

Abstract
Crucial transitions in cancer—including tumor initiation, local expansion, metastasis, and therapeutic resistance—involve complex interactions between cells within the dynamic tumor ecosystem. Transformative single-cell genomics technologies and spatial multiplex in situ methods now provide an opportunity to interrogate this complexity at unprecedented resolution. The Human Tumor Atlas Network (HTAN), part of the National Cancer Institute (NCI) Cancer Moonshot Initiative, will establish a clinical, experimental, computational, and organizational framework to generate informative and accessible three-dimensional atlases of cancer transitions for a diverse set of tumor types. This effort complements both ongoing efforts to map healthy organs and previous large-scale cancer genomics approaches focused on bulk sequencing at a single point in time. Generating single-cell, multiparametric, longitudinal atlases and integrating them with clinical outcomes should help identify novel predictive biomarkers and features as well as therapeutically relevant cell types, cell states, and cellular interactions across transitions. The resulting tumor atlases should have a profound impact on our understanding of cancer biology and have the potential to improve cancer detection, prevention, and therapeutic discovery for better precision-medicine treatments of cancer patients and those at risk for cancer.
Cancer forms and progresses through a series of critical transitions—from pre-malignant to malignant states, from locally contained to metastatic disease, and from treatment-responsive to treatment-resistant tumors (Figure 1). Although specifics differ across tumor types and patients, all transitions involve complex dynamic interactions between diverse pre-malignant, malignant, and non-malignant cells (e.g., stroma cells and immune cells), often organized in specific patterns within the tumor microenvironment. Morphological, genetic, and epigenetic diversity of pre-malignant and malignant cells, even from the same tumor, is crucial for cancer development, adaptation to new metastatic sites, and resistance to treatment. Interactions with non-malignant cells in the tumor microenvironment also play critical roles in driving disease progression. Tumor cells can remodel their environment to promote their growth, enable tissue invasion to nearby or distant sites, or evade immune clearance. Conversely, diverse immune and stromal cells can either restrict or promote tumor growth and progression depending on the context.
Figure 1. Crucial Transitions in Cancer.
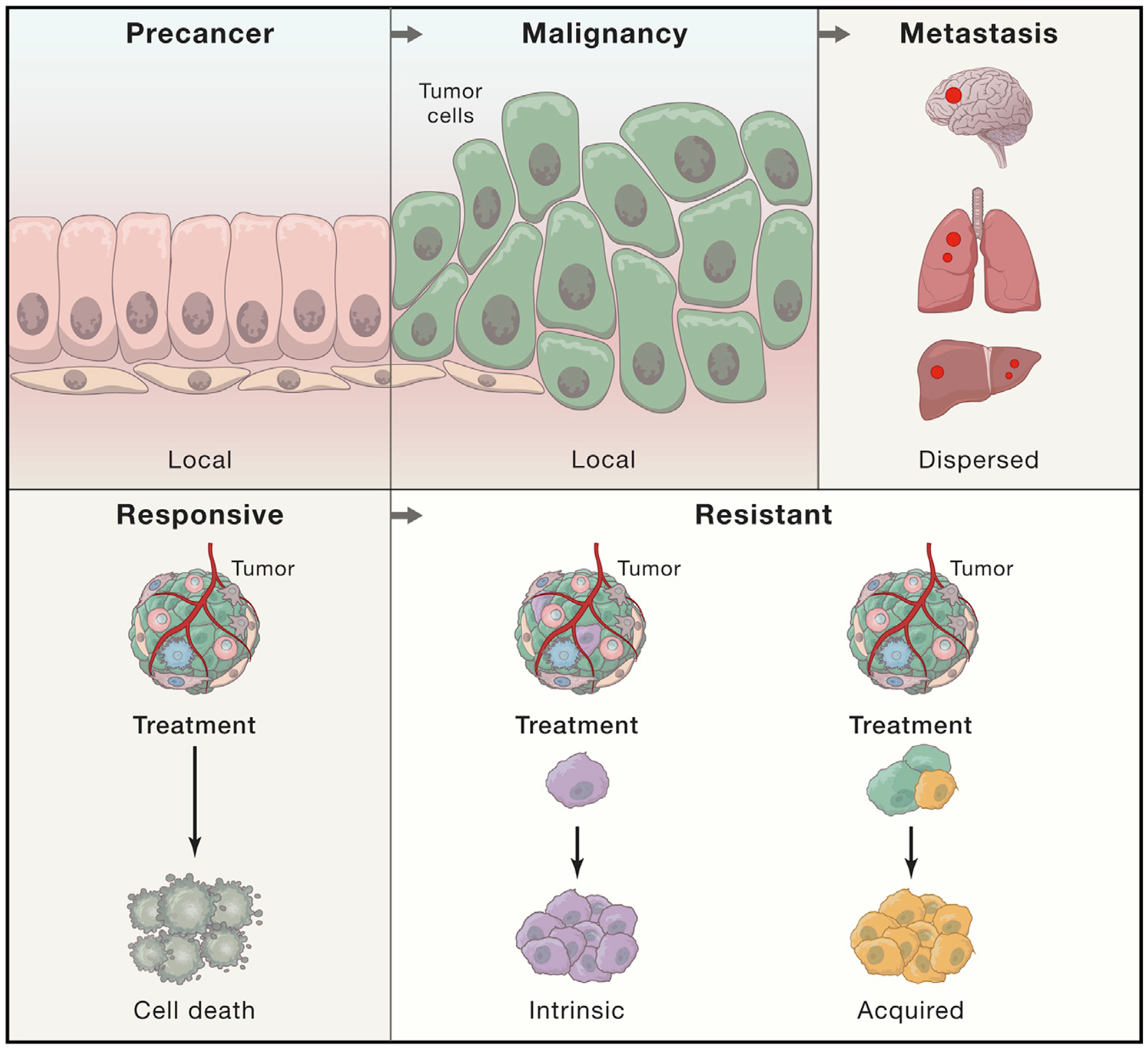
HTAN aims to generate 3D atlases of three critical transitions in cancer: tumor initiation (from pre-cancerous lesions to local malignancy), expansion (from local malignancy to metastasis), and progression to a therapy-resistant state through intrinsic (purple) or acquired (yellow) resistance mechanisms. These transitions involve complex interactions between pre-malignant, malignant, and/or non-malignant cells within the tumor ecosystem.
The genomic revolution in cancer has led to the identification and pursuit of numerous genetic drivers of malignancy (The Cancer Genome Atlas Research Network et al., 2013; The International Cancer Genome Consortium, 2010), but these efforts have relied, by necessity, on bulk profiling of advanced tumors, most commonly at a single point in time, with limited information about patient treatment and outcomes (Figure 2). This has made it difficult to capture the intricate cellular, spatial, and temporal dimensions of tumorigenesis and their role in disease progression and dissemination. Recent advances in single-cell and multiplexed spatial analysis of tissue allow us to interrogate this complexity at unprecedented resolution (Angelo et al., 2014; Buenrostro et al., 2015; Chen et al., 2016a; Chen et al., 2016b; Du et al., 2019; Gaublomme et al., 2019; Gierahn et al., 2017; Giesen et al., 2014; Goltsev et al., 2018; Habib et al., 2017; Kang et al., 2018; Lee et al., 2014; Lin et al., 2018; Macosko et al., 2015; Moffitt et al., 2016a; Moffitt et al., 2016b; Shalek et al., 2013; Ståhl et al., 2016; Stoeckius et al., 2018; Trombetta et al., 2014; Vickovic et al., 2016). As a result, it is now possible to systematically identify subcellular structures, cell types, cell states, and different genetic clones in a tumor and to relate them spatially to each other and to the overall tumor. This should allow us to better understand tumor evolution and heterogeneity with the promise of improved diagnostics and therapeutics.
Figure 2. HTAN Is Complementary to Previous Large-Scale Cancer Genomics Initiatives and Ongoing Atlas Efforts.
