

Abstract
Antimicrobial proteins such as S100A12 and S100A8/A9 are highly expressed and secreted by neutrophils during infection and participate in human immune response by sequestering transition metals. At neutral pH, S100A12 sequesters Zn2+ with nanomolar affinity, which is further enhanced upon calcium binding. We investigated the pH-dependence of human S100A12 zinc sequestration by using Co2+ as a surrogate. Apo-S100A12 exhibits strong Co2+ binding between pH 7.0 – 10.0 that progressively diminishes as the pH is reduced to 5.3. Ca2+-S100A12 can retain nanomolar Co2+ binding up to pH 5.7. NMR measurements revealed that calcium binding does not alter the sidechain protonation of the Co2+/Zn2+ binding histidines. Instead, the calcium-mediated modulation is achieved by restraining pH-dependent conformational changes to EF loop 1, which contains Co2+/Zn2+ binding Asp25. This calcium-induced enhancement of Co2+/Zn2+ binding may assist in promotion of antimicrobial activities in humans by S100 proteins during neutrophil activation at sub-neutral pH conditions.
Keywords: NMR spectroscopy, nutritional immunity, S100A12, S100 metalloproteins, Zinc sequestration
Graphical Abstract
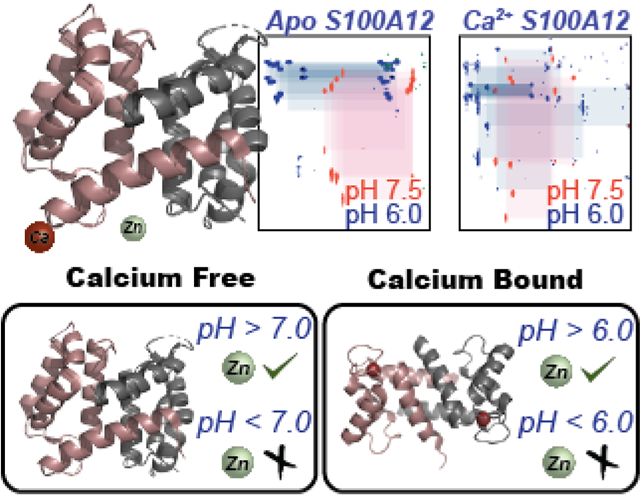
Calcium enhances the viable pH-range for metal sequestration mediated antimicrobial activities by S100A12.
Introduction
Metal sequestration mediated antimicrobial functions in the innate immune system have been identified for a subset of S100 family of proteins namely, S100A7, A100A8, S100A9, and S100A12. Intracellularly, the large family of S100 proteins is known for functions that depend on the ability of these proteins to bind to calcium ions and each member of the S100 family contains two Ca2+ binding EF hand motifs.[1] S100A7, S100A8, S100A9 and S100A12 bind to transition metal ions upon secretion into the extracellular space during infection.[2] While S100A8/A9 heterodimer sequesters Fe2+, Mn2+, Zn2+ and Ni2+, [3] homodimeric forms of S100A7 and S100A12 are responsible for Zn2+ homoeostasis.[2b, 4] Sequestration by S100 proteins limits the availability transition metal ion nutrients to pathogens and gives rise to the antimicrobial activities of S100A7, S100A8, S100A9 and S100A12. Primarily expressed in neutrophils, S100A12 is a key component of the human innate response. Overexpression of S100A12 (also known as Calgranulin C) has been correlated with several diseases. For example, elevated serum level of S100A12 have been reported during severe bacterial infection, in patients suffering from cystic fibrosis, and neurodegenerative,[5] metabolic and neoplastic disorders.[6] In vivo, S100A12, which inhibits the growth of the pathogens in vitro by chelating Zn2+ ions, is induced during H. pylori infection in humans.[7]
X-ray crystallography studies have shown that zinc binds a homodimeric S100A12 via a His3Asp motif that is comprised of two histidine residues from one monomer (His85 and His89) and a histidine and an aspartic acid residue (His15 and Asp25) from the second monomer (Figure 1A). The symmetric interface between the two monomeric units of S100A12 results in two His3Asp binding sites per homodimer.[8] Human S100A12 exhibits nanomolar binding affinity for Zn2+ at neutral pH-values allowing the protein to perform its antimicrobial activities efficiently and calcium binding further enhances the Zn2+ binding affinity of S100A12.[4d] Calcium binding to S100A12 is afforded by two EF hand motifs that coordinate Ca2+ in pentagonal bipyramidal configuration.[9] The C-terminal 12 residue canonical EF hand motif (EF hand II) binds to Ca2+ with high affinity (Kd ~ 10−5 to 10−7 M), while the N-terminal pseudo EF hand motif (EF hand I), composed of 14 residues has lower Ca2+ affinity (Kd ~ 10−3 to 10−4 M).[6] Notably, residue D25 belonging to EF hand I, is also a part of the His3Asp Zn2+ binding scaffold and calcium binding may alter the metal coordination geometry via this residue allowing for the association between calcium and zinc binding affinity of S100A12. Crystal structures of the apo and Ca2+ bound protein[8–9] have demonstrated that Ca2+ binding to the apo protein introduces a major structural rearrangement to helix III but minor changes to helix I and IV that contain the transition metal binding residues (Figure 1C). These structures do not fully explain the origin of previously reported association of Ca2+ and Zn2+ binding affinities.
Figure 1.
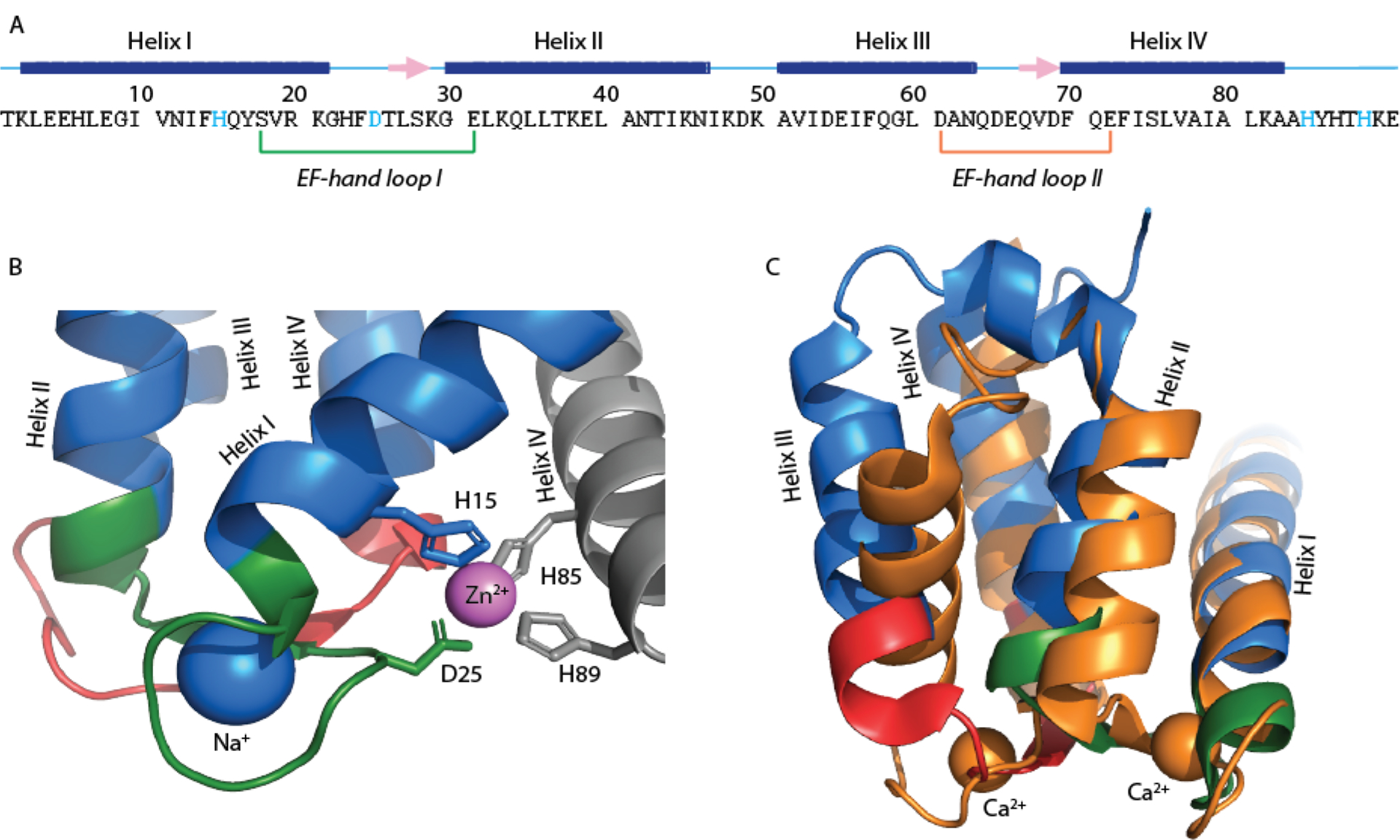
A) The primary sequence of human S100A12 showing Zn2+ binding residues (colored cyan) and EF hand loops. The secondary structure elements are presented above the sequence and displayed as blue bar for helices, pink arrows for β-strand and cyan line for loops; B) an expansion of Zn2+ bound S100A12 crystal structure showing the zinc binding site (PDB: 2WCB). The two monomers are colored blue and gray. The zinc and sodium ions are colored purple and blue, respectively and; C) an overlay of the crystal structures of apo S100A12 (PDB: 2WCF, blue) and Ca2+ bound S100A12 (PDB: 1E8A, orange). The EF-hand loops are colored using the same color scheme as in (A).
