

Abstract
Presently, the SARS-CoV-2 (COVID-19) pandemic has been spreading throughout the world. Some drugs such as lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir have been recommended for COVID-19 treatment by some researchers, but these drugs were not effective enough against this virus. This study based on in silico approaches was aimed to increase the anti-COVID-19 activities of these drugs by using caulerpin and its derivatives as an adjunct drug against SARS-CoV-2 receptor proteins: the SARS-CoV-2 main protease and the SARS-CoV-2 spike protein. Caulerpin exhibited antiviral activities against chikungunya virus and herpes simplex virus type 1. Caulerpin and some of its derivatives showed inhibitory activity against Alzheimer’s disease. The web server ANCHOR revealed higher protein stability for the two receptors with disordered score (< 0.6). Molecular docking analysis showed that the binding energies of most of the caulerpin derivatives were higher than all the suggested drugs for the two receptors. Also, we deduced that inserting NH2, halogen, and vinyl groups can increase the binding affinity of caulerpin toward 6VYB and 6LU7, while inserting an alkyl group decreases the binding affinity of caulerpin toward 6VYB and 6LU7. So, we can modify the inhibitory effect of caulerpin against 6VYB and 6LU7 by inserting NH2, halogen, and vinyl groups. Based on the protein disordered results, the SARS-CoV-2 main protease and SARS-CoV-2 spike protein domain are highly stable proteins, so it is quite difficult to unstabilize their integrity by using individual drugs. Also, molecular dynamics (MD) simulation indicates that binding of the combination therapy of simeprevir and the candidate studied compounds to the receptors was stable and had no major effect on the flexibility of the protein throughout the simulations and provided a suitable basis for our study. So, this study suggested that caulerpin and its derivatives could be used as a combination therapy along with lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir for disrupting the stability of SARS-CoV2 receptor proteins to increase the antiviral activity of these drugs.
Keywords: SARS-CoV-2, Caulerpin, Simeprevir, Molecular docking, ANCHOR and MD simulation
Introduction
Currently, the world suffers from the spreading of a novel SARS-coronavirus called COVID-19 or SARS-Cov-2, and unfortunately, it is now a global epidemic. The record of the first cases with COVID-19 infections was in Wuhan, Hubei Province, China, at the end of December 2019, and then the infections spread worldwide [1]. On April 9, 2020, the total number of confirmed cases of COVID-19 exceeded one and a half million with more than 88,000 deaths worldwide. There are four genera of CoVs—α, β, γ, and δ—and the SARS-CoV-2 belongs to β-coronavirus [1, 2]. The genetic sequence of the 2019 novel coronavirus is similar to the genetic sequence of SARS-CoV more than that of MERS-CoV [3]. The most common symptoms of SARS-CoV-2 infection are lymphopenia, fever, dry cough, and fatigue [4], but it may lead to severe pneumonia and pulmonary edema [5]. Until now, there is no specific effective drug against SARS-CoV-2, but some previous studies reported that chloroquine and remdesivir inhibited SARS-CoV-2 and the possibility of using these drugs in the treatment of COVID-19 [6]. The genome encodes four structural proteins of the coronavirus, which are spike glycoprotein (S), small envelope protein (E), matrix glycoprotein (M), and nucleocapsid protein (N) [1]. The human angiotensin-converting enzyme 2 (hACE2) is an important SARS-CoV-2 receptor which is found in the cells of lung tissue [7], and the SARS-CoV-2 (S) glycoprotein enters cells by using this receptor. Among the functional coronavirus proteins is 3CLpro, which is essential for transcription, processing, synthesis and modification of RNA, and replication of the virus [8]. Natural sources of active constituents are more preferable in the treatment than chemically synthesized drugs because most synthesized drugs have unwanted side effects on human health [9]. Most natural compounds have various and effective biological activities such as antimicrobial, anticancer, anti-inflammatory, and antidiabetic [10]. Caulerpin, a low toxic bisindole alkaloid, is a more common compound of the genus Caulerpa of green macroalgae and it was isolated from Caulerpa racemosa and the red alga Chondrus armatus [11]. Caulerpin is characterized by a variety of biological activities being an antitumor [12], growth regulator [13], antidiabetic, anticancer, antilarvicidal, antitubercular, antimicrobial, antiviral, spasmolytic, antinociceptive, plant growth regulator [14–17], and anti-inflammatory [18]. Caulerpin exhibited antiviral activities against chikungunya virus [19] and herpes simplex virus type 1 [11]. Caulerpin and some of its derivatives showed inhibitory activity against Alzheimer’s disease [20]. In this study, we aim to evaluate the inhibitory effect of caulerpin and its analogs against the SARS-CoV-2 main protease Mpro (PDB ID: 6LU7) protease and the SARS-CoV-2 spike protein Sp (PDB ID: 6VYB) by molecular docking analysis. Also, we study the effect of side chain replacement of caulerpin on its inhibitory effect compared with some used drugs which are currently used as SARS-CoV-2 inhibitors such as lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir. Also, drug-likeness model score and ADMET properties were computed and analyzed. The protein disordered results from ANCHOR was computed to show the stability of the SARS-CoV-2 main protease and the SARS-CoV-2 spike protein domain. Also, a combination therapy between caulerpin and its derivatives along with lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir for disrupting the stability of SARS-CoV-2 receptor proteins to increase the antiviral activity of these drugs was studied in this work. Also, molecular dynamics (MD) simulation was performed to study the binding of the combination therapy of simeprevir and the candidate studied compounds to the receptors and the stability of these complexes throughout the simulations in the presence of water.
Materials and methodology
Ligand preparation
The structures of all the studied compounds were downloaded in SDF format and shown in Fig. 1, and their SMILES are displayed in Table 1. The structures of all the studied compounds were further refined in ChemDraw3D Ultra to avoid any repetition, and energy minimization was carried out on all the studied compounds using Molecular Mechanics 2 (MM2) force field method before docking. The structures of lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir were obtained from the PubChem database. The structural optimization was carried out using MM2 force field. Afterwards, the structures were converted into pdbqt format by using the AutoDockTools 1.5.6 software.
Fig. 1.