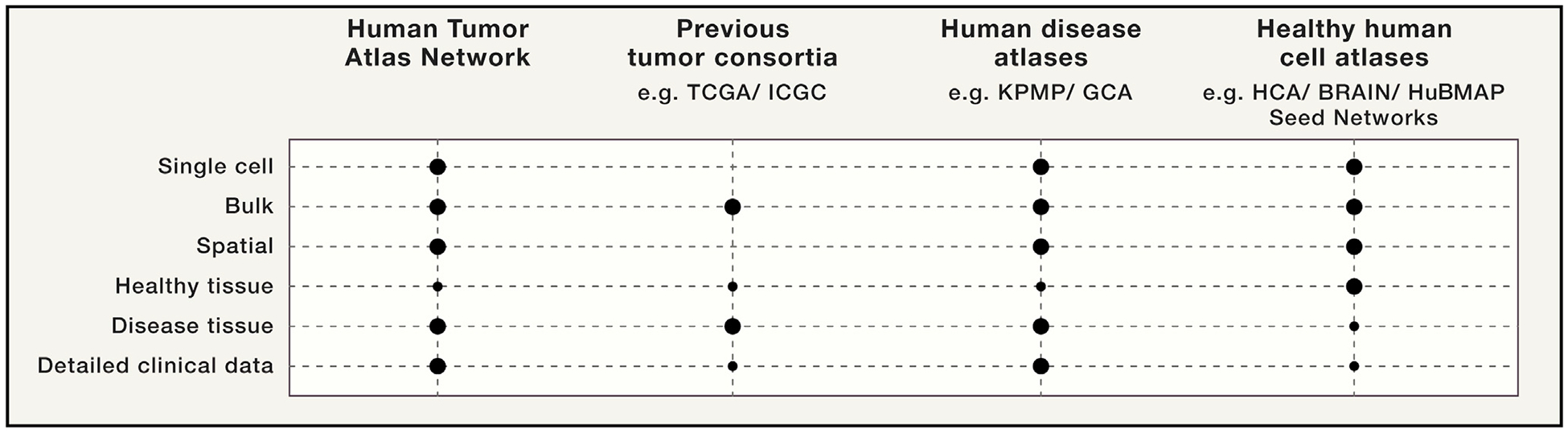
HTAN will illuminate aspects of malignancy that could not be fully addressed by previous large-scale cancer genomics programs and is complementary to ongoing atlas building efforts across healthy and disease tissues.
The Human Tumor Atlas Network (HTAN) was established by the National Cancer Institute (NCI) as part of the Cancer Moon-shot Initiative, tasked with the ambitious goal of making a decade’s worth of progress in cancer prevention, diagnosis, and treatment in just 5 years (https://ccr.cancer.gov/research/cancer-moonshot). HTAN will generate three-dimensional (3D) atlases of cancer transitions for a diverse set of pre-cancers and established tumors. Transitions will span tumor evolution from pre-malignancy to malignancy, from primary tumor to metastasis, and from pre-treatment to post-therapeutic response (Figure 1). Analogous to geographical information systems such as Google Maps, these interactive atlases should depict evolving tumor ecosystems in multiple dimensions across space and time to enable generalization and abstraction from individual tumor instances to their overall principles. The hope is that visualizing the structure, composition, and multiscale interactions at distinct points in tumor evolution will help identify novel predictive biomarkers and therapeutically relevant cell types, cell states, and cellular interactions and suggest new avenues for effectively targeting them. To construct even preliminary atlases, we must overcome substantial clinical, experimental, computational, and organizational challenges. To maximize the atlases’ impact and applicability, we also plan to develop robust yet flexible strategies for data integration, visualization, and sharing.
Here, we introduce the concept of a dynamic, multiparametric 3D tumor atlas, its potential to affect basic and translational research, and the experimental and computational strategies for its construction. We also discuss HTAN’s organization and opportunities for engagement and collaboration with complementary initiatives.
3D Atlases of Critical Transitions in Cancer
We envision tumor atlases as comprehensive, generalized catalogs of cell states, types, and programs and cell-state transitions and that these will incorporate the physical positions of tumor cells in relation to each other, the supporting stroma, and the extracellular matrix. To encompass the heterogeneous nature of tumors within and across patients, our atlases will generalize underlying features and programs that are unique or shared by multiple tumors and relate these molecular features to functional and clinical data elements and patient outcomes. HTAN atlases will represent different tumor types, disease sites, genders, and patient ethnicities. Because mechanisms of tumorigenesis are diverse and not all tumor types or metastatic sites can be sampled in the same way, the information available in specific atlases will vary.
Broadly, first-generation atlases are likely to comprise interactive 2D and 3D visualizations of sets of similar tumors and associate key molecular and cellular features with clinical data elements and patient outcomes (Figure 3). The Cancer Genome Atlas (TCGA) identified cancer drivers by finding mutations that significantly recur in tumors against a background of variation between individual patients while accounting for the context of characteristic histologic tumor features and limited clinical information (The Cancer Genome Atlas Research Network et al., 2013). Second-generation tumor atlases will need to achieve a similar level of abstraction from multi-parametric datasets to meaningfully represent the combined genetic, molecular, ultrastructural, cellular, and histological features that characterize a specific tumor stage and type. To accomplish this goal, HTAN tumor-atlas generation is expected to involve five interdependent steps: (1) collection of longitudinal data from diverse modalities across multiple spatial scales ranging from subcellular (<250 nm) to cell-cluster (~50 μm) resolution; (2) basic processing and quality control of each data modality to ensure accuracy and reproducibility; (3) identification of cell types, states, and positions for annotating tumor composition; (4) identification of features for describing cell-cell interactions, intercellular communication, cell neighborhoods, and mesoscale spatial motifs; and (5) integration of experimental and clinical datasets into a comprehensive atlas (Figure 3).
Figure 3. Building 3D Human Tumor Atlases.
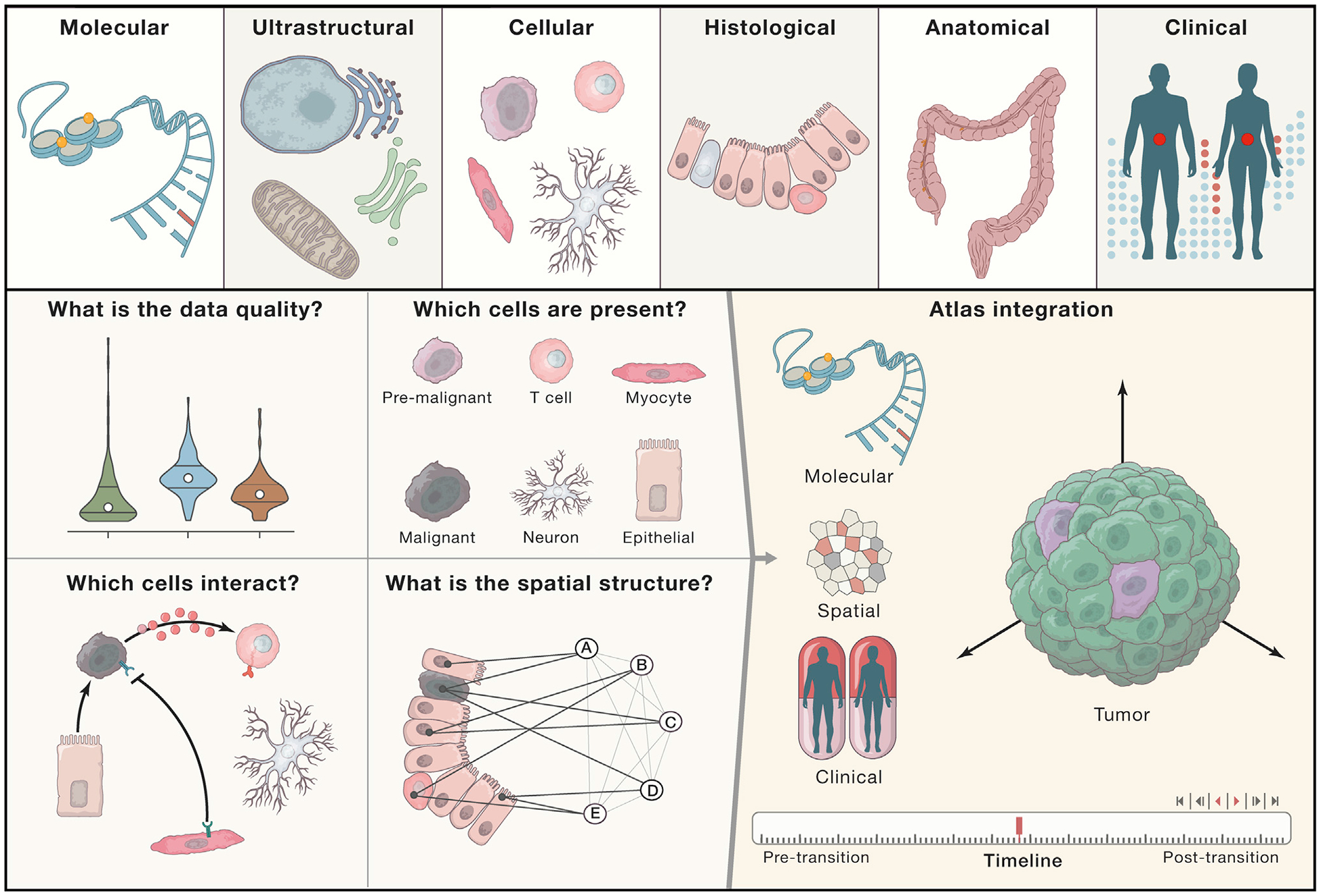
HTAN centers will measure data modalities at multiple scales of resolution, from molecular to ultrastructural to cellular to histological to anatomical (when possible), and will collect relevant clinical information from patients with tumors under study. These modalities will be used for profiling samples according to the capabilities of each HTAN center. Most centers will use both molecular and spatial profiling methods to generate data. Data that pass HTAN-defined basic processing and quality control will be utilized for interrogating cell-type composition, cell-cell interactions, and spatial structures (left panels). Atlases will then be constructed through the integration of these data modalities measured across time (right panel).
