

Abstract
BRCA1- and BRCA2-associated hereditary breast and ovarian cancer syndromes are among the best-known and most extensively studied hereditary cancer syndromes. Nevertheless, many patients who proved negative at BRCA genetic testing bring pathogenic mutations in other suppressor genes and oncogenes associated with hereditary breast and/or ovarian cancers. These genes include TP53 in Li–Fraumeni syndrome, PTEN in Cowden syndrome, mismatch repair (MMR) genes in Lynch syndrome, CDH1 in diffuse gastric cancer syndrome, STK11 in Peutz–Jeghers syndrome, and NF1 in neurofibromatosis type 1 syndrome. To these, several other genes can be added that act jointly with BRCA1 and BRCA2 in the double-strand break repair system, such as PALB2, ATM, CHEK2, NBN, BRIP1, RAD51C, and RAD51D. Management of primary and secondary cancer prevention in these hereditary cancer syndromes is crucial. In particular, secondary prevention by screening aims to discover precancerous lesions or cancers at their initial stages because early detection could allow for effective treatment and a full recovery. The present review aims to summarize the available literature and suggest proper screening strategies for hereditary breast and/or ovarian cancer syndromes other than BRCA.
1. Introduction
In hereditary cancer syndromes (HCSs), inherited mutations lead to an increased risk of developing certain tumors, frequently at an earlier age than in the rest of the population [1]. Elevated cancer risk is usually due to a mutation in a single gene involved in cell cycle regulation or in DNA damage repair mechanisms (Figure 1). The most widely known HCSs include hereditary breast and ovarian cancer syndromes due to mutations in the BRCA1/2 genes [2, 3], Li–Fraumeni syndrome due to mutations in TP53 [4], Cowden syndrome due to mutations in PTEN [5], Lynch syndrome, in which mutations in the DNA mismatch repair system are involved [6, 7], diffuse gastric cancer syndrome caused by CDH1 gene mutation [8], Peutz–Jeghers syndrome caused by mutations in the STK11 [9] gene, and neurofibromatosis type 1 syndrome caused by NF1 mutations [10]. Additionally, pathogenic alterations in PALB2 [11], ATM [12], CHEK2 [13], and NBN [14] are correlated with an increased risk for breast cancer and/or other cancers, whereas other genes such as BRIP1, RAD51C, and RAD51D are associated with an increased ovarian cancer risk [15].
Figure 1.
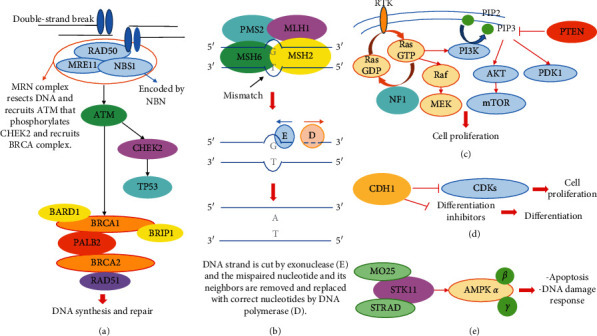
Molecular pathways involved in hereditary cancer risk. Susceptibility genes described in the text are reported in bold. G: guanine, T: thymine, A: adenine, E: exonuclease, D: DNA polymerase, RTK: receptor tyrosine kinase, PIP2: phosphatidylinositol 4,5-bisphosphate, PIP3: phosphatidylinositol (3,4,5)-trisphosphate, GDP: guanosine diphosphate, GTP: guanosine triphosphate, CDKs; cyclin-dependent kinases, and AMPK: 5′ adenosine monophosphate-activated protein kinase. (a) Homologus recombination (HR), (b) mismatch repair (MMR), (c) PTEN and NF1 pathways, (d) CDH1 pathway, and (e) STK11 pathway.
Management of cancer prevention is crucial in HCSs. Cancer prevention can be divided into primary and secondary strategies [16–23]. The aim of the primary prevention is to avoid cancer development by strategies including health counselling and education, environmental controls, prophylactic surgery, and chemoprevention. Secondary prevention by screening aims to discover precancerous lesions or cancers at their initial stages because early detection could allow for an effective treatment and full recovery. Strategies of primary and secondary cancer prevention are well established in the setting of BRCA-associated breast and ovarian cancer. For all other syndromes, on the other hand, the most appropriate screening protocol is still debated.
This review aims to summarize the available literature and suggest proper screening strategies for hereditary breast and/or ovarian cancer syndromes other than those associated with BRCA mutations.
2. Li–Fraumeni Syndrome
Li–Fraumeni syndrome is a rare autosomal dominant cancer predisposition syndrome that involves a germline mutation of the tumor protein 53 (TP53 gene) [4]. The estimated prevalence of pathogenetic germline TP53 mutations ranges from 1/10,000 to 1/25,000 in the UK and is estimated at 1/20,000 in the US [24]. The lifetime cancer risk in individuals with Li–Fraumeni syndrome is ≥70% for men and ≥90% for women [25]. Five cancer types account for the majority of Li–Fraumeni tumors: adrenocortical carcinomas, breast cancer, central nervous system tumors, osteosarcomas, and soft-tissue sarcomas [26]. Individuals with Li–Fraumeni syndrome are also at an increased risk of developing hematologic tumors (leukaemia and lymphomas), gastrointestinal cancers, gynecological tumors, and melanoma [4].
Surveillance recommendations for individuals with Li–Fraumeni syndrome are primarily based on the “Toronto protocol” [27]. For breast cancer, screening recommendations advise starting with clinical breast examination once in every 6–12 months from the age of 20. Annual breast MRI screening with contrast is suggested from 20 to 75 years of age. Given the increased sensitivity to ionizing radiation and the increased risk for radiation-induced malignancies in patients with germline pathogenic TP53 variants, there are concerns about the safety of repeated mammograms. There is no consensus in the literature, but in light of the limited additional sensitivity of mammography when MRI and alternating whole-body diffusion-weighted MRI are used, risks seem to outweigh benefits [28–30]. In case of family history of breast cancer diagnosed earlier than 20 years of age, breast MRI might start five years prior to the earliest age of diagnosis. Although there are no data regarding risk-reduction surgery in women with Li–Fraumeni syndrome, the option of risk-reducing bilateral mastectomy should be considered and discussed with female patients [6, 28]. Concerning gastrointestinal cancer, colonoscopy and upper endoscopy should be performed once in every 2–5 years starting from 25 years of age or five years prior to the earliest case of colorectal cancer in the family. Moreover, annual dermatologic examination is recommended from 18 years of age due to increased skin cancer risk, although less well-defined.
