditions, energy necessary to maintain myocardial contraction/relaxation cycle is derived from mitochondrial oxidation of long chain fatty acids (60–70%) and carbohydrates (20–30%)3,65,66. Coronary occlusion results in an immediate fall in coronary venous oxygen saturation, with a reduction in ATP production64. This causes a decline in regional contraction within several beats, reaching dyskinesis within 1 min67. As regional contraction ceases, there is a reduction in global LV contractility (dP/dt), a progressive rise in LV end-diastolic pressure and a fall in systolic pressure. AMPK therefore is activated in response to the decreased concentration of ATP in order to boost energy production and sustain the cardiomyocyte during this time of metabolic stress3,4. The magnitude of systemic hemodynamic changes varies with the severity of ischemia and the amount of the left ventricle subjected to ischemia. ST-segment changes on EKG develop within 2 min as efflux of potassium into the extracellular space reaches a critical level68. Irreversible injury begins after 20 min following a total occlusion but is delayed for up to 5 h following a partial occlusion69. The timing of administering a drug that activates AMPK is thus crucial, ideally as soon as possible following presentation.
7. AMPK in reperfusion injury
Following restoration of blood flow before irreversible injury, several types of RI have been observed in experimental animals that exacerbate myocardial infarction70. These consist of the following: (1) lethal reperfusion injury – reperfusion-induced death of cells that were still viable at the time of restoration of coronary blood flow; (2) vascular reperfusion injury – progressive damage to the microvasculature so that there is an expanding area of no reflow and loss of coronary vasodilatory reserve; (3) stunned myocardium – salvaged myocytes display a prolonged period of contractile dysfunction following restoration of blood flow because of abnormalities of intracellular metabolism leading to reduced energy production; and (4) reperfusion arrhythmias – bursts of ventricular tachycardia and, on occasion, ventricular fibrillation – that occur within seconds of reperfusion. The exact mechanism of RI remains incompletely understood, but is thought to include (i) cytosolic and mitochondrial Ca2+-overload, (ii) release of reactive oxygen species (ROS), (iii) changes in pH and intramitochondrial H+71, (iv) shift in substrate use, and (v) recruitment of inflammatory cells attacking viable myocytes. In concert, these derangements result in the formation of mitochondrial permeability transition pore (mPTP) upon reperfusion (Fig. 2). It spans both the inner and outer membranes and, when open, causes collapse of the transmembrane voltage gradient C and uncoupling of the mitochondria. Pore formation would prevent mitochondrial ATP production at reperfusion, a time when the ischemic but still salvageable myocytes would need it the most72. We now know that AMPK is stimulated by all of these changes. AMPK is activated by CaMKK-β in response to (i)73 and is redox-sensitive to (ii) and (iii)74. Decreased ATP:AMP (iv) and TAK1 in response to inflammatory cytokines (v) also activate AMPK. In addition, AMPK seems to inhibit the opening of mPTP and reduce infarct size in animal models75. Transgenic mice heart expressing a kinase dead form of AMPK demonstrated impaired recovery of left ventricular contractility and increased apoptosis on reperfusion11. A group of protective kinases, referred to as reperfusion injury salvage kinases (RISK), were found to somehow prevent transition pore formation at reperfusion. This comprises phosphatidylinositol 3-kinase (PI3K), Protein kinase B (Akt) and p42/p44 extra-cellular signal-regulated kinases (Erk 1/2). Even though AMPK has not been identified as a member of the RISKs, many of the agents that activate AMPK were also found to activate the RISK pathway including metformin, pioglitazone, statins and adenosine76. While autophagy in myocardial ischemia is cardioprotective as a means to conserve energy, continued autophagy upon reperfusion seems to be harmful with a shift more towards programmed cell death59. This however was shown to be due to beclin 1, independent of AMPK77.
Figure 2.
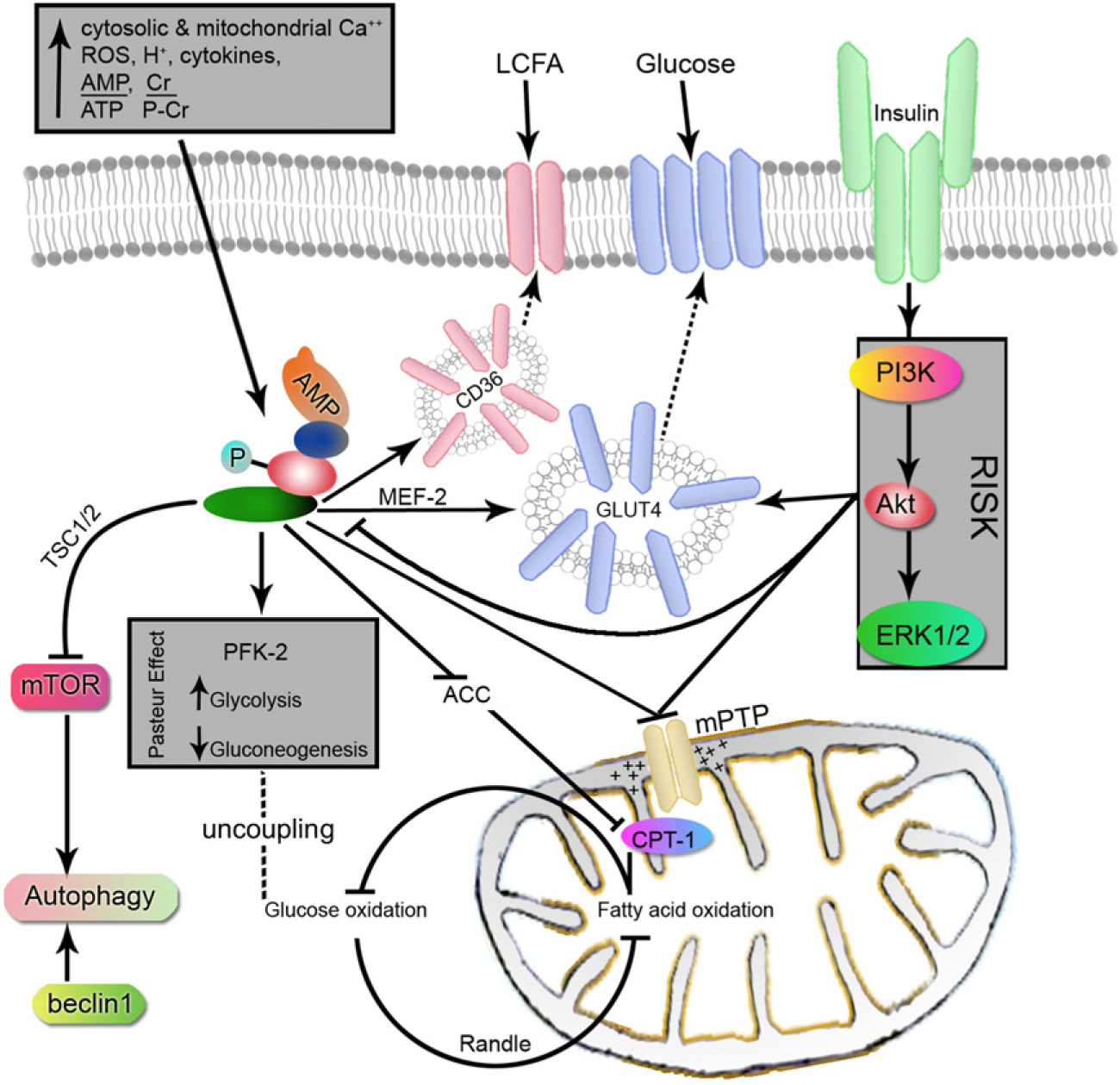