Structures of all the studied compounds (the red rectangles represented side chain replacement) (1–20)
Table 1.
SMILES of all the studied compounds (1–20)
| Compounds | SMILES |
|---|---|
| 1 | [H]N1c2c(\[c]([H])=C(/c3N([H])c4c([H])c([H])c([H])c([H])c4-c3\[c]([H])=C2\C(=O)OC([H])([H])[H])C(=O)OC([H])([H])[H])-c2c([H])c([H])c([H])c([H])c12 |
| 2 | [H]N1c2c(\[c]([H])=C(/c3N([H])[c]4=[c](c([H])c([H])c([H])c4[H])-c3\[c]([H])=C2\C(F)=O)C(F)=O)-[c]2=[c]1c([H])c([H])c([H])c2[H] |
| 3 | [H]N([H])C(=O)C1=[c]([H])/c-2c(N([H])[c]3=[c]-2c([H])c([H])c([H])c3[H])\C(=[c]([H])/c-2c\1N([H])[c]1=[c]-2c([H])c([H])c([H])c1[H])C(=O)N([H])[H] |
| 4 | [H]N([H])C(=O)C1=[c]([H])/c-2c(N([H])[c]3=[c]-2c([H])c([H])c([H])c3[H])\C(=[c]([H])/c-2c\1N([H])[c]1=[c]-2c([H])c([H])c([H])c1[H])C(F)=O |
| 5 | [H]C([H])=C([H])C([H])([H])C([H])([H])OC(=O)C1=[c]([H])/c-2c(N([H])[c]3=[c] 2c([H])c([H])c([H])c3[H])\C(=[c]([H])/c2c\1N([H])[c]1=[c]2c([H])c([H])c([H])c1[H])C(=O)OC([H])([H])C([H])([H])C([H])=C([H])[H] |
| 6 | [H]c1c([H])c([H])[c]-2=[c](Oc3c-2\[c]([H])=C(/c2O[c]4=[c](c([H])c([H])c([H])c4[H])-c2\[c]([H])=C3\C(=O)OC([H])([H])[H])C(=O)OC([H])([H])[H])c1[H] |
| 7 | [H]c1c([H])c([H])[c]-2=[c](c1[H])N(c1c-2\[c]([H])=C(/c2N([c]3=[c](c([H])c([H])c([H])c3[H])-c2\[c]([H])=C1\[c](=O):[o]C([H])([H])[H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H])[c](=O):[o]C([H] |
| 8 | [H]N1c2c(\[c]([H])=C(/c3N([H])c4c([H])c([H])c([H])c(Cl)c4-c3\[c]([H])=C2\C(=O)OC([H])([H])[H])C(=O)OC([H])([H])[H])-c2c([H])c([H])c([H])c([H])c12 |
| 9 | [H]N1c2c(\[c]([H])=C(/c3N([H])c4c([H])c([H])c([H])c(c4-c3\[c]([H])=C2\C(=O)OC([H])([H])[H])C([H])([H])[H])C(=O)OC([H])([H])[H])-c2c([H])c([H])c([H])c([H])c12 |
| 10 | [H]N1c2c(\[c]([H])=C(/c3N([H])c4c([H])c([H])c([H])c(c4-c3\[c]([H])=C2\C(=O)OC([H])([H])[H])C([H])([H])[H])C(=O)OC([H])([H])[H])-c2c1c([H])c([H])c([H])c2C([H])([H])[H] |
| 11 | [H]N1c2c(\[c]([H])=C(/c3N([H])[c]4=[C](=[c]([H])c(Cl)c([H])c4Cl)c3\[c]([H])=C2\C(=O)OC([H])([H])[H])C(=O)OC([H])([H])[H])-[c]2=[c]1c(Cl)c([H])c(Cl)c2[H] |
| 12 | [H]Oc1c([H])c([H])[c]2=[c](c1[H])-c1c(N2[H])\C(=[c]([H])/c-2c(N([H])[c]3=[c]-2c([H])c([H])c([H])c3[H])\C(=[c]1\[H])C(=O)OC([H])([H])[H])C(=O)OC([H])([H])[H] |
| 13 | [H]Oc1c([H])c([H])c([H])[c]-2=[c]1N([H])c1c-2\[c]([H])=C(/c2N([H])[c]3=[c](c([H])c([H])c([H])c3[H])-c2\[c]([H])=C1\C(=O)OC([H])([H])[H])C(=O)OC([H])([H])[H] |
| 14 | [H]C([H])=C([H])C([H])C(=O)C1=[c]([H])/c-2c(N([H])[c]3=[c]-2c([H])c([H])c([H])c3[H])\C(=[c]([H])/c-2c\1N([H])[c]1=[c]-2c([H])c([H])c([H])c1[H])C(=O)OC([H])([H])[H] |
| 15 | [H]N1c2c(\[c]([H])=C(/c3N([H])[c]4=[c](c([H])c([H])c([H])c4[H])-c3\[c]([H])=C2\C(=O)OC([H])([H])[H])C(=O)OC([H])([H])C([H])([H])C([H])([H])C([H])([H])[H])-[c]2=[c]1c([H])c([H])c([H])c2[H] |
| 16 | [H]N1c2c(\[c]([H])=C(/c3N([H])[c]4=[c](c([H])c([H])c([H])c4[H])-c3\[c]([H])=C2\C(=O)OC([H])([H])C([H])([H])C([H])([H])C([H])([H])[H])C(=O)OC([H])([H])C([H])([H])C([H])([H])C([H])([H])[H])-[c]2=[c]1c([H])c([H])c([H])c2[H] |
| 17 | [H]N1c2c(\[c]([H])=C(/c3N([H])c4c(c([H])c([H])c([H])c4C([H])([H])[H])-c3\[c]([H])=C2\C(=O)OC([H])([H])[H])C(=O)OC([H])([H])[H])-c2c([H])c([H])c([H])c(c12)C([H])([H])[H] |
| 18 | [H]C([H])=C([H])C([H])C(=O)C1=[c]([H])/c-2c(N([H])[c]3=[c]-2c([H])c([H])c([H])c3[H])\C(=[c]([H])/c-2c\1N([H])[c]1=[c]-2c(c([H])c([H])c1[H])C([H])([H])[H])C(F)=O |
| 19 | [H]N1c2c(\[c]([H])=C(/c3N([H])[c]4=[c](-c3\[c]([H])=C2\C(F)=O)c(c([H])c([H])c4[H])C([H])([H])[H])C(=O)OC([H])([H])C([H])([H])C([H])([H])C([H])([H])[H])-[c]2=[c]1c([H])c([H])c([H])c2[H] |
| 20 | [H]C([H])=C([H])C([H])C(=O)C1=[c]([H])/c-2c(N([H])[c]3=[c]-2c([H])c([H])c([H])c3[H])\C(=[c]([H])/c-2c\1N([H])[c]1=[c]-2c([H])c([H])c([H])c1[H])C(I)=O |
Protein preparation
The 3D crystal structure of the SARS-CoV-2 main protease Mpro (PDB ID: 6LU7) and the cryo-electron microscopic structure of the SARS-CoV-2 spike protein Sp (PDB ID: 6VYB) were taken from the PDB (Protein Data Bank) site. Small molecules were removed from the crystal structures of 6LU7 by using the BIOVIA Discovery Studio software [21].
Molecular docking
Polar hydrogens and Kollman charges were added to the protein and a pdbqt format file was generated by using the AutoDockTools 1.5.6 software. The protein was prepared using the protein preparation wizard of AutoDockTools 1.5.6. Polar hydrogens and Kollman charges were added to the protein and a pdbqt format file was generated by using AutoDockTools. All water molecules were deleted from 6LU7 and 6VYB. The torsions for the ligands were set by detecting the roots in AutoDockTools 1.5.6 followed by setting aromaticity criteria of 7.5. We defined a grid size with 60 Å × 60 Å × 60 Å for two receptors and the Lamarckian genetic algorithm (LGA) was assigned to carry out the molecular docking process, as described in this study [22]. To validate the docking protocol, bound ligand inhibitor N3 coordinates in the crystal complex of 6LU7 were removed and the bond orders were checked. Then, we performed the docking studies of N3 inside 6LU7 to validate the docking protocol. Once docking is done, the best pose was selected based on binding energy, ligand–receptor interactions, and active site residues. Then, the docked pose with that of the co-crystallized structure was simply aligned and then root mean square deviation (RMSD) was calculated lower than 1.0 Å. For docking calculations, Gasteiger partial charges were assigned to the tested derivatives and inhibitor N3, and nonpolar hydrogen atoms were merged. All torsions were allowed to rotate during docking. The standard docking protocol for rigid and flexible ligand docking consisted of 10 independent runs per ligand, using an initial population of 50 randomly placed individuals, with 2.5 × 106 energy evaluations, a maximum number of 27,000 iterations, a mutation rate of 0.02, a crossover rate of 0.80, and an elitism value of 1. The probability of performing a local search on an individual in the population was 0.06, using a maximum of 300 iterations per local search. After docking, the 10 solutions were clustered into groups with RMS deviations lower than 1.0 Å. The clusters were ranked by the lowest energy representative of each cluster. The results obtained from the docking process were visualized using the BIOVIA Discovery Studio software.
Multiple ligand simultaneous docking