HTAN atlases will be constructed around clinical transitions for multiple adult and pediatric malignancies (Figures 1 and 4)—although not all transitions can feasibly be studied in all tumor types. Because understanding tissue architecture is a major goal, HTAN will focus mostly on solid tumors, particularly those that represent critical unmet needs in oncology. These correspond to tumor types with poor prognosis, including triple-negative breast cancer, high-grade glioma, glioblastoma, high-risk neuroblastoma, pediatric sarcoma, high-risk acute lymphoblastic leukemia, and pancreatic ductal adenocarcinoma; pre-malignancies in breast, lung, hematologic, prostate, and colorectal cancers and cutaneous melanoma (both hereditary and sporadic); primary and metastatic lung and pancreatic cancer; and drug-resistant metastatic breast cancer, metastatic melanoma, and metastatic colorectal carcinoma (Figure 4).
Figure 4. Key Tumors Studied by the Consortium.
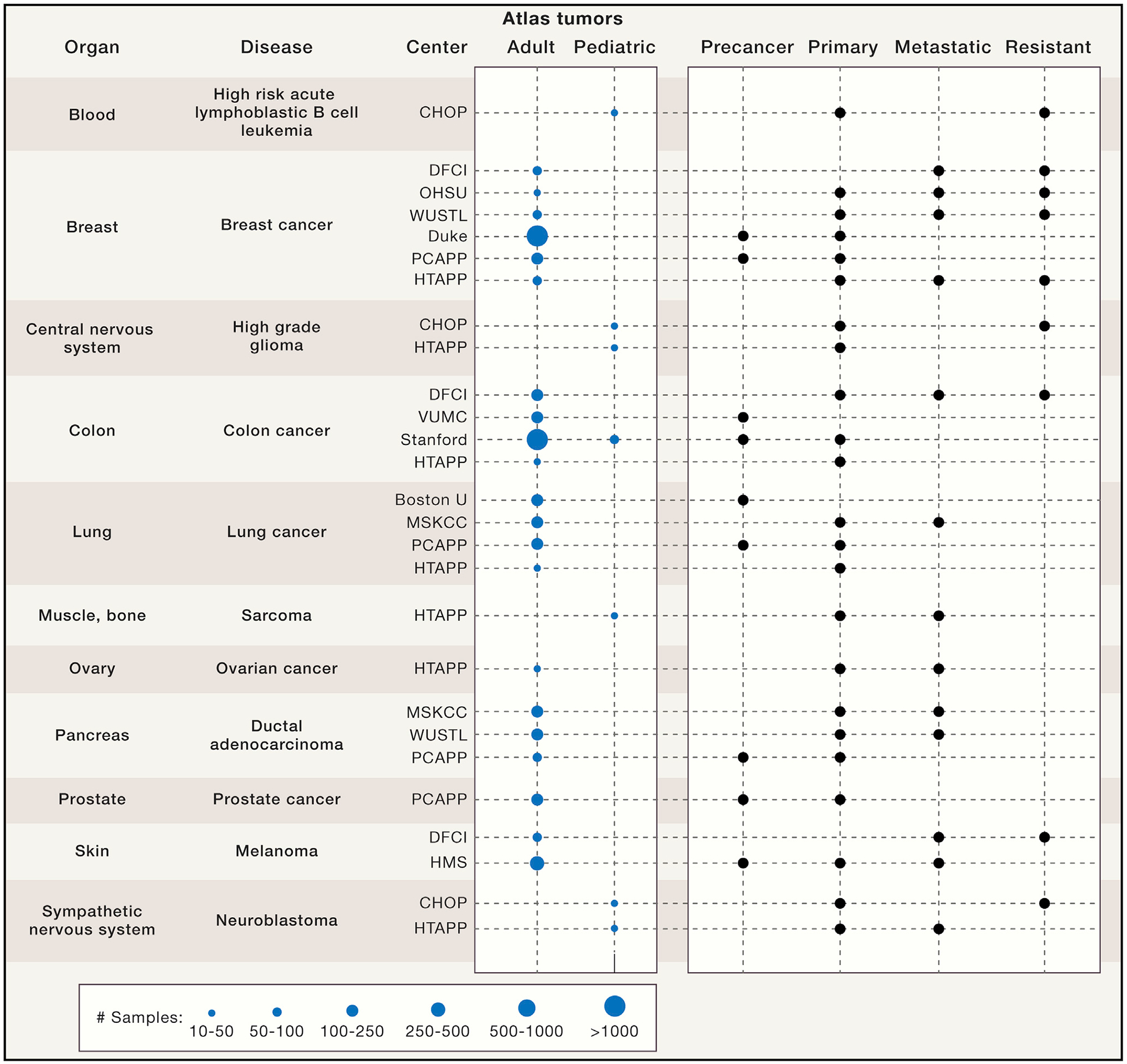
HTAN centers will generate 3D atlases of human tumors spanning different tissue types across adult and pediatric tumors from patients with pre-cancer, primary tumors, and metastases, as well as resistant tumors before and after treatment. Projected ranges for the number of samples to be profiled over the 5-year HTAN period are depicted for each center and tumor type. In some cases, multiple samples will be profiled from the same patient. HTAN includes the following centers: Children’s Hospital of Philadelphia (CHOP), Dana-Farber Cancer Institute (DFCI), Oregon Health & Science University (OHSU), Washington University in St. Louis (WUSTL), Duke University School of Medicine, Pre-Cancer Atlas Pilot Project (PCAPP), Human Tumor Pilot Project (HTAPP), Vanderbilt University Medical Center (VUMC), Stanford University, Boston University (BU), Memorial Sloan Kettering Cancer Center (MSCKK), and Harvard Medical School (HMS).
Once constructed, HTAN atlases should facilitate clinical predictions by using features or biomarkers—such as molecular levels, cellular composition, or in situ structural and molecular patterns—that correlate with and ideally predict relevant clinical transitions, including treatment response (Figure 5A). Moreover, multiparametric insights from atlases are expected to guide future investigations into the basic biological processes that underlie malignant transformation (Figure 5B). A user should be able to query an atlas by using data from a new specimen without the need for the full battery of measurements (Figure 5C). For example, one could use a new specimen measured only with hematoxylin and eosin (H&E) staining or single-cell RNA-seq (scRNA-seq) to obtain predictions for other layers (e.g., the location of specific cells and molecules in the H&E stain or the origin of scRNA-seq profiles within a histology section). As a result, one mode of testing can connect genomics and histopathology and help to glean a wealth of spatiomolecular information. Finally, HTAN efforts should inform future charting efforts by the broader scientific community; drive the development of image quantification analytics such as TCGA did for genome-scale sequencing analysis; help integrate data across tumors, healthy tissue, and other disease states; and enable additional tumor specimens to be compared with this reference dataset or added to it with relative ease.
Figure 5. What We Can Learn from the Atlases and How to Query Them.