Illustration of the role of AMPK during reperfusion. AMPK activation in early reperfusion increases the uptake of glucose and long chain fatty acids (LCFA) into the cell. This coincides with increased anaerobic glycolysis and fatty acid oxidation for ATP production. In the absence of insulin, fatty acids are preferred over glucose for oxidation resulting in the uncoupling of end products of glycolysis. Both AMPK and RISK function to prevent the opening of mPTP and loss of the electrochemical gradient within the mitochondria. Autophagy is turned on initially by AMPK inhibition of mTOR and later by beclin 1.
Needless to say that the graveness of reperfusion injury is proportional to the duration of ischemia. The effectiveness of agents directed against reperfusion injury rapidly declines the later they are administered after reperfusion; eventually, no beneficial effect is detectable in animal models after 45 to 60 min of reperfusion has elapsed75.
8. Pharmacological agents
A very interesting combination of drugs has emerged that utilize the AMPK pathway. There are two groups of antihyperglycemics: metformin and glitazones (TZDs), an adenosine analogue tested in bypass surgery, a drug formerly used in sepsis, a thienopyridone and statins, which are the only drugs currently approved for acute coronary syndrome. Cannabinoids, present in marijuana have also been found, in a small study, to stimulate AMPK in the heart and hypothalamus of rats78.
8.1. Metformin
First used in 1957, it is the most commonly used drug for treatment of type 2 diabetes. The activation of AMPK by metformin is mediated indirectly through increased AMP:ATP ratio. This is hypothesized to occur through weak inhibition of complex I of the respiratory chain and a relative fall in cellular ATP concentration79. This inhibits AMPK dephosphorylation thereby potentiating its phosphorylation by the upstream kinase LKB180. Adenine nucleotide-independent mechanisms have also been postulated but poorly understood81,82. The cardioprotective actions of metformin through AMPK are not solely limited to its ability to reduce myocardial infarct size, as demonstrated by Calvert et al.83 with the acute administration before ischemia or at reperfusion. The dose used for AMPK activation of 125 μg/kg79 was much less than the maximum therapeutic dose of 2550 mg84. A very recent study has shown the cardioprotective benefits of metformin extend beyond acute coronary syndrome (ACS) to cardiac transplantation. Murine hearts treated with metformin perioperatively were found to have significantly better graft beating scores and less luminal narrowing, an effect that lasted more than 50 day post surgery79. This is believed to be mainly due to decreased RI, apoptosis and cardiac allograft vasculopathy secondary to AMPK activation. Chronic metformin therapy in diabetic patients is also known to improve lipoprotein profiles, reduce oxidative stress, and improve vascular stability85. Dogs with tachycardia-induced heart failure were found to have improved left ventricular function with chronic AMPK activation with metformin or AICAR86. The metformin dose used there was 100 mg/kg/day, similar to the therapeutic dose for diabetes. Advantages of metformin use are its good bioavailability and relatively safe profile. The dose for optimal AMPK activation is yet to be determined. An interesting question arises when considering metformin in ameliorating ischemia/reperfusion injury in the setting of ACS. Would diabetic patients already on chronic metformin therapy (assuming AMPK activation) benefit from a ‘booster’ dose of metformin as much as naive patients?
8.2. Glitazones (TZDs)
Rosiglitazone and pioglitazone are insulin sensitizers. Pioglitazone is the only drug remaining in this class that may be prescribed without restriction. TZDs bind to the nuclear membrane receptors PPARγ (peroxisome proliferator-activated receptors gamma) and affect DNA expression. Like metformin, TZDs have been found to increase the AMP:ATP ratio leading to AMPK activation87. PPARγ agonists further activate AMPK through increased expression of adiponectin88,89. Most experiments have been done using rosiglitazone as it is the most selective and potent PPARγ agonist in its class90. Rosiglitazone treatment also has been shown to reduce myocardial apoptosis and infarction size post RI by restoring the balance between the pro-apoptotic and antiapoptotic mitogen-activated protein kinases (MAPKs), increasing phosphatidylinositol-3-kinase (PI3K)-Akt phosphorylation, and inhibiting p42/44 MAPK91.