Multiple ligand simultaneous docking (MLSD) method simulates the interaction of multiple ligands inside the receptor. The multiple ligands can be a mixture of substrates or cofactors. The present implementation of MLSD assumes the algorithms and scoring function of AutoDock4 and helps in investigating the interaction between multiple ligands with the target receptor. In setting the dock parameters for MLSD, inhibitor molecules were read separately using AutoDockTools (ADT) and saved with appropriate torsions and charges as .PDBQT files. Meanwhile, the receptor molecule was also read and a dock parameter file (.dpf) was generated. With individual dock parameter files of the inhibitor molecules, the substrate’s dpf was merged into one single file to run MLSD simulation. Once prepared with the merged dock parameter file, docking begins with the random initialization of the population. MLSD stands separate from single ligand docking where different conformations of multiple ligands can be run simultaneously. To achieve this, the program loads the .PDBQT files of both substrate and inhibitor to perform simulation. Each ligand will be randomly initialized with its own set of state variables attaining specific configuration, ligand center, torsion tree, and a group of atomic coordinates. Standard LGA procedure and the pseudo-Solis and Wets method were applied for energy minimization. After each generation of genetic operations, the MLSD program maps the genotype of a ligand back to phenotype so that each ligand has its own phenotype, and the coordinates of all the parameters set in the docking MLSD protocol and the validation of the docking method were done according to this study [23].
Analysis of drug-likeness and ADMET properties of all the studied bioactive compounds
The drug-likeness prediction of all the studied bioactive compounds was carried out by Lipinski filter (http://www.scfbio-iitd.res.in/software/drugdesign/lipinski.jsp), according to which an orally active drug should comply with a minimum of four of the five laid down criteria for drug-likeness, namely: molecular mass, cLogP, hydrogen donor and acceptor, and molar refractive index [24]. Furthermore, pharmacokinetic properties like absorption, distribution, metabolism, excretion, and toxicity of all the studied compounds were predicted utilizing the admetSAR database (http://lmmd.ecust.edu.cn/admetsar1/predict) [25]. Also, all the studied compounds were submitted to an in silico molecular properties of descriptors and prediction of bioactivity score based on Lipinski’s rule of five using the Molinspiration server (http://www.molinspiration.com).
Molecular dynamics simulation
The structures of the highest binding complexes obtained from the molecular docking study of simeprevir + compound 7–6LU7, simeprevir + compound 19–6LU7, simeprevir + compound 7–6VYP, and simeprevir + compound 7–6VYP complexes were prepared for MD simulation using standard dynamic cascade implicit in Discovery Studio. The MD simulation of the studied complexes was carried out at 10 ns using CHARMm force field for all atoms in the complex. The simulation started by solvating the complex in triclinic box using the TIP3P water model. The counter ions were added to neutralize the system. In the 6LU7 complexes, the studied systems were solvated in 6506 water molecules and neutralized by 20 sodium and 17 chloride as counter ions, while in the state of 6VYP complexes, the studied systems were solvated in 46,502 water molecules and neutralized by 128 sodium and 123 chloride as counter ions. Periodic boundary conditions were used. Throughout the simulation, each complex system is maintained at the temperature of 300 K with constant pressure. Energy minimization was done for 50,000 steps. The trajectories were collected for every nanosecond to get insights into the interactions at the atomistic level. All MD protocol was carried out according to this study [26]. The complexes resulting from MD simulation were analyzed for RMSD and root mean square fluctuation (RMSF). Also, the interaction energy was calculated to gain insight into the importance of electrostatic and van der Waals contributions in the formation of complexes. The calculation of interaction energy was carried out also by Discovery Studio on the last 1 ns obtained from the MD of the system.
Determination of protein stability
The stability of protein receptors was decided using the web server (http://iupred.enzim.hu and http://iupred.elte.hu) algorithm IUPred2 and ANCHOR that were assigned for this step. The FASTA files of the protein receptors of SARS-CoV-2 main protease (PDB ID: 6LU7) containing 306 residues and SARS-CoV-2 spike protein domain (PDB ID: 6VYB) with 975 residues were uploaded on the web server for protein disorders estimation [27, 28]. The predication mechanism of the IUPred algorithm depends on the energy estimation approach at a low-resolution statistical potential to discover the ability of amino acid pairs to produce contacts and examine as globular protein structures [29]. The statistical potential computes the energy for all residues related to its interactions with other structure-contacting residues in the state of known structure. The total stabilizing energy of the system can be calculated by the sum of residue-level energy and intrachain interactions in protein structure energy. Consequently, this novel technique has been established to decide these energies are exactly from the amino acid sequence of unknown structure as shown in this study [30]. Similar to IUPred, ANCHOR also utilizes the energy calculation method for identifying the disordered binding sites. In spite of the general disorder tendency, two additional terms have also been inserted into this method, to calculate energy based on the interaction with a globular protein and with the disturbed sequence [31].
Results and discussion
Prediction of protein stability
The diagram taken from the web server (https://iupred2a.elte.hu) (Fig. 2) after downloading the FASTA file of each protein receptor produced a score less than 0.6 for all residues of 6LU7 as shown in Fig. 2a and of 6VYB as shown in Fig. 2b, showing that the reliability of residues in selected protein receptors of SARS-CoV-2 is very high [32, 33]. So, SARS-CoV-2 main protease and SARS-CoV-2 spike protein are extremely stable proteins, so it is very difficult to unstabilize the stability of these proteins through using individual drugs. Hence, individual drugs like lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir may not be able to disrupt the stability of SARS-CoV-2 main protease (PDB ID: 6LU7) and SARS-CoV-2 spike protein domain (PDB ID: 6VYB).
Fig. 2.

