

SUMMARY
Autism is a clinically heterogeneous neurodevelopmental disorder characterized by impaired social interactions, restricted interests and repetitive behaviors. Despite significant advances in the genetics of autism, understanding how genetic changes perturb brain development and impact clinical symptoms remains elusive. Here, we present a multiplex human pluripotent stem cell (hPSC) platform, in which 30 isogenic disease lines are pooled in a single dish and differentiated into prefrontal cortex (PFC) lineages to efficiently test early-developmental hypotheses of autism. We define subgroups of autism mutations that perturb PFC neurogenesis and are correlated to abnormal WNT/βcatenin responses. Class 1 mutations (8/27) inhibit, while Class 2 mutations (5/27) enhance PFC neurogenesis. Remarkably, autism patient data reveal that individuals carrying subgroup-specific mutations differ clinically in their corresponding language acquisition profiles. Our study provides a framework to disentangle genetic heterogeneity associated with autism and points toward converging molecular and developmental pathways of diverse autism-associated mutations.
Graphical Abstract
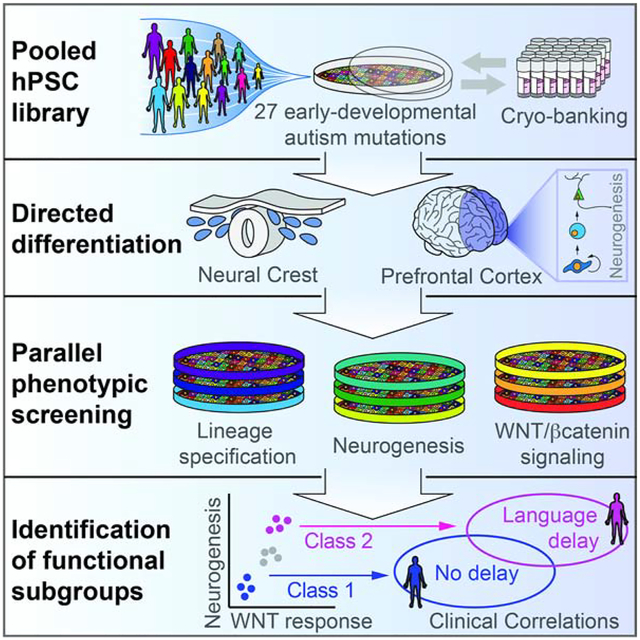
eTOC
Large-scale sequencing studies have identified hundreds of autism-associated genes, yet a systematic understanding of their functional consequences during human neurodevelopment remains elusive. Studer and colleagues develop a pooled human pluripotent stem cell-based phenotypic screening platform, and identify subgroups of autism genes with convergent molecular, developmental, and clinical associations.
INTRODUCTION
The immense genetic diversity of autism (De Rubeis et al., 2014; Durand et al., 2007; Glessner et al., 2009; Iossifov et al., 2015; Iossifov et al., 2014; Jamain et al., 2003; Krumm et al., 2015; Ronemus et al., 2014; Sanders et al., 2015; Sebat et al., 2007) presents a major challenge to our understanding of the disease and to the future development of novel therapeutic strategies. This challenge reflects a broader limitation in studying human disorders, as most experimental models fail to capture the genetic heterogeneity and cell type specific vulnerability characteristic of complex disease (McClellan and King, 2010).
Neuroimaging and neuropathology studies show frequent alterations in prefrontal cortex (PFC) growth and neurogenesis in autism patients (Courchesne et al., 2011; Courchesne et al., 2007; Hazlett et al., 2017; Stoner et al., 2014). In addition, bioinformatic approaches indicate that autism-associated genes interact with transcriptional networks of the frontal cortex and cerebellum (Willsey et al., 2013), and segregate into two temporal categories with peak expression at post-conception week (PCW) 8–20 or shortly after birth. The early category of genes is associated with transcription and chromatin remodeling while the latter category of genes is associated with synapse development and function.
The question of whether a given autism mutation directly perturbs PFC growth and neurogenesis can be studied using traditional animal models. For multi-gene disorders however, the functional characterization of dozens of genes in animal or cell-based models remains challenging and is typically restricted to resource intensive settings such as large-scale consortia (Sweet, 2017). Human pluripotent stem cells (hPSCs), which provide access to disease-relevant human tissue through high-quality differentiation protocols (Sterneckert et al., 2014), offer a promising alternative for modeling complex disorders and to test early-developmental hypotheses of autism (Courchesne et al., 2007; De Ferrari and Moon, 2006; Ernst, 2016; Kalkman, 2012; Krumm et al., 2014; Mariani et al., 2015; Packer, 2016a, b). In particular, the advent of facile genome editing technologies such as CRISPR/Cas9 make it theoretically possible to study tens or hundreds of disease-associated mutations. However, the laborious nature of studying individual mutations in hPSCs, concerns about line-to-line variability and marked cellular heterogeneity remain major stumbling blocks that need to be overcome for the routine study of complex neuropsychiatric disease in hPSCs.
RESULTS
hPSC-based multiplex platform for functional interrogation of 30 isogenic lines in parallel
Here, we designed an hPSC-based multiplex platform in which multiple disease lines are pooled and differentiated into disease-relevant cell types (Figure 1A). The rationale for this approach, which was pioneered in cancer cell lines (Birsoy et al., 2014; Yu et al., 2016), is to simultaneously increase experimental throughput in both the number of genotypes and phenotypes that can be studied while also reducing inter-line assay variability. Phenotypes for each line are discerned by measuring changes in the relative allele frequency over time (e.g. growth phenotype (Birsoy et al., 2014)) or across physically separated phenotypic cell fractions (e.g. cell-state or drug-response (Yu et al., 2016)). Allele frequencies are measured using droplet digital PCR (ddPCR) due to its high sensitivity and reproducibility (Hindson et al., 2013). We reliably determined genome frequency of a given line after dilution of > 1:7,000 (Figure 1B). The multiplex platform was next validated with a physical separation-based assay in which a pool of 8 hPSC lines was segregated based on the level of CTNNB1 protein expression using fluorescence activated cell sorting (FACS) (Figure 1C, left panel). As expected, the CTNNB1-knockout line was enriched in the CTNNB1-low fraction, while all other 7 lines were enriched in the CTNNB1-high fraction (Figure 1C, right panel).
Figure 1. Design and validation of hPSC-based multiplex analysis platform.