The association between TZDs and heart failure is well recognized as a class effect3. Fluid retention is mediated through increased sodium reabsorption of the renal PPARγ-dependent pathway in the collecting tubules. In 2007, a meta-analysis by Nissen et al.92 suggested that use of rosiglitazone in type 2 diabetes was associated with a significant increase in the risk of myocardial infarction and with an increase in the risk of death from cardiovascular causes that had borderline significance. Although the method and statistical analysis used in this study have been criticized, subsequent meta-analyses showed similar concerns93. In spite of these concerns, one can argue that acute rosiglitazone administration may prove beneficial in ACS through AMPK activation regardless of the implicating long-term increased mortality3,64. Unpublished results from our lab suggest that the benefit of acute rosiglitazone therapy during myocardial ischemia is even more pronounced in diabetic mice.
8.3. Activated protein C (APC)
APC is a vitamin K-dependent plasma serine protease that down-regulates clotting and inflammatory pathways94. Because of such properties, APC was approved in septic shock in 2001 before a Cochrane review a decade later showed no significant mortality benefit95. In the interim it was found that APC exerts cardioprotective effects by decreasing apoptosis in cardiomyocytes and inhibiting expression of inflammatory mediators after myocardial ischemia through AMPK. The activation of cardiac AMPK by APC was mediated through the APC receptors: endothelial protein C receptor (EPCR) and protease activated receptor-1 (PAR-1), and was dependent on CaMKK-β. Moreover, APC decreased inflammatory signaling pathways JNK and NF-κB, and down-regulated the expression of inflammatory cytokines in ischemic hearts. These cardioprotective effects of APC were abrogated in AMPKα2 knockout mice31.
8.4. 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR)
AICAR is a drug that is metabolized inside cells into ZMP (5-aminoimidazole-4-carboxamide-riboside), an analogue of AMP96. ZMP activates AMPK by direct and allosteric activation although less effective than AMP17. Balschi et al.97 demonstrated the AICAR-dependant phosphorylation of AMPK in isolated mouse heart. Interestingly AICAR made it to phase III trials, as early as 1994 in (Coronary Artery Bypass Graft) CABG surgery, long before our current understanding of the AMPK pathway. This was simply because of the rationale that increased levels of adenosine potentiates vasodilatation, preconditioning and inhibits inflammation98. RED-CABG in 2009, the last phase III trial involving AICAR, was terminated early when an interim analysis showed low probability of success. Limitations to its use include the requirement for intravenous infusion and variable potency. It also causes bradycardia and can lead to hypoglycemia when administered intravenously. One interesting conflict is that AICAR has been shown to induce protein kinase B (Akt), a potent activator of mTORC1, in an AMPK-independent manner99. Activation of mTORC1 inhibits autophagy which has been shown to play a role in ameliorating cardiac ischemic injury.
8.5. A-769662
A cell-permeable small organic compound was identified as an effective AMPK activator by screening a chemical library of over 70,000 compounds followed by lead optimization100. First found to have beneficial effects on metabolism in db/db mice, A-769662 was shown to activate AMPK both allosterically and by inhibiting dephosphorylation of AMPK on Thr172, analogous to the effects of AMP101. Similarly macrophage migration inhibitory factor (MIF), a pleiotropic cytokine that is released by ischemic cardiomyocytes was found to exert cardioprotective effects by activating AMPK10,12. While developing small molecule antagonists of the MIF receptor for immunomodulatory applications102, the compound MIF20 was uncovered that significantly augmented AMPK phosphorylation in the presence of MIF (unpublished data). The clinical benefit of those two compounds however remains largely investigational.
8.6. Statins
HMG-CoA reductase inhibitors have been shown to have cardiovascular benefits in patients with and without diabetes beyond their lipid-lowering effects. Actually the pleiotropic benefits of statins extend to patients without cardiovascular disease and with normal cholesterol levels103,104. Statins activate AMPK in the myocardium and endothelial cells by mechanisms that are not fully understood. Up-regulation of eNOS by atorvastatin in endothelial cells of mice without altering AMP:ATP ratio suggests another indirect mechanism of AMPK activation105. Rossoni et al.106 have shown that acute simvastatin-mediated relaxation of healthy endothelial cells of rats was mediated via AMPK and eNOS phosphorylation. This effect was totally blunted after adding the AMPK inhibitor, Compound C106.
9. AMPK and diabetes
Type 2 diabetes mellitus is a fasting hyperglycemic state of insulin resistance leading to impaired peripheral glucose utilization. A key contributing factor to these abnormalities is the failure of insulin to suppress gluconeogenesis and hepatic glucose production. AMPK activity is thought to be impaired in diabetes as AMPK stimulation in animal models has been shown to improve glycemia. This was also established using AMPK activators other than the antidiabetic drugs: metformin and glitazones107,108. Increasing glucose uptake into the cells and suppressing endogenous glucose production and lipolysis suggest AMPK activators as a possible new class of antidiabetic agents.
10. Insulin/AMPK interaction
The use of insulin for ACS seemed logical in 1963 by Sodi-Pallares, with the intention of facilitating potassium flux in the ischemic myocardium, the so-called polarizing therapy. Glucose and potassium were later added to counteract the effects of large dose insulin to become known as GIK therapy109. With the identification of the insulin signaling pathway, insulin was found to have comprehensive anti-inflammatory, antioxidant and antiapoptotic properties through PI3K and Akt (RISK pathway) independent of AMPK110. Insulin also suppressed the high blood fatty acid levels commonly observed in patients with ACS. Insulin-mediated activation of PI3K-Akt led to activation of endothelial nitric oxide synthase (eNOS) which inhibited mPTP opening in cardiomyocyte mitochondria. Yet the promising non-metabolic effects of insulin failed to translate to significant mortality benefit in a trial comprising 20,000 patients with myocardial infarction (MI)111. One possible explanation is the dynamic interaction between insulin and AMPK during ischemia then reperfusion in the heart. Insulin activates Akt which directly phosphorylates AMPKα1/α2 on Ser485/491 preventing AMPKK from phosphorylating AMPKα at its primary activation site, Thr172.112 This is believed to be beneficial as decreased AMPK-mediated oxidation of fatty acids relieved the inhibition of glucose oxidation mainly at the level of pyruvate dehydrogenase. This rechanneled the end products of glycolysis into aerobic respiration with subsequent decrease in the detrimental protons and lactate accumulation. However this cardioprotective effect demonstrated in isolated hearts was abolished in the presence of palmitate, simulating the high free circulating fatty acid levels present during reperfusion113; an effect that was observed at an insulin concentration of 100 nM but not, the near-physiological, 0.6 nM. Interestingly palmitate seems to also activate PP2A which dephosphorylates and inactivates AMPK114. This appears to be in response to negative feedback by ceramide, a second messenger in the sphingomyelin signaling pathway. Added to that, heparin that is conventionally used in ACS, stimulates lipoprotein lipase increasing further FFA levels and favoring their oxidation. More studies are needed with different levels of insulin to better understand the relationship between anabolic insulin and catabolic AMPK.