Prediction of protein disorder using the IUPred web server for a SARS-CoV-2 main protease (PDB ID: 6LU7) and b SARS-CoV-2 spike protein domain (PDB ID: 6VYB) receptors
Molecular docking
The 2D interactions of all the studied compounds inside 6LU7 and 6VYB are shown in Figs. 3, 4, 5, and 6 and Figs. 7, 8, 9, and 10, respectively. Table 2 displays the molecular docking analysis results for all the studied compounds and some proposed antiviral drugs against 6LU7 and 6VYB. The molecular docking analysis showed that compounds 3, 4, 7, 10, 12, 16, 18, and 19 exhibited the highest binding energy (− 10.88, − 10.61, − 10.59, − 10.50361, − 11.38, − 10.67, − 11.44, and − 11.81 kcal/mol, respectively) compared to all the chemical drugs lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir as shown in Table 2 in the state of 6LU7. The studied compounds that exhibited the highest binding energy interact with amino acid residuals of 6LU7 through different types of interactions such as hydrogen bond, halogen, pi-alkyl, pi-sigma, and van der Waals. Compound 7 interacts with ILE 152 and PHE 8 residuals of 6LU7 through pi-alkyl, with TYR 154 through hydrogen bond interaction, and with ASP 155 and LYS 12 through van der Waals interaction as shown in Fig. 4. Compound 10 interacts with TRP 218 and LEU 220 residuals of 6LU7 through pi-alkyl and with ARG 217 through hydrogen bond interaction as shown in Fig. 4. Compound 19 interacts with MET 276 and LEU 286 residuals of 6LU7 through pi-alkyl and with ASN 277 through van der Waals interaction as shown in Fig. 6. Compound 18 interacts with LYS 97 and PRO 99 residuals of 6LU7 through pi-alkyl and with PRO 96 through halogen interaction as shown in Fig. 6, while compound 3 interacts with LIE 152 and PRO 9 residuals of 6LU7 through pi-alkyl and with PHE 294, VAL 297, and SER 301 through van der Waals interaction and also with ASP 153 and ARG 298 through hydrogen bond interaction as shown in Fig. 3. The molecular docking analysis showed that compounds 7, 10, 11, 14, 17, 18, 19, and 20 exhibited the highest binding energy (− 12.04, − 10.8, − 10.22, − 10.61, − 10.82, − 9.45, − 11.02, and − 11.78 kcal/mol, respectively) compared to all the chemical drugs lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir as shown in Table 2 in the state of 6VYB. Also, the studied compounds which exhibited the highest binding energy interact with amino acid residuals of 6LU7 through different types of interactions such as hydrogen bond, halogen, pi-alkyl, pi-sigma, and van der Waals. Compound 7 interacts with TYR 612 and ARG 319 residuals of 6VYB through hydrogen bond interaction, with GLN 321 and VAL 620 through van der Waals interaction, and also with PHE 318 through pi-pi-T–shaped interaction as shown in Fig. 8. Compound 10 interacts with LEU 303 and GLN 957 residuals of 6VYB through hydrogen bond interaction and also with LYS 304 and TYR 313 through pi-alkyl interactions as shown in Fig. 8. Compound 19 interacts with SER 596 residual of 6LU7 through hydrogen bond interaction and also with LYS 310 and GLN 314 and 613 through van der Waals interaction as shown in Fig. 10, while compound 20 interacts with LYS 964, LEU 48, and SER 305 residuals of 6LU7 through hydrogen bond interaction and with ASN 860 through van der Waals interactions as shown in Fig. 10. In general, the binding affinity of the studied compounds in the state of 6VYB is larger than that of 6LU7. Also, we found that compounds 7, 10, and 19 have the same behavior against 6VYB and 6LU7. So, most of the studied compounds can act as 6VYB and 6LU7 inhibitors, especially compounds 7, 10, and 19.
Fig. 3.