As regards many of the other cancers associated with Li–Fraumeni syndrome, early symptom-based detection is quite difficult. General recommendations include complete physical examination (including blood pressure evaluation, full neurologic exams, assessment of growth, sudden weight gain or loss, Cushingoid appearance, or signs of virilization in children) once in every 3–4 months until the age of 18 and then once in every six months. Annual whole-body diffusion-weighted MRI could allow for early detection of adrenocortical carcinomas and sarcomas, based on the results of multiple international trials [27, 31, 32]. As far as the central nervous system is concerned, the Toronto protocol with modifications [27] recommends annual brain MRI: first, MRI with contrast and then without contrast if previous MRI is normal and no new abnormality has been detected, in order to minimize the potential for gadolinium accumulation in the basal ganglia in individuals undergoing multiple enhanced MRIs [28]. Periodic blood tests can be considered in those at increased risk for myelodysplastic syndrome or leukaemia due to prior cancer treatments [28].
3. Cowden Syndrome
Cowden syndrome is the most prevalent PTEN hamartoma tumor syndrome associated with multiple hamartomatous and/or cancerous lesions in the skin, mucous membranes, thyroid, breast, endometrium, kidney, and brain [33]. Affected individuals usually have macrocephaly, trichilemmomas, and papillomatous papules, and the syndrome becomes apparent by the late 20s [5]. The estimated incidence of Cowden syndrome is 1/200,000, but it is likely to be underestimated due to the difficulties of making a clinical diagnosis of the disease [34]. Cowden syndrome is an autosomal dominant disorder due to germline PTEN mutation in 80% of cases [35].
The lifetime risk of developing breast cancer is 85%, with an average age at diagnosis between 38 and 46 years [5]. NCCN guidelines [6] recommend clinical breast examination once in every six months beginning at 25 years of age and annual mammogram and breast MRI screening with contrast starting at 30–35 years of age. However, screening should start 5–10 years prior to the earliest case of breast cancer in the family. Although there are no data regarding risk-reduction surgery in women with Cowden syndrome, the option of risk-reducing bilateral mastectomy should be considered.
The lifetime risk for thyroid cancer (usually follicular, rarely papillary) is approximately 35% [36]. Annual thyroid ultrasound from the time of diagnosis, including childhood, should be performed according to NCCN recommendations [6].
The risk for endometrial cancer may be close to 28% [36]. There are no data on screening for endometrial cancer. Routine transvaginal ultrasound has low sensitivity and specificity, especially in premenopausal women, whereas endometrial biopsy is highly sensitive and specific, but invasive. Therefore, screening with endometrial biopsy once in every 1–2 years may be considered, while hysterectomy should be discussed on a case-by-case basis, according to NCCN guidelines [6].
Half as many individuals with Cowden syndrome have adenomatous or hyperplastic colorectal polyps associated with early-onset (<50 years of age) colorectal cancer in 13% of patients [37]. Routine colonoscopy should be performed from the age of 35 once in every five years or more frequently, if the patient is symptomatic or polyps are found. However, screening should start 5–10 years before the age of the earliest case of colorectal cancer in the family.
Renal carcinoma may be present in up to 30% of patients. Melanoma skin cancer is also increased in patients with Cowden disease and may occur in 5% of patients [38]. Yearly to biennial renal imaging (preferably through CT or MRI) beginning at the age of 40 is recommended to screen renal cell carcinoma, while yearly dermatologic evaluation could help to detect early melanoma.
Brain tumors as well as vascular malformations occasionally affect individuals with Cowden syndrome. Cerebellar dysplastic gangliocytoma (Lhermitte-Duclos disease), a rare central nervous system tumor, can also be found in Cowden syndrome. However, the risk of developing these conditions is not well defined [39]. In the presence of neurological symptoms, especially in children, assessment of psychomotor abilities and brain MRI should be performed [6].
4. Lynch Syndrome
Lynch syndrome is caused by a germline mutation in one of four DNA mismatch repair genes (MLH1, MSH2, MSH6, or PMS2) [40] or deletions in the EPCAM gene resulting in MSH2 silencing [41]. The estimated population frequency is 1 : 370 to 1 : 2,000 in Western populations [42]. Lynch syndrome is characterized by an increased lifetime risk for colorectal cancer (48–57% vs. 4.5%), endometrial cancer (43–57% vs. 2.7%), and other cancers including stomach (up to 13%), ovary (up to 24%), small bowel, hepatobiliary tract, urinary tract, brain, and skin [43].
Guidelines for cancer screening in patients with Lynch syndrome have been proposed by several groups including the American College of Gastroenterology, United States Multi-Society Task Force on Colorectal Cancer [44], European Hereditary Tumor Group [45], American Society of Clinical Oncology [46], and National Comprehensive Cancer Network [6].
Colonoscopy is recommended once in every 1–2 years starting from 20 to 25 years of age or 2–5 years before than the youngest diagnosis age in the family. Moreover, chromoendoscopy is a promising technique that could facilitate the detection of lesions and flat adenomas [47].
Regarding gynaecologic cancers, lifetime risk varies according to mutated gene and patient's age [48]. Transvaginal ultrasound and serum CA-125 testing were shown to be neither sufficiently sensitive nor specific to warrant a routine recommendation for early detection of endometrial and ovarian cancers. However, they may be required at clinicians' discretion in assessing tumor risk on a case-by-case basis [6]. Annual endometrial biopsy can be used as a screening tool for endometrial cancer because of its high sensitivity and sensibility [6]. Total hysterectomy is an option that may be considered to reduce the risk of endometrial cancer in women with Lynch syndrome; likewise, bilateral salpingo-oophorectomy may reduce the incidence of ovarian cancer [49]. Since there is no effective screening for gynaecologic cancers, women should be educated on relevant symptoms such as abnormal uterine bleeding, pelvic or abdominal pain, bloating, dyspepsia, or increased urinary frequency or urgency.