11. Non-pharmacological AMPK activators
11.1. Caloric restriction
One independent factor increasing AMPK activity is caloric restriction (CR)115. CR being defined as moderate restriction (20–40%) of caloric intake compared to ad libitum feeding. CR has shown promising results in increasing longevity and reducing the risk factors for cardiovascular disease, diabetes and cancer116. The exact mechanism of AMPK activation remains elusive, but the general belief is that CR simulates an energy-deficient state that is sensed by AMPK as an increase in the AMP:ATP ratio. Resveratrol, a small polyphenol present in red grapes, was found to have CR-like metabolic effects primarily through activation of AMPK117. While utilizing CR as an inducer of AMPK seems impractical in the setting of acute myocardial infarction, it is nonetheless a weak one. On a clinical note, current medical practices entail that most patients admitted for ACS and planned intervention be in a fasting state to reduce the risk of aspiration. Further research in CR is needed to be able to answer when to resume feeds in those patients and how aggressive we should be controlling glucose levels in the diabetic subgroup.
11.2. Preconditioning and human in vivo model for reperfusion injury
Charles Murry, a fellow then in Duke University laboratory, first demonstrated the principle of preconditioning (PC) in canine hearts in 1986. He reported that subjecting the heart to four cycles of 5-min coronary occlusion/5-min reperfusion would make the heart very resistant to infarction from a subsequent lethal ischemic insult119. This seems to be the strongest endogenous protective mechanism of the heart. Later PC was divided into early and late phases, the latter mediated by upregulation of cardioprotective genes by mechanisms that are still unclear. This involved the upregulation of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX2), aldose reductase, 12-Lipoxygenase (12-LO) and Mn-superoxide dismutase (Mn-SOD) leading to enhanced production of NO and PGI2/PGE2 and removal of oxygen free radicals. One established link is that PC activates AMPK and upregulates GLUT4 expression in a PKC-dependent manner120. Others include activation of the RISK pathway and eventual prevention of mPTP formation. The clinical applicability of ischemic PC remains limited because of the obligate need to initiate it before the onset of ischemia which is seldom possible to predict. However, several strategies using AMPK activators have been developed that can be applied before or at the time of reperfusion which mimic ischemic PC.
Worth mentioning is the in vivo model of skeletal muscle preconditioning in humans by Rongen et al.121 from the Netherlands. The value of such experiment is that it was the first one to demonstrate preconditioning in humans with fairly reproducible results. There, ischemia was induced in non-dominant hands by inflating a blood pressure cuff to 200 mmHg followed by intermittent hand gripping till exhaustion for 10 min followed by reperfusion. Annexin A5 scintigraphy was used as a marker for apoptosis. Ischemia–reperfusion injury was then quantified as the percentage difference in uptake between experimental and control hands. Preconditioning was simulated by 10 min of forearm ischemia and a 10-min period of reperfusion without simultaneous exercise. Pharmacological preconditioning was done with intra-arterial adenosine infusion with similar results. Using the same model in subjects with metabolic syndrome, Rongen et al.122 have demonstrated improved reperfusion injury in a crossover study of 4 mg bid of AMPK inducer rosiglitazone vs. placebo for 8 weeks.
Numerous papers have been published worldwide showing promising results regarding utilizing AMPK phosphorylation in ameliorating ischemia/reperfusion injury on various animal models10,14,31,64. Only a handful of these have translated to human research. This in part is due to variations in the protocols used and reproducibility of the published results. Some even argue that using animal models is misleading with the conviction that human myocardium is biochemically more tolerant to ischemia than animal models. If 20 min of total occlusion is how long it takes for irreversible myocardial injury to occur in humans, how much time of acute occlusion should be simulated in disease-free coronaries with no collaterals of mice, rabbits or pigs corrected for their relative metabolisms? This has lead to the Consortium for preclinicAl assESsment of cARdioprotective therapies (CAESAR), an initiative from the NHLBI aimed at the preclinical research118. The plan of which is to perform systematic preclinical testing of cardioprotective therapies using standardized protocols performed by blinded investigators and analyzed by a single statistical core as done for randomized, multicenter clinical trials. Pharmacological agents showing consistent results and statistical power will be promoted to clinical trials.
12. Conclusions
The actions of activated AMPK previously described have generated many questions regarding its potential role in cardiovascular disease. AMPK activation during myocardial ischemia might indeed protect the heart against injury. Additional tremendous applications can include adjuvant therapy to cardiac bypass surgery, protecting donor hearts before transplant and treatment of acute ischemic strokes. We now know that activated AMPK effectively preconditions the heart, improves glucose uptake, glycoysis and fatty acid oxidation to provide ATP. It also stimulates autophagy and inhibits cholesterol and glycogen synthesis to conserve energy during ischemic stress. Other benefits to AMPK activation during ischemia extend beyond cardiomyocytes to improve endothelial metabolism, function and angiogenesis. How much of this response is beneficial and how much is harmful is not clear. How is the cellular response to physiological stress different from infarction? To what degree can we extrapolate the cellular responses in physiological stress situations as exercise and caloric restriction to pathological situations as myocardial infarction without making it harmful? Since AMPK is rapidly inactivated during reperfusion, sustained iatrogenic AMPK activation may prove detrimental. Continued ‘switched on’ autophagy would eventually lead to cell death. Favoring fatty acid over glucose oxidation would exaggerate the uncoupled ATP production from glycolysis leading to increased proton and lactate production, cardiac inefficiency and contractile dysfunction.