2D interaction of the studied compounds (1–5) inside 6LU7
Fig. 4.

2D interaction of the studied compounds (6–10) inside 6LU7
Fig. 5.

2D interaction of the studied compounds (11–15) inside 6LU7
Fig. 6.

2D interaction of the studied compounds (16–20) inside 6LU7
Fig. 7.

2D interaction of the studied compounds (1–5) inside 6VYB
Fig. 8.

2D interaction of the studied compounds (6–10) inside 6VYB
Fig. 9.

2D interaction of the studied compounds (11–15) inside 6VYB
Fig. 10.

2D interaction of the studied compounds (16–20) inside 6VYB
Table 2.
Molecular docking analysis of the studied compounds (1–20) and some antiviral drugs against 6LU7 and 6VYB
| Ligand | 6LU7 | 6VYB |
|---|---|---|
| Binding energy (ΔG) kcal/mol | Binding energy (ΔG) kcal/mol | |
| 1 | − 8.77 | − 8.73 |
| 2 | − 10.33 | − 9.39 |
| 3 | − 10.88 | − 9.28 |
| 4 | − 10.61 | − 9.18 |
| 5 | − 10.25 | − 9.26 |
| 6 | − 9.60 | − 9.00 |
| 7 | − 10.59 | − 12.04 |
| 8 | − 10.42 | − 9.30 |
| 9 | − 8.53 | − 8.63 |
| 10 | − 10.50 | − 10.87 |
| 11 | − 8.86 | − 10.22 |
| 12 | − 11.38 | − 9.13 |
| 13 | − 8.14 | − 8.16 |
| 14 | − 9.72 | − 10.61 |
| 15 | − 8.57 | − 8.395 |
| 16 | − 10.67 | − 9.00 |
| 17 | − 9.62 | − 10.82 |
| 18 | − 11.44 | − 9.45 |
| 19 | − 11.81 | − 11.02 |
| 20 | − 9.97 | − 11.78 |
| Lopinavir | − 8.93 | − 8.23 |
| Amprenavir | − 8.69 | − 7.47 |
| Chloroquine | − 8.11 | − 7.10 |
| Hydroxychloroquine | − 8.43 | − 7.85 |
| Simeprevir | − 10.46 | − 9.45 |
Effect of side chain on binding energy (side chain contribution in binding energy)
In this section, we summarize the effect of side chain replacement on the binding energy of all the studied compounds against 6VYB and 6LU7 as shown in Table 3. The study on the effect of side chain on binding energy is very important in this work to summarize the ability of different functional groups in binding energy contribution. As shown in Table 3, inserting NH2, halogen, and vinyl groups can increase the binding affinity of caulerpin toward 6VYB and 6LU7 due to their high electronegativity, which can yield more different interactions with active sites of receptors. These interactions lead to an increase in the binding energy between these ligands inside receptors, while inserting an alkyl group decreases the binding affinity of caulerpin toward 6VYB and 6LU7 as shown in Table 3, due to their low electronegativity, which can yield less different interactions than the previous groups with active sites of receptors. These interactions lead to a decrease in the binding energy between these ligands inside the receptors. The effect of side chain on binding energy in this study is in good agreement with this previous study [20]. Because there is a simple difference between side chains of all derivatives in this study (there are high similarities between all studied compounds in their structures in which their backbones are the same), the contribution of substituents in binding energy is a simple difference between the main scaffold and substituted derivatives. So, we can modify the inhibitory effect of caulerpin against 6VYB and 6LU7 by inserting NH2, halogen, and vinyl groups.
Table 3.
Effect of side chain on binding energy obtained from docking the studied compounds inside 6LU7and 6VYB