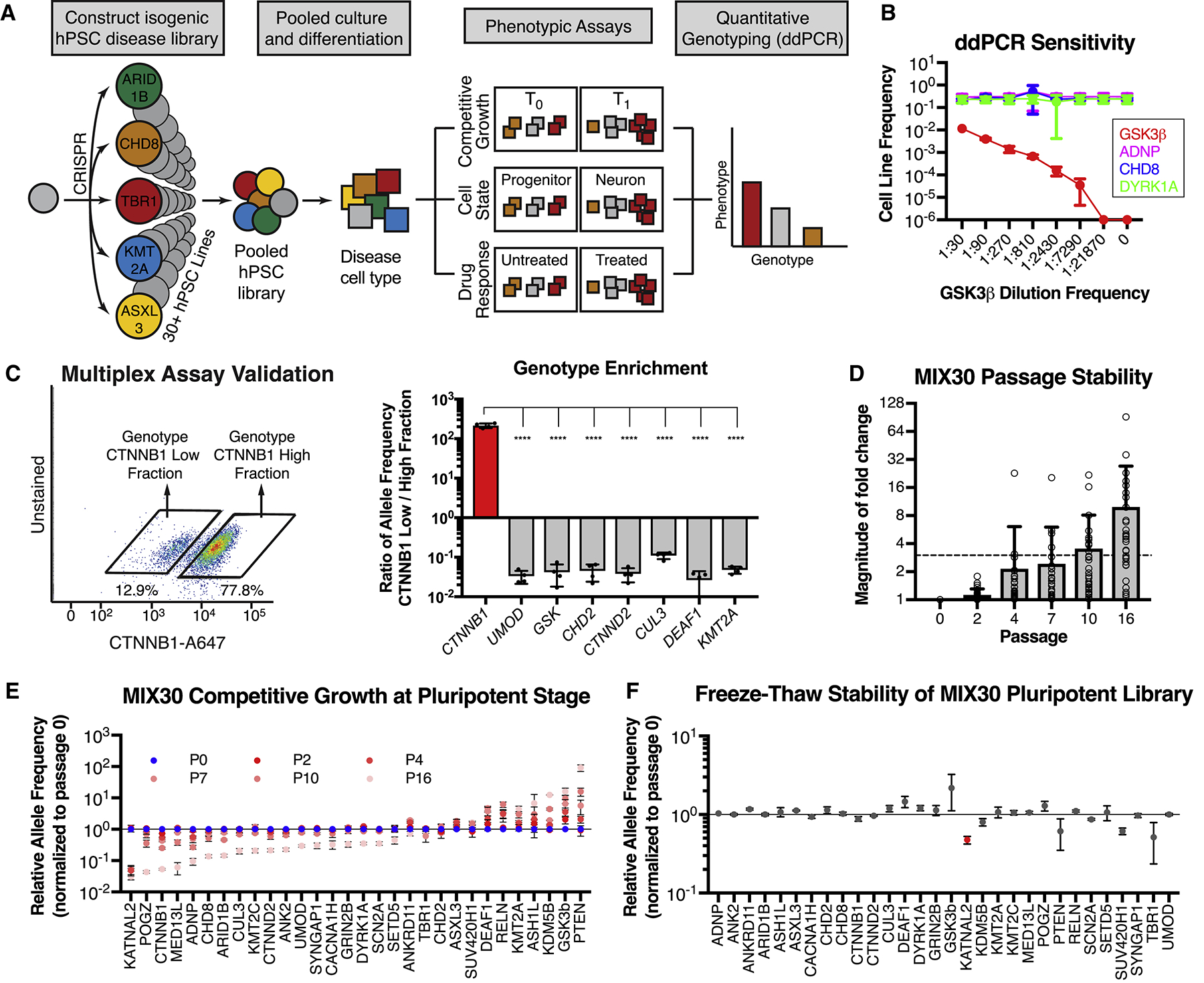
(A) Multiplex assay design. Individual disease-associated hPSC lines are generated using CRISPR/Cas9, pooled, and differentiated into a disease-relevant tissue. The pooled differentiation can be assayed for growth, cell-state, or drug-response phenotypes by determining relative allele frequencies for each line in comparison to an internal standard (negative control). For example, growth phenotypes are determined by measuring changes in allele frequency over time (T1 vs T0). Cell-state phenotypes are determined by measuring differences in allele frequency between physically separated populations (e.g. neurons versus progenitors separated via fluorescence activated cell sorting (FACS) or magnetic sorting (MACS), cells exposed (or not) to a given drug or cells separated based on migratory potential). Drug response phenotypes are determined by measuring differences in allele frequency between treated and untreated pools. Allele frequencies are measured using ddPCR. (B) ddPCR is a sensitive and accurate method to measure allele frequency, as determined by a serial dilution assay. ddPCR could detect the GSK3β line within a 4-line mixture until it reached a frequency between 1:7290 and 1:21870, using a read depth of ~15,000. n = 3 dilution series, mean ± S.D. (C) Validation of multiplex assay. A pool of 8 hPSC lines, including CTNNB1, UMOD, and GSK3β mutant lines, was separated into CTNNB1-low and CTNNB1-high expressing fractions using intracellular FACS with a CTNNB1 antibody. Each fraction was genotyped with ddPCR to calculate relative allele frequencies (allele frequency in sorted fraction / allele frequency in unsorted fraction). The CTNNB1 mutant line was enriched in the CTNNB1-low fraction while all other lines were depleted in this fraction. Graph depicts mean ± S.D. dots represent individual multiplex assays. One-way ANOVA followed by Dunnett’s test. **** p < 0.0001. n = 4 independent trials. (D) Average magnitude of allele frequency fold change in MIX30 library, normalized to passage 0, remained < 3x for 7 passages. Individual data points represent average fold change per line across MIX30 pools (n = 3). Mean ± S.D., dots represent individual lines within pooled cultures. (E) Competitive growth dynamics of all lines in MIX30 library at pluripotency stage. Lines with selective growth advantage (e.g. PTEN and GSK3β) appear to suppress growth of most other lines by passage 16. n = 3 MIX30 pools, mean ± S.E.M. (F) Allele frequencies measured one day after thawing are largely unaffected by freeze-thaw cycle. Graph depicts mean ± S.E.M, two-sided t-test using Benjamini-Hochberg method for multiple comparison. Red indicates line with significantly reduced allele frequency (FDR < 0.05). n = 3 MIX30 pools. ddPCR, droplet digital PCR. See also Figure S1 and Table S1.