If one may formulate a drug for the treatment of myocardial infarction with our current understanding of cellular metabolism, it would be a single-use drug that can be given within the first hour of suspected ACS. It would have the same potency to directly activate AMPK as Compound A-769662, with a similar safety and bioavailability as metformin. It would be a drug selective to AMPK unlike AICAR which seems to inhibit autophagy through other pathways, but has a similar half-life as to not persist long after reperfusion. It would be one that does not cross the blood brain barrier as to not affect hypothalamic AMP-activated protein kinase. And finally it would be a drug that allows fatty acid and glucose oxidation to go hand in hand like perhaps an AMPK activator and just the right amount of insulin.
Ultimately, manufacturing such a drug for clinical use must involve our pharmaceutical friends which, understandably are not excited about this as clinicians are. With the survival rate for U.S. patients hospitalized with MI being approximately 95% using conventional therapy, pharmaceutical companies are less interested in investing into a new single-use drug that must be administered in a timely fashion prior to reperfusion. The market for chronic treatments to survivors of MI is much broader and much more lucrative. Furthermore, enthusiasm for development of such a drug with so many downstream effects would be dampened by the required expense of the clinical trials that would be necessary to obtain approval. Hopefully with the amount of research being invested in cardiac cell metabolism and the CAESAR initiative the pharmaceutical companies will soon pick up the scent.
Acknowledgements
This work was supported by grant awarded by American Heart Association SDG0835169N and grant awarded by American Diabetes Association Basic Science 11-BS-92 to JL.
References
- 1.Mellbin LG, Bjerre M, Thiel S, Hansen TK. Complement activation and prognosis in patients with type 2 diabetes and myocardial infarction: a report from the digami 2 trial. Diabetes Care 2012;35:911–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thiele H, Wohrle J, Hambrecht R, Rittger H, Birkemeyer R, Lauer B, et al. Intracoronary versus intravenous bolus abciximab during primary percutaneous coronary intervention in patients with acute ST-elevation myocardial infarction: a randomised trial. Lancet 2012;379:923–31. [DOI] [PubMed] [Google Scholar]
- 3.Morrison A, Li J. PPAR gamma and AMPK—advantageous targets for myocardial ischemia/reperfusion therapy. Biochem Pharmacol 2011;82:195–200. [DOI] [PubMed] [Google Scholar]
- 4.Young LH. AMP-activated protein kinase conducts the ischemic stress response orchestra. Circulation 2008;117:832–40. [DOI] [PubMed] [Google Scholar]
- 5.Young LH. A crystallized view of AMPK activation. Cell Metab 2009;10:5–6. [DOI] [PubMed] [Google Scholar]
- 6.Hardie DG. Role of AMP-activated protein kinase in the metabolic syndrome and in heart disease. FEBS Lett 2008;582:81–89. [DOI] [PubMed] [Google Scholar]
- 7.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 2011;25: 1895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hardie DG. Signal transduction: how cells sense energy. Nature 2011;472:176–7. [DOI] [PubMed] [Google Scholar]
- 9.Hardie DG. Cell biology. Why starving cells eat themselves. Science 2011;331:410–1. [DOI] [PubMed] [Google Scholar]
- 10.Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 2008;451:578–82. [DOI] [PubMed] [Google Scholar]
- 11.Russell RR 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest 2004;114:495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, et al. Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation 2010;122:282–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Ma H, Tong C, Zhang H, Lawlis GB, Li Y, et al. Overnutrition and maternal obesity in sheep pregnancy alter the JNK-IRS-1 signaling cascades and cardiac function in the fetal heart. FASEB J 2010;24:2066–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med 2005;11:1096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao P, Wang J, Ma H, Xiao Y, He L, Tong C, et al. A newly synthetic chromium complex-chromium (D-phenylalanine)(3) activates AMP-activated protein kinase and stimulates glucose transport. Biochem Pharmacol 2009;77:1002–10. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Coven DL, Miller EJ, Hu X, Young ME, Carling D, et al. Activation of AMPK alpha- and gamma-isoform complexes in the intact ischemic rat heart. Am J Physiol Heart Circ Physiol 2006;291:H1927–34. [DOI] [PubMed] [Google Scholar]
- 17.Baron SJ, Li J, Russell RR 3rd, Neumann D, Miller EJ, Tuerk R, et al. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res 2005;96:337–45. [DOI] [PubMed] [Google Scholar]
- 18.Kemp BE, Oakhill JS, Scott JW. AMPK structure and regulation from three angles. Structure 2007;15:1161–3. [DOI] [PubMed] [Google Scholar]
- 19.Chopra I, Li HF, Wang H, Webster KA. Phosphorylation of the insulin receptor by AMP-activated protein kinase (AMPK) promotes ligand-independent activation of the insulin signalling pathway in rodent muscle. Diabetologia 2012;55:783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young LH, Li J, Baron SJ, Russell RR 3rd. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc Med 2005;15:110–8. [DOI] [PubMed] [Google Scholar]
- 21.Light PE. Familial Wolff-Parkinson-White syndrome: a disease of glycogen storage or ion channel dysfunction? J Cardiovasc Electrophysiol 2006;17:S158–61. [DOI] [PubMed] [Google Scholar]
- 22.da Silva Xavier G, Leclerc I, Varadi A, Tsuboi T, Moule SK, Rutter GA. Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochem J 2003;371:761–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardie DG, Carling D, Carlson M. The AMP-activated/snf1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem 1998;67:821–55. [DOI] [PubMed] [Google Scholar]