Given the high concentration of calcium (~ millimolar) in the extracellular space, upon secretion into the blood serum and while performing its antimicrobial functions of zinc sequestration, S100A12 is expected to be Ca2+ bound. Neutrophils, that primarily express S100A12, are activated during extracellular acidosis.[10] Under these conditions, which are associated with inflammatory reactions against pathogens, sub-neutral pH values for the blood serum (5.5–7.0) have been reported. Extracellular acidosis also induces a transient increase in intracellular Ca2+ concentrations.[10b] Taking all these findings into consideration, it is reasonable to suggest that during infection and upon secretion into the extracellular space S100A12 experiences sub-neutral pH conditions and is Ca2+ bound. Although metal binding affinities of S100A12 have been measured at neutral pH values, Zn2+ binding to S100A12 under such physiologically relevant conditions of pH 5.5 – 7.0 have not been investigated.
Here, we investigated the pH dependence of Zn2+ sequestration by S100A12 by monitoring Co2+binding as a surrogate ion. We show that S100A12 transition metal affinity is significantly reduced between the pH of 5.3 – 7.0, and can be restored upon Ca2+ binding, suggesting that the Zn2+ homeostasis mediated antimicrobial activities of S100A12 will be diminished in this pH range. We present NMR studies of apo and Ca2+-S100A12 under various pH conditions to investigate the origin of the loss of Co2+/Zn2+ binding in apo S100A12 at acidic pH and the preservation of transition metal binding affinity by Ca2+-S100A12 under identical conditions. Previously sub-nanomolar Zn2+ binding affinity at pH 7.5 have been reported for human S100A12,[4d] making it difficult to appreciate the role of Ca2+ in S100A12 antimicrobial functions. However, our results indicate that under the physiologically relevant sub-neutral pH conditions, expected for activated neutrophils, S100A12 cannot efficiently sequester Zn2+ to kill pathogens without the aid of calcium ions. Thus, the high Ca2+ serum concentrations are imperative for the functions of S100A12 in the human innate immune response. Our studies also underscore the importance of in vivo and in vitro investigations of other prominent members of the human innate response, such as S100A8/A9 heterodimer, whose metal sequestration based antimicrobial activities may be similarly modulated by pH and calcium binding.
Results
pH variations do not disrupt the tertiary structures of apo and Ca2+-S100A12.
To investigate the pH dependence of S100A12 antimicrobial activities, we first determined if the protein remains in a well-folded and in soluble conformation under variable pH conditions examined in this work. 1H-15N HSQC measurements were performed for pH values ranging from 4.5 – 10.4 in increments of 0.5 pH points. For clarity Figure 2 shows an overlay of HSQC spectra acquired at pH values of 4.5, 6.0 and 8.3 for apo and Ca2+-S100A12. For all pH conditions, the backbone amide nuclei exhibited well-distributed resonances with sufficiently narrow line widths, indicating that the 3D structure of the protein remains folded in this pH range. 88 out of 91 expected backbone resonances were detected for the apo protein, while the calcium bound form exhibited 89 well-resolved signals. The backbone assignments for 86 residues in the apo-protein were made with the aid of 1H, 13C, 15N heteronuclear experiments, as reported by us previously.[11] For calcium bound S100A12, triple-resonance experiments were conducted to confirm the reported resonance assignments in the literature[12] and a near complete agreement was observed providing resonance assignments for 83 out of 91 residues.
Figure 2.
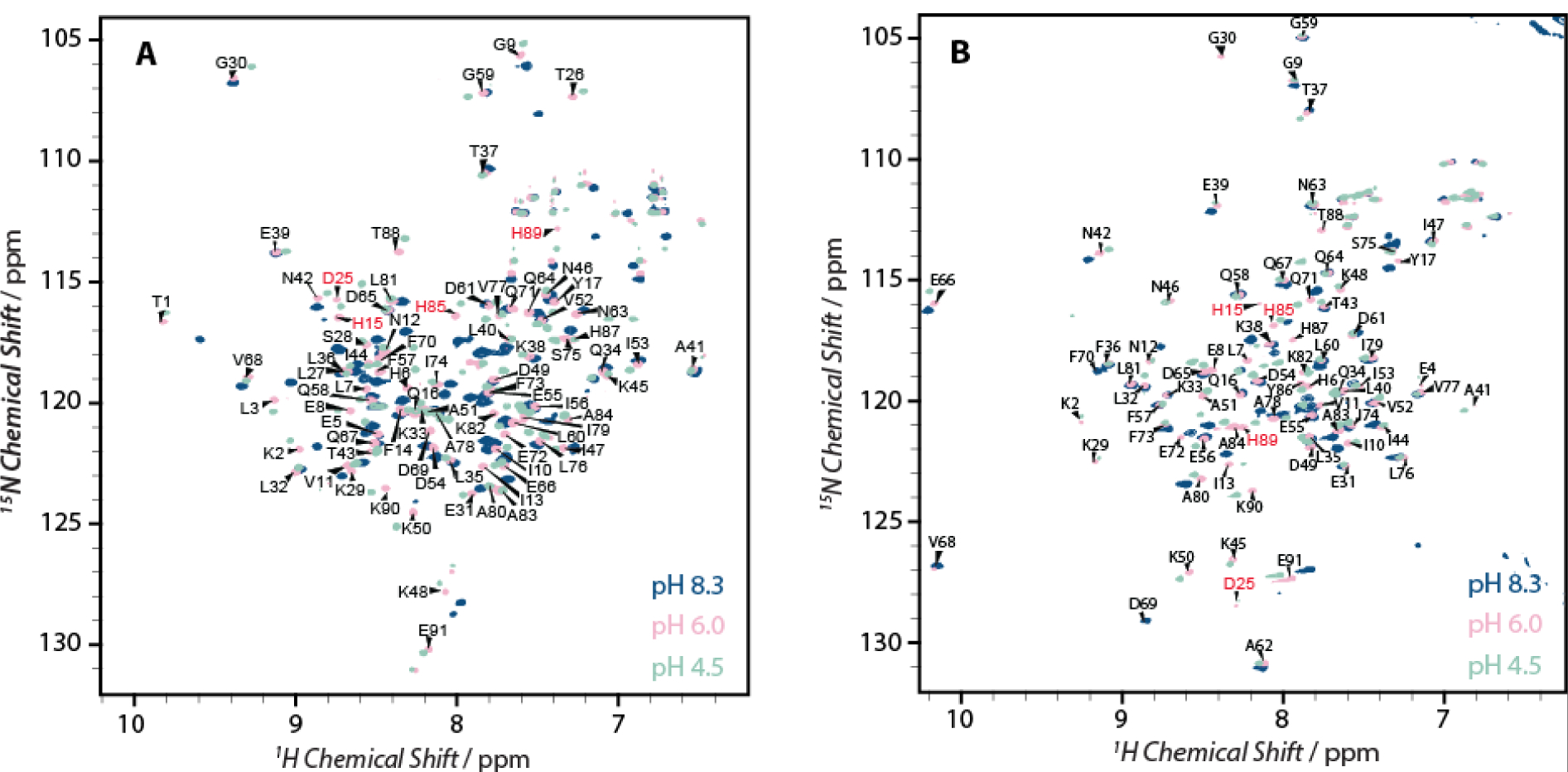
Overlay of 14.1 T 1H-15N HSQC spectra of the S100A12 at pH 8.3 (blue), 6.0 (pink) and 4.5 (light green) for A) apo-protein and, B) calcium bound S100A12. The residues involved in His3Asp Zn2+ binding motif are labeled in red.
X-ray crystallography and NMR studies have demonstrated that calcium binding to apo-S100A12 introduces significant conformational rearrangement to the tertiary structure of the protein, particularly in helices II and III (Figure 1B).[9, 12] This structural rearrangement is evident in the 1H-15N HSQC spectra of Ca2+-S100A12 which shows major perturbations in the amide resonances compared to the apo-protein. 1H-15N HSQC spectra of Ca2+-S10A12 acquired in the pH range of 4.5 to 8.3 also demonstrate that calcium binding is retained under these conditions (Figure S1), as resonances corresponding to the apo form were not detected. For both apo- and Ca2+-S100A12, significant pH dependent perturbations were observed for several residues in the 1H-15N HSQC spectra. Large perturbations in the backbone amide (NH and HN) resonances may originate from variations in the local electronic environment of these nuclei. Such changes may be introduced due to the protonation/deprotonation of sidechains of charged residues or conformational variations with pH. Although Zn2+ binds at the interface of the homodimer, no detectable differences between the two monomers of the homodimer have been noted in the crystal structures and, previous and current NMR studies. Therefore, in this report, the NMR data is reported in terms of the monomeric protein.
Transition metal binding to apo protein is inhibited at low pH.