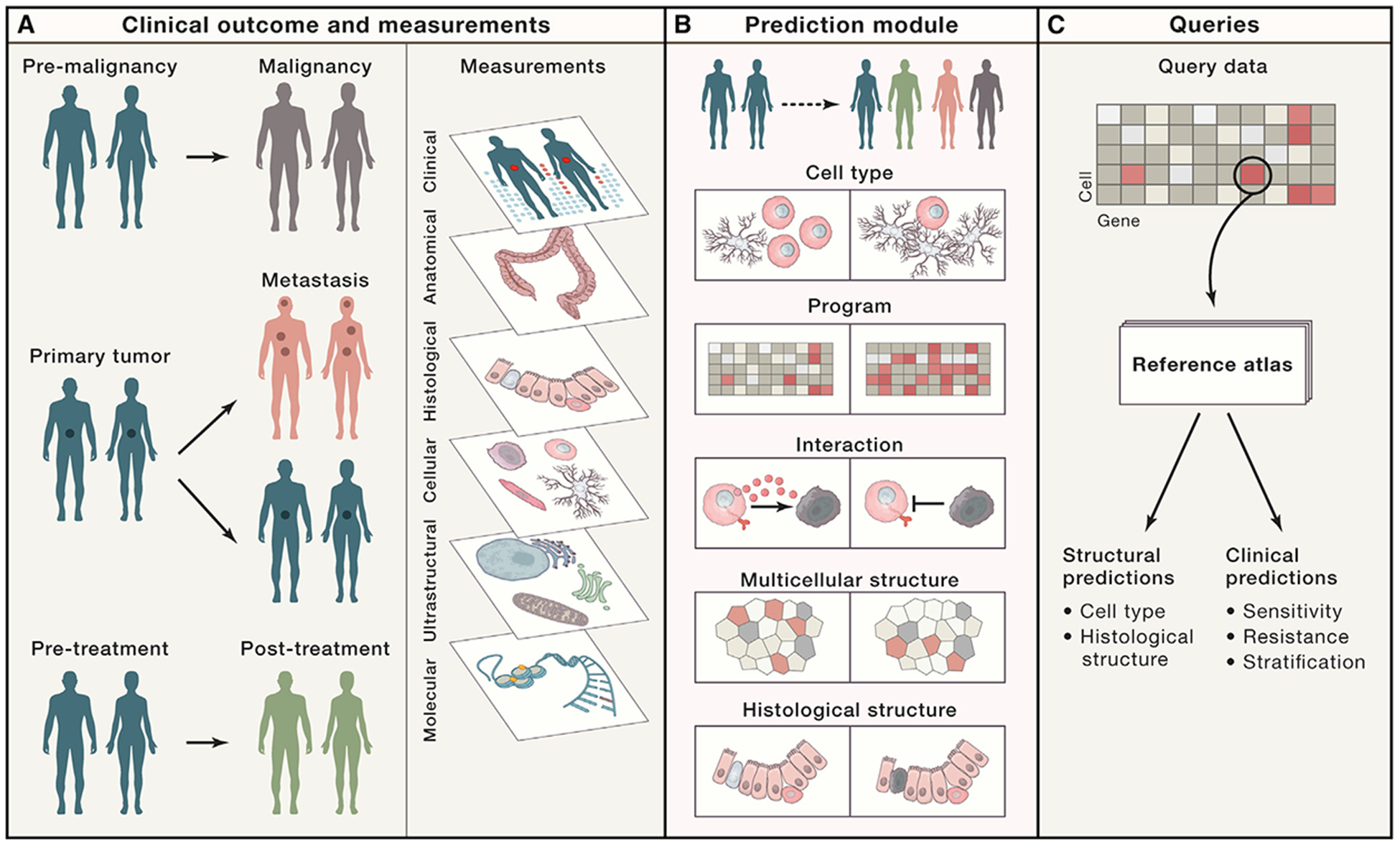
(A and B) HTAN centers will combine clinical outcome and measurement data to (A) capture shared and unique characteristics and features across tumors—or subsets of tumors—and (B) associate them with “structural” features such as genes, molecules, cells, cellular interactions and structures, and histology. (C) By identifying features that correlate with clinical transitions and disease states, responses to treatment, and/or structural and molecular traits, tumor atlases will facilitate clinical and structural predictions according to query datasets.
The Promise of Tumor Atlases for Improving Clinical Care
HTAN atlases will illuminate at least three translational aspects of malignancy that could not be fully addressed by previous large-scale cancer genomics programs (e.g., TCGA and the International Cancer Genome Consortium [ICGC]) (Figure 2). First, the tumor microenvironment and its constituent cellular interactions—accessible to HTAN through spatial and single-cell approaches—represent remarkable targeting opportunities for therapy (Binnewies et al., 2018; Galon and Bruni, 2019; Sharma et al., 2017). Second, whereas most previous efforts focused on primary tumors with limited treatment and outcome data (Beane et al., 2019; Shain et al., 2015), HTAN emphasizes longitudinal sampling (including pre-cancers, advanced tumors, metastases, and treatment responses) alongside collection of comprehensive clinical data. Together, these will inform detection, prevention, and treatment strategies (Srivastava et al., 2018). Third, the integration of spatial methods will help link cancer genomics and histopathology, the two primary means of diagnosing cancer and informing therapy. As a result, predictive biomarkers based on HTAN’s integrative, multimodal analyses could outperform genetic or histological biomarkers alone. For example, they might include compositions and arrangements of cancer and non-cancer cell neighborhoods or spatial heterogeneity in transcriptional, epigenetic, or mutational states along with features from H&E and immunohistochemistry staining. Machine-learning (ML) approaches could make it possible to relate such features to more traditional measures of tumor state, thus bridging the gap between established diagnostic methods and spatiomolecular information.
In the context of pre-malignancies, HTAN-defined biomarkers could help stratify lesions that are likely to progress and thus benefit from therapy, and they could inform positive prognoses for which treatments can be reduced or avoided. Moreover, they will help users to identify and assess the efficacy of new prevention strategies, enable early detection, and potentially promote more effective and less invasive screening contexts. In cancers such as in situ ductal carcinoma, the most common histologically recognized form of early breast cancer, HTAN-identified genomic alterations and cellular compositions associated with progression and outcome could inform prognosis and intervention strategies. Similar approaches can be taken in colorectal and skin cancers.
For established cancers, HTAN data could help to identify patients at risk for local invasion or metastasis, facilitate the detection of recurrence, and distinguish between aggressive and slow-growing tumors. Cancer models, signatures, and bio-markers based on atlas data are further expected to define targets for intervention and to predict treatment responses and potential resistance mechanisms, including the role of the tumor microenvironment. Genomics alone cannot reveal all aspects of complex resistance mechanisms. HTAN atlases of cancer transitions will help to define the range and spatial distribution of clonal variation within or across tumors and could guide treatment design for tumors that are likely to progress. For example, adjuvant therapy is currently effective in only 5%–20% of common breast, colon, and lung cancers, and predictive markers for invasion and metastasis could obviate the need for adjuvant therapy in a substantial subset of patients. Another example is the Immunoscore (Galon and Bruni, 2019), which accounts for the spatiotemporal interplay of numerous different immune cell types in the tumor and is used for predicting relapse in early-stage colon cancer.
All HTAN atlases will provide spatially resolved immunopheno-types and are therefore likely to inform response and resistance to immuno-oncology drugs. Knowing which cells express immune checkpoint receptors and their ligands, and where in a tumor juxtracrine signaling can occur, is particularly helpful for the design of new combination therapies. For example, a resistance program expressed by malignant cells, associated with T cell exclusion and cold immune niches, predicts clinical responses to anti-PD-1 therapy in melanoma and can be reversed by CDK4/6 inhibitors (Jerby-Arnon et al., 2018).
HTAN atlases should allow for more precise treatment of cancer patients or those with increased cancer risk and the tailoring of first-line therapies to reduce the risk of resistance and recurrence. They will also help in the development of new strategies for patients whose tumors are unlikely to respond to existing treatments.
How to Build a Tumor Atlas
Because building 3D tumor atlases requires the integration of clinical, experimental, computational, and organizational frameworks, HTAN efforts span a wide range of activities from sample collection and tool development for cellular and spatial profiling to data analysis, atlas visualization, and querying. HTAN is committed to openly disseminating the clinical data, experimental methods, computational tools, standards, and multimodal data that it generates through these efforts.
Biospecimen Collection and Clinical Annotation
To ensure the broadest impact and usage, HTAN will obtain high-quality samples from ethnically diverse populations. Samples will be extensively annotated so that they capture detailed and harmonized clinical data elements, including demographic information and treatment history. Most samples will be prospectively collected, and in select cases, multisite sampling will be used for acquiring matched combinations of normal tissues, pre-cancer lesions, primary tumors, and metastases. To study resistance to therapy, HTAN will profile pre- and post-treatment samples in several tumor types (Figure 4), including, in some cases, matched pre- and post-samples from the same patient.
To study the transition from pre-cancer to cancer, HTAN investigators will leverage retrospectively banked frozen or formalin-fixed paraffin-embedded (FFPE) biopsies or tissue re-sections procured from primary tumors with adjacent pre-cancer components not accessible in screening settings. They will also prospectively collect longitudinal samples of advanced or adjacent pre-cancerous lesions. Where possible, longitudinal cohorts will follow cases where pre-invasive lesions progress to carcinoma.
To study the metastatic transition, HTAN will procure biopsies and surgical resections from primary and metastatic sites. A proportion of sampled cases that return with recurrent cancer will enable the capture of matched metastatic tissue via biopsy or resection. Single-cell profiling can be applied for small biopsy specimens, and most imaging modalities currently work well with needle cores, although fine-needle aspirates typically produce too much tissue distortion. To study the transition to therapeutic resistance, HTAN investigators will sample lesions both before and after treatment. Prospective longitudinal sampling will also be supplemented by rapid autopsies, which enable extensive sampling of primary and disseminated tumors at multiple sites from the same individual (Iacobuzio-Donahue et al., 2019).
High-resolution single-cell and spatial assays require appropriate quality metrics for diverse sample types (resections, biopsies, and body fluids) and processing methods (fresh, frozen, and fixed) and harmonized collection of metadata. HTAN investigators will address several key challenges during specimen collection, including (1) the documentation of pre-analytic variables, such as the time from sample collection to profiling as well as photographic documentation of the samples; (2) stream lined handling of fresh, frozen, and fixed tissues by testing, benchmarking, and sharing protocols and standard operating procedures (SOPs); (3) the manipulation of a single tumor specimen for multiple assays by prioritizing technologies depending on sample size; (4) longitudinal sampling from the same individuals; and (5) obtaining cases that represent the spectrum of disease from a range of possible samples to profile.