Regarding gastric, duodenal, and more distant small bowel cancer, there is insufficient evidence to recommend surveillance [50], except for individuals with relevant family history of these tumors [51]. Besides, esophagogastroduodenoscopy extended to the duodenum or into the jejunum once in every 3–5 years starting from 40 years of age should be considered in case of mutation in MLH1, MSH2, or EPCAM [45]. Considering that infection with Helicobacter pylori is a cause of gastric cancer, testing and treating for this bacterium is suggested [46].
There is no clear evidence to support screening for urinary tract cancer, except for individuals with a family history of urothelial cancer or MSH2 mutation who may benefit from annual urinalysis beginning at 30–35 years of age [6]. The International Cancer of the Pancreas Screening (CAPS) Consortium recommends screening for pancreatic cancer in patients with Lynch syndrome and one first-degree relative with pancreatic cancer [52]. Nonetheless, no protocol for pancreatic cancer screening has been established yet. The NCCN panel therefore recommends MRI and endoscopic ultrasonography as screening modalities to be performed at high-volume centres with multidisciplinary teams and preferably in a research protocol [6]. By reason of the increased risk for brain cancer, in addition, annual physical and neurologic examination from 25 to 30 years of age may be considered, although no data support this practice [6].
Some studies have shown that mutations in MLH1 and MSH2, and less frequently in PMS2 and MSH6, could be associated with increased breast cancer risk [53–55]. Nevertheless, no specific recommendations for breast screening in women with Lynch syndrome have been made available so far, beyond those offered to the average risk population [6]. Finally, a study suggested an increased risk for prostate cancer in men with Lynch syndrome [56]. However, there is no sufficient evidence to recommend different prostate cancer screening from the rest of the population [6].
5. Diffuse Gastric Cancer Syndrome
Hereditary diffuse gastric cancer is a cancer susceptibility syndrome defined by the early onset of diffuse gastric cancer with or without lobular breast cancer. It is mainly caused by germline mutations in the epithelial cadherin (CDH1) gene. A most serious problem is that genetic diagnosis remains unknown in up to 60% of patients [57]. The risk for symptomatic gastric cancer, occurring by the age of 80, ranged between 67 and 70% in men and 56 and 83% in women, whereas the risk for breast cancer among women, especially the lobular phenotype, amounted to 52% [8].
Prophylactic total gastrectomy is strongly recommended between 18 and 40 years of age [58]. Screening by esophagogastroduodenoscopy with multiple random biopsy once in every 6–12 months should be reserved to patients who cannot undergo prophylactic total gastrectomy, since upper endoscopy may not detect early precursor lesions [59].
In women, finally, annual mammogram with consideration of breast MRI with contrast beginning at the age of 30 (or prior to that, with a family history of breast cancer before the age of 25) is recommended by NCCN guidelines [6]. However, given the high lifetime risk and the low sensitivity of mammography for lobular breast cancer, the added value of MRI over mammography seems high in this situation [60]. Risk-reducing mastectomy may be discussed with these carriers, depending on family history [6].
6. Peutz–Jeghers Syndrome
Germline pathogenic alterations in STK11 are associated with Peutz–Jeghers syndrome. This is an autosomal dominant disorder characterized by hamartomatous gastrointestinal polyps, mucocutaneous pigmentation, and an increased risk of colorectal, gastric, pancreatic, gallbladder, small bowel, gynaecologic (uterus, cervix, and ovary), breast, testicular, and lung cancers [9].
Regarding the risk of colorectal, gastric, and small bowel cancers, colonoscopy, upper endoscopy, and capsule endoscopy should be recommended once in every 2–3 years, starting from the late teens [61]. Moreover, the American College of Gastroenterology recommends magnetic resonance cholangiopancreatography with contrast or endoscopic ultrasound once in every 1–2 years from 30 years of age, in order to detect early pancreatic cancer [62].
For breast cancer, annual mammogram and breast MRI screening with contrast should be recommended from 25 years of age. In women, transvaginal ultrasound, serum CA-125, and pelvic exam with Pap smear should be proposed annually beginning at 18 years of age. No data on the benefit of risk-reducing mastectomy are available, so that this procedure may be considered based on family history [6].
In males, annual testicular exam and, subsequently, ultrasound in case of symptomaticity or abnormality on exam are suggested from birth to the teen years [62].
7. Neurofibromatosis Type 1
Pathogenic variants of NF1 cause neurofibromatosis type 1. This is an autosomal dominant HCS associated with increased risk for nervous system tumors (especially malignant peripheral nerve sheath tumors), gastrointestinal stromal tumors, and breast cancer [10].
The American Academy of Paediatrics and the American College of Medical Genetics and Genomics have published guidelines for children and adult surveillance [63, 64]. Annual physical examination, annual ophthalmologic examination in children (less frequently in adults), regular developmental assessment in children, regular blood pressure monitoring, and MRI for followup of clinically suspected intracranial tumors and other internal tumors are recommended. Additionally, annual mammography, possibly associated with breast MRI, is suggested between 30 and 50 years of age [65]. After 50 years of age, breast cancer risk in women with NF1 mutation becomes similar to that of the rest of the population. Breast MRI could therefore be discontinued [66], while mammography can be performed at longer intervals. No data on the benefit of risk-reducing mastectomy are available, so that this procedure may be considered based on family history [6].
8. Other Breast and Ovarian Cancer Predisposition Genes
In addition to the known high-penetrance pathogenic variants of BRCA1/2, mutations in other intermediate or low-penetrant genes can increase the risk of breast and/or ovarian cancer. According to a retrospective analysis, these mutations account for 7.4% of patients who met the NCCN criteria for BRCA1/2 mutation test [6]. The most common are PALB2, ATM, and CHEK2 [67].