A key feature of the multiplex platform is the ability to model complex, multi-gene disorders in a single experiment. Toward this end, we used CRISPR/Cas9 to construct an isogenic disease library of high-confidence autism mutations from a 46XY founder hPSC line (Figure S1A, Table S1). Frameshift mutations were introduced into the specific exons in which indels have been identified in patients (De Rubeis et al., 2014; Iossifov et al., 2014). This strategy was designed to phenocopy patient mutations, though it is possible that frameshifts at different places in the same exon can result in different phenotypes. Genes were selected based on a high confidence score in the SFARI Gene Database and further filtered for genes with early expression during cortical development in vivo (BrainSpan - https://www.brainspan.org/) and in vitro (Cortecon (van de Leemput et al., 2014)). The resulting library was comprised of 27 autism lines and was enriched for genes related to transcription/chromatin-remodeling (Parikshak et al., 2013). While all mutations selected for hPSC engineering were based on mimicking the specific mutations of patients with autistic traits, some of those patients may suffer from broader developmental defects that could also contribute to in vitro disease phenotypes. As a negative control line, we targeted the intron of a gene that is not expressed in neural tissue (UMOD). Two WNT/βcatenin-related positive controls (CTNNB1 and GSK3β) were also included.
We established three independent 30-line mixtures by pooling all lines at the pluripotent stage (MIX30A, B, C) (Figure S1B). Initial pools were generated from the same isogenic clones so that we could assess the amount of variability introduced by the pooling approach. MIX30 pools retained expression of pluripotency genes (Figure S1C, OCT4+/SSEA+ 89.7% ± 1.1%) and all individual lines were well represented at the pluripotent stage (Figure S1D). Spontaneous TP53 mutations (Merkle et al., 2017) were not detected by ddPCR at levels above background in any of the 12 lines examined (Figure S1E) or by Sanger sequencing of the TP53 DNA binding domain (data not shown).
With our gene targeting approach, for any one locus, we were largely able to only generate either monoallelic or biallelic frameshift indels, but not both. Thus, while autism patients harbor heterozygous mutations, our library consists of both monoallelic and biallelic mutant lines (Table S1). The efficiency of gene editing in bi-allelic lines was significantly higher than that in mono-allelic lines (p = 0.023) (Figure S1F). This could be due to differences in gRNA efficiency or to selection against bi-allelic mutations in monoallelic lines. The efficiency of gRNAs that induced mono-allelic vs. bi-allelic changes is not predicted to be different (Figure S1G). Furthermore, genes targeted in mono-allelic lines were not more likely to be essential genes or intolerant to gene mutations (Figure S1H,I).
Allele frequencies were roughly stable at the pluripotent stage, with less than 3-fold average change through seven passages (Figure 1D,E). Still, growth differences between lines at the pluripotent stage will eventually cause some lines to lose representation or unduly reduce assay sensitivity. Therefore, we implemented a cryopreservation strategy for maintaining sufficient stocks of the original pools, and limited cell expansion of the pools at pluripotent stage to fewer than five passages prior to initiating differentiation. Allele frequencies remained largely unaffected by freeze-thaw cycles (Figure 1F), allowing for expansion, quality-control, and long-term storage of each of the three pools. Stringent quality-control for pluripotent marker expression, genomic integrity and stable allele frequencies during pluripotency growth and freeze/thaw cycles is particularly essential when confirming suitability of control clones (e.g. UMOD) as those drive overall data quantification.
Derivation of human prefrontal cortex-like tissue from hPSCs
A second key feature of the multiplex platform is that it utilizes hPSCs, which can differentiate into nearly any human cell type, and thus offers great flexibility with respect to modeling genetic variants in a disease-appropriate cellular context. Since the PFC is a major locus of autism pathology (Willsey et al., 2013), we devised a strategy utilizing timed exposure to FGF8, a classic organizer of anterior cortical development in vivo (Fukuchi-Shimogori and Grove, 2001), to pattern cortical progenitors to a PFC-like identity (Figure 2A). By day 18, PFC cultures were composed of near homogenous neuroepithelial precursors of dorsal telencephalic identity (Figure 2B, Figure S2A) and expressed high levels of the frontal cortex marker SP8 but low levels of the occipital cortex (OCC) marker COUPTF1 (O’Leary et al., 2007) (Figure 2B, Figure S2B). The BrainSpan transcriptional atlas (https://www.brainspan.org) was used to define 14 gene transcripts that are differentially expressed between human fetal PFC and OCC at PCW8 (Figure S2C). qRT-PCR analysis revealed a high correlation of PFC versus OCC marker enrichment between in vivo and in vitro derived tissue (Figure 2C, R2 = 0.6191, p = 0.0008). This finding was confirmed by RNA-seq analysis which showed strong correlation between in vivo and in vitro expression when examining the top 200 PFC and OCC enriched genes (Figure 2D–G, Table S2).