Co2+ binds to the His3Asp zinc binding motif and exhibits an electronic transition at 556 nm.[4d] We utilized this optical transition as a probe to monitor Zn2+ binding to S100A12 at various pH-values. At neutral pH conditions, zinc binds to S100A12 with nanomolar binding affinity.[4d] Addition of 1 equivalent of cobalt to S100A12 at pH 7.0 yielded the 556 nm optical transition, which was in quantitative agreement with the amount of the protein used. A pH-titration was conducted by gradually increasing the pH of the solution to 10.4, while monitoring the 556 nm absorbance, as shown in Figure 3 (orange trace). In this pH-range of 7.0 – 10.4, no change in the 556 nm absorbance was observed, demonstrating that the metal binding affinity remains unchanged under neutral to alkaline pH conditions. Incremental lowering of pH from 7.0 resulted in gradual loss of the 556 nm absorbance between the pH of 5.3 – 7.0. At pH values of 3.4 – 5.3 negligible absorbance was observed, suggesting complete loss of metal binding in these conditions. These results indicate that although S100A12 is a high affinity Zn2+ chelator at neutral and alkaline pH values, its metal binding efficacy diminishes dramatically at sub-neutral pH.
Figure 3.
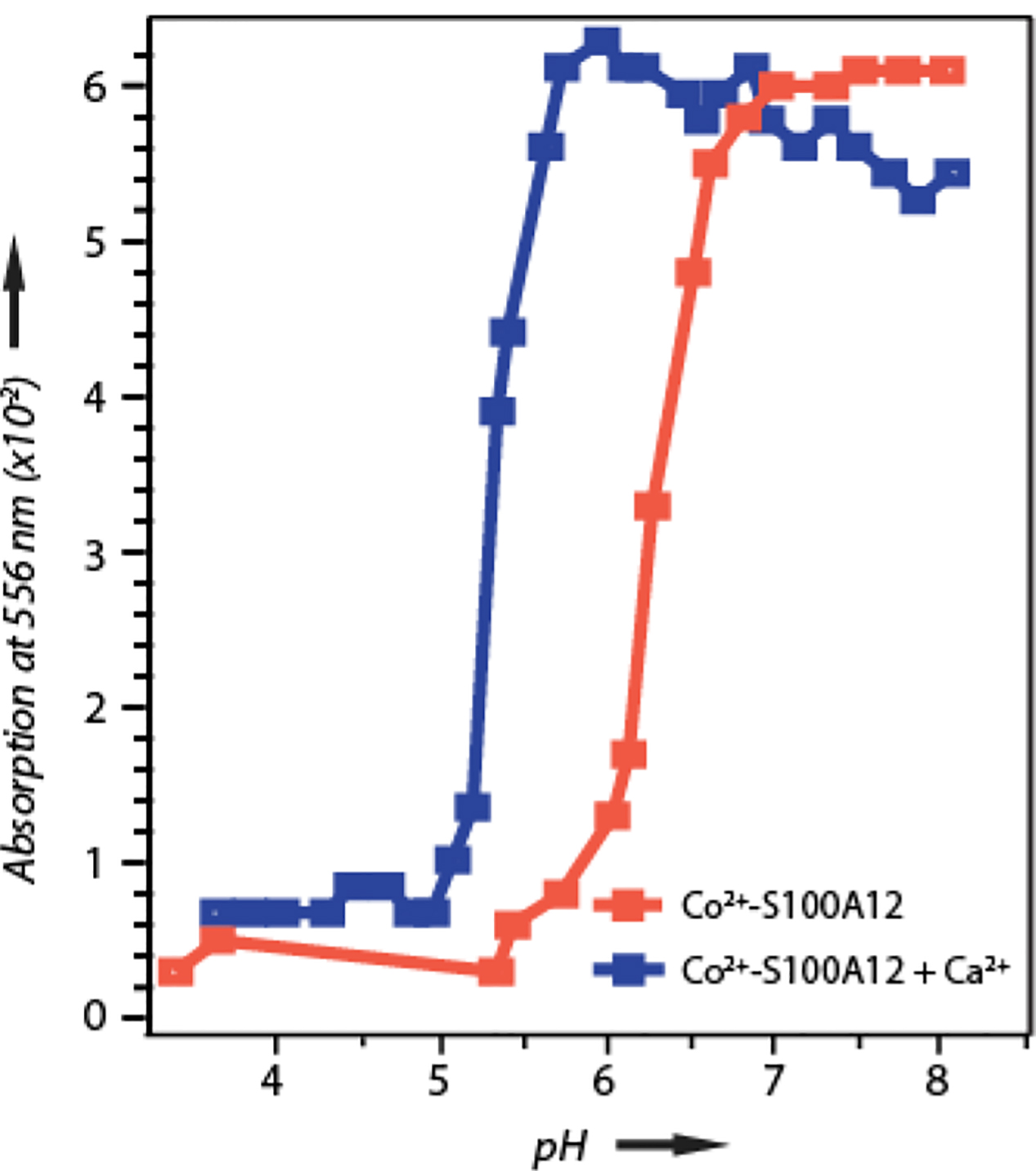
pH dependence of Co2+ binding to S100A12 in the absence (orange) and presence (blue) of Ca2+ shown with a plot of absorbance at 556 nm versus pH. At pH values greater than 7.3 minor protein precipitation in the calcium bound S100A12 was observed that interfered with the baselines of the absorption spectra, giving rise to irregular absorption maxima under these conditions.
Calcium binding restores the metal affinity at low pH.
Calcium binding to S100A12 enhances the ability of S100A12 to bind to Zn2+.[4d] Similar to the apo protein, the pH dependence zinc binding to Ca2+ loaded S100A12 was monitored using Co2+ as surrogate to Zn2+. However, unlike the apo protein, minor amount of precipitation at pH values greater than 8.0 was observed, presumably from insoluble calcium salts. This precipitation interfered with the baseline of the absorption spectra, as shown in Figure 3 (blue trace). Therefore, we will focus our attention on the results of pH titration below pH 7.0. In the presence of Ca2+, the optical absorption spectrum of Co2+ -S100A12 remains the same as the apo protein and addition of one equivalent Co2+ to S100A12 at pH 7.0 gives a 556 nm optical transition that is in quantitative agreement with the amount of protein used. As shown in Figure 3 (blue trace), the pH titration of Ca2+-S100A12 revealed that the protein retains Co2+ binding up to pH 5.7 in the presence of calcium. These results demonstrate that the viable pH range for transition metal binding is broadened in the presence of calcium and unlike the apo-protein that exhibits loss in metal binding below pH 7.0, Ca2+-S100A12 can retain Co2+ binding down to pH 5.7. Below pH 5.7, the calcium loaded protein starts to exhibit diminished Co2+ affinity and a gradual loss in Co2+ binding was observed in the pH range of 5.0 – 5.7. At pH values below 5.0, negligible metal binding was observed in the presence of 1 equivalent Co2+.
Sidechain protonation of one or more of the three histidine residues (H15, H85 and H89) responsible for metal binding could be a likely origin of the observed pH-dependent loss in metal binding. The imidazole ring of histidine residues in polypeptide chains has three tautomers: two neutral tautomeric forms (Nδ1H and Nε2H) and the protonated imidazolium state. The pKa of titratable nitrogens in the histidine imidazole ring can greatly depend on the local protein environment. For instance, histidine in small peptides exhibit pKa of ~ 6, while in protein environment the pKa values can range between 3–10.[13] Metal ligation to histidine residue takes place via imidazole nitrogens, protonation of these atoms can prevent metal binding. In S100A12, calcium binding may alter the pKa’s of histidine residues, facilitating enhanced Co2+/Zn2+ binding at lower pH compared to the apo-protein. If the loss in Co2+ binding at low pH-values originates from the protonation of coordinating histidine residues, then the pH titration curves in Figure 3 suggests average pKa values of 6.3 and 5.2 for apo- and Ca2+ -S100A12, respectively. This may indicate that although the Ca2+ and Zn2+/Co2+ binding sites are structurally distinct; calcium binding may modulate Zn2+ binding to S100A12 by altering the apparent pKa values of metal binding histidines. To test this hypothesis, the protonation states of histidine residues were determined for apo- and Ca2+-S100A12 using NMR spectroscopy at biologically pertinent pH values of 6.0 and 7.5 and, the results are described below.
Protonation state of histidine residues in apo and Ca2+-S100A12.
Although crystal structures of apo and Ca2+ bound S100A12 have been reported,[8–9] most crystal structures lack the resolution to decipher the protonation state of titratable amino acids. We utilized NMR spectroscopy to observe the protonation state of metal binding histidines in S100A12. Due to fast exchange with solvent water molecules, protons bonded to imidazole nitrogens are not observed in 1H-15N HSQC NMR spectra. Non-exchangeable aromatic protons (Hδ2 and Hε1) attached to Cδ2 and Cε1 carbon atoms can be observed through 2JN-H coupling in long range 1H-15N HMQC correlation spectra.[14] The cross-peak pattern and intensities in long range 1H-15N HMQC NMR spectra[14–15] can be used to probe the tautomeric states of histidine residues as shown in Figure S2. In the protonated imidazolium form of histidine, Nε2 and Nδ1 nuclei exhibit chemical shifts of ~173 and 176 ppm, respectively. In the neutral form, the Nε2H tautomer has Nε2 and Nδ1 chemical shifts ~164 and 249 ppm, while this pattern is reversed in the Nδ1H tautomer.[14–15]
The polypeptide chain of S100A12 contains six histidine residues. While His15, His85 and His89 form the His3Asp Zn2+/Co2+ binding motif, H6, H23 and H87 do not participate in metal binding. Figure 4A shows 1H-15N HMQC spectra of apo-S100A12 at pH 6.0 and 7.5. At pH 7.5, histidine sidechain resonances originating from four out of six histidines are observed. The loss of resonances from the remaining two residues is presumably due to signal broadening, which may result from fast chemical or conformational exchange. The cross-peak pattern and the Nδ1 chemical shifts around 220 ppm for the four observed histidines suggest they exist in the neutral the Nε2H tautomeric form. In contrast, when the pH is reduced to 6.0, resonances originating from all six histidine residues are detected.15N chemical shift between 170–190 ppm suggest that these residues exist in protonated imidazolium form. No evidence of neutral tautomers for any of the histidine residues was observed at pH 6.0.