8.1. PALB2
It is estimated that 0.6%–3% of patients with breast cancer harbour a mutation in PALB2 (partner and localizer of BRCA2) [11, 15, 68]. Moreover, women carrying pathogenetic variants of PALB2 have a 35% lifetime risk to develop breast cancer by 70 years of age. The higher the number of relatives affected, the higher the risk [69]. Breast ultrasound and MRI are recommended yearly from 25 to 29 years of age, alternating once in every six months. Annual mammogram and breast MRI screening are alternatively recommended once in every six months, starting at 30 until 65 years of age [6].
Some studies highlight a possible association between PALB2 mutations and ovarian cancer. Recently, PALB2 has also been reported to be a new pancreatic cancer susceptibility gene [70]. However, the associated risks are unclear and not well-estimated. Furthermore, no effective screening method is available for ovarian or pancreatic cancer. Screening and/or risk-reducing surgery should be individualized based on familial history [71].
8.2. ATM, CHEK2, NBN, and BARD1
Individuals carrying heterozygous pathogenic variants in ATM have a 33% cumulative lifetime risk for breast cancer by 80 years of age [12]. Mammogram with consideration of breast MRI is recommended yearly from 40 years of age [6]. No data are available on the benefit of risk-reducing mastectomy, so that this procedure may be considered based on family history [6]. ATM heterozygous pathogenic variants have been reported in some cases of familial ovarian [15], pancreatic [72], and prostate [73] cancer. Screening for pancreatic and ovarian cancers in carriers of ATM pathogenic variants is not recommended in the absence of familial antecedents, while men should be encouraged to participate in prostate cancer screening [6]. Homozygous or compound heterozygous ATM mutations cause ataxia telangiectasia, a syndrome characterized by progressive cerebellar ataxia, oculomotor apraxia, immunodeficiency, and general increased risk of malignancies [74].
The rate of CHEK2 germline mutation is higher in Northern European countries than in Mediterranean ones. Certain mutations in the CHEK2 gene (c.1100delC and I157T) are associated with increased breast cancer risk, with a cumulative lifetime risk ranging from 28% to 37% depending on family history [13, 75]. Mammogram and breast MRI once a year start at 40 years of age [6]. No data are available on the benefit of risk-reducing mastectomy, so that this procedure may be considered based on family history [6]. Within families carrying pathogenic CHEK2 variants, there is also an increased risk of other malignancies including colon, prostate, kidney, bladder, and thyroid cancers [76], with the vast majority of data for the c1100delC variant. Colonoscopy once in every five years, beginning from 40 years of age or 10 years earlier than the age of diagnosis for any first-degree relative with colorectal cancer, is recommended in individuals carrying CHEK2 mutations [6]. Currently, there are no specific medical management guidelines to address the possible risk of developing prostate, kidney, bladder, and thyroid cancer in these individuals.
Individuals with slavic founder heterozygous NBN mutation 675del5 have an increased risk of developing numerous types of cancer, including breast (up to 30% at 80 years of age) and ovarian cancer. Moreover, an unestimated increased risk of prostate cancer at 80 years of age is also apparent in men [77]. The presence of biallelic hypomorphic NBN mutations leads to the Nijmegen breakage syndrome, a rare autosomal recessive syndrome of chromosomal instability mainly characterized by microcephaly at birth, combined immunodeficiency, and predisposition to malignancies. Approximately 40% of the affected patients develop a malignancy before the age of 21 [14]. In slavic mutation carriers, breast MRI is recommended yearly from 40 years of age, whereas no recommendations are provided for ovarian and prostate cancer screening [6]. No data are available on the benefit of risk-reducing mastectomy, so that this procedure may be considered based on family history [6].
Deleterious BARD1 germline variants are significantly associated with early-onset breast cancer, according to recent studies [78, 79]. On the grounds of these data, intensified breast cancer screening programs should be offered to women carrying pathogenic BARD1 gene variants. However, the starting age and the frequency of mammogram and/or breast MRI have not been established yet.
8.3. BRIP1, RAD51C, and RAD51D
Mutations in BRIP1, RAD51C, or RAD51D are associated with an increased risk of developing ovarian cancer. The prevalence rate of BRIP1, RAD51C, or RAD51D pathogenic variants is about 1% in women with ovarian cancer [15]. Nevertheless, there are no data supporting screening for ovarian cancer. Transvaginal ultrasound and serum CA-125 testing have not been shown to be sufficiently sensitive or specific, even in the setting of women at high risk of ovarian cancer due to an inherited mutation [6]. NCCN guidelines recommend that risk-reducing salpingo-oophorectomy should be considered beginning at 45–50 years of age [6]. At present, BRIP1, RAD51C, or RAD51D are not associated with an increased risk for breast cancer [7].
9. Conclusions
In the last years, a large number of large case-control studies have shown the correlation between mutations in some genes and an increased risk of developing breast and/or ovarian cancer, explaining tumor recurrence in those families where mutations in BRCA1/2 were not found. However, the lifetime risk in case of low-penetrant genes has not been defined yet and additional prospective studies are needed to establish a more customised screening program for carriers. Summary of the recommendations for each predisposition gene discussed in the present review is reported in Table 1.
Table 1.
Summary of the recommendations for each predisposition gene.