Figure 2. Deriving human prefrontal cortex-like tissue from hPSCs.
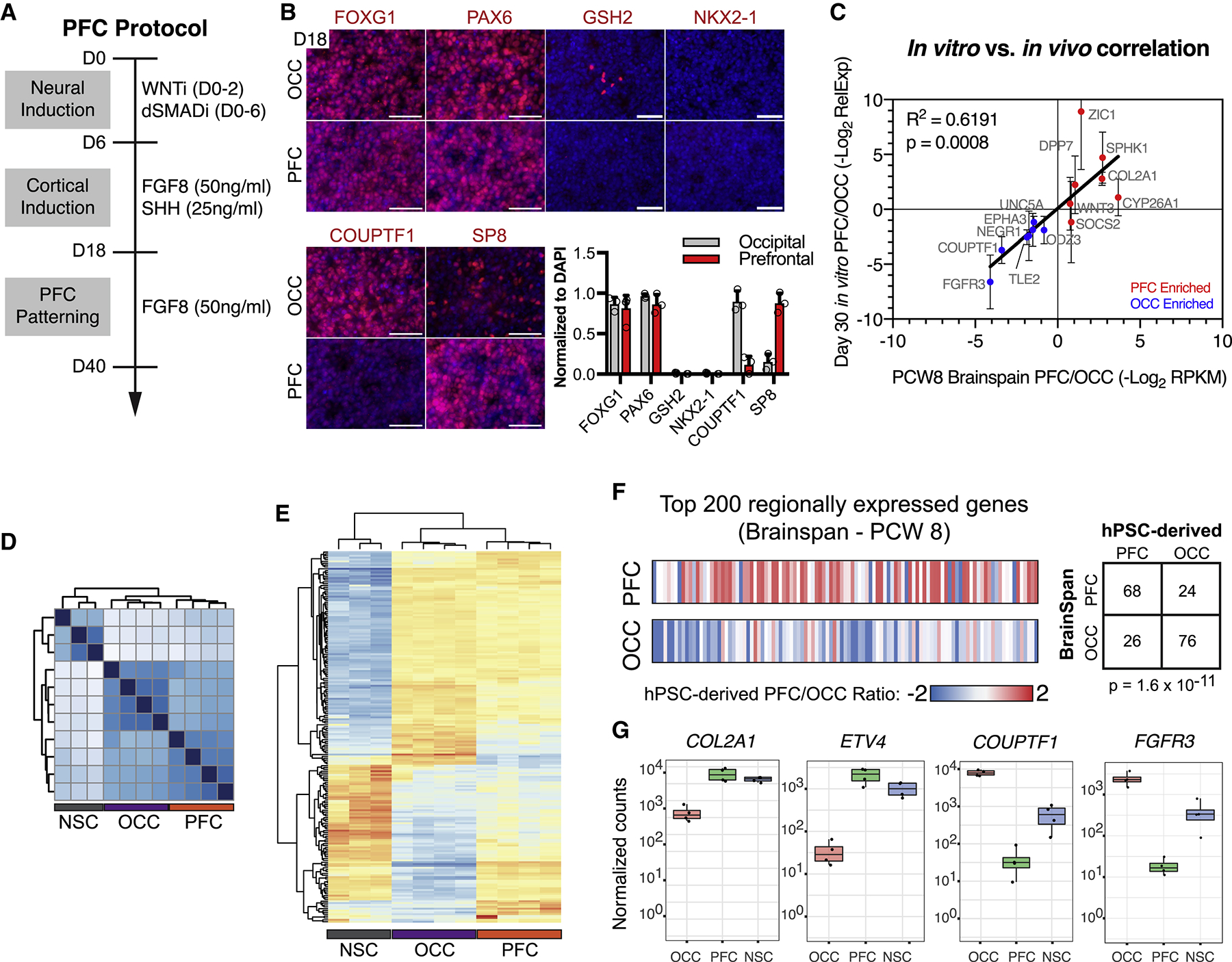
(A) Schematic illustration for hPSC-derived PFC cultures. (B) PFC and OCC cultures express appropriate regional markers, assessed by immunocytochemistry. FOXG1+/PAX6+/GSH2−/NKX2.1- indicates general cortical identity. FOXG1 (PFC, 81.6±16.1%; OCC, 86.5±8.5%), PAX6 (PFC, 86.5±11.2%; OCC, 96.9±3.3%), GSH2 (PFC, 0.0±0.0%; OCC, 1.0±0.9%) and NKX2.1 (PFC, 0.0±0.0%; OCC, 0.1±0.1%). SP8 and COUPTF1 are PFC and OCC markers, respectively. SP8 (PFC, 88.0±12.4%; OCC, 15.0±10.0%), COUPTF1 (PFC, 12.0±10.8%; OCC, 90.3±12.9%). Graph depicts mean ± S.D., dots represent individual differentiations. n = 3 differentiations. (C) Differential transcript expression of 14 genes between human fetal PFC and OCC, defined using BrainSpan transcriptional atlas (see methods), were highly correlated in vitro and in vivo, R2 = 0.6191, p = 0.0008; n = 5 differentiations, mean ± S.D. (D) Unsupervised clustering of RNA-seq data from NSC, OCC, and PFC cultures. (E) Unsupervised clustering of differentially expressed genes between NSC, OCC, and PFC cultures. (F) Ratio of hPSC-derived PFC to OCC gene expression for the top 200 genes more highly expressed in human fetal PFC versus OCC (top row) and the top 200 genes more highly expressed in human fetal OCC versus PFC (bottom row). Chi-squared p = 1.6×10−11, n = 4 PFC and OCC differentiations each. (G) Four examples of genes with differential gene expression from RNA-seq analysis. dSMADi, dual-SMAD inhibitors SB431542 and LDN193189; PCW, post-conception week; PFC, prefrontal cortex; OCC, occipital cortex; NSC, neural stem cell; WNTi, tankyrase inhibitor XAV939. Scale bars = 100 μm. See also Figure S2 and Table S2.
After establishing regional identity, we next sought to identify specific neurogenic cell-types within PFC cultures relevant to autism (Courchesne et al., 2011; Courchesne et al., 2007; Stoner et al., 2014). Neurons (DCX+) are born from multipotent cortical neural stem cells (SOX2+) or from proneural intermediate progenitor cells (IPCs, TBR2+) (Figure S2D). Immunocytochemistry and FACS-analysis revealed the presence of all three cell-types in our PFC culture system (Figure S2E,F). Characterization of neuronal subtype identity revealed the presence of primarily deep layer V and VI cortical projection neurons, with few callosal, upper-layer or subpallial cells (Figure S2G).
Do autism mutations perturb PFC neurogenesis?
To test the impact of autism mutations on PFC neurogenesis, we differentiated MIX30 pools into PFC-like tissue (Figure S3A–C) and used FACS to isolate bulk (All), neural stem cell (SOX2+), IPC (TBR2+), and neuronal (DCX+) fractions (Figure 3A, Figure S3D,E). A pilot study was performed in parallel using pools of 8 lines to determine if the multiplex approach could reliably detect neurogenic phenotypes. Indeed, the GSK3β positive control line among the pool of 8 lines showed an expected neurogenic phenotype (Figure S3C), in agreement with studies in the mouse (Ahn et al., 2014; Marcus et al., 1998). Examination of the MIX30 pools showed robust and comparable efficiencies of neural induction across all autism lines (Figure 3B). However, despite efficient cortical specification, remarkably, 13/27 autism lines showed abnormal PFC neurogenesis as assessed by significant changes in either neuronal production (DCX/SOX2 ratio) or neural stem cell enrichment (SOX2/All ratio) (Figure 3C, Table S3). Eight lines (ANKRD11, ASH1L, ASXL3, CUL3, DEAF1, KDM5B, KMT2C, and RELN) showed neural stem cell enrichment and decreased neuronal output. In addition, seven of these eight lines also exhibited increased IPC production (Figure 3D), suggesting a block in cell-cycle exit. An additional five lines (CHD8, DYRK1A, GRIN2B, KMT2A, and SUV420H1) showed neural stem cell depletion and increased neuronal output (Figure 3C). While there was no correlation between early neural growth and PFC growth across MIX30 lines (Figure 3E, p = 0.31), there was a highly significant negative correlation of hits between PFC neurogenesis and PFC growth (Figure 3F, r = −0.792, p = 0.0001). The high hit rate of mutations affecting PFC neurogenesis versus the lack of any hits affecting neural induction (Figure 3G) supports the specificity of autism mutations in perturbing PFC development. Overall, these data illustrate that autism mutations fall into distinct functional subgroups that coordinately dysregulate proliferation and neuronal differentiation, and thus nominate PFC neural precursor cells as a convergently dysregulated cell-type in autism. We designate mutations that inhibit PFC neurogenesis as Class 1, while mutations that lead to precocious PFC neurogenesis as Class 2 (Figure 3H). Interestingly, at least 5/8 Class 1 mutations are known to regulate Polycomb signaling, a pathway previously implicated in NSC self-renewal and neurogenic competency (Corley and Kroll, 2015; Testa, 2011).
Figure 3. Multiplex analysis reveals functional subgroups of autism-associated mutations that dysregulate PFC neurogenesis.