Figure 4.
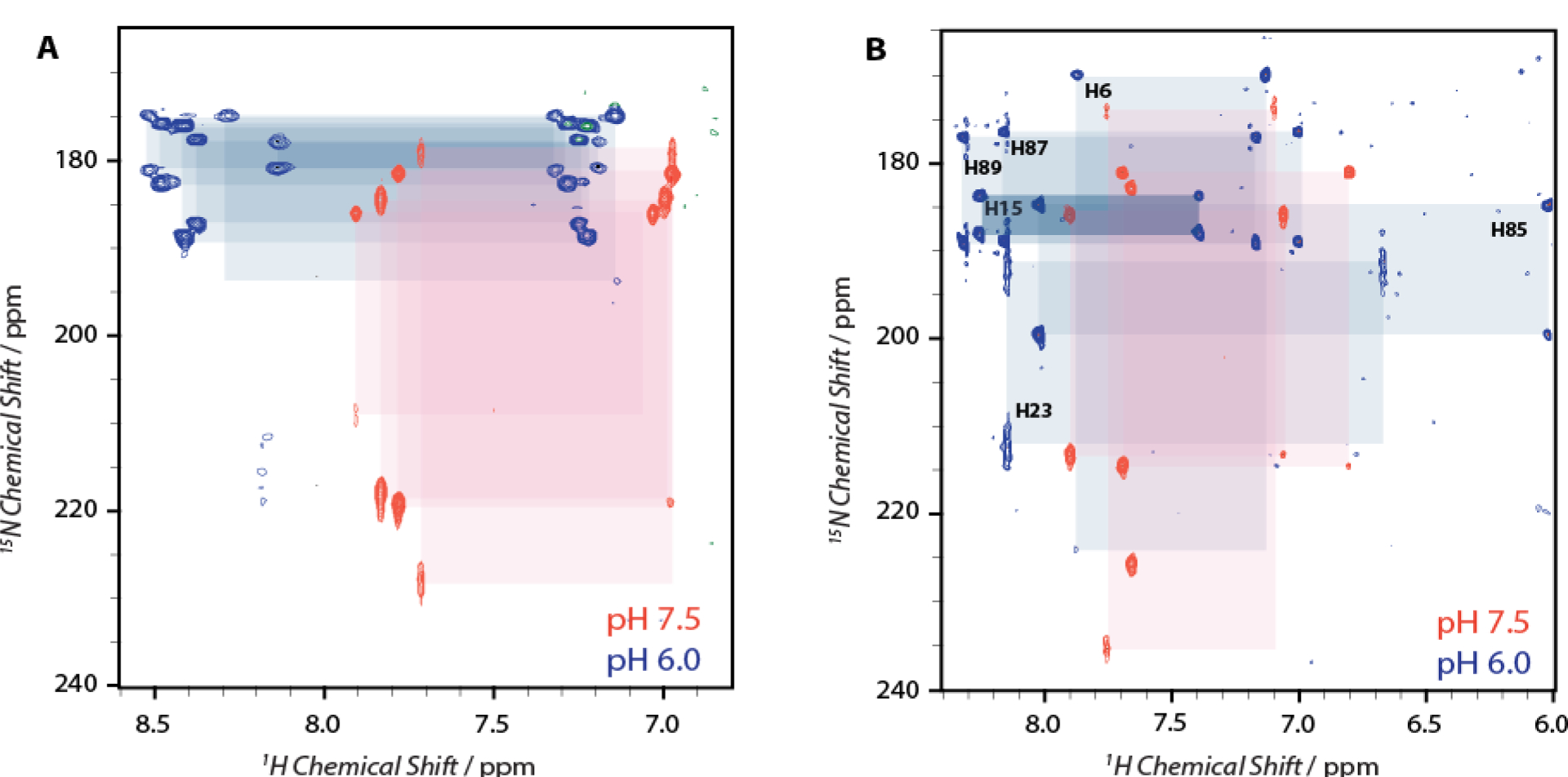
1H-15N HMQC spectra showing the tautomeric forms of histidine residues in (A) apo (16.8 T) and; (B) calcium bound S100A12 (14.1 T) at pH 7.5 (orange) and 6.0 (blue). The cross-peak patterns belonging to a single residue are marked with colored boxes.
Similar to the apo protein at pH 7.5, the 1H-15N HMQC spectrum of calcium bound S100A12 shows cross-peak patterns corresponding to four out of six histidine residues, while at pH 6.0, all of the six histidine residues can be detected (Figure 4B). At pH 7.5, all observed histidine residues are present in the neutral Nε2H tautomeric state. Interestingly, unlike the apo state, the cross-peak patterns at pH 6.0 indicate that only four histidines exist in the protonated form while two are present as Nε2H tautomers. In order to establish the identity of these histidines, additional 3D experiments were performed that allowed for the assignments of resonances in the 1H-15N HMQC spectrum and site-specific identification of histidine protonation states in Ca2+ bound S100A12 at pH 6.0 (vide infra). Based on these assignments, the metal binding histidine residues (H15, H85 and H89), along with H87 exist in the protonated form at pH 6.0 (Figure 4B). Residue H6 bears the Nε2H tautomeric state, while the protonation state of H23 cannot be unambiguously established. The signals originating from H23 exhibit line broadening, which may originate from an equilibrium between the protonated and neutral tautomers. Taken together, the histidine residues of the His3Asp motif are all protonated at pH 6.0, both in the presence and absence of calcium, implying that Ca2+ binding does not alter the sidechain pKa of the metal binding amino acids. These results indicate that the modulation of Zn2+ binding by calcium is not regulated by the variations in the electronic environments of the histidine residues leading to change in the apparent pKa values, in the presence and absence of calcium.
Sidechain resonance assignment for Ca2+-S100A12 at pH 6.0.
3D HNCACB, HN(CO)CACB and 2D (HB)CB(CGCDCE)HE and (HB)CB(CGCD)HD measurements were performed in order to assign sidechain resonances of histidine residues observed in 1H-15N HMQC spectra (Figure 5). HNCACB, HN(CO)CACB spectra allowed to establish a correlation between the backbone amide and the Cβ nuclei (Figure 5C), while (HB)CB(CGCDCE)HE and (HB)CB(CGCD)HD experiments provided correlation between Cβ and Hδ2/Hε1 nuclei (Figure 5B). Subsequently, with the aid of previously made backbone assignments,[11] the sidechain resonances in 1H-15N HMQC spectra were assigned. The separation between the resonances from H87 and H89 was not readily discernible in the spectra. These resonances were indirectly assigned with the assistance of the 1H-15N NOESY-HSQC spectrum (Figure 5D). The assignments of resonances in 1H-15N HMQC spectrum, along with the tautomeric state of histidine residues, are reported in Table 1. Attempts were made to utilize a similar strategy to assign histidine residues in apo-(pH 6.0 and 7.5) and Ca2+-S100A12 at pH 7.5. However, unambiguous assignments cannot be made for the above states of the protein due to lack of resolution in the indirect 13C (Cβ) dimension of 2D (HB)CB(CGCDCE)HE and (HB)CB(CGCD)HD experiments. Although assignments are not available for sidechain resonances of the apo-protein at pH 6.0 and 7.5 and, for Ca2+-S100A12 at pH 7.5, we note that all histidine residues in the polypeptide chain (with and without calcium) at pH 7.5 exit in the neutral Nε2H tautomeric state and in the protonated state at pH 6.0. Therefore, albeit a lack of availability of site-specific assignments, our data suggests that calcium binding does not affect protonation of the metal binding histidines, which are present in the neutral and the protonated forms at pH 7.5 and 6.0, respectively, both in the presence and the absence of calcium.
Figure 5.