Analysis of the drug-likeness model score and ADMET properties of all the studied bioactive compounds
The drug-likeness model scores for all the studied compounds were computed using the Molinspiration server (http://www.molinspiration.com) and their values are presented in Table 4. Compounds having zero or negative value should not be considered as drug-like. The maximum drug-likeness score was found out to be 0.94 for compound 7 followed by compounds 10 and 19 with a drug-likeness score of 0.88 and 0.87, respectively, suggesting that these compounds have a better chance to be developed as drug leads. The ADMET properties such as absorption, distribution, metabolism, excretion, and toxicity of all the studied compounds were predicted using the admetSAR database (http://lmmd.ecust.edu.cn/admetsar1/predict). The database supports ADMET profiles which involve some features to study the ability of the studied compounds to act as drug leads such as blood–brain barrier (BBB) penetration, human intestinal absorption (HIA), Caco-2 cell permeability, cytochrome P450 (CYP) inhibitory promiscuity, AMES toxicity, carcinogenicity, and rat acute toxicity LD50 which are calculated and displayed in Table 5. As shown in Table 5, all the studied compounds may cross the BBB and are absorbed in the human intestine (HIA) showing permeability for Caco-2 cells, whereas compound 5 showed negative result for BBB, HIA, and Caco-2 cell permeability. CYP is a group of isozymes containing the metabolism of drugs, steroids, fatty acids, bile acids, and carcinogens. The results indicate that these studied compounds are nonsubstrate and noninhibitor of CYP enzymes [34]. In terms of AMES toxicity, all the studied compounds were observed to be nontoxic. The carcinogenicity model indicated a noncarcinogenic nature of all the studied compounds. Rat acute toxicity LD50 of all the studied compounds was found between 1.84 and 2.55 mol/kg. The finding strongly provides the ability of most studied compounds to act as a drug, except compounds 3, 5, and 9 as shown in Table 5.
Table 4.
Prediction of drug-likeness model score of all the studied compounds (1–20)
| Compounds | Drug-likeness model score by MolSoft |
|---|---|
| 1 | 0.69 |
| 2 | 0.55 |
| 3 | − 0.68 |
| 4 | 0.81 |
| 5 | − 0.83 |
| 6 | 0.21 |
| 7 | 0.94 |
| 8 | 0.90 |
| 9 | − 0.97 |
| 10 | 0.88 |
| 11 | 0.23 |
| 12 | 0.44 |
| 13 | 0.53 |
| 14 | 0.39 |
| 15 | 0.48 |
| 16 | 0.63 |
| 17 | 0.70 |
| 18 | 0.69 |
| 19 | 0.74 |
| 20 | 0.87 |
Table 5.
Prediction of ADMET descriptors of all the studied compounds (1–20)
| Compounds | BBB | HIA | Caco-2 permeability | CYP inhibitory promiscuity | AMES toxicity | Carcinogenicity | Rat acute toxicity LD50 (mol/kg) |
|---|---|---|---|---|---|---|---|
| 1 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 1.84 |
| 2 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.21 |
| 3 | BBB− | HIA− | Caco2− | Low | Nontoxic | Noncarcinogenic | 2.34 |
| 4 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.56 |
| 5 | BBB− | HIA− | Caco2− | Low | Nontoxic | Noncarcinogenic | 2.19 |
| 6 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.28 |
| 7 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.26 |
| 8 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.79 |
| 9 | BBB− | HIA− | Caco2− | Low | Nontoxic | Noncarcinogenic | 2.55 |
| 10 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.13 |
| 11 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.15 |
| 12 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.36 |
| 13 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.41 |
| 14 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.43 |
| 15 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.46 |
| 16 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.09 |
| 17 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.13 |
| 18 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.16 |
| 19 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.03 |
| 20 | BBB+ | HIA+ | Caco2+ | Low | Nontoxic | Noncarcinogenic | 2.25 |
The molecular properties of descriptors and prediction of bioactivity score of all the studied compounds were analyzed based on Lipinski’s rule of five using the Molinspiration server (http://www.molinspiration.com) and their values are given in Tables 6 and 7, respectively. All theoretical background of these calculations was carried out according to this study [35]. Lipinski’s rule of five is commonly used in the development and drug design to expect oral bioavailability of drug molecules. Lipinski’s rule was established based on five rules to compute the ability of the compound to act as an orally active drug. So, the orally active drug must have no more than one violation of the following standards: (i) octanol/water partition coefficient (logP), which measured the lipophilicity of a molecule must be not greater than five; (ii) a molecular weight (MW) less than 500 Da; (iii) not more than five hydrogen bond donors (nON); (iv) not more than 10 hydrogen bond acceptors (nOHN); and (v) topological polar surface area (TPSA) below the limit of 160 Å. As shown in Table 6, only compounds 4, 6, 7, 10–13, and 19 did not violate any of Lipinski’s rule of five. TPSA measures the bioavailability of the drug molecule and is closely related to the hydrogen bonding potential of a compound. The TPSA of the studied compounds was observed in the range of 36.46–108.38 Å and is well below the limit of 160 Å. The number of rotatable bonds (nRot) measures conformational stability which was found to be acceptable for most of the studied compounds. All calculated values for the studied compounds are less than 10, so these compounds are conformationally stable. It can be predicted that only compounds 4, 6, 7, 9, 11, 12, 13, and 19 obeyed Lipinski’s rule of five and are likely to be orally active. The bioactivity scores of all the studied compounds for drug targets are presented in Table 7. A molecule having a bioactivity score more than 0.00 is most likely to display significant biological activities; values − 0.50 to 0.00 are assumed to be moderately active, while values less than − 0.50 are presumed to be inactive. As shown in Table 7, compounds 7, 8, 10, 13, 19, and 20 exhibit significant biological activities, while other compounds show moderate biological activities.
Table 6.
Prediction of the molecular properties of descriptors of all the studied compounds (1–20)
| Compounds | LogP | TPSA | natom | MW | nON | nOHN | nVio | nRot | Volume |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 5.10 | 84.42 | 30 | 398.48 | 6 | 2 | 1 | 4 | 346.46 |
| 2 | 5.37 | 65.22 | 28 | 374.35 | 4 | 2 | 1 | 2 | 305.02 |
| 3 | 3.05 | 91.75 | 28 | 371.37 | 6 | 6 | 1 | 2 | 317.94 |
| 4 | 4.41 | 36.46 | 29 | 382.14 | 5 | 4 | 0 | 2 | 311.59 |
| 5 | 6.93 | 84.19 | 36 | 478.55 | 6 | 2 | 1 | 10 | 436.00 |
| 6 | 5.92 | 78.38 | 30 | 400.14 | 6 | 2 | 0 | 11 | 344.55 |
| 7 | 4.30 | 88.99 | 32 | 451.05 | 7 | 4 | 0 | 12 | 371.57 |
| 8 | 5.73 | 84.19 | 31 | 432.86 | 6 | 2 | 1 | 4 | 359.99 |
| 9 | 4.50 | 86.15 | 31 | 412.11 | 6 | 2 | 0 | 4 | 363.97 |
| 10 | 5.91 | 84.19 | 32 | 426.08 | 6 | 2 | 1 | 4 | 379.91 |
| 11 | 4.91 | 86.22 | 31 | 446.19 | 6 | 2 | 0 | 4 | 344.88 |
| 12 | 4.60 | 104.42 | 31 | 414.42 | 7 | 3 | 0 | 4 | 354.48 |
| 13 | 4.84 | 104.42 | 31 | 428.33 | 6 | 3 | 0 | 4 | 366.71 |
| 14 | 5.71 | 74.96 | 31 | 408.46 | 5 | 2 | 1 | 5 | 365.44 |
| 15 | 6.54 | 84.19 | 33 | 440.50 | 6 | 2 | 1 | 7 | 396.86 |
| 16 | 7.98 | 84.19 | 36 | 482.58 | 6 | 2 | 1 | 10 | 447.27 |
| 17 | 5.91 | 84.19 | 32 | 426.47 | 6 | 2 | 1 | 4 | 379.58 |
| 18 | 6.42 | 65.72 | 31 | 410.45 | 4 | 2 | 1 | 4 | 361.39 |
| 19 | 4.26 | 74.96 | 33 | 442.49 | 5 | 2 | 0 | 6 | 392.81 |
| 20 | 6.74 | 65.72 | 30 | 504.33 | 4 | 2 | 2 | 4 | 363.89 |
Table 7.
Prediction of the bioactivity score of all the studied compounds (1–20)
| Compounds | GPCR | ICM | KI | NRL | PI | EI |
|---|---|---|---|---|---|---|
| 1 | 0.02 | 0.06 | − 0.33 | 0.08 | − 0.04 | 0.18 |
| 2 | 0.17 | 0.07 | − 0.22 | 0.23 | 0.07 | 0.27 |
| 3 | − 0.84 | − 1.94 | − 1.53 | − 1.65 | − 0.55 | − 1.11 |
| 4 | − 0.29 | − 0.14 | − 0.12 | − 0.28 | − 0.09 | − 0.33 |
| 5 | − 0.26 | − 0.13 | − 0.09 | − 0.30 | − 0.08 | − 0.40 |
| 6 | 0.84 | 0.94 | 0.53 | 0.65 | 0.55 | 0.11 |
| 7 | 0.38 | 0.22 | 0.04 | 0.36 | 0.25 | 0.30 |
| 8 | 0.41 | 0.28 | 0.09 | 0.48 | 0.29 | 0.15 |
| 9 | − 0.30 | − 0.28 | − 0.08 | − 0.32 | − 0.21 | − 0.34 |
| 10 | 0.60 | 1.95 | 2.62 | 3.04 | 3.15 | − 1.56 |
| 11 | − 0.65 | − 0.39 | − 0.91 | − 0.04 | 0.33 | 0.36 |
| 12 | − 0.61 | − 0.31 | − 0.81 | − 0.08 | 0.30 | 0.32 |
| 13 | 0.25 | − 0.19 | − 0.88 | − 0.06 | 0.35 | 0.38 |
| 14 | − 0.65 | − 0.44 | − 0.94 | − 0.04 | 0.33 | 0.39 |
| 15 | − 0.65 | − 0.41 | − 0.77 | − 0.04 | 0.28 | 0.41 |
| 16 | − 0.65 | − 0.32 | − 0.76 | − 0.04 | 0.35 | 0.36 |
| 17 | − 0.55 | − 0.32 | − 0.77 | − 0.06 | 0.30 | 0.45 |
| 18 | − 0.51 | − 0.35 | − 0.98 | − 0.09 | 0.29 | 0.34 |
| 19 | 0.43 | 0.29 | 0.07 | 0.46 | 0.27 | 0.18 |
| 20 | 0.14 | 0.04 | − 0.51 | 0.73 | 0.07 | 0.51 |
GPCR GPCR ligand, ICM ion channel modulator, KI kinase inhibitor, NRL nuclear receptor ligand, PI protease inhibitor, EI enzyme inhibitor
Combination therapy
The global energy of interacted molecules was associated with free binding energy and their higher negative value explains higher binding probability [36]. Based on the molecular docking study, it was noticed that the predicted antiviral activity of most of the caulerpin derivatives against SARS-CoV-2 infection targeting remarkable COVID-19 main protease and S-receptor binding domain are larger than those of all drugs in this study especially compounds 7, 10, and 19. In this section, we study combination therapy of compounds yielding the highest binding in molecular docking together with lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir for disrupting the stability of SARS-CoV-2 main protease and SARS-CoV-2 spike proteins. As shown in the “Introduction,” all compounds under investigation in this study showed inhibitory activity against Alzheimer’s disease [20], so all compounds are safe and nontoxic for biological applications. So, we can use them in combination therapy along with lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir drugs which are actually used in the treatment of COVID-19 patients. The obtained binding energy values of compounds 7, 10, and 19 are higher than those of the drugs lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir against 6VYB and 6LU7. These results of molecular docking indicated that compounds 7, 10, and 19 as adjunct drugs could be potent antiviral molecules along with lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir or other antiviral conventional drugs for disruption of the integrity of SARS-CoV-2 protein receptors. Simeprevir produces the highest binding energy toward 6VYB and 6LU7 compared with other drugs. Figures 11 and 12 show the schematic presentation of combination therapy of simeprevir with compounds 7, 10, and 19 suppressing the activity of the main protease and receptor binding protein which plays an essential role in the completion of the SARS-CoV-2 life cycle, respectively.
Fig. 11.