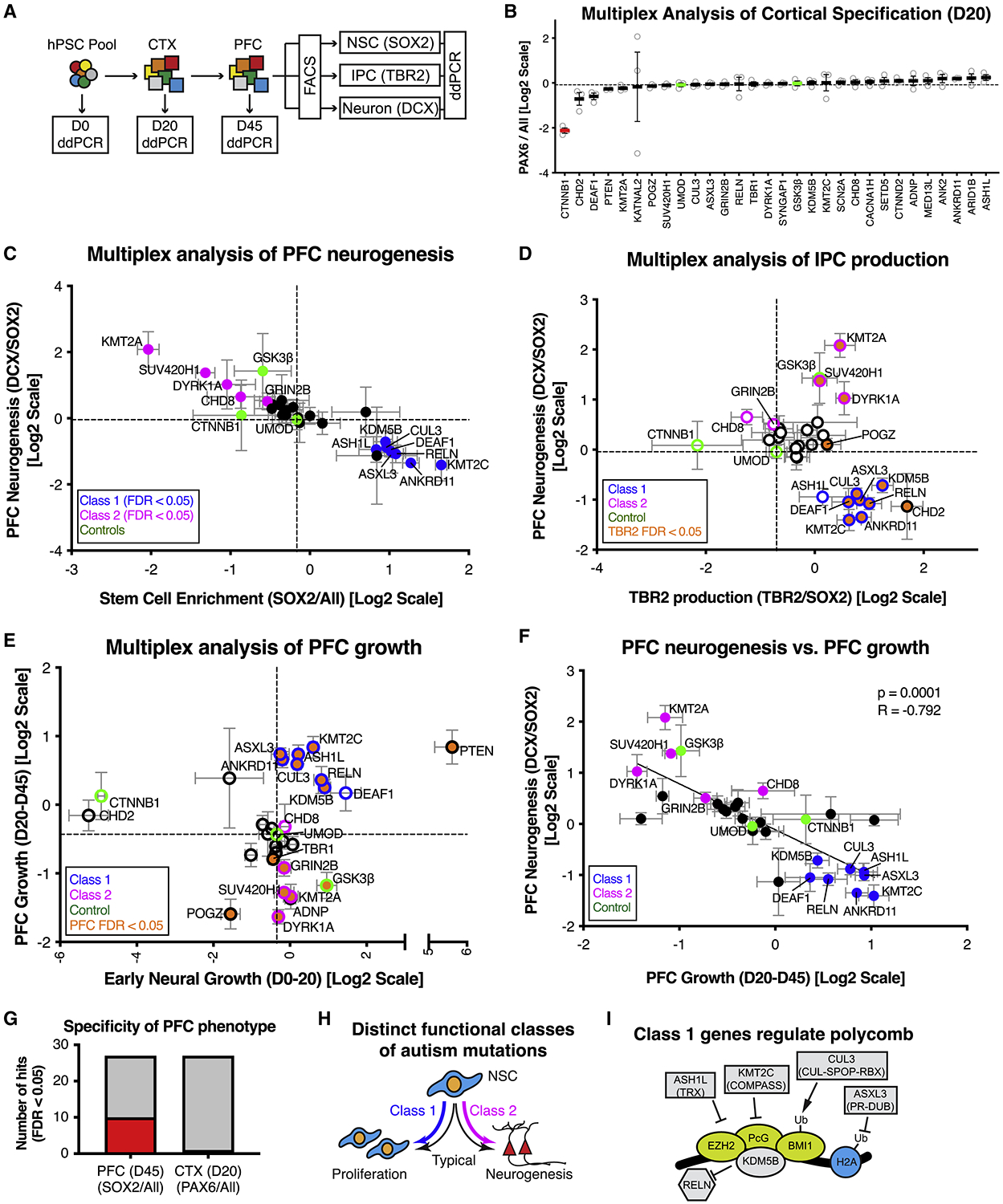
(A) Schematic illustration of multiplex strategy to test autism mutations for alterations in PFC growth and neurogenesis. Growth phenotypes are determined by measuring changes in allele frequency from D0 to D20 (early neural growth), and D20 and D45 (PFC growth). PFC neurogenesis phenotypes are determined by measuring changes in allele frequency between NSC (SOX2), IPC (TBR2), and Neuron (DCX) sorted fractions. (B) Multiplex assay testing each autism line for cortical specification. Average relative cell line enrichment in PAX6+ fraction, relative to an unsorted day 20 MIX30 fraction (ANOVA p < 0.0001). Red bars indicate cell lines with significant increases or decreases in enrichment score compared to UMOD (FDR < 0.05). Graph depicts mean ± S.E.M., dots represent individual differentiations. FDR calculated using Welch ANOVA with post-hoc comparison to UMOD, p values corrected using Benjamini, Krieger, Yekutieli method. n = 3 differentiations. (C) Scatter plot of multiplex neurogenesis assay showing changes in neuronal production (DCX/SOX2 ratio) and stem cell enrichment (SOX2/All ratio). Class 1 mutations (8/27, blue: FDR < 0.05) exhibit decreased neuronal production or increased stem cell enrichment. Class 2 mutations (5/27, magenta: FDR <0.05) exhibit increased neuronal production or decreased stem cell enrichment. n = 5 differentiations from three MIX30 pools. (D) Scatter plot of multiplex neurogenesis assay showing changes in IPC production (TBR2/SOX2 ratio) correlated with PFC neurogenesis phenotypes (DCX/SOX2 ratio), normalized to a negative control (UMOD). 7/8 Class 1 mutations (blue edge) exhibit increased IPC production (orange, FDR<0.05), while Class 2 (magenta edge) mutations are variable in IPC production phenotypes. n ≥ 3 differentiations from at least two MIX30 pools. (E) Scatter plot of multiplex assay showing competitive growth phenotypes during early neural (D20/D0 ratio) and PFC growth phases (D45/D20 ratio). 7/8 Class 1 mutations (blue edge) show increased PFC growth (orange, FDR<0.05) while 4/5 Class 2 mutations (magenta edge) exhibit decreased PFC growth (orange, FDR<0.05). n = 5 differentiations from three MIX30 pools. (F) Significant negative correlation between PFC neurogenesis and PFC growth parameters (r = −0.792, p=0.0001). For all scatter plots, FDR calculated using Welch ANOVA with post-hoc Benjamini, Krieger, Yekutieli method for multiple comparisons. Dotted lines demarcate negative control UMOD. (G) 10/27 cell lines exhibited a stem cell enrichment phenotype (SOX2/All) during PFC development at day 45, while 0/27 lines exhibited a neural induction phenotype (PAX6/All) during an earlier cortical development phase at day 20. Red and grey bars indicate number of cell lines with positive and negative phenotypes respectively. (H) Summary of class-specific PFC development phenotypes. NSC behavior is characterized by a balance between proliferation and neurogenesis (black arrows). Autism mutations skew this balance toward proliferation (Class 1, blue arrow) or neurogenesis (Class 2, magenta arrow). (I) Five of eight class 1 genes are known regulators of polycomb signaling. ASH1L is a trithorax group protein (Gregory et al., 2007), and KMT2C is a member of the COMPASS complex (Piunti and Shilatifard, 2016). ASXL3 is part of the Polycomb repressive deubiquitinase complex (Srivastava et al., 2016). CUL3 regulates polycomb through ubiquitination (Hernandez-Munoz et al., 2005). KDM5B occupies over 50% of polycomb sites (Schmitz et al., 2011). In addition, DEAF1 mutant mice have a homeotic transformation phenotype (Hahm et al., 2004). All error bars are mean ± S.E.M. Internal standards are colored green. CTX, cerebral cortex; D, day; ddPCR, droplet digital PCR; FACS, fluorescent activated cell sorting; hPSC; human pluripotent stem cell; NSC, neural stem cell; PFC, prefrontal cortex; IPC, intermediate progenitor cell. See also Figure S3 and Table S3.