Cellular and Spatial Profiling
Most HTAN centers have adopted a two-pronged approach that pairs the collection of single-cell profiles from dissociated specimens (transcriptome, multiplexed protein, genome-wide chromatin accessibility, or methylation) with spatially resolved multiplexed assays for RNAs or proteins in tissue (Figure 3 and Box 1). The two types of approaches are complementary: whereas single-cell profiling methods often lack spatial resolution, spatially resolved methods currently either are low in sample throughput or multiplex fewer measurements. Some centers will record anatomical information longitudinally by using magnetic resonance imaging (MRI), computed tomography, or positron emission tomography (PET), and some will record tissue structure at very high resolution by using serial electron microscopy. Bulk genomic, epigenomic, proteomic, metabolomic, and lipidomic profiling will facilitate the integration of existing data from prior consortia (The Cancer Genome Atlas Research Network et al., 2013; The International Cancer Genome Consortium, 2010; Rudnick et al., 2016). Among the key challenges for HTAN is the development of techniques for single-cell and multiplex spatial profiling of specimens in FFPE blocks (Foley et al., 2019). In particular, most clinical samples of pre-cancers are preserved as FFPE blocks for diagnosis, but FFPE reduces the availability of macromolecules for genomic assays. Additional challenges include the need to develop, deploy, and disseminate new methodologies in a rapidly changing technology landscape, optimize tumor-type-specific protocols, and obtain multiple data types from the same sample. Benchmarking efforts across the network will help tackle these challenges. Such efforts include a series of trans-network projects across multiple centers with the goal of improving SOPs to maximize data reproducibility.
Box 1. Toolbox Used for Building 3D Tumor Atlases across Scales.
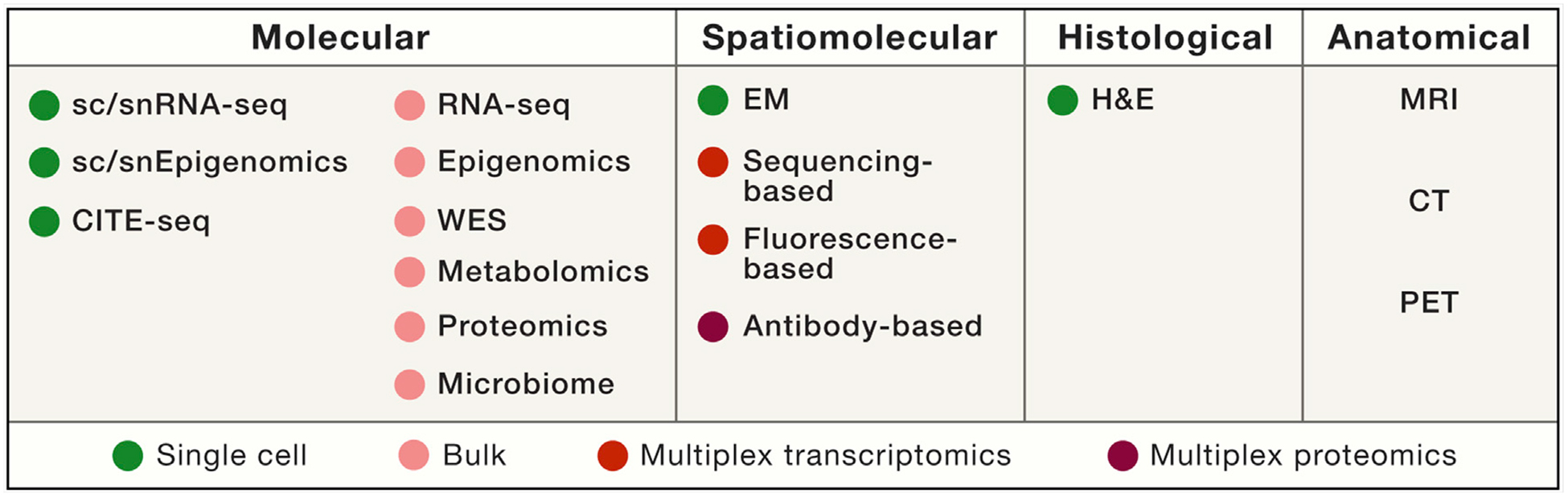
The HTAN toolbox includes molecular, spatiomolecular, histological, and anatomical profiling.
Molecular Profiling
Single-Cell Profiling
(1) Single-cell or single-nucleus RNA sequencing (sc/snRNA-seq). Massively parallel sc/snRNA-seq methods allow quick and cost-effective profiling of gene expression from tens of thousands of single cells or nuclei (Habib et al., 2017; Macosko et al., 2015). For snRNA-seq, frozen tissue samples can be accrued over time and then multiplexed for reducing costs and limiting potential batch effects (Gaublomme et al., 2019; Kang et al., 2018). HTAN centers will use several profiling methods, including approaches based on plates (Patel et al., 2014; Shalek et al., 2013; Tirosh et al., 2016a; Trombetta et al., 2014; Wallrapp et al., 2017), droplets (Habib et al., 2017; Macosko et al., 2015), microwells (Gierahn et al., 2017), and combinatorial indexing (Rosenberg et al., 2018; Vitak et al., 2017). This methodological breadth ensures that the optimal platform can be used by a given center according to considerations such as tissue type, accessibility, required scale, and level of expertise. (2) To examine gene regulation, HTAN centers will also profile epigenomic features such as chromatin accessibility and DNA methylation by using single-cell epigenomics approaches (Buenrostro et al., 2015). (3) HTAN centers will also quantify proteins from single cells by using cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) (Stoeckius et al., 2017) to confirm and complement the sc/snRNA-seq data.
Bulk Profiling
For a subset of samples, HTAN researchers will apply bulk characterization methods, including (1) whole-exome sequencing to characterize the mutations, copy-number alterations, and clones in the tumor; (2) bulk RNA-seq to provide transcriptome annotations and help determine whether any major cell subset is disproportionally lost in dissociation; (3) epigenomic profiling as a control for all the cell types present in a sample; (4) proteomics measurements by reverse-phase protein array and mass spectrometry; and (5) metabolomics. Many of these assays have been used for profiling tumors in other consortia and will facilitate the integration of existing legacy data with HTAN data.
Spatiomolecular Profiling
HTAN centers will deploy multiplexed in situ RNA and protein assays that could rely on imaging, sequencing, spatial coding, and computational inference. These technologies fall into two broad categories: (1) approaches that quantify specific nucleic acids (Chen et al., 2015; Chen et al., 2016a; Moffitt et al., 2016a; Moffitt et al., 2016b) or proteins (Angelo et al., 2014; Giesen et al., 2014; Goltsev et al., 2018) by using targeted nucleic acid probes or antibodies, respectively, and (2) those that profile RNA by using sequencing (Lee et al., 2014; Ståhl et al., 2016; Vickovic et al., 2016). These methods have a trade-off between multiplexing capacity and spatial resolution, and currently most genome-wide methods do not reach single-cell resolution. In some cases, sub-cellular profiling will be performed.
Histological and Anatomical Profiling
Hematoxylin and eosin (H&E) staining of tissue sections will inform the histology of the tumor tissue and will be complemented by longitudinal but lower-resolution technologies, such as MRI and PET, for anatomical imaging. Together, these methods will provide information about tissue architecture and a connection to common assays deployed in clinical care.
Data Analysis and Atlas Building
Computational analysis is a fundamental component of HTAN tumor-atlas construction and iteratively guides study and experimental design. HTAN researchers will employ computational approaches to map between cellular and spatial profiles and multiscale histological and anatomical structures. They will further integrate these with clinical data across time to generate a coherent atlas that is broadly accessible and can be dynamically queried by the scientific community. HTAN researchers will also aim to predict the functional impact of cell-cell interactions and test these predictions with functional validation studies. A particular challenge in the assembly of tumor atlases is the seemingly idiosyncratic nature of each tumor. However, akin to the discovery of recurring driver mutations in cancer genome atlases, the goal of a tumor cell atlas is to find recurrent higher-order features by using computational and statistical means to identify common attributes across tumors. At the same time, it will also be important to identify unique tumor features and to connect both common and unique features to clinical outcomes.
The integration of multiple data modalities constitutes a major frontier in computational biology (Achim et al., 2015; Durruthy-Durruthy et al., 2014; Satija et al., 2015). Progress on similar problems in other computational domains could provide guidance (Jha et al., 2018; Ma et al., 2019; Wang et al., 2016). For example, new ML approaches leverage the unifying concept that cells lie on a low-dimensional manifold defined by related biological features (genome, epigenome, transcriptome, proteome, cell neighborhoods, etc.) and that similar cells neighbor each other (Argelaguet et al., 2019). Deep-learning approaches in particular are well suited to mapping this manifold because they do not require the important features to be defined beforehand, provide a natural means of integrating different feature types (e.g., single-cell data described as vectors and spatial data described as images), and are highly scalable (Amodio and Krishnaswamy, 2018; Liu et al., 2019; Lopez et al., 2019).