| Predisposition genes | Cancer risk | Lifetime risk | Surveillance |
|---|---|---|---|
| High-penetrance genes for breast and/or ovarian cancer | |||
|
| |||
| TP53 | Adrenocortical gland | 6–13% [25] | Ultrasound of abdomen and pelvis: every 3–4 mos, birth to age 18 yrs [27] |
| Breast | 54% [25] | Clinical breast examination: every 6–12 mos, age ≥ 20 yrs | |
| Breast MRI screening with contrast (with or without mammogram): annually, age 20–75 yrs [27]∗ | |||
| Central nervous system | 6–19% [25] | Neurologic exam: annually, all ages | |
| Brain MRI: annually [27] | |||
| Sarcomas | 5–22% [25] | Whole-body MRI: annually, all ages | |
| Ultrasound of abdomen and pelvis: annually, age ≥18 yrs [27] | |||
| Hematologic tumors | NA | Periodic blood test if increased risk for myelodysplastic syndrome or leukaemia [28] | |
| Gastrointestinal system | NA | Upper endoscopy and colonoscopy: every 2–5 yrs, age ≥25 yrs [27] | |
| Skin | NA | Dermatologic exam: annually, age ≥18 yrs [27] | |
|
| |||
| PTEN | Breast | 85% [5] | Clinical breast examination: every 6 mos, age ≥ 25 yrs |
| Mammogram and breast MRI with contrast: annually, age 30–75 yrs [6]∗ | |||
| Thyroid | 35% [36] | Ultrasound of thyroid: annually, all ages [6] | |
| Endometrium | 28% [36] | Endometrial biopsy: every 1–2 yrs [6]∗ | |
| Colon and rectum | 9% [36] | Colonoscopy: every 5 yrs, age ≥ 35 yrs [6] | |
| Kidney | 30% [36] | CT or MRI of abdomen: every 1–2 yrs, age ≥ 40 yrs [6] | |
| Melanoma | 5% [38] | Dermatologic exam: annually, age ≥18 yrs [38] | |
|
| |||
| CDH1 | Stomach | 56–83% [8] | Upper endoscopy: every 6–12 mos, age ≥ 18 yrs [59]∗ |
| Breast | 52% [8] | Mammogram and breast MRI with contrast: annually, age ≥ 30 yrs [6]∗ | |
|
| |||
| STK11 | Colon and rectum | 39% [9] | Colonoscopy: every 2–3 yrs, age ≥ 18 yrs [61] |
| Stomach | 29% [9] | Upper endoscopy: every 2–3 yrs, age ≥ 18 yrs [61] | |
| Small bowel | 13% [9] | Capsule endoscopy: every 2–3 yrs, age ≥ 18 yrs [61] | |
| Pancreas | 11–36% [9] | MR cholangiopancreatography with contrast or endoscopic ultrasound: every 1–2 yrs, age ≥ 30 yrs [62] | |
| Breast | 32–54% [9] | Clinical breast examination: every 6 mos, age ≥ 20 yrs | |
| Mammogram and breast MRI with contrast: annually, age ≥ 25 yrs [6]∗ | |||
| Ovary, cervix, and uterus | 9–21% [9] | Transvaginal ultrasound, serum CA 125, pelvic exam with pap smear: annually, age ≥ 18 yrs [6] | |
| Testis | 9% [9] | Testicular exam: annually, until 18 yrs [62] | |
| Lung | 7–17% [9] | Not recommended | |
| Low-/moderate-penetrance genes for breast and/or ovarian cancer | |||
|
| |||
| PALB2 | Breast | 35% [69] | Mammogram and breast MRI with contrast: annually, age ≥ 30 yrs [6]∗ |
| Ovary, pancreas | NA | Not recommended | |
|
| |||
| CHEK2 | Breast | 28–37% [13, 75] | Mammogram and breast MRI with contrast: annually, age ≥ 40 yrs [6]∗ |
| Colon | NA | Colonoscopy: every 5 yrs, age ≥ 40 yrs [6] | |
| Prostate, kidney, bladder, and thyroid | NA | Not recommended | |
|
| |||
| NBN (675del5) | Breast | Up to 30% [77] | Breast MRI with contrast: annually, age ≥ 40 yrs [6]∗ |
| Ovary and prostate | NA | Not recommended | |
|
| |||
| MLH1, MSH2, MSH6, PMS2, EPCAM | Colon and rectum | 48–57% [43] | Colonoscopy: every 1-2 yrs, age ≥ 20–25 yrs [6] |
| Endometrium | 43–57% [43] | Not recommended∗ | |
| Ovary | Up to 24% [43] | Not recommended∗ | |
| Stomach, small bowel | 4–13% [43] | Upper endoscopy: every 3–5 yrs, age ≥ 40 yrs if relevant family history or mutation in MLH1, MSH2 or EPCAM [45, 51] | |
| Hepatobiliary tract | Up to 4% [43] | In research protocol [6] | |
| Urinary tract | Up to 25% [43] | Urinalysis: annually, age ≥ 30–35 yrs if relevant family history or MSH2 mutation [6] | |
| Brain | 1–4% [43] | Physical and neurologic examination: annually, age ≥ 25–30 yrs [6] | |
| Breast (MSH2, MLH1, PMS2, or MSH6) | 11–18% [53–55] | Not recommended | |
| Prostate | NA | Not recommended | |
|
| |||
| ATM | Breast | 33% [12] | Mammogram with consideration of breast MRI with contrast: annually, age ≥ 40 yrs [6]∗ |
| Ovary, prostate, and pancreas | NA | Not recommended | |
| BRIP1, RAD51C, RAD51D | Ovary | Up to 10% [15] | Not recommended∗ |
|
| |||
| NF1 | Nervous system | 8–16% [64] | Physical and eye examination: annually, every age [63, 64] |
| Breast | 17% [64] | Mammogram and breast MRI with contrast: annually, age 30–50 yrs [65]∗ | |
| BARD1 | Breast | NA | Not recommended |
NA = not available, ∗Risk-reducing surgery can be considered based on type of mutation and family history.