Schematic presentation for the combination therapy of simeprevir with compounds 7, 10, and 19 suppressing the activity of 6LU7 that plays an essential role in the completion of the SARS-CoV-2 life cycle
Fig. 12.

Schematic presentation for the combination therapy of simeprevir with compounds 7, 10, and 19 suppressing the activity of 6VYB that plays an essential role in the completion of the SARS-CoV-2 life cycle
Molecular dynamics simulation
To confirm the docking results and get more insight into the stability of the ligand–protein complex, MD simulations were carried out for the highest binding energy of simeprevir with compounds 7 and 19 inside 6VYB and 6LU7 receptor complexes in the solvated states at 10 ns as shown in Fig. 13. The results of MD simulations have been examined on the basis of RMSD and RMSF values as a function of time.
Fig. 13.

MD snapshots of solvated a 6LU7 and b 6VYB receptors in complex with simeprevir + compound 7 during molecular dynamics simulations at 10 nm, in which the line ribbon represented 6LU7 (a) and 6VYB (b), the yellow atoms represented compound 7 in the bottom view and simeprevir in the top view, the red and white colors represented oxygen and hydrogen atoms of water molecules, respectively, and finally blue and green colors represented sodium and chloride, respectively, as counter ions
Root mean square deviation
To examine the change in the protein dynamics and the conformational stability of the protein–ligand complexes, the protein complexed with the studied compounds were subjected to 10 ns MD simulations. Standard dynamics cascade module implicit in the DS software was employed to measure the RMSD and RMSF. The RMSD measures the direct changes in the protein from the initial coordinates. The RMSD values of the protein backbone in complex with the potential inhibitors were computed with respect to the initial structure as a frame reference (0 to 10 ns). The RMSD values steadily increased from 0 to 4 ns and reached equilibration after that throughout the simulation period. The RMSD values for all the studied complexes showed oscillations between 2 and 4 ns indicating that the studied compounds were adapting another conformation within the binding pocket as shown in Fig. 14. The average RMSD values for the last 1 ns for all the studied systems were 1.15 ± 0.04, 1.74 ± 0.08, 1.53 ± 0.03, and 1.30 ± 0.12 for simeprevir + compound 7–6LU7, simeprevir + compound 19–6LU7, simeprevir + compound 7–6VYP, and simeprevir + compound 7–6VYP complexes, respectively.
Fig. 14.
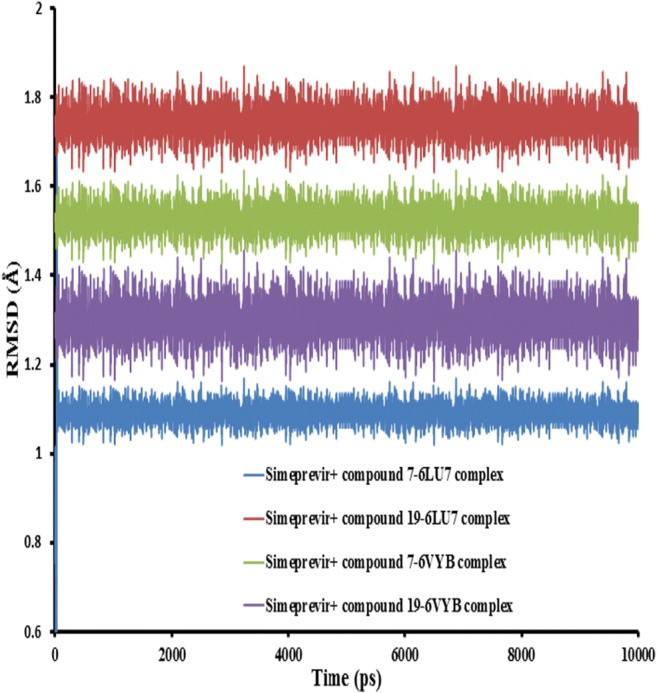
Plot of root mean square deviation (RMSD) of receptor backbone vs. simulation time for solvated 6LU7 and 6VYB receptors in complex with simeprevir + compounds 7 and 19 during molecular dynamics simulations at 10 nm
Lower RMSD value of the complex indicates its stability with combination therapy of simeprevir and the two candidate compounds and provided a suitable basis for our study.
Root mean square fluctuation
RMSF was measured with respect to the backbone atom of each amino acid residue, and the plot of RMSF was used to depict the fluctuations at the residue level. The RMSF plot as shown in Figs. 15 and 16 of solvated 6LU7 and 6VYB receptors, receptively, in complex with simeprevir + compounds 7 and 19 during 10 ns molecular dynamics simulations at 10 nm exhibited a similar trend of residue fluctuation profile for both free receptors (6LU7 and 6VYB) and the complexes with a low average RMSF. This trend in the RMSF plot for the complex indicates that binding of combination therapy of simeprevir and the two candidate compounds to the receptors was stable and had no major effect on the flexibility of the protein throughout the simulations. To explore more insights on the local protein flexibility, the time average of RMSF values of the 300 amino acids of 6LU7 and 975 amino acids of 6VYB receptors in the presence of the inhibitor (simeprevir + compounds 7 and 19) over the simulation period was calculated. The RMSF values for the three complexes suggested that the following residues ALA 206, VAL 204, and LEU 205 showed less fluctuation in all complexes in the state of 6LU7 and ALA27, TYR28, and THR29 in the state of 6VYB as shown in Figs. 15 and 16, respectively. The average RMSF values were 0.55 ± 0.07, 0.69 ± 0.09, and 0.87 ± 0.07 Å for 6LU7, simeprevir + compound 7–6LU7, and simeprevir + compound 19–6LU7 complexes, respectively, and 0.35 ± 0.03, 0.41 ± 0.07, and 0.58 ± 0.05 Å for 6VYB, simeprevir + compound 7–6VYB, and simeprevir + compound 19–6VYB complexes, respectively.
Fig. 15.
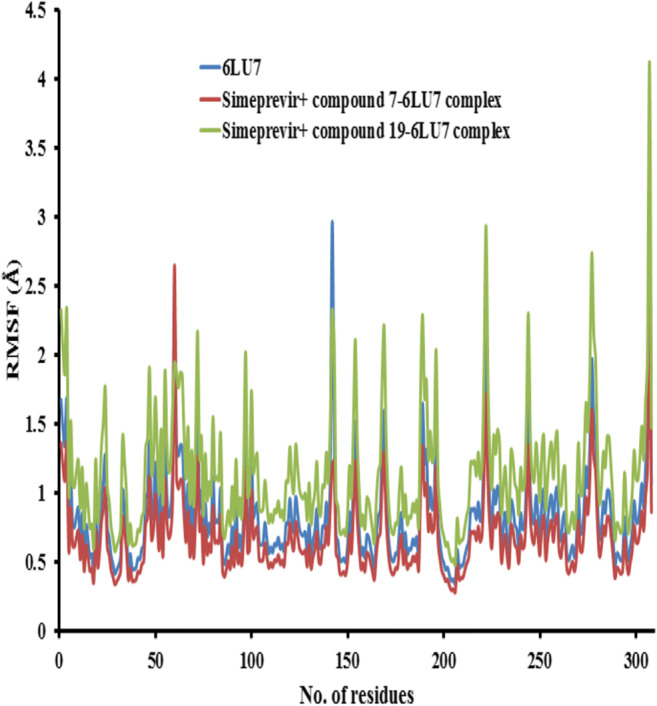
The root mean square fluctuation (RMSF) values of 6LU7 protease alone and in complex with simeprevir + compounds 7 and 19 were plotted against residue numbers
Fig. 16.
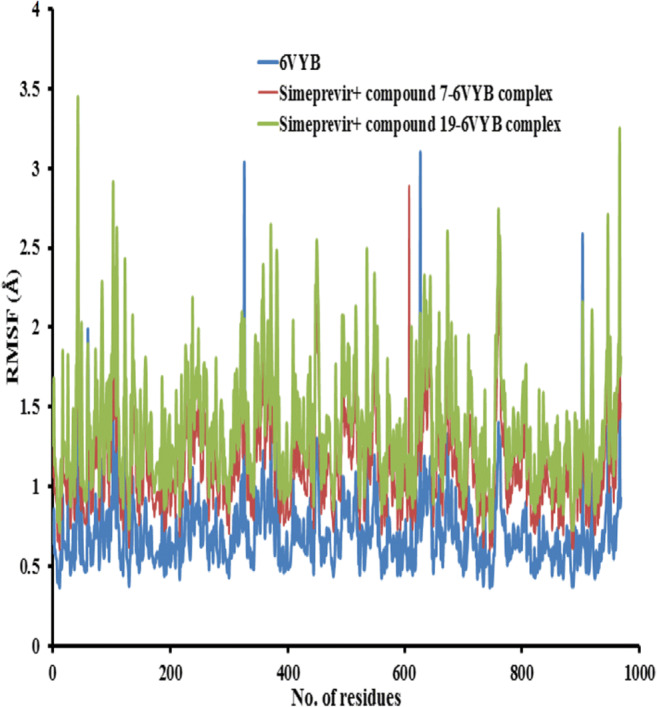
The root mean square fluctuation (RMSF) values of 6VYB receptor alone and in complex with simeprevir + compounds 7 and 19 were plotted against residue numbers
Conclusion
Numerous antiviral or other conventional drugs are being examined against COVID-19, but there are still no positive results yet. This study has emphasized to know the exact reason, by considering the two receptor proteins of SARS-CoV-2: SARS-CoV-2 main protease (PDB ID: 6LU7) and SARS-CoV-2 spike protein domain (PDB ID: 6VYB), why FDA (Food and Drug Administration)-approved drugs or other conventional drugs are not working against SARS-CoV-2. The protein disordered results from ANCHOR showed that the SARS-CoV-2 main protease and the SARS-CoV-2 spike protein domain are highly stable proteins, so it is quite difficult to unstabilize the integrity of these proteins by using individual drugs. The molecular docking analysis revealed that most of the studied compounds can act as 6VYB and 6LU7 inhibitors, especially compounds 7, 10, and 19. Also, we deduced that inserting NH2, halogen, and vinyl groups can increase the binding affinity of caulerpin toward 6VYB and 6LU7, while inserting an alkyl group decreases the binding affinity of caulerpin toward 6VYB and 6LU7. So, we can modify the inhibitory effect of caulerpin against 6VYB and 6LU7 by inserting NH2, halogen, and vinyl groups. The drug-likeness model score suggested that most of the studied compounds, especially 7, 10, and 19, have a better chance to be developed as drug leads. The ADMET properties strongly provide the ability of most studied compounds to act as a drug, except compounds 3, 5, and 9. The molecular properties of descriptors and prediction of bioactivity score confirmed that compounds 7, 10, and 19 did not violate any of Lipinski’s rule of five and exhibit significant biological activities. Based on protein disordered results, we study combination therapy for compounds yielding the highest binding in molecular docking along with lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir for disrupting the stability of SARS-CoV-2 main protease and SARS-CoV-2 spike proteins. The RMSD of the receptor–ligand complexes of both receptors has maintained stability at around 2 Å and the RMSD of the three drugs complexed with the protein is in the favorable range within 1.2 Å and has remained stable during the simulations. The backbone atoms of the complex and free receptor show similar RMSF, indicating the stability of the combination therapy between the studied compounds and simeprevir inside 6LU7 and 6VYB receptors. Hence, the combination of the studied compounds and simeprevir is highly effective against SARS-CoV-2 protease, and these drugs can be explored further for drug repurposing against the successful inhibition of COVID-19. Finally, the antiviral activity of lopinavir, simeprevir, hydroxychloroquine, chloroquine, and amprenavir could be increased against SARS-CoV-2 by using caulerpin and its derivatives as combination therapy.