HTAN atlases will provide benchmarking data for ML algorithms currently in development (Goodfellow et al., 2017) to improve performance on tasks such as identifying cell-type and spatial features associated with clinical characteristics. The accuracy of ML-based cell-type calling can be tested through comparison of assignments made from H&E or immunohistochemistry with HTAN analysis of multiplexed imaging data and spatially resolved RNA expression data (Figure 5). These methods will be designed to help pathologists make more accurate assessments. We envision HTAN data as a treasure trove for the development of algorithmic approaches to mining spatially resolved ‘omic data, which could have potentially far-reaching implications in tumor biology. We expect the final atlas for each tumor type to consist of a set of recurrent structured features that characterize cell-type composition, cell states, cell-cell interactions, cell neighborhoods, and histological modules, as well as their relation to key temporal transitions.
Integrated atlas data will be made accessible through a dedicated HTAN portal, which will consist of a federated set of services and platforms spanning multiple technologies and tools. Initially, the platform will federate and extend the capabilities of the Sage Synapse platform, the cBioPortal for Cancer Genomics (https://www.cbioportal.org/; Gao et al., 2013), and the Cancer Digital Slide Archive. New computational tools developed by HTAN will also be integrated into the portal as they mature.
Challenges and Possible Solutions
Building tumor atlases poses several overarching challenges. First, samples representative of multiple stages of tumor progression will typically come from different patients and thus require us to infer pathways and mechanisms of progression computationally.
Second, atlases will have heterogenous datasets, reflecting differences that are technical (e.g., sample processing times), biological (e.g., diverse patient groups, cancer tissue origin, and disease status), or in profiling modalities (e.g., single-cell or single-nucleus RNA-seq [sc/snRNA-seq], assay for transposase-accessible chromatin using sequencing [ATAC-seq], and distinct spatial profiling methods). Technical hurdles can be addressed with both batch correction methods developed for bulk genomics data (Büttner et al., 2019; Johnson et al., 2007) and newer methods developed for single-cell genomics (Luecken and Theis, 2019). Merging samples with biological differences is more complicated because methods for merging these data-sets must balance between recognizing the cell types and states that are common across datasets and not overcorrecting or losing important biological differences (Barkas et al., 2019; Korsunsky et al., 2019; Lopez et al., 2019; Stuart et al., 2019; Welch et al., 2019). Trans-network efforts will help to improve the needed cell-type calling and alignment.
Third, assessing the predictive potential of HTAN requires a statistical power analysis for the number of individuals, cells, and fields of view needed for each tumor type. Although single-cell profiling studies might collect fewer tumors than TCGA, the single-cell resolution addresses compositional confounders (Smillie et al., 2019) and thus helps to reveal disease mechanisms. The power of HTAN datasets will increase by integration with prior bulk profiling studies in TCGA and ICGC through deconvolution (Jerby-Arnon et al., 2018; Tirosh et al., 2016a; Tirosh et al., 2016b) as well as by transfer learning for training predictive models while borrowing parameters from a model that performs well on a similar task (Azizi et al., 2018; Wang et al., 2019; Yosinski et al., 2014).
Fourth, HTAN will need heterogenous data types (Box 1 and Figure 3) along with clinical data (Figure 3) in one coherent model. This can benefit from ML approaches, such as multido-main translation (Yang and Uhler, 2019), that rely on the assumption that all observations (e.g., expression, epigenomics, and histology) should reflect a single latent space and that learn a joint embedding of all data modalities, either unsupervised or supervised with clinical data. The result will be a general model that aims to predict—on the basis of cellular compositions, states, interactions, and histological organization—the relevant clinical outcomes.
Fifth, there is an explosion in technological development, particularly for spatial assays measuring RNA and protein and for multiomic measurements, many of which have not been applied to tumor tissue. HTAN trans-network efforts will test and benchmark new technologies to assess the extent to which they can be used for profiling clinical samples.
HTAN as a Community Resource
To ensure that its efforts have the broadest impact, HTAN is committed to open-access publication and the sharing of data, clinical information, metadata, experimental protocols, and computational tools with the broader scientific community. These commitments are consistent with the ambitious goals of the NCI Cancer Moonshot data-sharing and open-access publication policy (https://www.cancer.gov/research/key-initiatives/moonshot-cancer-initiative/funding/public-access-policy).
All HTAN-generated data will be available through a dedicated cloud-enabled data portal provided by the HTAN Data Coordination Center (DCC); this portal will have proper access controls as required by patient privacy concerns and be based on findable, accessible, interoperable, and reusable (FAIR) data principles and standards compatible with other Cancer Moonshot Initiatives (Wilkinson et al., 2016). The HTAN data portal will be directly available to the international scientific community and will facilitate the transfer of HTAN data to existing and planned data nodes within the wider NCI Cancer Research Data Commons ecosystem. Data tables will also be accessible and queryable with the use of NCI Cloud Resources. HTAN strives to provide compatibility with other relevant platforms, such as the Human Cell Atlas (HCA) Data Coordination Platform and the Human Bio-molecular Atlas Program (HuBMAP) High-Performance Integrated Virtual Environment portal. To maximize the accessibility and interoperability of HTAN data and tools, the DCC will work in collaboration with HTAN centers to develop and unify annotation standards for clinical data elements used across HTAN and to develop and unify metadata on biospecimens and assay-specific data types. Application program interfaces will ensure inter-operability of data-validation and -visualization tools, and the availability of source code on GitHub will make it possible for others to build on HTAN technology.
Given the diverse data types and dynamic technology landscape, HTAN centers will both build data pipelines based on existing algorithms and leverage new tools developed by both consortium members and the broader scientific community as they arise. They will also develop analytical tools and fast, interactive, multiscale visualization algorithms that integrate molecular and spatial data. For example, one HTAN center has already released web-based and desktop tools for rapid zooming and panning across multichannel tumor images obtained by cyclic immunofluorescence (CyCIF, https://www.cycif.org; Krueger et al., 2020; Lin et al., 2015; Lin et al., 2018).
Just as importantly, HTAN will share experimental protocols and SOPs by using both open-access platforms such as protocols.io (Teytelman et al., 2016) and hands-on training by dedicated teams of HTAN researchers. To better disseminate knowledge about complex, multistep laboratory procedures, one HTAN pilot project (see below) has developed a “specialized work acquisition teams” model in which a group of experts from one laboratory travels to another to work side by side to fully, accurately, and quickly convey their experimental and computational know-how.
Who Is Part of HTAN?
HTAN currently consists of a network of ten interdisciplinary research centers—five focused on pre-cancers and five studying more advanced tumors—working together along with the DCC. Each center focuses on one or more aspects of specific tumor types and transitions (Figure 4) and employs a subset of the experimental and analytical tools of the HTAN toolbox (Box 1). The centers coordinate through a joint steering committee with four working groups (policy, clinical and biospecimen, molecular characterization, and data analysis working groups). More information on funded HTAN research projects can be found on the NCI Cancer Moonshot website (https://www.cancer.gov/research/key-initiatives/moonshot-cancer-initiative/implementation/human-tumor-atlas).
HTAN members are actively engaged with other complementary initiatives both at the NIH—including HuBMAP (HuBMAP Consortium, 2019), the Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Initiative (Ecker et al., 2017), the NCI Information Technologies for Cancer Research (ITCR), the Cancer Systems Biology Consortium (CSBC), and the Cancer Research Data Commons—and internationally, such as the HCA (Regev et al., 2017; Rozenblatt-Rosen et al., 2017) and the Global Alliance for Genomics and Health. HTAN will interact and synergize with these efforts to obtain a broader and more complete picture of the dynamic molecular and cellular states that drive cancers, their therapeutic responses, and/or resistance. The 3D maps of normal human organs are expected to serve as particularly valuable references for corresponding tumor atlases (Figure 2). Collaboration with atlas-building consortia for other diseases will further serve to address overlapping experimental and computational challenges (Figure 2). Likewise, work in the CSBC could provide much-needed experimental data for multiscale computational modeling of tumor biology.
In consideration of the challenges that HTAN faces, two feasibility pilot projects were initiated prior to its launch: the Pre-Cancer Atlas Pilot Project (PCAPP) and the Human Tumor Atlas Pilot Project (HTAPP). These projects were designed to develop and advance the standardization of key protocols, procedures, metadata schemata, and tools for use by HTAN. Data and protocols generated by the pilot projects are being shared both within HTAN and with the broader community. For example, PCAPP has generated SOPs and guidelines for generating FFPE blocks for optimal spatial profiling, an imaging panel for pre-cancer samples, and protocols for low-input profiling of DNA and RNA from small FFPE samples (Foley et al., 2019). HTAPP has developed a sc/snRNA-seq toolbox for systematic processing of various fresh and frozen tumor samples (Slyper et al., 2019) and a cloud analysis pipeline (Li et al., 2019).