Acknowledgments
This work was supported by the Angela Serra Association for Cancer Research that paid the article processing charges.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Garber J. E., Offit K. Hereditary cancer predisposition syndromes. Journal of Clinical Oncology. 2005;23(2):276–292. doi: 10.1200/jco.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 2.Hall J., Lee M., Newman B., et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250(4988):1684–1689. doi: 10.1126/science.2270482. [DOI] [PubMed] [Google Scholar]
- 3.Wooster R., Neuhausen S., Mangion J., et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265(5181):2088–2090. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- 4.Schneider K., Zelley K., Nichols K. E., et al. Li-fraumeni syndrome. In: Adam M. P., Ardinger H. H., Pagon R. A., et al., editors. GeneReviews® [Internet] Seattle, WA, USA: University of Washington; 1999. https://www.ncbi.nlm.nih.gov/books/NBK1311/ [PubMed] [Google Scholar]
- 5.Eng C. PTEN hamartoma tumor syndrome. In: Adam M. P., Ardinger H. H., Pagon R. A., et al., editors. GeneReviews® [Internet] Seattle, WA, USA: University of Washington; 2001. https://www.ncbi.nlm.nih.gov/books/NBK1488/ [PubMed] [Google Scholar]
- 6.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) 2019. Jen kintown, PA, USA: National Comprehensive Cancer Network; 2019. Genetic/familial high-risk assessment: colorectal. [DOI] [PubMed] [Google Scholar]
- 7.Suszynska M., Klonowska K., Jasinska A. J., Kozlowski P. Large-scale meta-analysis of mutations identified in panels of breast/ovarian cancer-related genes - providing evidence of cancer predisposition genes. Gynecologic Oncology. 2019;153(2):452–462. doi: 10.1016/j.ygyno.2019.01.027. [DOI] [PubMed] [Google Scholar]
- 8.Kaurah P., MacMillan A., Boyd N., et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA. 2007;297(21):2360–2372. doi: 10.1001/jama.297.21.2360. [DOI] [PubMed] [Google Scholar]
- 9.McGarrity T. J., Amos C. I., Baker M. J. Peutz-jeghers syndrome. In: Adam M. P., Ardinger H. H., Pagon R. A., et al., editors. GeneReviews® [Internet] Seattle, WA, USA: University of Washington; 2001. https://www.ncbi.nlm.nih.gov/books/NBK1266/ [PubMed] [Google Scholar]
- 10.Friedman J. M. Neurofibromatosis 1. In: Adam M. P., Ardinger H. H., Pagon R. A., et al., editors. GeneReviews® [Internet] Seattle , WA, USA: University of Washington; 1998. https://www.ncbi.nlm.nih.gov/books/NBK1109/ [Google Scholar]
- 11.Couch F. J., Shimelis H., Hu C., et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncology. 2017;3(9):1190–1196. doi: 10.1001/jamaoncol.2017.0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marabelli M., Cheng S.-C., Parmigiani G. Penetrance ofATMGene mutations in breast cancer: a meta-analysis of different measures of risk. Genetic Epidemiology. 2016;40(5):425–431. doi: 10.1002/gepi.21971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cybulski C., Wokołorczyk D., Jakubowska A., et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. Journal of Clinical Oncology. 2011;29(28):3747–3752. doi: 10.1200/jco.2010.34.0778. [DOI] [PubMed] [Google Scholar]
- 14.Chrzanowska K. H., Gregorek H., Dembowska-Bagińska B., Kalina M. A., Digweed M. Nijmegen breakage syndrome (NBS) Orphanet Journal of Rare Diseases. 2012;7(13) doi: 10.1186/1750-1172-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu H.-M., Li S., Black M. H., et al. Association of breast and ovarian cancers with predisposition genes identified by large-scale sequencing. JAMA Oncology. 2019;5(1):51–57. doi: 10.1001/jamaoncol.2018.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toss A., Venturelli M., Molinaro E. Hereditary pancreatic cancer: a retrospective single-center study of 5143 italian families with history of BRCA-related malignancies. Cancers. 2019;11(2):p. E193. doi: 10.3390/cancers11020193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grandi G., Sammarini M., Chiara Del Savio M., Toss A., Facchinetti F. Combined hormonal contraceptives in BRCA gene mutation carriers: why not? The European Journal of Contraception & Reproductive Health Care. 2019;24(6):417–419. doi: 10.1080/13625187.2019.1647335. [DOI] [PubMed] [Google Scholar]
- 18.Cortesi L., Canossi B., Battista R., et al. Breast ultrasonography (BU) in the screening protocol for women at hereditary-familial risk of breast cancer: has the time come to rethink the role of BU according to different risk categories? International Journal of Cancer. 2019;144(5):1001–1009. doi: 10.1002/ijc.31794. [DOI] [PubMed] [Google Scholar]
- 19.Cortesi L., De Matteis E., Toss A., et al. Evaluation of transvaginal ultrasound plus CA-125 measurement and prophylactic salpingo-oophorectomy in women at different risk levels of ovarian cancer: the modena study group cohort study. Oncology. 2017;93(6):377–386. doi: 10.1159/000479155. [DOI] [PubMed] [Google Scholar]
- 20.Toss A., Grandi G., Cagnacci A., et al. The impact of reproductive life on breast cancer risk in women with family history or BRCA mutation. Oncotarget. 2017;8(6):9144–9154. doi: 10.18632/oncotarget.13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Razzaboni E., Toss A., Cortesi L., et al. Acceptability and adherence in a chemoprevention trial among women at increased risk for breast cancer attending the modena familial breast and ovarian cancer center (Italy) The Breast Journal. 2013;19(1):10–21. doi: 10.1111/tbj.12045. [DOI] [PubMed] [Google Scholar]
- 22.Patel V. L., Busch E. L., Friebel T. M., et al. Association of genomic domains in BRCA1 and BRCA2 with prostate cancer risk and aggressiveness. Cancer Research. 2020;80(3):624–683. doi: 10.1158/0008-5472.CAN-19-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nielsen S. M., Eccles D. M., Romero I. L, et al. Genetic testing and clinical management practices for variants in non-BRCA1/2 breast (and breast/ovarian) cancer susceptibility genes: an international survey by the evidence-based Network for the interpretation of germline mutant alleles (ENIGMA) clinical working group. JCO Precision Oncology. 2018;2 doi: 10.1200/PO.18.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamihara J., Schneider K. Li-Fraumeni syndrome. 2014. https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=196.