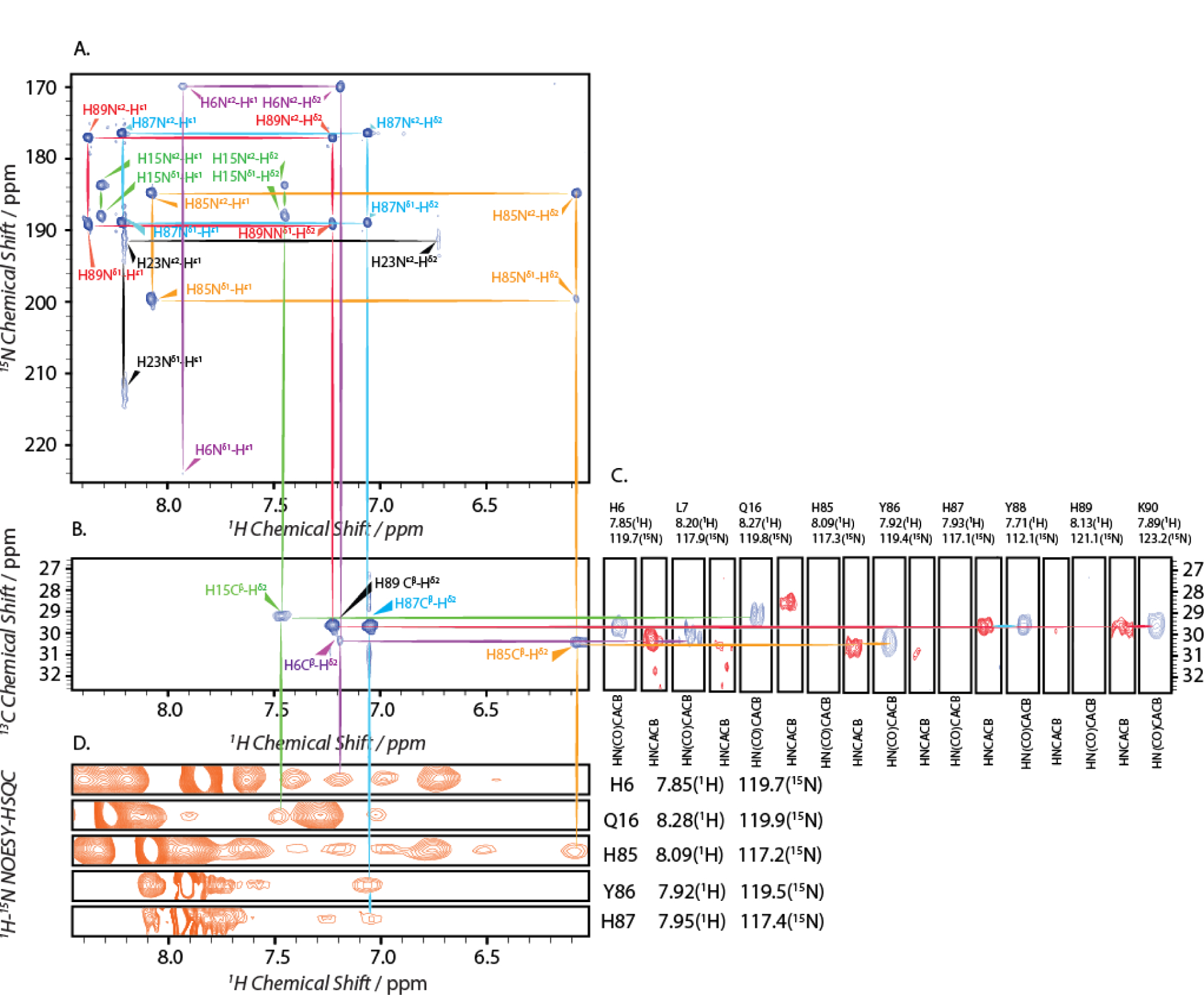
2D and 3D 1H, 15N, 13C experiments conducted for the assignments of histidine sidechain nuclei of calcium bound S100A12 at pH 6.0. A) 1H-15N HMQC; B) 2D 1H-13C (HB)CB(CDCGCD)HD; C) a strip plot of the 3D HN(CO)CACB (blue) and HNCACB (red) spectra; D) a strip plot of the 1H-15N NOESY-HSQC (orange) spectrum. Resonances associated with a residue are labeled and marked with same color across all spectra.
Table 1.
Sidechain resonance assignment of histidine residues in calcium bound S100A12 at pH 6.0.
| Residue | Nε2 (ppm) | Nδ1 (ppm) | Hεl (ppm) | Hδ2 (ppm) | Tautomeric state |
|---|---|---|---|---|---|
| H6 | 169.9 | 224.2 | 7.93 | 7.19 | Nε2H tautomer |
| H15 | 183.7 | 187.9 | 8.31 | 7.45 | protonated |
| H23 | 191.2 | 212.2 | 8.20 | 6.73 | N/Aa |
| H85 | 184.7 | 199.6 | 8.08 | 6.02 | protonated |
| H87 | 176.4 | 188.9 | 8.22 | 7.06 | protonated |
| H89 | 177.1 | 189.2 | 8.38 | 7.23 | protonated |
cannot be determined
pH dependent chemical shift perturbations in apo and Ca2+-S100A12.
To quantitatively assess the changes in the backbone conformation due to variations in pH, Cα and NH chemical shifts in the presence and absence of calcium were monitored at multiple pH conditions. While several factors effecting the local electronic environment of the backbone amide nitrogen may give rise to perturbation in NH chemical shift, 13Cα chemicals shifts correlate strongly with protein backbone conformation.[16] Backbone chemical shift assignments for apo- and Ca2+-S100A12 at pH 6.0 and 7.0, were made with the aid of standard triple-resonance experiments (see Experimental Section), and the calculated chemical shift perturbations (CSP) are displayed in Figure 6. These two pH values for CSP calculations were chosen because the loss in Co2+ binding was observed in the pH range of 5.3 – 7.0 for the apo-protein, while calcium restored the metal binding affinity up to pH 5.7. Therefore, the pH span of 6.0–7.0 represents the conditions under which the apo-protein exhibits a loss in metal binding, while the calcium loaded protein retains its metal chelation.
Figure 6.
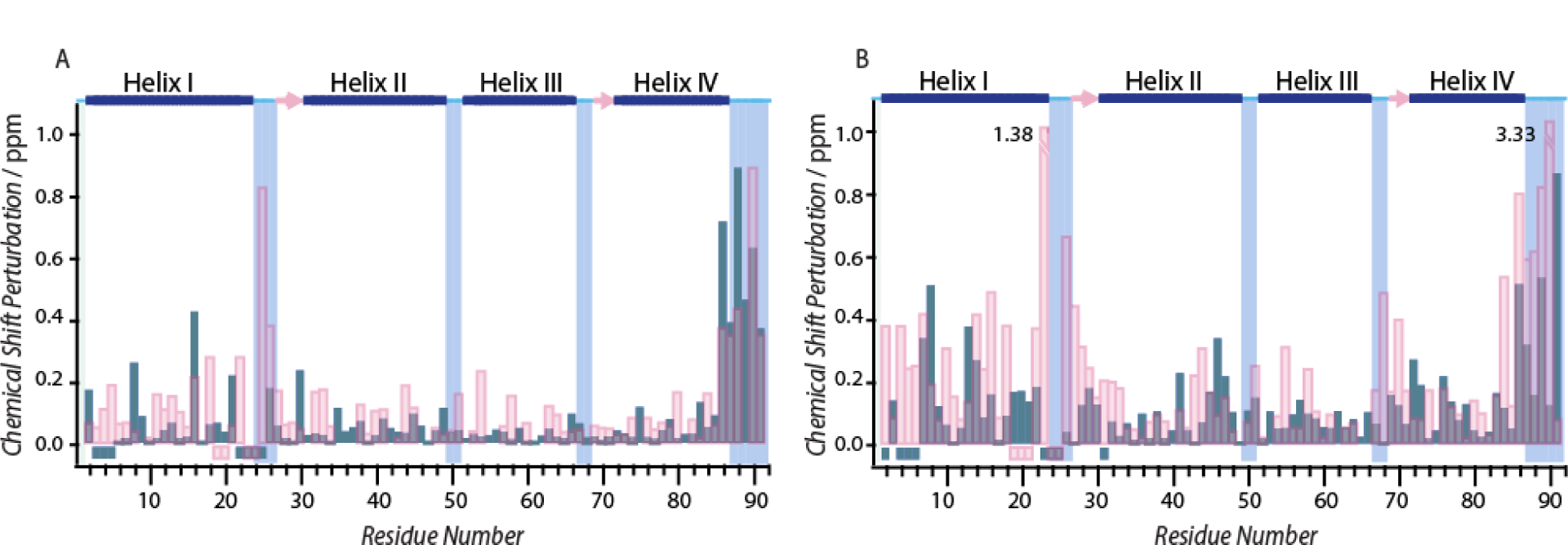
Site specific absolute values of chemical shift perturbation between pH 6.0 and 7.0 for (A) Cα and, (B) NH nuclei in the absence (pink) or presence (blue) of calcium ion. The negative values represent residues for which resonance assignments are not available. The secondary structure of the apo-protein determined from the crystal structure (PDB code 2WCF) is displayed at the top: α-helices (blue bars), β-strands (pink arrows), and loop regions (cyan lines). The values 1.38 and 3.33 in (B) refer to CSPs that are larger than the displayed range of the plot.
Majority of residues in the apo-protein did not exhibit large CSPs for Cα and NH nuclei. However, CSPs greater than 0.2 and 0.4 ppm for Cα and NH nuclei, respectively, were observed for the residues in the C-terminal helix IV. Notably, this region of the protein contains the three metal binding histidine residues. Additionally, the residues (residues 22 – 29) in the loop preceding helix II also demonstrate large CSPs. These residues belong to the EF hand-1 of the polypeptide chain (Figure 1), which bears the Co2+/Zn2+ binding D25 and participates in calcium binding. These results indicate that the conformation around the His3Asp motif (H15, D25, H85 and H89), along with the EF hand −1 loop, is affected upon pH variation. As in the apo-protein, while minor CSPs were observed for majority of the polypeptide chain in Ca2+-S100A12, helix IV shows large CSPs of magnitude similar to that of the apo-form. However, in contrast to the apo-protein, the residues in EF hand-1 loop, which contains D25 do not experience significant CSP. We note that although complete resonance assignments for all the residues in the EF hand-1 are not available, the conclusions above can be made as majority of residues have been assigned.