Outlook
By building a framework for integrating cellular and spatial molecular profiling of tumors during key clinical transitions, HTAN provides an opportunity to refine our fundamental concepts of malignancy and is poised to have a profound impact on translational medicine (Figure 6). HTAN atlases span multiple scales, and we expect their high-resolution view of clinically relevant transitions to highlight new aspects of the development and evolution of malignancy, metastatic disease, therapeutic response, and resistance—including both tumor-specific and universal mechanisms.
Figure 6. HTAN Will Have a Profound Impact on Cancer Biology and Medicine.
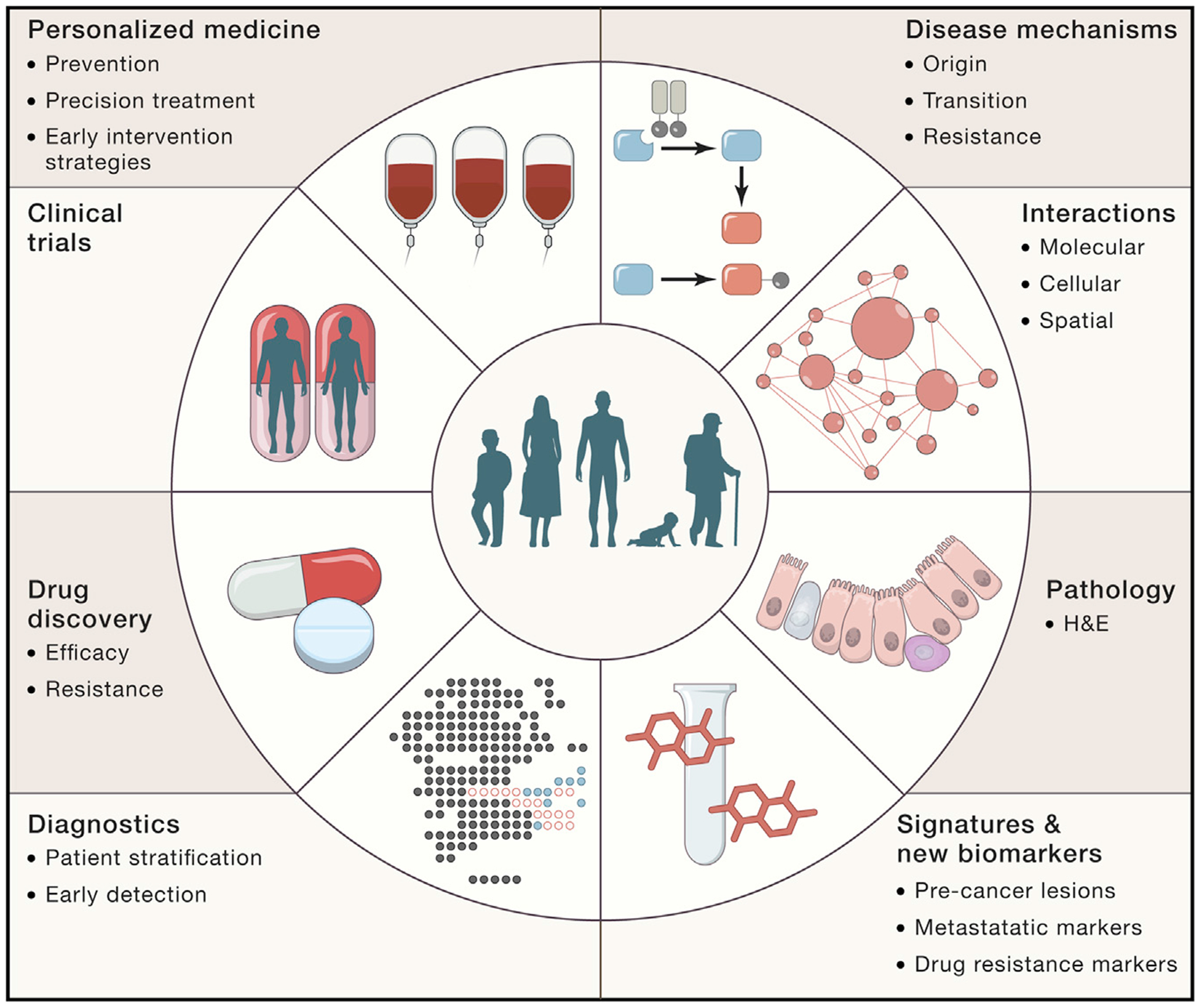
The translational promise of a tumor cell atlas ranges from basic understanding of disease mechanisms, diagnosis, prognosis, treatment monitoring, drug development, biomarker discovery, and patient stratification and will ultimately facilitate an era of precision medicine.
In pre-cancers, HTAN should help identify the genetic, epigenetic, and environmental factors involved in the earliest steps of malignancy, such as non-cell-autonomous factors distinguishing successful from unsuccessful immune surveillance. In later stage cancers, atlases should help us to understand the difference between immune-infiltrated and cold tumors; the drivers of metastasis, which could be more readily discernable with spatial data than with purely genomic data; and the impact of tumor heterogeneity and ecosystems on therapeutic response and resistance. A better understanding of these mechanisms will inform new predictive models and prognostic biomarkers, signatures, and diagnostics that can ultimately be deployed at point of care to improve the outcomes of cancer patients. It will also improve the development of preventive strategies, therapeutic agents, and drug combinations that can effectively target these processes at multiple steps in the malignant process.
HTAN atlases will be shared community resources that accelerate both exploratory and hypothesis-driven research. The network will provide protocols, software, and best-practice guidelines to promote the development and deployment of technologies that we believe will have a profound impact on the study of human tumors, including open standards in histology and histopathology. HTAN’s collaborative nature will establish a model within our scientific community for the integration of efforts and data. We hope that HTAN will give rise to a foundational resource that will improve the understanding, diagnosis, monitoring, and treatment of cancer patients.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jennifer Beane-Ebel for critical reading of the manuscript. This work is supported by the National Cancer Institute Cancer Moonshot under awards U24 CA233243, U2C CA233195, U2C CA233238, U2C CA233254, U2C CA233262, U2C CA233280, U2C CA233284, U2C CA233285, U2C CA233291, U2C CA233303, and U2C CA233311. The Lung Pre-Cancer Atlas is partly supported by funding from Janssen Research & Development LLC (principal investigator [PI]: S.A.M.), Johnson & Johnson (PI: S.A.M.), and a Stand Up To Cancer (SU2C)-LUNGevity-American Lung Association Lung Cancer Interception Dream Team Translational Cancer Research Grant (SU2C-AACR-DT23-17 to S.M.D. and A.E.S.). SU2C is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. The Human Tumor Atlas Pilot Project is supported by federal funds from the NIH National Cancer Institute (task order no. HHSN261100039 under contract no. HHSN261201500003I). A.R. is an investigator of the Howard Hughes Medical Institute. A.R. and O.R.R. are supported by the Klarman Cell Observatory at the Broad Institute. D.S. is funded by an Early Postdoc Mobility fellowship (no. P2ZHP3_181475) from the Swiss National Science Foundation.
DECLARATION OF INTERESTS
A.R. is a founder of and equity holder in Celsius Therapeutics, an equity holder in Immunitas Therapeutics, and a scientific advisory board (SAB) member of Syros Pharmaceuticals, ThermoFisher Scientific, Asimov, and NeoGene Therapeutics. A.K.S. has received compensation for consulting for and being on the SAB of Honeycomb Biotechnologies, Cellarity, Cogen Therapeutics, and Dahlia Biosciences. O.R.R., A.K.S., and A.R. are co-inventors on patent applications filed by the Broad Institute to inventions relating to single-cell genomics, such as in PCT/US2018/060860 and US provisional application no. 62/745,259. J.W.G. has licensed technologies to Abbott Diagnostics and Danaher and has ownership positions in PDX Pharmaceuticals and Convergent Genomics. He serves as an advisor to New Leaf Ventures and receives private-sector research support from Zeiss, ThermoFisher, Danaher, Micron, PDX Pharmaceuticals, and Quantitative Imaging. C.I.D. receives research support from Bristol-Myers Squibb. S.S. is a consultant for RareCyte, Inc. A.S. is an employee of Johnson & Johnson. S.A.M. has commercial research grants from Johnson & Johnson. P.K.S. is a member of the SAB or board of directors of and has equity in Glencoe Software and RareCyte Inc., which create software and instruments for tissue imaging.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.03.053.