Importantly, the PFC neurogenesis phenotypes did not correlate with other biologically unrelated assays including hPSC growth (Figure S3G), early neural growth (Figure S3H), or cortical patterning (Figure S3I), further demonstrating specificity of the observed results. Interestingly, hPSC growth did correlate with early neural, but not PFC growth, suggesting that early neural and PFC lineages are driven by distinct developmental programs (Figure S3J,K).
Validation of multiplex neurogenesis assays
To validate these findings, we performed a second set of multiplex experiments using a new hPSC pool (MIX32) composed of 13 independently established clones, 13 matched original clones as well as additional controls (Figure 4A). We aimed to replicate the predominant phenotype of the 13 original clones. PFC neurogenesis phenotypes from the original MIX30 pool were highly correlated to phenotypes of the independent clones in the new MIX32 validation pool (r = 0.797, p =0.0006) (Figure 4B), confirming that the observed phenotypes are largely due to the effects of introduced CRISPR mutations. Qualitatively, 9/13 phenotypes replicated using an FDR cutoff of 0.05 for MIX30 phenotypes. However, 11/13 lines replicated when the FDR cutoff of MIX30 phenotypes was increased to 0.01 (Figure 4C), as would be expected from increasing statistical stringency. Further testing of the CHD8 independent clone, which did not replicate, using a single genotype assay ultimately did show an increase in PFC neurogenesis thus matching the original phenotype (Figure 4D). Pool-specific effects, with partial suppression of phenotypes in either the MIX30 or MIX32 pool were observed for ADNP, CHD8, and KDM5B lines which may be due to pool-specific, cell non-autonomous effects or due to effects related to assay sensitivity.
Figure 4. Validation of multiplex neurogenesis assay.
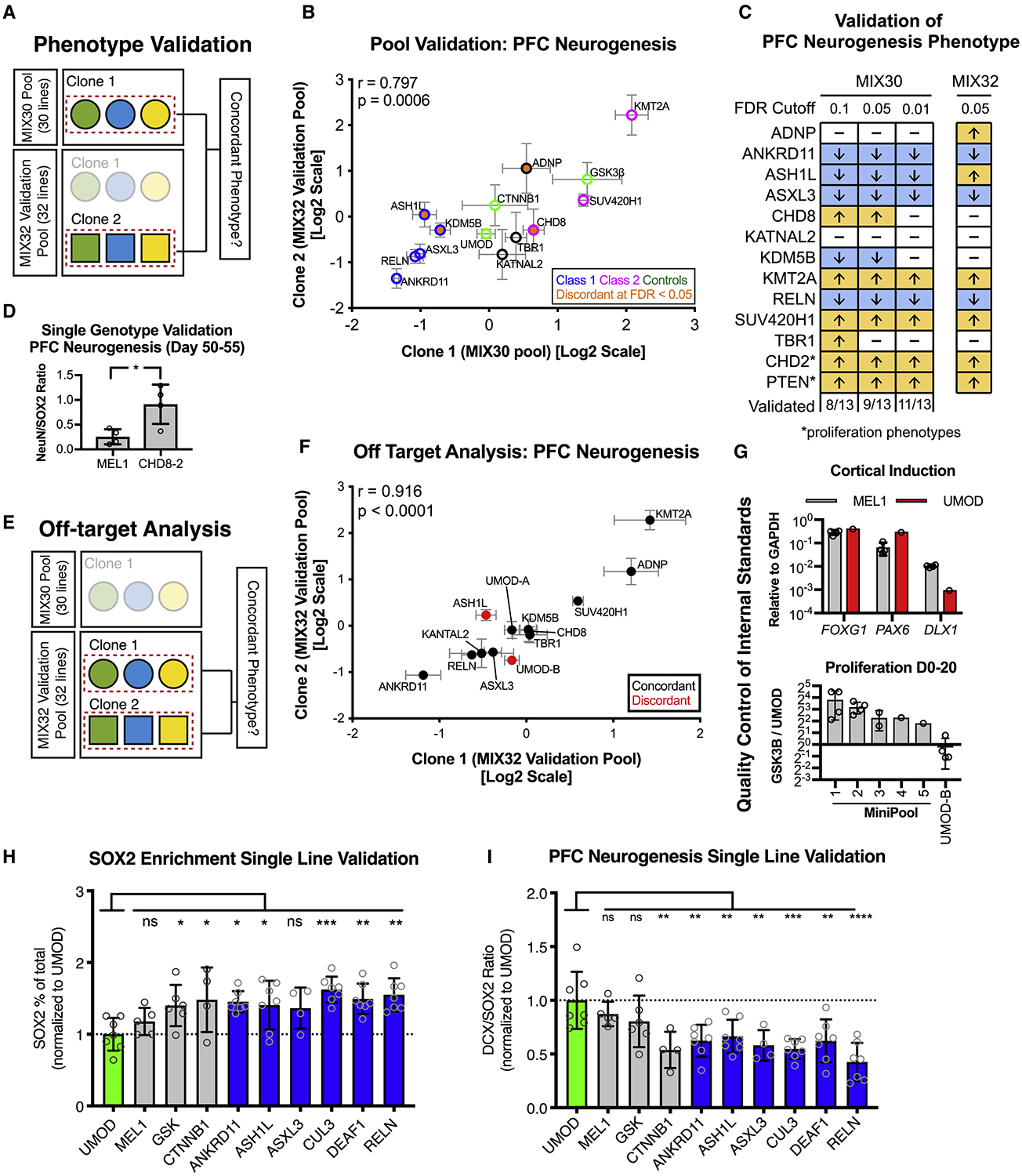
(A) Strategy for phenotypic validation using the MIX32 validation pool. The validation pool contains 13 pairs of autism clones, the first clone of each pair was from the original MIX30 pool, while the second was an independent clone. 9 independent clones were generated with distinct guide RNAs. A pool-based validation was performed in which the phenotype from the original MIX30 pool (clone 1) was compared to the phenotype of the independent clone (clone 2) from the validation pool. Pairs that showed discordance could then be tested in single line assays. (B) Scatter plot showing comparison of phenotypes between clone 1 (MIX30 pool) and clone 2 (validation pool). Overall correlation r = 0.797, p = 0.0006. Graph depicts mean ± S.E.M. Test pool n = 5 differentiations from three MIX30 pools, Validation pool n = 4 differentiations from three MIX32 pools. (C) Schematic comparing PFC neurogenesis phenotypes from the original MIX30 pool to the validation pool. Proliferation phenotypes are compared for CHD2 and PTEN. 9/13 clones validated using an FDR cut-off of 0.05. Increasing MIX30 FDR stringency to 0.01 increases validation rate to 11/13. Arrows represent direction of phenotype. FDR calculated using Welch ANOVA with post-hoc comparisons to UMOD, p-values corrected for multiple comparisons using Benjamini, Krieger, Yekutieli method. Test pool n = 5 differentiations from three MIX30 pools, Validation pool n = 4 differentiations from three MIX32 pools. (D) Testing of CHD8 clone 2 in a single genotype PFC differentiation shows increased ratio of NeuN/SOX2 compared to MEL1 control at day 45. Graph depicts mean±S.D., dots represent individual differentiations. Two-sided student t-test. * p = 0.022. MEL1 n = 4 differentiations, CHD8-2 n = 4 differentiations. (E) Strategy to quantify off-target rate in the MIX30 pool. A validation pool was generated that contains 15 pairs of clones, 13 autism-associated and 2 control pairs. One clone of each pair was from the original MIX30 pool, while the second clone of each pair is an independent clone. 