- 24.Kim AS, Miller EJ, Wright TM, Li J, Qi D, Atsina K, et al. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol 2011;51:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dolinsky VW, Dyck JR. Role of AMP-activated protein kinase in healthy and diseased hearts. Am J Physiol Heart Circ Physiol 2006;291:H2557–69. [DOI] [PubMed] [Google Scholar]
- 26.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2003;2:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakamoto K, Zarrinpashneh E, Budas GR, Pouleur AC, Dutta A, Prescott AR, et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am J Physiol Endocrinol Metab 2006;290:E780–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altarejos JY, Taniguchi M, Clanachan AS, Lopaschuk GD. Myocardial ischemia differentially regulates LKB1 and an alternate 5’-AMP-activated protein kinase kinase. J Biol Chem 2005;280:183–90. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR 3rd, Young LH. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res 2005;97:872–9. [DOI] [PubMed] [Google Scholar]
- 30.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 2005;280:29060–6. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Yang L, Rezaie AR, Li J. Activated protein c protects against myocardial ischemic/reperfusion injury through AMP-activated protein kinase signaling. J Thromb Haemost 2011;9:1308–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He BJ, Joiner ML, Singh MV, Luczak ED, Swaminathan PD, Koval OM, et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med 2011;17:1610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, et al. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest 2011;121:3277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem 2006;281:25336–43. [DOI] [PubMed] [Google Scholar]
- 35.Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, et al. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med 2000;6:556–63. [DOI] [PubMed] [Google Scholar]
- 36.Goldstein BJ, Scalia RG, Ma XL. Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med 2009;6:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Tao L, Yuan Y, Lau WB, Li R, Lopez BL, et al. Cardioprotective effect of adiponectin is partially mediated by its AMPK-independent antinitrative action. Am J Physiol Endocrinol Metab 2009;297:E384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 2000;20:1595–9. [DOI] [PubMed] [Google Scholar]
- 39.Budanov AV, Karin M. P53 target genes sestrin1 and sestrin2 connect genotoxic stress and MTOR signaling. Cell 2008;134:451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore F, Weekes J, Hardie DG. Evidence that AMP triggers phosphorylation as well as direct allosteric activation of rat liver AMP-activated protein kinase. A sensitive mechanism to protect the cell against ATP depletion. Eur J Biochem 1991;199:691–7. [DOI] [PubMed] [Google Scholar]
- 41.Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci 2010;67:3407–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Hu X, Selvakumar P, Russell RR 3rd, Cushman SW, Holman GD, et al. Role of the nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation in heart muscle. Am J Physiol Endocrinol Metab 2004;287:E834–41. [DOI] [PubMed] [Google Scholar]
- 43.Bogan JS, Hendon N, McKee AE, Tsao TS, Lodish HF. Functional cloning of tug as a regulator of GLUT4 glucose transporter trafficking. Nature 2003;425:727–33. [DOI] [PubMed] [Google Scholar]
- 44.Gong H, Xie J, Zhang N, Yao L, Zhang Y. MEF2a binding to the GLUT4 promoter occurs via an AMPKalpha2-dependent mechanism. Med Sci Sports Exerc 2011;43:1441–50. [DOI] [PubMed] [Google Scholar]
- 45.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol 2000;10:1247–55. [DOI] [PubMed] [Google Scholar]
- 46.Marsin AS, Bouzin C, Bertrand L, Hue L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J Biol Chem 2002;277:30778–83. [DOI] [PubMed] [Google Scholar]
- 47.Hardie DG. The AMP-activated protein kinase pathway-new players upstream and downstream. J Cell Sci 2004;117:5479–87. [DOI] [PubMed] [Google Scholar]
- 48.Heather LC, Clarke K. Metabolism, hypoxia and the diabetic heart. J Mol Cell Cardiol 2011;50:598–605. [DOI] [PubMed] [Google Scholar]
- 49.Hue L, Taegtmeyer H. The randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 2009;297:E578–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 2011;13:376–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bultot L, Guigas B, Von Wilamowitz-Moellendorff A, Maisin L, Vertommen D, Hussain N, et al. AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem J 2012;443:193–203. [DOI] [PubMed] [Google Scholar]
- 52.Randriamboavonjy V, Isaak J, Fromel T, Viollet B, Fisslthaler B, Preissner KT, et al. AMPK alpha2 subunit is involved in platelet signaling, clot retraction, and thrombus stability. Blood 2010;116: 2134–40. [DOI] [PubMed] [Google Scholar]
- 53.Inoki K, Kim J, Guan KL. AMPK and MTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol 2012;52:381–400. [DOI] [PubMed] [Google Scholar]
- 54.Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, et al. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. Elegans. Curr Biol 2007;17:1646–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sengupta A, Molkentin JD, Paik JH, DePinho RA, Yutzey KE. FOXO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J Biol Chem 2011;286: 7468–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 2003;100:8466–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen M, Wang Y, Qu A. PGC-1 alpha accelerates cytosolic Ca2+ clearance without disturbing Ca2+ homeostasis in cardiac myocytes. Biochem Biophys Res Commun 2010;396:894–900. [DOI] [PubMed] [Google Scholar]
- 58.Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 2009;20:98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takagi H, Matsui Y, Hirotani S, Sakoda H, Asano T, Sadoshima J. AMPK mediates autophagy during myocardial ischemia in vivo. Autophagy 2007;3:405–7. [DOI] [PubMed] [Google Scholar]
- 60.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 2005;6:439–48. [DOI] [PubMed] [Google Scholar]