- 25.Mai P. L., Best A. F., Peters J. A., et al. Risks of first and subsequent cancers amongTP53mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer. 2016;122(23):3673–3681. doi: 10.1002/cncr.30248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guha T., Malkin D., Inherited “. TP53 mutations and the Li-Fraumeni syndrome. Cold Spring Harb Perspect Med. 2017;7(4) doi: 10.1101/cshperspect.a026187.a026187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villani A., Shore A., Wasserman J. D., et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. The Lancet Oncology. 2016;17(9):1295–1305. doi: 10.1016/s1470-2045(16)30249-2. [DOI] [PubMed] [Google Scholar]
- 28.Kratz C. P., Achatz M. I., Brugières L., et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clinical Cancer Research. 2017;23(11):e38–e45. doi: 10.1158/1078-0432.ccr-17-0408. [DOI] [PubMed] [Google Scholar]
- 29.Gondim G. R. M., Formiga M. N. C., Castro D. G., et al. Adjuvant radiation therapy in patients with breast cancer and Li-Fraumeni syndrome: oncologic results and incidence of second neoplasms. Journal of Clinical Oncology. 2018;36(15) doi: 10.1200/jco.2018.36.15_suppl.e12589. [DOI] [Google Scholar]
- 30.Heymann S., Delaloge S., Rahal A., et al. Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiation Oncology. 2010;5:104. doi: 10.1186/1748-717x-5-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ballinger M. L., Best A., Mai P. L., et al. Baseline surveillance in Li-Fraumeni syndrome using whole-body magnetic resonance imaging. JAMA Oncology. 2017;3(12):1634–1639. doi: 10.1001/jamaoncol.2017.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villani A., Tabori U., Schiffman J., et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. The Lancet Oncology. 2011;12(6):559–567. doi: 10.1016/s1470-2045(11)70119-x. [DOI] [PubMed] [Google Scholar]
- 33.Pilarski R. Cowden syndrome: a critical review of the clinical literature. Journal of Genetic Counseling. 2009;18(1):13–27. doi: 10.1007/s10897-008-9187-7. [DOI] [PubMed] [Google Scholar]
- 34.Pilarski R., Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. Journal of Medical Genetics. 2004;41(5):323–326. doi: 10.1136/jmg.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hobert J. A., Eng C. PTEN hamartoma tumor syndrome: an overview. Genetics in Medicine. 2009;11(10):687–694. doi: 10.1097/gim.0b013e3181ac9aea. [DOI] [PubMed] [Google Scholar]
- 36.Tan M.-H., Mester J. L., Ngeow J., Rybicki L. A., Orloff M. S., Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clinical Cancer Research. 2012;18(2):400–407. doi: 10.1158/1078-0432.ccr-11-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heald B., Mester J., Rybicki L., Orloff M. S., Burke C. A., Eng C. Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology. 2010;139(6):1927–1933. doi: 10.1053/j.gastro.2010.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garofola C., Gross G. P. StatPearls [Internet] Treasure Island, FL, USA: StatPearls Publishing; 2020. Cowden disease (multiple hamartoma syndrome) https://www.ncbi.nlm.nih.gov/books/NBK525984/20. [Google Scholar]
- 39.Pilarski R., Burt R., Kohlman W., Pho L., Shannon K. M., Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. JNCI Journal of the National Cancer Institute. 2013;105(21):1607–1616. doi: 10.1093/jnci/djt277. [DOI] [PubMed] [Google Scholar]
- 40.Boland C. R., Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073–2087. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kempers M. J., Kuiper R. P., Ockeloen C. W., et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. The Lancet Oncology. 2011;12(1):49–55. doi: 10.1016/s1470-2045(10)70265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hampel H., de la Chapelle A. How do we approach the goal of identifying everybody with Lynch Syndrome? Familial Cancer. 2013;12(2):313–317. doi: 10.1007/s10689-013-9611-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kohlmann W., Gruber S. B. Lynch syndrome. In: Adam M. P., Ardinger H. H., Pagon R. A., et al., editors. GeneReviews® [Internet] Seattle, WA, USA: University of Washington; 2004. https://www.ncbi.nlm.nih.gov/books/NBK1211/ [Google Scholar]
- 44.Giardiello F. M., Allen J. I., Axilbund J. E., et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US multi-society Task Force on colorectal cancer. American Journal of Gastroenterology. 2014;109(8):1159–1179. doi: 10.1038/ajg.2014.186. [DOI] [PubMed] [Google Scholar]
- 45.Vasen H. F. A., Blanco I., Aktan-Collan K., et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62(6):812–823. doi: 10.1136/gutjnl-2012-304356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stoffel E. M., Mangu P. B., Gruber S. B., et al. Hereditary colorectal cancer syndromes: American society of clinical Oncology clinical practice guideline endorsement of the familial risk-colorectal cancer: European society for medical Oncology clinical practice guidelines. Journal of Clinical Oncology. 2015;33(2):209–217. doi: 10.1200/jco.2014.58.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haanstra J. F., Kleibeuker J. H., Koornstra J. J. Role of new endoscopic techniques in Lynch syndrome. Familial Cancer. 2013;12(2):267–272. doi: 10.1007/s10689-013-9610-6. [DOI] [PubMed] [Google Scholar]
- 48.Møller P., Seppälä T., Bernstein I., et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66(3):464–472. doi: 10.1136/gutjnl-2015-309675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmeler K. M., Lynch H. T., Chen L.-m., et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. New England Journal of Medicine. 2006;354(3):261–269. doi: 10.1056/nejmoa052627. [DOI] [PubMed] [Google Scholar]
- 50.Renkonen-Sinisalo L., Sipponen P., Aarnio M., et al. No support for endoscopic surveillance for gastric cancer in hereditary non-polyposis colorectal cancer. Scandinavian Journal of Gastroenterology. 2002;37(5):574–577. doi: 10.1080/00365520252903134. [DOI] [PubMed] [Google Scholar]
- 51.Clinical Kim J., Braun D., Ukaegbu C., et al. Factors associated with gastric cancer in individuals with Lynch syndrome. Clinical Gastroenterology and Hepatology. 2019;S1542-3565(19):30745–30751. doi: 10.1016/j.cgh.2019.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canto M. I., Harinck F., Hruban R. H., et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013;62(3):339–347. doi: 10.1136/gutjnl-2012-303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harkness E. F., Barrow E., Newton K., et al. Lynch syndrome caused byMLH1mutations is associated with an increased risk of breast cancer: a cohort study. Journal of Medical Genetics. 2015;52(8):553–556. doi: 10.1136/jmedgenet-2015-103216. [DOI] [PubMed] [Google Scholar]