Secondary structure propensities of apo- and Ca2+-S100A12.
pH induced changes in the secondary structure of the protein may give rise to the observed CSPs in Figure 6. To investigate the pH dependence on the secondary structure, SSPs were calculated for S100A12 at pH 6.0 and 7.0 in the presence and absence of calcium. These SSP values are plotted against the residue numbers and displayed in Figure 7. As shown in Figure 7A and B, no major pH dependent variations in SSPs values was observed for the four helices in apo and calcium loaded protein, suggesting that the pH variations do not alter the major secondary structure composition in S100A12. Although the SSPs are not affected by pH, a comparison between the SSPs of the apo and Ca2+ bound S100A12 reveals that the secondary structure of loop (residue 22–29) belonging to EF hand-1 is different in the two forms of the protein. In the apo state, this region exists in coil-like secondary structure while upon calcium binding, these residues engage in a β-strand conformation. Below, based on these overall findings, we discuss the role of the EF hand-1 loop residues and the secondary structure employed by them towards calcium modulated zinc binding to S100A12.
Figure 7.
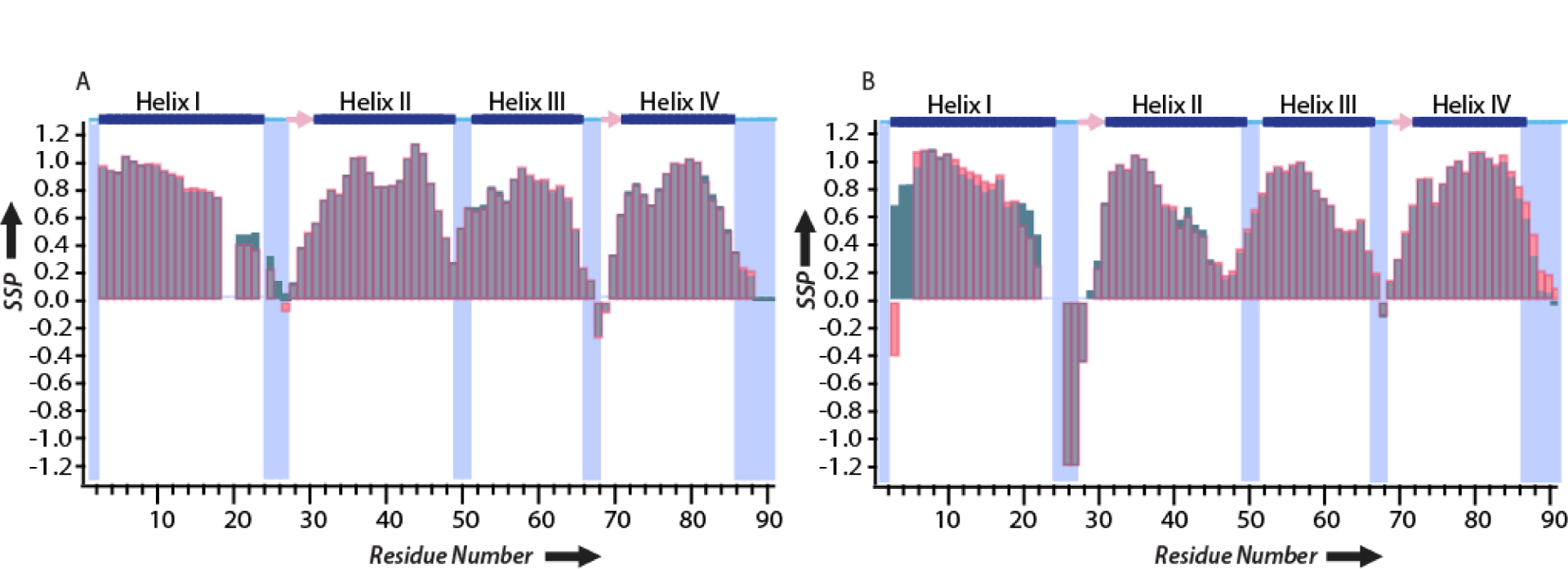
SSP based Cα and NH chemical shifts at pH 7.0 (red) and 6.0 (blue) for (A) Apo-S100A12 and, (B) Ca2+-S100A12. The secondary structure of the apoprotein determined from the crystal structure (PDB code 2WCF) is displayed at the top: α-helices (blue bars), β-strands (pink arrows), and loop regions (cyan lines).
Discussion
1H-15N HSQC spectra of S100A12 show noticeable variations in the amide backbone chemical shifts throughout the polypeptide even with minor pH changes (Figure S3). These CSPs indicate that S100A12 undergoes pH-dependent conformational changes. In addition, HSQC titrations also reveal that in the pH range of 4.5 – 8.0, the tertiary structure of the protein remains folded both in the absence and presence of calcium. Although, near neutral pH values best mimic the most cellular conditions, studies in the literature have shown that human neutrophils, which constitutively express and secrete S100A12 are activated during extracellular acidosis.[10] Under these conditions, the pH of the extracellular milieu can drop to pH 5.5. Therefore, metal sequestration properties of S100A12 pertaining to its biological functions of nutritional immunity need to be investigated at sub-neutral pH conditions. S100A12 is a high efficiency Zn2+ chelator, exhibiting nanomolar binding affinity at pH 7.0, which is further enhanced upon calcium binding.[4d] Our results have demonstrated that although S100A12 is structurally resistant to pH variations (in the range of pH 4.5 – 10.4), the metal chelation efficiency of the apo-protein dramatically reduces in the pH range of 5.3 – 7.0. However, calcium binding can retain Zn2+ chelation down to pH 5.7 and below this pH the calcium bound protein also shows loss in metal binding.
Given the strong nanomolar zinc binding affinity of apo-S100A12 at neutral pH conditions, it is difficult to evaluate the biological role of calcium binding towards antimicrobial activities of S100A12. Due to the high concentration of Ca2+ in the extracellular space, upon secretion, calcium bound S100A12 and not its apo form is expected participate in Zn2+ homeostasis. Here we present a plausible explanation for the relevance of calcium binding towards metal sequestration functions to S100A12. We propose that upon neutrophil activation initiated by extracellular acidosis, S100A12 secreted into the serum experiences sub-neutral pH conditions between pH 5.5 – 6.0. At this pH values, the apo protein is not an efficient Zn2+ chelator. However, due to the abundance of Ca2+ in the extracellular space, S100A12 is loaded with calcium upon secretion. Unlike the apo-protein, Ca2+-S100A12 can efficiently bind Zn2+ in the pH of 5.5 – 6.0, allowing S100A12 to perform its antimicrobial functions.
S100A8/A9 and S100A12 together compose ~ 50% of the total cytoplasmic proteins in neutrophils. S100A8/A9 heterodimer binds to various transition metal ions via a His3Asp motif, identical to S100A12, and a His6 site. The His3Asp binding motif of S100A8/A9 bears remarkable similarities to S100A12 including this site sharing the Asp residue with Ca2+ binding EF hand 1 of the dimer.[17] Also, similar to S100A12, S100A8/A9 exhibits nanomolar Zn2+ binding affinity which is enhanced upon Ca2+ binding.[3a] Given such commonalities between Zn2+ sequestration of these two components of the human immune response, we suggest that pH variations may also regulate the transition metal binding to S100A8/A9 with dependence similar to S100A12. Therefore, the pH dependence and Ca2+ mediated enhancement of Zn2+ sequestration presented here may represent a general mechanism employed by neutrophils during the activation of these immune cells to excecute antimicrobial functions. Below, based on the results presented here, we discuss the mechanism of calcium mediated modulation of Zn2+ binding to S100A12 as the pH is varied.
The pH dependence of Co2+ binding to S100A12 and modulation of viable metal binding pH range by calcium as shown in Figure 2 represents apparent Co2+ binding affinities in the absence and presence of calcium. We consider and discuss two plausible explanations for the observed tuning of Co2+ binding by calcium. Firstly, it is conceivable that Co2+/Zn2+ Kd of the His3Asp motif does not change with pH; instead the differences in the apparent Kd observed in the pH titration curves with and without calcium originates from the variations in the sidechain pKa values of metal binding histidine residues. Due to the protonatable sidechains of the H15, H85 and H89 residues, the Co2+/Zn2+ binding motif in S100A12 may be sensitive to pH changes particular because pKa values of 3–10 are common for histidine sidechains in protein environments. Binding of calcium may not affect the inherent Kd of the His3Asp motif but alter the pKa values of histidine sidechains, resulting differential protonation of coordinating histidine residues and loss in the observed binding affinities between apo and Ca2+-S100A12. However, the tautomeric states derived from 1H-15N HMQC measurements do not support this hypothesis. At pH 7.5, all observed histidine residues in 1H-15N HMQC spectra exist in neutral tautomeric form for both apo and calcium bound S100A12 (Figure 4). The pH titration curves in Figure 2, if indicative of histidine pKa values suggest pKa of 6.3 and 5.2, respectively for S100A12 in the absence and presence of calcium. At pH 6.0, assuming pKa of 6.3, the concentration of the neutral and the protonated tautomer should be near equal in the apo protein. However, 1H-15N HMQC spectrum of apo-S100A12 (Figure 4A) demonstrates that all histidines exist in the protonated tautomeric form with no evidence of neutral tautomer.