Acknowledgments
H.R. Abd El-Mageed is grateful to Dr. F.M. Mustafa (Chemistry Department, Faculty of Sciences, Beni-Suef University, Beni Suef, Egypt) for her continuous assistance, moral and scientific support, and fruitful discussions.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.de Oliveira MD, de Oliveira KM. Comparative computational study of SARS-CoV-2 receptors antagonists from already approved drugs. Cep. 2020;8:7. [Google Scholar]
- 2.Shen K, Yang Y, Wang T, Zhao D, Jiang Y, Jin R, Zheng Y, Xu B, Xie Z, Lin L, Shang Y. Diagnosis, treatment, and prevention of 2019 novel coronavirus infection in children: experts’ consensus statement. World J Pediatr. 2020;7:1–9. doi: 10.1007/s12519-020-00343-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang T, Du Z, Zhu F, Cao Z, An Y, Gao Y, Jiang B. Comorbidities and multi-organ injuries in the treatment of COVID-19. Lancet. 2020;395:52. doi: 10.1016/S0140-6736(20)30558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Yu T. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;3:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, Guo L, Guo R, Chen T, Hu J, Xiang Z. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11:1–2. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M (2020) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B 10:766–788 [DOI] [PMC free article] [PubMed]
- 9.Boxall ABA. The environmental side effects of medication. EMBO Rep. 2004;5:1110–1116. doi: 10.1038/sj.embor.7400307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Angiolella L, Sacchetti G, Efferth T (2018) Antimicrobial and antioxidant activities of natural compounds. Evid Based Complement Alternat Med:1–3. 10.1155/2018/1945179 [DOI] [PMC free article] [PubMed]
- 11.Macedo NRPV, Ribeiro MS, Villaça RC, Ferreira W, Pinto AM, Teixeira VL, Cirne-Santos C, Paixão ICNP, Giongo V. Caulerpin as a potential antiviral drug against herpes simplex virus type 1. Rev Bras Farmacogn Braz J Pharmacogn. 2012;22:861–867. [Google Scholar]
- 12.Ayyad SEN, Badria FA. Caulerpin, an antitumor indole alkaloid from Caulerpa racemosa. Alex J Pharm Sci. 1994;8:217. [Google Scholar]
- 13.Xu XH, Su JG. The separation, identification and bioassay of caulerpin. Zhongshan Daxue Xuebao Ziran Kexueban. 1996;35:64–66. [Google Scholar]
- 14.Librán-Pérez M, Pereiro P, Figueras A, Novoa B. Antiviral activity of palmitic acid via autophagic flux inhibition in zebrafish (Danio rerio) Fish Shellfish Immunol. 2019;95:595–605. doi: 10.1016/j.fsi.2019.10.055. [DOI] [PubMed] [Google Scholar]
- 15.Nagappan T, Vairappan CS. Nutritional and bioactive properties of three edible species of green algae, genus Caulerpa (Caulerpaceae) J Appl Phycol. 2014;26(2):1019–1027. [Google Scholar]
- 16.Mangala SW, Govenkar B. Constituents of Chondria armata. Phytochemistry. 2000;54(8):979–981. doi: 10.1016/s0031-9422(00)00078-9. [DOI] [PubMed] [Google Scholar]
- 17.Lucena A, et al. The bisindole alkaloid caulerpin, from seaweeds of the genus Caulerpa, attenuated colon damage in murine colitis model. Mar Drugs. 2018;16(9):318. doi: 10.3390/md16090318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Souza ÉT, Pereira de Lira D, Cavalcanti de Queiroz A, Costa da Silva DJ, Bezerra de Aquino A, Mella C, Eliane A, Prates Lorenzo V, De Miranda GE, Araújo-Júnior D, Xavier J. The antinociceptive and anti-inflammatory activities of caulerpin, a bisindole alkaloid isolated from seaweeds of the genus Caulerpa. MarDrugs. 2009;7:689–704. doi: 10.3390/md7040689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esteves PO, de Oliveira MC, de Souza BC, Cirne-Santos CC, Laneuvlille VT, Palmer Paixão IC. Antiviral effect of caulerpin against chikungunya. Nat Prod Commun. 2019;1:1934578X19878295. [Google Scholar]
- 20.Lorenzo VP, Barbosa Filho JM, Scotti L, Scotti MT. Combined structure-and ligand-based virtual screening to evaluate caulerpin analogs with potential inhibitory activity against monoamine oxidase B. Rev Bras. 2015;25:690–697. [Google Scholar]
- 21.Dassault Systèmes BIOVIA Discovery studio modeling environment, release 2017. Dassaul Systèmes, San Diego
- 22.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raghavendra S, Rao Aditya SJ, Kumar V, Ramesh CK Multiple ligand simultaneous docking (MLSD): a novel approach to study theeffect of inhibitors on substrate binding to PPO. Comput Biol Chem. 10.1016/j.compbiolchem.2015.09.008 [DOI] [PubMed]
- 24.Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 25.Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52:3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 26.Chen X-R, Wang X-T, Hao M-Q, Zhou Y-H, Cui W-Q, Xing X-X, Xu C-G, Bai J-W, Li Y-H Homology modeling and virtual screening to discover potent inhibitors targeting the imidazole glycerophosphate dehydratase protein in Staphylococcus xylosus. Front Chem 5:98. 10.3389/fchem.2017.00098 [DOI] [PMC free article] [PubMed]
- 27.Mészáros B, Erdős G, Dosztányi Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018;2:329–337. doi: 10.1093/nar/gky384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dosztányi Z (2018) Prediction of protein disorder based on IUPred. Protein Sci. 10.1002/pro.3334 [DOI] [PMC free article] [PubMed]
- 29.Thomas PD, Dill KA. An iterative method for extracting energy-like quantities from protein structures. Proc Natl Acad Sci. 1996;93:11628–11633. doi: 10.1073/pnas.93.21.11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dosztanyi Z, Csizmok V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 2005;347:827–839. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 31.Mészáros B, Simon I, Dosztányi Z (2009) Prediction of protein binding regions in disordered proteins. PLoS Comput Biol 5(5). 10.1371/journal.pcbi.1000376 [DOI] [PMC free article] [PubMed]
- 32.Ortega JT, Serrano ML, Pujol FH, Rangel HR. Unrevealing sequence and structural features of novel coronavirus using in silico approaches: the main protease as molecular target. EXCLI J. 2020;19:400. doi: 10.17179/excli2020-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hwang WC, Lin Y, Santelli E, Sui J, Jaroszewski L, Stec B, Farzan M, Marasco WA, Liddington RC. Structural basis of neutralization by a human anti-severe acute respiratory syndrome spike protein antibody, 80R. J Biol Chem. 2006;28:34610–34616. doi: 10.1074/jbc.M603275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes ZE, Walsh TR. Non-covalent adsorption of amino acid analogues on noble-metal nanoparticles: influence of edges and vertices. Phys Chem Chem Phys. 2016;18:17525–17533. doi: 10.1039/c6cp02323a. [DOI] [PubMed] [Google Scholar]
- 35.Ahmed, S. A., Rahman, A. A., Elsayed, K. N., Abd El-Mageed, H. R., Mohamed, H. S., & Ahmed, S. A. (2020). Cytotoxic activity, molecular docking, pharmacokinetic properties and quantum mechanics calculations of the brown macroalga Cystoseira trinodis compounds. Journal of Biomolecular Structure and Dynamics, (just-accepted), 1-31. [DOI] [PubMed]
- 36.Srivastava AK, Tewari M, Shukla HS, Roy BK (2015) In silico profiling of the potentiality of curcumin and conventional drugs for CagA oncoprotein inactivation. Arch Pharm (Weinheim). 10.1002/ardp.201400438 [DOI] [PubMed]