9 independent clones were generated with distinct guide RNAs, 6 independent clones were generated with the same guide RNA. Off-target effects were determined by assessing phenotypic concordance using the multiplex PFC neurogenesis assay (see Fig. 2a). (F) Scatter plot showing comparison of phenotypes between clone pairs in validation pool. Overall correlation r = 0.916, p < 0.0001. (G) Description of quality control to ensure that internal standard is suitable controls. Upper panel, cortical patterning of the UMOD is comparable to that of MEL1. MEL1 n = 3 differentiation, UMOD n = 1 differentiation. Lower panel, proliferation of the UMOD line was compared to the positive control line GSK3β, showing an expected lower proliferation rate than GSK3β across 5 mini-pools. As an example of the importance of quality control measures, the UMOD-B clone from the MIX32 pool, which showed off-target effect, would not have passed quality control as it did not show a lower proliferation rate than GSK3β. Minipool 1, n = 4; minipool 2, n = 4; minipool 3, n = 2; minipool 4, n = 1; minipool 5, n =1; UMOD-B, n = 4 differentiations. (H) Percent of SOX2 cells per differentiation, normalized to UMOD. 5/6 class 1 genes (blue) showed the expected increase in SOX2 percentage, while MEL1 was similar to UMOD. ANOVA p = 0.0025\. (I) DCX/SOX2 ratio for each line, normalized to UMOD. 6/6 class 1 genes (blue) showed the expected increase in DCX/SOX2 ratio, while MEL1 was similar to UMOD. ANOVA p < 0.0001. Graphs depict mean ± S.D., dots represent individual differentiations. Comparisons made using one-way ANOVA with post-hoc comparisons to UMOD, corrected using Dunnett test. Samples scaled to group mean. For all panels, UMOD, n = 7; MEL1, n = 5; GSK3β, n = 6; CTNNB1, n = 4; ANKRD11, n = 7; ASH1L, n = 7; ASXL3, n = 4; CUL3, n = 7; DEAF1, n = 7; RELN, n =7 differentiations. FACS, fluorescent activated cell sorting. See also Figure S4.
To measure the actual off-target rate of multiplex neurogenesis phenotypes, we designed another validation assay using the MIX32 pool. Since the MIX32 pool contains pairs of independently generated mutant lines, established using either identical or independent gRNAs, we could compare mutant pairs within the same pool to remove any pool-specific effect and isolate off-target effects in clones (Figure 4E). Among the 15 pairs of lines in MIX32, the observed validation rate was 8/9 for pairs targeted using distinct gRNAs and 5/6 for pairs targeted with the same gRNA. Across all clones, the correlation rate for phenotypes was very high (p < 0.0001, r=0.916) (Figure 4F). The two discordant clone pairs could be due to off-target effects, culture-induced genetic mutations or simply due to limitations in the sensitivity of the pooling approach. Importantly, this analysis revealed an off-target effect in a third UMOD control line (UMOD-B). Unlike the original UMOD clone in MIX30, the UMOD-B clone was not subjected to rigorous quality control prior to inclusion in the study. For example, whereas the original UMOD clone consistently proliferated at a lower rate than the GSK3β line across five pools, the UMOD-B clone proliferated at the same rate as the GSK3β line (Figure 4G). This observation highlights the importance of stringent quality control in all hPSC lines but particularly for those serving as internal standards.
PFC neurogenesis phenotypes were also validated using single genotype differentiations for six Class 1 lines (Figure 4H,I, Figure S4A,B). Furthermore, studies of neurogenesis in animal models confirm the overall findings for many of the genes including ANKRD11 (Gallagher et al., 2015), ARID1B (Jung et al., 2017), CHD2 (Kim et al., 2018; Shen et al., 2015), CHD8 (Durak et al., 2016; Gompers et al., 2017), DYRK1A (Arranz et al., 2019; Kurabayashi and Sanada, 2013), KMT2A (Huang et al., 2015), and RELN (Deguchi et al., 2003; Hammond et al., 2010; Johnson et al., 2015; Lakoma et al., 2011; Nowakowski et al., 2017). A final validation question is how pool size and composition impact gene-specific phenotypes. Correlation of MIX30 neurogenesis phenotypes with data from pilot studies that used 8-line mixtures (MIX8) demonstrated overall reproducibility and stability of PFC neurogenesis phenotypes to changes in pool size (Figure S4C–F).
Do autism mutations dysregulate WNT/βcatenin signaling?