Similarly, based on the postulated pKa value of 5.2 for metal binding histidines in Ca2+-S100A12, the neutral tautomer should be ca. six times greater in concentrations compared to the protonated tautomer at pH 6.0. The restoration of metal binding ability at this pH value in the presence of calcium could be justified if the neutral tautomer is the major species in Ca2+-S100A12 at pH 6.0. 1H-15N HMQC measurements disprove this hypothesis and demonstrate that similar to the apo form, all metal binding histidine residues in Ca2+-S100A12 exist as protonated tautomer. These experiments indicate that the inflection point observed in the pH dependent metal binding curves does not reflect protonation of coordinating histidine residues, which is also not the origin of the loss in metal binding.
A second plausible explanation may be that calcium binding alters the inherent binding affinity of the Zn2+ motif resulting in the observed differences in metal binding in the presence and absence of calcium. Such variations may originate from calcium induced structural variations to the His3Asp binding scaffold. To evaluate this hypothesis, we analyzed residue specific pH dependent CSPs in S100A12 in the absence and presence of calcium. The pH values of 6.0 and 7.0 were chosen to examine the CSPs because the apo protein shows gradual loss in Co2+ binding in this range, while Ca2+-S100A12 retains its bound cobalt ion. Furthermore, based on the reported studies of extracellular acidosis triggered neutrophil activation,[10] this pH range is also biologically relevant. Large CSPs were observed in protein samples prepared in the absence and presence of calcium for most of the residues in helix IV, which contains two out of the three Co2+/Zn2+ binding histidines. In addition to this, similar magnitudes of CSPs for helix IV residues were observed in both apo and Ca2+-S100A12, including metal binding H85 and H89 residues. Significant Cα and NH CSPs suggest that these perturbations originate from pH induced conformational changes and that calcium binding does not modulate the conformation of this region of S100A12.
A noticeable difference in the CSPs for apo and Ca2+-S100A12 can be observed in the residues belonging to the calcium binding EF hand 1 motif. As shown in Figure 6, the residues of this loop exhibit minor pH-dependent CSPs upon calcium binding. Contrary to the calcium loaded protein, the EF hand-1 loop shows large perturbations as the pH is varied in the apo protein. This 14 residue EF hand is the non-canonical, S100-specific calcium binding motif found in this family of proteins.[18] Asp25, which participates in Co2+/Zn2+ binding, belongs to this loop and calcium may introduce structural restraints to this residue. Our results reveal that calcium binding minimizes pH induced conformational perturbations to the EF hand 1 loop, which may in turn, provide the structural stability to the His3Asp motif via Asp25 thereby regulating the pH dependence of S100A12 metal binding.
SSP analysis showed that while the pH induced conformational changes do not alter the secondary structure of the protein, calcium binding promotes a β-strand conformation for residues D25-L27 of the EF hand 1 loop. These residues occupy a coil-like configuration in the apo protein. The β-strand conformation of these loop residues upon calcium binding is not disrupted by pH variations (Figure 7) and may play a critical role by introducing structural constraints needed to provide improved zinc affinity. The coil-like configuration in the apo protein, on the other hand, is susceptible to pH induced conformational changes, as demonstrated by the CSPs in Figure 6. Therefore, we posit that the calcium modulated zinc affinity of S100A12 is executed via the EF hand 1 loop residues, which in the absence of calcium, occupies a flexible coil conformation that is susceptible to pH variations. Calcium binding induces β-strand conformation providing resistance towards pH mediated structural variations and expands the viable Zn2+ binding pH range to sub-neutral conditions. These results explain the relevance of calcium binding to S100A12 and its role in zinc sequestration and antimicrobial functions of S100A12. While the apo protein has high Zn2+ affinity at neutral pH, the abundance of calcium in the extracellular milieu may assist in S100A12 antimicrobial functions at the sites of infection by extending the viable pH range for zinc sequestration during extracellular acidosis. We have recently reported on structural perturbations induced upon divalent metal ion (Zn2+, Ca2+ and, Zn2+/Ca2+) binding to apo-S100A12 at pH 7.0.[11] Upon Zn2+ binding, S100A12 forms oligomeric assemblies that are not amenable to standard solution NMR methodologies owing to their large molecular weights. To overcome this hindrance, we utilized solid state NMR spectroscopy to demonstrate Zn2+ binding introduces distinct structural changes in helix II of the polypeptide chain. With the results reported herein, pH dependent solid state NMR studies on Zn2+ bound assemblies of S100A12 may provide additional insights into structural perturbations with pH variations and such studies will be undertaken in the future.
Conclusion
S100A12 is a high affinity Zn2+ chelating protein at neutral pH conditions. Zn2+ binding to S100A12 allows the protein to participate in the innate immune response via zinc sequestration mediated antimicrobial functions. Although calcium binding to the two EF hand loops of S100A12 increases its Zn2+ affinity, the mechanism of calcium mediated zinc sequestration is not completely known. Here, we demonstrate that while apo-S100A12 exhibits strong Zn2+ binding at neutral pH conditions, the protein gradually loses its Zn2+ affinity as the pH is reduced below 7.0. Calcium binding to S100A12 extends the viable pH range for high affinity Zn2+ binding to pH 5.7. These results indicate that calcium binding not only improves S100A12 Zn2+ affinity, it also allows the protein to efficiently perform zinc sequestration at sub-neutral pH conditions. We propose that given the already high Zn2+ affinity at neutral pH, the biologically relevant role of calcium binding is to extend the viable zinc binding pH range of S100A12 to sub-neutral conditions which may be generated during extracellular acidosis and that such dependence may also be exhibited by other antimicrobial innate immune proteins, particularly S100A8/A9 heterodimer. We demonstrate that the loss of zinc binding below pH 7.0 for the apo protein does not originate from the protonation of metal binding histidines. Instead our results indicate the extended pH range for zinc binding originates when calcium ligation introduces β-strand conformation to S100 specific EF hand loop, providing resistance from pH induced conformational changes.
Experimental Section
S100A12 expression and purification.
Human S100A12 (UniProt ID P80511) cDNA was synthesized and subcloned into the pET-41a(+) vector by GenScript followed by transformation to BL21(DE3) E.coli competent cells. The E.coli cell culture induced from a single colony of obtained from plasmid transformation was incubated at 37 °C with shaking at 200 rpm in Luria Broth (LB) or M9 minimal medium which contained U-15N NH4Cl or both U-15N NH4Cl and U-13C6 D-glucose. After induction by 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), the cell culture in M9 minimal medium was allowed to grow at 18 °C, while cell culture in LB was allowed to grow at 37 °C for 4 hours. Cells were collected by centrifugation at 4,000 rpm at 4 °C for 20 minutes. The harvested cell pellet was resuspended in lysis buffer (50 mM Tris-HCl, pH 8.3). The resuspended cells were sonicated with a microtip for 20 min of processing time with 5 seconds on and one second off. After sonication, cell lysate was centrifuged at 17,000 rpm at 4 °C for 40 minutes. The supernatant was collected and injected into a diethylaminoethanol (DEAE) anion exchange column pre-equilibrated with loading buffer (50 mM Tris-HCl, pH 8.3). For purification with the DEAE column, a gradient of 0–10% B was gradually applied over a volume of 50 mL (Eluent A, 50 mM Tris-HCl, pH 8.3; Eluent B, 50 mM Tris-HCl, 1 M NaCl, pH 8.3) and fractions were collected. Fractions containing S100A12 were determined by SDS-PAGE gel electrophoresis, combined and concentrated for further purification. To ensure generation of apo state of the protein upon purification, 25 mM ethylenediaminetetraacetic acid (EDTA) was added to remove divalent metal ions followed by dialysis and buffer exchange to remove excess EDTA. In the next purification step, size exclusion chromatography (GE Healthcare Life Science, HiLoad 16/600 Superdex 75 prep grade) was performed on a column pre-equilibrated with 20 mM HEPES pH 7.5, 150 mM NaCl. Fractions containing S100A12 were pooled and SDS-PAGE gels were used to evaluate the purity of the protein. The protein concentration was determined by observing the 280 nm absorbance (ε = 2980 M−1 cm−1). The amino acid sequence of the purified protein was verified as reported previously.[11]
UV-Vis spectroscopy.
The samples for UV-Vis spectroscopy were prepared at 0.1 mM or 0.2 mM S100A12 concentration in 20 mM HEPES, 150 mM NaCl, pH 7.5 buffer. The buffer pH was raised with 0.1 M Sodium Carbonate – Sodium Bicarbonate buffer up to pH 10.4, or lowered up to pH 3.5 using dilute HCl. For UV-Vis measurements on Ca2+-S100A12, 20 equivalents of calcium were added. Co2+-S100A12 complex was detected by monitoring the absorbance at 556 nm and quantified using previously reported ε = 820 M−1cm−1 (for S100A12 dimers).[4d]
NMR sample preparation and NMR spectroscopy.
Uniformly 15N and/or 13C labeled samples for solution NMR measurements were prepared in buffer containing 20 mM MES, 150 mM NaCl at pH 6.0 or 20 mM HEPES, 150 mM NaCl, pH 7.5 with 10% D2O and 200 μM 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS). NMR samples were prepared at 0.7 mM protein concentration. The pH of the buffer during pH titration experiments was adjusted using NaOH or HCl.