REFERENCES
- Achim K, Pettit JB, Saraiva LR, Gavriouchkina D, Larsson T, Arendt D, and Marioni JC (2015). High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat. Biotechnol 33, 503–509. [DOI] [PubMed] [Google Scholar]
- Amodio M, and Krishnaswamy S (2018). MAGAN: aligning biological manifolds. arXiv arXiv:1803.00385. [Google Scholar]
- Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, Levenson RM, Lowe JB, Liu SD, Zhao S, et al. (2014). Multiplexed ion beam imaging of human breast tumors. Nat. Med 20, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argelaguet R, Arnol D, Bredikhin D, Deloro Y, Velten B, Marioni JC, and Stegle O (2019). MOFA+: a probabilistic framework for comprehensive integration of structured single-cell data. bioRxiv. 10.1101/837104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkas N, Petukhov V, Nikolaeva D, Lozinsky Y, Demharter S, Khodosevich K, and Kharchenko PV (2019). Joint analysis of heterogeneous single-cell RNA-seq dataset collections. Nat. Methods 16, 695–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beane JE, Mazzilli SA, Campbell JD, Duclos G, Krysan K, Moy C, Perdomo C, Schaffer M, Liu G, Zhang S, et al. (2019). Molecular subtyping reveals immune alterations associated with progression of bronchial pre-malignant lesions. Nat. Commun 10, 1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner M, Miao Z, Wolf FA, Teichmann SA, and Theis FJ (2019). A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 16, 43–49. [DOI] [PubMed] [Google Scholar]
- Chen F, Tillberg PW, and Boyden ES (2015). Optical imaging. Expansion microscopy. Science 347, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Wassie AT, Cote AJ, Sinha A, Alon S, Asano S, Daugharthy ER, Chang JB, Marblestone A, Church GM, et al. (2016a). Nanoscale imaging of RNA with expansion microscopy. Nat. Methods 13, 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Shishkin AA, Zhu X, Kadri S, Maza I, Guttman M, Hanna JH, Regev A, and Garber M (2016b). Evolutionary analysis across mammals reveals distinct classes of long non-coding RNAs. Genome Biol. 17, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Lin JR, Rashid R, Maliga Z, Wang S, Aster JC, Izar B, Sorger PK, and Santagata S (2019). Qualifying antibodies for image-based immune profiling and multiplexed tissue imaging. Nat. Protoc 14, 2900–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durruthy-Durruthy R, Gottlieb A, Hartman BH, Waldhaus J, Laske RD, Altman R, and Heller S (2014). Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell 157, 964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker JR, Geschwind DH, Kriegstein AR, Ngai J, Osten P, Polioudakis D, Regev A, Sestan N, Wickersham IR, and Zeng H (2017). The BRAIN Initiative Cell Census Consortium: lessons learned toward generating a comprehensive brain cell atlas. Neuron 96, 542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley JW, Zhu C, Jolivet P, Zhu SX, Lu P, Meaney MJ, and West RB (2019). Gene expression profiling of single cells from archival tissue with laser-capture microdissection and Smart-3SEQ. Genome Res. 29, 1816–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, and Bruni D (2019). Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov 18, 197–218. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaublomme JT, Li B, McCabe C, Knecht A, Yang Y, Drokhlyansky E, Van Wittenberghe N, Waldman J, Dionne D, Nguyen L, et al. (2019). Nuclei multiplexing with barcoded antibodies for single-nucleus genomics. Nat. Commun 10, 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierahn TM, Wadsworth MH 2nd, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Love JC, and Shalek AK (2017). Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 14, 395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schüffler PJ, Grolimund D, Buhmann JM, Brandt S, et al. (2014). Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422. [DOI] [PubMed] [Google Scholar]
- Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, Black S, and Nolan GP (2018). Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow I, Bengio Y, and Courville A (2017). Deep Learning (MIT Press; ). [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium HuBMAP (2019). The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Cancer Genome Consortium (2010). International network of cancer genome projects. Nature 464, 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobuzio-Donahue CA, Michael C, Baez P, Kappagantula R, Hooper JE, and Hollman TJ (2019). Cancer biology as revealed by the research autopsy. Nat. Rev. Cancer 19, 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AH, Anand S, Singh M, and Veeravasarapu V (2018). Disentangling factors of variation with cycle-consistent variational auto-encoders. Proceedings of the Springer International Publishing 15th European Conference, September 8–14, 2018, Part III. [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, Wan E, Wong S, Byrnes L, Lanata CM, et al. (2018). Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol 36, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger R, Beyer J, Jang WD, Kim NW, Sokolov A, Sorger PK, and Pfister H (2020). Facetto: Combining Unsupervised and Supervised Learning for Hierarchical Phenotype Analysis in Multi-Channel Image Data. IEEE Trans. Vis. Comput. Graph 26, 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, Terry R, Jeanty SS, Li C, Amamoto R, et al. (2014). Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Gould J, Yang Y, Sarkizova S, Tabaka M, Ashenberg O, Rosen Y, Slyper M, Kowalczyk MS, Villani A-C, et al. (2019). Cumulus: a cloud-based data analysis framework for large-scale single-cell and single-nucleus RNA-seq. bioRxiv. 10.1101/823682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Fallahi-Sichani M, and Sorger PK (2015). Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun 6, 8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Izar B, Wang S, Yapp C, Mei S, Shah PM, Santagata S, and Sorger PK (2018). Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7, e31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Huang Y, Singh R, Vert J-P, and Stafford Noble W (2019). Jointly embedding multiple single-cell omics measurements. bioRxiv. 10.1101/644310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R, Nazaret A, Langevin M, Samaran J, Regier R, Jordan M, and Yosef N (2019). A joint model of unpaired data from scRNA-seq and spatial transcriptomics for imputing missing gene expression measurements. arXiv, arXiv:1905.02269 [Google Scholar]
- Luecken MD, and Theis FJ (2019). Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol 15, e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Jiang W, Jie Z, Jiang Y, and Liu W (2019). Matching image and sentence with multi-faceted representations. IEEE Trans. Circ. Syst. Video Tech 10.1109/TCSVT.2019.2916167. [DOI] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Hao J, Bambah-Mukku D, Lu T, Dulac C, and Zhuang X (2016a). High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. Proc. Natl. Acad. Sci. USA 113, 14456–14461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Hao J, Wang G, Chen KH, Babcock HP, and Zhuang X (2016b). High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl. Acad. Sci. USA 113, 11046–11051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, et al. (2017). The Human Cell Atlas. eLife 6, e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W, et al. (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O, Stubbington MJT, Regev A, and Teichmann SA (2017). The Human Cell Atlas: from vision to reality. Nature 550, 451–453. [DOI] [PubMed] [Google Scholar]
- Rudnick PA, Markey SP, Roth J, Mirokhin Y, Yan X, Tchekhovskoi DV, Edwards NJ, Thangudu RR, Ketchum KA, Kinsinger CR, et al. (2016). A description of the Clinical Proteomic Tumor Analysis Consortium (CPTAC) common data analysis pipeline. J. Proteome Res 15, 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, et al. (2015). The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med 373, 1926–1936. [DOI] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, et al. (2013). Sin gle-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, Smillie CS, Smith-Rosario G, Wu J, Dionne D, et al. (2019). A single-cell and single-nucleus RNA-seq toolbox for fresh and frozen human tumors. bioRxiv. 10.1101/761429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, et al. (2019). Intraand Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 178, 714–730.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Ghosh S, Kagan J, and Mazurchuk R (2018). The making of a PreCancer Atlas: promises, challenges, and opportunities. Trends Cancer 4, 523–536. [DOI] [PubMed] [Google Scholar]
- Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, et al. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82. [DOI] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, Smibert P, and Satija R (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck W.r., Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902. https://www.ncbi.nlm.nih.gov/pubmed/31178118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teytelman L, Stoliartchouk A, Kindler L, and Hurwitz BL (2016). Protocols.io: virtual communities for protocol development and discussion. PLoS Biol. 14, e1002538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016a). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. (2016b). Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta JJ, Gennert D, Lu D, Satija R, Shalek AK, and Regev A (2014). Preparation of single-cell RNA-seq libraries for next generation sequencing. Curr. Protoc. Mol. Biol 107, 4.22.1–4.22.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickovic S, Ståhl PL, Salmén F, Giatrellis S, Westholm JO, Mollbrink A, Navarro JF, Custodio J, Bienko M, Sutton LA, et al. (2016). Massive and parallel expression profiling using microarrayed single-cell sequencing. Nat. Commun 7, 13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitak SA, Torkenczy KA, Rosenkrantz JL, Fields AJ, Christiansen L, Wong MH, Carbone L, Steemers FJ, and Adey A (2017). Sequencing thousands of single-cell genomes with combinatorial indexing. Nat. Methods 14, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, Hofree M, Cuoco MS, Rodman C, Farouq D, et al. (2017). The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Yan X, Lee H, and Livescu K (2016). Deep Variational Canonical Correlation Analysis. arXiv, arXiv:161003454. [Google Scholar]
- Wang J, Agarwal D, Huang M, Hu G, Zhou Z, Ye C, and Zhang NR (2019). Data denoising with transfer learning in single-cell transcriptomics. Nat. Methods 16, 875–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, and Stuart JM (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, and Macosko EZ (2019). Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177, 1873–1887.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten J-W, da Silva Santos LB, Bourne PE, et al. (2016). The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3, 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KD, and Uhler C (2019). Multi-domain translation by learning un-coupled autoencoders. arXiv, arXiv:1902.03515 [Google Scholar]
- Yosinski J, Clune J, Bengio Y, and Lipson H (2014). How transferable are features in deep neural networks? arXiv, arXiv:1411.1792. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.