- 54.Goldberg M., Bell K., Aronson M., et al. Association between the Lynch syndrome gene MSH2 and breast cancer susceptibility in a Canadian familial cancer registry. Journal of Medical Genetics. 2017;54(11):742–746. doi: 10.1136/jmedgenet-2017-104542. [DOI] [PubMed] [Google Scholar]
- 55.Roberts M. E., Jackson S. A., Susswein L. R., et al. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genetics in Medicine. 2018;20(10):1167–1174. doi: 10.1038/gim.2017.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haraldsdottir S., Hampel H., Wei L., et al. Prostate cancer incidence in males with Lynch syndrome. Genetics in Medicine. 2014;16(7):553–557. doi: 10.1038/gim.2013.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan R. Y. C., Ngeow J. Hereditary diffuse gastric cancer: what the clinician should know. World Journal of Gastrointestinal Oncology. 2015;7(9):153–160. doi: 10.4251/wjgo.v7.i9.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seevaratnam R., Coburn N., Cardoso R., et al. A systematic review of the indications for genetic testing and prophylactic gastrectomy among patients with hereditary diffuse gastric cancer. Gastric Cancer. 2012;15(1):S153–S163. doi: 10.1007/s10120-011-0116-3. [DOI] [PubMed] [Google Scholar]
- 59.Lim Y. C., di Pietro M., O’Donovan M., et al. Prospective cohort study assessing outcomes of patients from families fulfilling criteria for hereditary diffuse gastric cancer undergoing endoscopic surveillance. Gastrointestinal Endoscopy. 2014;80(1):78–87. doi: 10.1016/j.gie.2013.11.040. [DOI] [PubMed] [Google Scholar]
- 60.Johnson K., Sarma D., Hwang E. S. Lobular breast cancer series: imaging. Breast Cancer Research. 2015;17(1):p. 94. doi: 10.1186/s13058-015-0605-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dunlop M. G. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polypolis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut. 2002;51(5):V21–v27. doi: 10.1136/gut.51.suppl_5.v21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Syngal S., Brand R. E., Church J. M., Giardiello F. M., Hampel H. L., Burt R. W. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. American Journal of Gastroenterology. 2015;110(2):223–262. doi: 10.1038/ajg.2014.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller D. T., Freedenberg D., Schorry E., et al. Health supervision for children with neurofibromatosis type 1. Pediatrics. 2019;143(5) doi: 10.1542/peds.2019-0660. [DOI] [PubMed] [Google Scholar]
- 64.Stewart D. R., Korf B. R., Nathanson K. L., Stevenson D. A., Yohay K. Care of adults with neurofibromatosis type 1: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG) Genetics in Medicine. 2018;20(7):671–682. doi: 10.1038/gim.2018.28. [DOI] [PubMed] [Google Scholar]
- 65.Evans D. G. Are we ready for targeted early breast cancer detection strategies in women with NF1 aged 30-49 years? American Journal of Medical Genetics Part A. 2012;158A(12):3054–3055. doi: 10.1002/ajmg.a.35585. [DOI] [PubMed] [Google Scholar]
- 66.Seminog O. O., Goldacre M. J. Age-specific risk of breast cancer in women with neurofibromatosis type 1. British Journal of Cancer. 2015;112(9):1546–1548. doi: 10.1038/bjc.2015.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kurian A. W., Hughes E., Handorf E. A., et al. Breast and ovarian cancer penetrance estimates derived from germline multiple-gene sequencing results in women. JCO Precision Oncology. 2017;1(1):1–12. doi: 10.1200/po.16.00066. [DOI] [PubMed] [Google Scholar]
- 68.Casadei S., Norquist B. M., Walsh T., et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Research. 2011;71(6):2222–2229. doi: 10.1158/0008-5472.can-10-3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Antoniou A. C., Casadei S., Heikkinen T., et al. Breast-cancer risk in families with mutations in PALB2. New England Journal of Medicine. 2014;371(6):497–506. doi: 10.1056/nejmoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jones S., Hruban R. H., Kamiyama M., et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324(5924):p. 217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tung N., Domchek S. M., Stadler Z., et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nature Reviews Clinical Oncology. 2016;13(9):581–588. doi: 10.1038/nrclinonc.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shindo K., Yu J., Suenaga M., et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. Journal of Clinical Oncology. 2017;35(30):3382–3390. doi: 10.1200/jco.2017.72.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pilié P. G., Johnson A. M., Hanson K. L., et al. Germline genetic variants in men with prostate cancer and one or more additional cancers. Cancer. 2017;123(20):3925–3932. doi: 10.1002/cncr.30817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chun H. H., Gatti A. R. Ataxia-teleangiectasia, an evolving phenotype. DNA Repair. 2004;3(8-9):1187–1196. doi: 10.1016/j.dnarep.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 75.Weischer M., Bojesen S. E., Ellervik C., Tybjærg-Hansen A., Nordestgaard B. G. CHEK2∗1100delC genotyping for clinical assessment of breast cancer risk: meta-analyses of 26,000 patient cases and 27,000 controls. Journal of Clinical Oncology. 2008;26(4):542–548. doi: 10.1200/jco.2007.12.5922. [DOI] [PubMed] [Google Scholar]
- 76.Cybulski C., Górski B., Huzarski T., et al. CHEK2 is a multiorgan cancer susceptibility gene. The American Journal of Human Genetics. 2004;75(6):1131–1135. doi: 10.1086/426403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Seemanová E., Jarolim P., Seeman P., et al. Cancer risk of heterozygotes with the NBN founder mutation. Journal of the National Cancer Institute. 2007;99(24):1875–1880. doi: 10.1093/jnci/djm251. [DOI] [PubMed] [Google Scholar]
- 78.Weber-Lassalle N., Borde J., Weber-Lassalle K., et al. Germline loss-of-function variants in the BARD1 gene are associated with early-onset familial breast cancer but not ovarian cancer. Breast Cancer Res. 2019;21(1):p. 55. doi: 10.1186/s13058-019-1137-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Suszynska M., Kluzniak W., Wokolorczyk D., et al. BARD1 is A Low/Moderate breast cancer risk gene: evidence based on an association study of the central European p.Q564X recurrent mutation. Cancers. 2019;11(6):p. 740. doi: 10.3390/cancers11060740. [DOI] [PMC free article] [PubMed] [Google Scholar]