NMR spectra were acquired at 14.1 T on a Varian Inova NMR spectrometer outfitted with an HCN cryoprobe or at the City University of New York’s Advanced Science Research Center at 16.8 T. The Larmor frequencies (at 14.1 T) were 599.93, 150.87 and 60.79 MHz for 1H, 13C and 15N, respectively, and the sample temperature was maintained at 25 °C or 37 °C. All NMR spectra were processed with NMRpipe[19] and analyzed with Sparky.[20] A forward linear prediction to twice the number of original data points followed by zero-filling to twice the total number of points was used for all experiments. 30°- and 60°-shifted sine bell apodization with water suppression function were used for both dimensions. Backbone assignments of Cα and NH were accomplished by 2D 1H-15N heteronuclear multiple-quantum coherence (HSQC),[21] 3D HNCACB[22] and HN(CO)CACB[23] experiments (all part of the Agilent BioPack library). 1H-15N heteronuclear multiple quantum coherence (HMQC)[21] experiment using a 22.4 ms delay period was employed to detect long range correlation between Nδ1/Nε2 and Hε1/Hδ2 histidine sidechain nuclei via 2JN-H couplings.[14] Histidine sidechain Hε1/Hδ2 assignments were made with the aid of 2D 1H-13C (HB)CB(CGCDCE)HE[24] and (HB)CB(CGCD)HD[24] correlation spectra that provided correlation between known Cβ chemical shifts and Hε1/Hδ2 nuclei.
Secondary structure propensity calculations.
The secondary structure propensity (SSP) of S100A12 was calculated by the SSP program[25] using the Cα and NH chemical shift as inputs. For each residue, the Cα and NH were combined to calculate a single score representing the expected fraction of α– or β– secondary structures. The α–, β– or random coil structure is predicted based on the proximity of the SSP value to 1.0, −1.0 or 0 respectively. For example, an SSP score of 1 represents a fully formed α structure and a score of −1 represents a fully formed β structure.
Supplementary Material
Acknowledgement
This work was supported by the City University of New York startup funds, the National Institutes of Health (R15GM131338) and PSC CUNY award (62234-00 50) to RG. ANJ acknowledges the support from the Rosemary O’ Halloran Scholarship at the College of Staten Island. NMR data presented herein were collected (in part) at the City University of New York’s Advanced Science Research Center (CUNY ASRC) Biomolecular NMR Facility and the New York Structural Biology Center. RG is a member of the New York Structural Biology Center. Data collected using 500 MHz spectrometer is supported by NIH grant S10OD016432.
Footnotes
Publisher's Disclaimer: This manuscript has been accepted after peer review and appears as an Accepted Article online prior to editing, proofing, and formal publication of the final Version of Record (VoR). This work is currently citable by using the Digital Object Identifier (DOI) given below. The VoR will be published online in Early View as soon as possible and may be different to this Accepted Article as a result of editing. Readers should obtain the VoR from the journal website shown below when it is published to ensure accuracy of information. The authors are responsible for the content of this Accepted Article.
References
- [1].Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL, Curr. Mol. Med 2013, 13, 24–57. [PMC free article] [PubMed] [Google Scholar]
- [2].a) Gilston BA, Skaar EP, Chazin, Science China: Life Sciences 2016, 59, 792–801; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zackular JP, Chazin WJ, Skaar EP, J. Biol. Chem 2015, 290, 18991–18998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Brophy MB, Hayden JA, Nolan EM, J. Am. Chem. Soc 2012, 134, 18089–18100; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP, Science 2008, 319, 962–965; [DOI] [PubMed] [Google Scholar]; c) Hayden JA, Brophy MB, Cunden LS, Nolan EM, J. Am. Chem. Soc 2012, 135, 775–787; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hayden JA, Brophy MB, Cunden LS, Nolan EM, J. Am. Chem. Soc 2013, 135, 775–787; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Nakashige TG, Zhang B, Krebs C, Nolan EM, Nat. Chem. Biol 2015, 11, 765; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Nakashige TG, Zygiel EM, Drennan CL, Nolan EM, J. Am. Chem. Soc 2017, 139, 8828–8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Gläser R, Harder J, Lange H, Bartels J, Christophers E, Schröder J-M, Nat. Immunol 2004, 6, 57; [DOI] [PubMed] [Google Scholar]; b) Lee KC, Eckert RL, J. Invest. Dermatol 2007, 127, 945–957; [DOI] [PubMed] [Google Scholar]; c) Kehl-Fie TE, Skaar EP, Curr. Opin. Chem. Biol 2010, 14, 218–224; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cunden LS, Gaillard A, Nolan EM, Chem. Sci 2016, 7, 1338–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Foell D, Seeliger S, Vogl T, Koch H, Maschek H, Harms E, Sorg C, Roth J, Thorax 2003, 58, 613–617; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Larsen A, Bronstein IB, Dahl O, Wentzel-Larsen T, Kristoffersen EK, Fagerhol MK, Scandinavian journal of immunology 2007, 65, 192–201; [DOI] [PubMed] [Google Scholar]; c) Kim MH, Choi YW, Choi HY, Myung KB, Cho SN, Br. J. Dermatol 2006, 154, 594–601; [DOI] [PubMed] [Google Scholar]; d) Buhimschi IA, Zhao G, Pettker CM, Bahtiyar MO, Magloire LK, Thung S, Fairchild T, Buhimschi CS, Am. J. Obstet. Gynecol 2007, 196, 181.e181–181.e113; [DOI] [PubMed] [Google Scholar]; e) Leclerc E, Fritz G, Vetter SW, Heizmann CW, Biochim. Biophys. Acta 2009, 1793, 993–1007. [DOI] [PubMed] [Google Scholar]
- [6].Pietzsch J, Hoppmann S, Amino acids 2009, 36, 381–389. [DOI] [PubMed] [Google Scholar]
- [7].Haley KP, Delgado AG, Piazuelo MB, Mortensen BL, Correa P, Damo SM, Chazin WJ, Skaar EP, Gaddy JA, Infect. Immun 2015, 83, 2944–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Moroz OV, Blagova EV, Wilkinson AJ, Wilson KS, Bronstein IB, J. Mol. Biol 2009, 391, 536–551. [DOI] [PubMed] [Google Scholar]
- [9].Moroz OV, Antson AA, Murshudov GN, Maitland NJ, Dodson GG, Wilson KS, Skibshøj I, Lukanidin EM, Bronstein IB, Acta Crystallogr., Sect. D: Biol. Crystallogr 2001, 57, 20–29. [DOI] [PubMed] [Google Scholar]
- [10].a) Trevani AS, Andonegui G, Giordano M, López DH, Gamberale R, Minucci F, Geffner JR, J. Immunol 1999, 162, 4849–4857; [PubMed] [Google Scholar]; b) Martínez D, Vermeulen M, Trevani A, Ceballos A, Sabatté J, Gamberale R, Álvarez ME, Salamone G, Tanos T, Coso A, J. Immunol 2006, 176, 1163–1171. [DOI] [PubMed] [Google Scholar]
- [11].Wang Q, Aleshintsev A, Bolton D, Zhuang J, Brenowitz MD, Gupta R, Biochemistry 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hung K-W, Hsu C-C, Yu C, Biomol J. NMR 2013, 57, 313–318. [DOI] [PubMed] [Google Scholar]
- [13].a) Bhattacharya S, Sukits SF, MacLaughlin KL, Lecomte J, Biophys. J 1997, 73, 3230–3240; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Takahashi T, Nakamura H, Wada A, Biopolymers 1992, 32, 897–909; [DOI] [PubMed] [Google Scholar]; c) Plesniak LA, Connelly GP, Wakarchuk WW, McIntosh LP, Protein Sci. 1996, 5, 2319–2328; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Day RM, Thalhauser CJ, Sudmeier JL, Vincent MP, Torchilin EV, Sanford DG, Bachovchin CW, Bachovchin WW, Protein Sci. 2003, 12, 794–810; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Schubert M, Poon DK, Wicki J, Tarling CA, Kwan EM, Nielsen JE, Withers SG, McIntosh LP, Biochemistry 2007, 46, 7383–7395; [DOI] [PubMed] [Google Scholar]; f) Singer UA, Forman-Kay JD, Protein Sci. 1997, 6, 1910–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pelton J, Torchia DA, Meadow N, Roseman S, Protein Sci. 1993, 2, 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Wasylishen RE, Tomlinson G, Can. J. Biochem 1977, 55, 579–582; [DOI] [PubMed] [Google Scholar]; b) Platzer G, Okon M, McIntosh LP, J. Biomol. NMR 2014, 60, 109–129. [DOI] [PubMed] [Google Scholar]
- [16].Spera S, Bax A, J. Am. Chem. Soc 1991, 113, 5490–5492. [Google Scholar]
- [17].Cunden LS, Nolan EM, Biochemistry 2018, 57, 1673–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhou Y, Yang W, Kirberger M, Lee HW, Ayalasomayajula G, Yang JJ, Proteins: Structure, Function, and Bioinformatics 2006, 65, 643–655. [DOI] [PubMed] [Google Scholar]
- [19].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, Biomol J. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- [20].Goddard T, Kneller D, University of California 2006. [Google Scholar]
- [21].Cavanagh J, Fairbrother WJ, Palmer III AG, Skelton NJ, Protein NMR spectroscopy: principles and practice, Elsevier, 1995. [Google Scholar]
- [22].Grzesiek S, Bax A, J. Mag. Res 1992, 99, 201–207. [Google Scholar]
- [23].Grzesiek S, Bax A, J. Am. Chem. Soc 1992, 114, 6291–6293. [Google Scholar]
- [24].Yamazaki T, Forman-Kay JD, Kay LE, J. Am. Chem. Soc 1993, 115, 11054–11055. [Google Scholar]
- [25].Marsh JA, Singh VK, Jia Z, Forman-Kay JD, Protein Sci. 2006, 15, 2795–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.