- 61.Kim J, Kundu M, Viollet B, Guan KL. AMPK and MTOR regulate autophagy through direct phosphorylation of ULK1. Nat Cell Biol 2011;13:132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (HATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011;331:456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang J, Li J. Activated protein C: a potential cardioprotective factor against ischemic injury during ischemia/reperfusion. Am J Transl Res 2009;1:381–92. [PMC free article] [PubMed] [Google Scholar]
- 64.Morrison A, Yan X, Tong C, Li J. Acute rosiglitazone treatment is cardioprotective against ischemia-reperfusion injury by modulating AMPK, Akt, and JNK signaling in nondiabetic mice. Am J Physiol Heart Circ Physiol 2011;301:H895–H902. [DOI] [PubMed] [Google Scholar]
- 65.Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation 2007;116:901–9. [DOI] [PubMed] [Google Scholar]
- 66.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovasc Res 2004;61:448–60. [DOI] [PubMed] [Google Scholar]
- 67.Hoole SP, White PA, Read PA, Heck PM, West NE, O’Sullivan M, et al. Coronary collaterals provide a constant scaffold effect on the left ventricle and limit ischemic left ventricular dysfunction in humans. J Appl Physiol 2012;112:1403–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chierchia S, Brunelli C, Simonetti I, Lazzari M, Maseri A. Sequence of events in angina at rest: primary reduction in coronary flow. Circulation 1980;61:759–68. [DOI] [PubMed] [Google Scholar]
- 69.Downey JM, Cohen MV. Reducing infarct size in the setting of acute myocardial infarction. Prog Cardiovasc Dis 2006;48:363–71. [DOI] [PubMed] [Google Scholar]
- 70.Piper HM, Garcia-Dorado D. Cardiac protection takes off. Cardiovasc Res 2009;83:163–4. [DOI] [PubMed] [Google Scholar]
- 71.Lemasters JJ. The mitochondrial permeability transition and the calcium, oxygen and pH paradoxes: one paradox after another. Cardiovasc Res 1999;44:470–3. [DOI] [PubMed] [Google Scholar]
- 72.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res 2003;60:617–25. [DOI] [PubMed] [Google Scholar]
- 73.Erickson JR, He BJ, Grumbach IM, Anderson ME. CaMKII in the cardiovascular system: sensing redox states. Physiol Rev 2011;91:889–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choi SL, Kim SJ, Lee KT, Kim J, Mu J, Birnbaum MJ, et al. The regulation of AMP-activated protein kinase by H2O2. Biochem Biophys Res Commun 2001;287:92–7. [DOI] [PubMed] [Google Scholar]
- 75.Paiva MA, Rutter-Locher Z, Goncalves LM, Providencia LA, Davidson SM, Yellon DM, et al. Enhancing AMPK activation during ischemia protects the diabetic heart against reperfusion injury. Am J Physiol Heart Circ Physiol 2011;300:H2123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a risk for cardioprotection. Heart Fail Rev 2007;12:217–34. [DOI] [PubMed] [Google Scholar]
- 77.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and beclin 1 in mediating autophagy. Circ Res 2007;100:914–22. [DOI] [PubMed] [Google Scholar]
- 78.Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, et al. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J Biol Chem 2005;280:25196–201. [DOI] [PubMed] [Google Scholar]
- 79.Chin JT, Troke JJ, Kimura N, Itoh S, Wang X, Palmer OP, et al. A novel cardioprotective agent in cardiac transplantation: metformin activation of AMP-activated protein kinase decreases acute ischemia-reperfusion injury and chronic rejection. Yale J Biol Med 2011;84:423–32. [PMC free article] [PubMed] [Google Scholar]
- 80.Hardie DG. Neither LKB1 nor AMPK are the direct targets of metformin. Gastroenterology 2006;131:974–5. [DOI] [PubMed] [Google Scholar]
- 81.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002;51:2420–5. [DOI] [PubMed] [Google Scholar]
- 82.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000;348:607–14. [PMC free article] [PubMed] [Google Scholar]
- 83.Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNos-mediated signaling. Diabetes 2008;57:696–705. [DOI] [PubMed] [Google Scholar]
- 84.Retnakaran R, Qi Y, Harris SB, Hanley AJ, Zinman B. Changes over time in glycemic control, insulin sensitivity, and beta-cell function in response to low-dose metformin and thiazolidine-dione combination therapy in patients with impaired glucose tolerance. Diabetes Care 2011;34:1601–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001;108:1167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sasaki H, Asanuma H, Fujita M, Takahama H, Wakeno M, Ito S, et al. Metformin prevents progression of heart failure in dogs: role of AMP-activated protein kinase. Circulation 2009;119:2568–77. [DOI] [PubMed] [Google Scholar]
- 87.Fryer LG, Parbu-Patel A, Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 2002;277:25226–32. [DOI] [PubMed] [Google Scholar]
- 88.Daimon M, Oizumi T, Saitoh T, Kameda W, Hirata A, Yama-guchi H, et al. Decreased serum levels of adiponectin are a risk factor for the progression to type 2 diabetes in the Japanese population: the funagata study. Diabetes Care 2003;26:2015–20. [DOI] [PubMed] [Google Scholar]
- 89.Guo Z, Xia Z, Yuen VG, McNeill JH. Cardiac expression of adiponectin and its receptors in streptozotocin-induced diabetic rats. Metabolism 2007;56:1363–71. [DOI] [PubMed] [Google Scholar]
- 90.Young PW, Buckle DR, Cantello BC, Chapman H, Clapham JC, Coyle PJ, et al. Identification of high-affinity binding sites for the insulin sensitizer rosiglitazone (BRL-49653) in rodent and human adipocytes using a radioiodinated ligand for peroxisomal proliferator-activated receptor gamma. J Pharmacol Exp Ther 1998;284: 751–9. [PubMed] [Google Scholar]
- 91.Kilter H, Werner M, Roggia C, Reil JC, Schafers HJ, Kintscher U, et al. The PPAR-gamma agonist rosiglitazone facilitates Akt rephosphorylation and inhibits apoptosis in cardiomyocytes during hypoxia/reoxygenation. Diabetes Obes Metab 2009;11:1060–7. [DOI] [PubMed] [Google Scholar]
- 92.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007;356:2457–71. [DOI] [PubMed] [Google Scholar]
- 93.Stulc T, Ceska R. Rosiglitazone in the prevention of diabetes and cardiovascular disease: dream or reality?. Med Sci Monit 2008;14:45–7. [PubMed] [Google Scholar]
- 94.Esmon CT. The protein C pathway. Chest 2003;124:26–32. [DOI] [PubMed] [Google Scholar]
- 95.Yang L, Bae JS, Manithody C, Rezaie AR. Identification of a specific exosite on activated protein C for interaction with protease-activated receptor 1. J Biol Chem 2007;282:25493–500. [DOI] [PubMed] [Google Scholar]
- 96.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells?. Eur J Biochem 1995;229:558–65. [DOI] [PubMed] [Google Scholar]
- 97.Zhang L, Frederich M, He H, Balschi JA. Relationship between 5-aminoimidazole-4-carboxamide-ribotide and AMP-activated protein kinase activity in the perfused mouse heart. Am J Physiol Heart Circ Physiol 2006;290:1235–43. [DOI] [PubMed] [Google Scholar]
- 98.Leung JM, Stanley T 3rd, Mathew J, Curling P, Barash P, Salmenpera M, et al. An initial multicenter, randomized controlled trial on the safety and efficacy of acadesine in patients undergoing coronary artery bypass graft surgery. Spi research group. Anesth Analg 1994;78:420–34. [DOI] [PubMed] [Google Scholar]
- 99.Jhun BS, Jin Q, Oh YT, Kim SS, Kong Y, Cho YH, et al. 5-aminoimidazole-4-carboxamide riboside suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of phosphatidylinositol 3-kinase/Akt activation in raw 264.7 murine macrophages. Biochem Biophys Res Commun 2004;318:372–80. [DOI] [PubMed] [Google Scholar]
- 100.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 2006;3:403–16. [DOI] [PubMed] [Google Scholar]
- 101.Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule a-769662, a member of the thienopyridone family. J Biol Chem 2007;282:32539–48. [DOI] [PubMed] [Google Scholar]
- 102.Jorgensen WL, Trofimov A, Du X, Hare AA, Leng L, Bucala R. Benzisothiazolones as modulators of macrophage migration inhibitory factor. Bioorg Med Chem Lett 2011;21:4545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Heart protection study collaborative group. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002;360:7–22.12114036 [Google Scholar]
- 104.Mora S, Ridker PM. Justification for the use of statins in primary prevention: an intervention trial evaluating rosuvastatin (jupiter)-can C-reactive protein be used to z target statin therapy in primary prevention?. Am J Cardiol 2006;97:33–41. [DOI] [PubMed] [Google Scholar]
- 105.Sun W, Lee TS, Zhu M, Gu C, Wang Y, Zhu Y, et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation 2006;114:2655–62. [DOI] [PubMed] [Google Scholar]
- 106.Rossoni LV, Wareing M, Wenceslau CF, Al-Abri M, Cobb C, Austin C. Acute simvastatin increases endothelial nitric oxide synthase phosphorylation via AMP-activated protein kinase and reduces contractility of isolated rat mesenteric resistance arteries. Clin Sci 2011;121:449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 2007;67:6745–52. [DOI] [PubMed] [Google Scholar]
- 108.Bergeron R, Previs SF, Cline GW, Perret P, Russell RR 3rd, Young LH, et al. Effect of 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese zucker rats. Diabetes 2001;50:1076–82. [DOI] [PubMed] [Google Scholar]
- 109.Dirksen MT, Laarman GJ, Simoons ML, Duncker DJ. Reperfusion injury in humans: a review of clinical trials on reperfusion injury inhibitory strategies. Cardiovasc Res 2007;74:343–55. [DOI] [PubMed] [Google Scholar]
- 110.Jonassen AK, Sack MN, Mjos OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and P70s6 kinase cell-survival signaling. Circ Res 2001;89:1191–8. [DOI] [PubMed] [Google Scholar]
- 111.Mehta SR, Yusuf S, Diaz R, Zhu J, Pais P, Xavier D, et al. Effect of glucose-insulin-potassium infusion on mortality in patients with acute ST-segment elevation myocardial infarction: the create-ecla randomized controlled trial. J Am Med Assoc 2005;293:437–46. [DOI] [PubMed] [Google Scholar]
- 112.Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, et al. Insulin antagonizes ischemia-induced thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of ser485/491. J Biol Chem 2006;281:5335–40. [DOI] [PubMed] [Google Scholar]
- 113.Folmes CD, Clanachan AS, Lopaschuk GD. Fatty acids attenuate insulin regulation of 5’-AMP-activated protein kinase and insulin cardioprotection after ischemia. Circ Res 2006;99:61–8. [DOI] [PubMed] [Google Scholar]
- 114.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2a by palmitate inhibits AMP-activated protein kinase. J Biol Chem 2007;282:9777–88. [DOI] [PubMed] [Google Scholar]
- 115.Canto C, Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiology 2011;26:214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fontana L, Partridge L, Longo VD. Extending healthy life span-from yeast to humans. Science 2010;328:321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Um JH, Park SJ, Kang H, Yang S, Foretz M, McBurney MW, et al. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 2010;59:554–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lefer DJ, Bolli R. Development of an nih consortium for preclinical assessment of cardioprotective therapies (caesar): a paradigm shift in studies of infarct size limitation. J Cardiovasc Pharmacol Ther 2011;16:332–9. [DOI] [PubMed] [Google Scholar]
- 119.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986;74:1124–36. [DOI] [PubMed] [Google Scholar]
- 120.Nishino Y, Miura T, Miki T, Sakamoto J, Nakamura Y, Ikeda Y, et al. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc Res 2004;61:610–9. [DOI] [PubMed] [Google Scholar]
- 121.Rongen GA, Oyen WJ, Ramakers BP, Riksen NP, Boerman OC, Steinmetz N, et al. Annexin a5 scintigraphy of forearm as a novel in vivo model of skeletal muscle preconditioning in humans. Circulation 2005;111:173–8. [DOI] [PubMed] [Google Scholar]
- 122.Rennings AJ, Meijer P, van Uden DJ, Tack CJ, Smits P, Boerman OC, et al. Rosiglitazone reduces ischaemia-reperfusion injury in patients with the metabolic syndrome. Eur Heart J 2010;31:983. [DOI] [PubMed] [Google Scholar]