In addition to probing developmental phenotypes related to cell fate specification and proliferation, our multiplex platform also allows us to evaluate the cell-type specific activity of key molecular pathways. The WNT/βcatenin pathway is a critical regulator of stem cell proliferation and neurogenesis during cortical development (Chenn, 2008; Hirabayashi et al., 2004; Munji et al., 2011) and is a central node among a network of autism-related genes (Gilman et al., 2011; Krumm et al., 2014; Packer, 2016a). We therefore tested autism lines for the ability to respond to WNT/βcatenin signaling by treating day 35 MIX30 PFC cultures with the GSK3α/β inhibitor CHIR99021 (3μM) for 10 days, using stem cell proliferation as an initial readout of WNT activity (Kim et al., 2009) (Figure 5A). Strikingly, 7/8 Class 1 mutations were hyporesponsive to CHIR-induced stem cell proliferation (Figure 5B). WNT hyporesponsiveness phenotypes were validated in single genotype assays demonstrating that 5/5 Class 1 lines have a blunted proliferation and cell-cycle re-entry response to CHIR99021 stimulation (Figure S5). Furthermore, 3/5 lines exhibited higher baseline proliferation than controls, which could account for their hyporesponsive phenotype. One possible explanation is that Class 1 gene mutations dysregulate transcriptional targets of WNT signaling that control basal neural stem cell proliferation and differentiation. It is interesting that inhibition of both GSK3 isoforms with CHIR99021 enhanced stem cell proliferation while GSK3β specific knock-out promoted neurogenesis. In accordance with this observation, previous studies have shown that knockout of both GSK3 isoforms is necessary to observe a strong cortical progenitor proliferation phenotype (Kim et al., 2009), while specific inhibition of GSK3β can promote cortical neurogenesis (Ahn et al., 2014).
Figure 5. Class-specific dysregulation of WNT signaling.
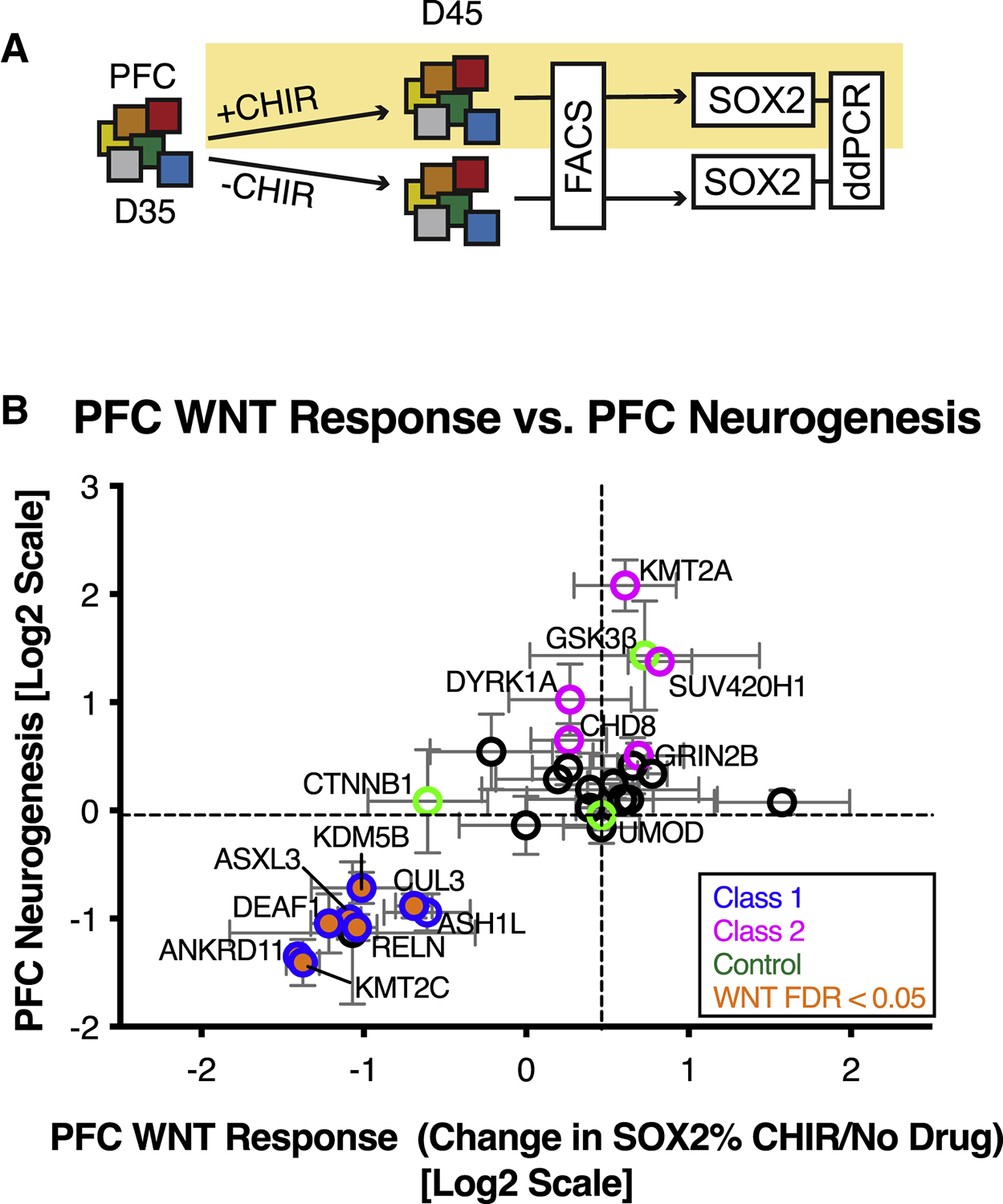
(A) Schematic illustration of multiplex strategy to test autism mutations for WNT/βcatenin response during PFC growth. D35 Pooled PFC cultures are treated with the CHIR99021 (GSK3 inhibitor, 3μM) for 10 days and compared to untreated cultures. Stem cell proliferation is used as a read-out of WNT activity. (B) Correlation of PFC WNT response with PFC neurogenesis phenotype. 7/8 Class 1 mutations (blue edge) are hyporesponsive to WNT signaling (orange, FDR < 0.05). FDR calculated using Welch ANOVA with post-hoc comparisons to UMOD, p-values corrected for multiple comparisons using Benjamini, Krieger, Yekutieli method n = 4 differentiations from three MIX30 pools. See also Figure S5 and Table S3.
Do autism mutations dysregulate neural crest development?
Lineage specificity of the WNT-response phenotype was assessed by differentiating the MIX30 library to CD49d+ cranial neural crest (CNC) precursors (Fattahi et al., 2016), whose specification is dependent on WNT/βcatenin activity (Dorsky et al., 1998) (Figure 6A, S6A–C). Class 1 mutations impaired CNC specification (Figure 6B), further supporting class-specific WNT dysregulation.
Figure 6. Class-specific dysregulation of neural crest development.
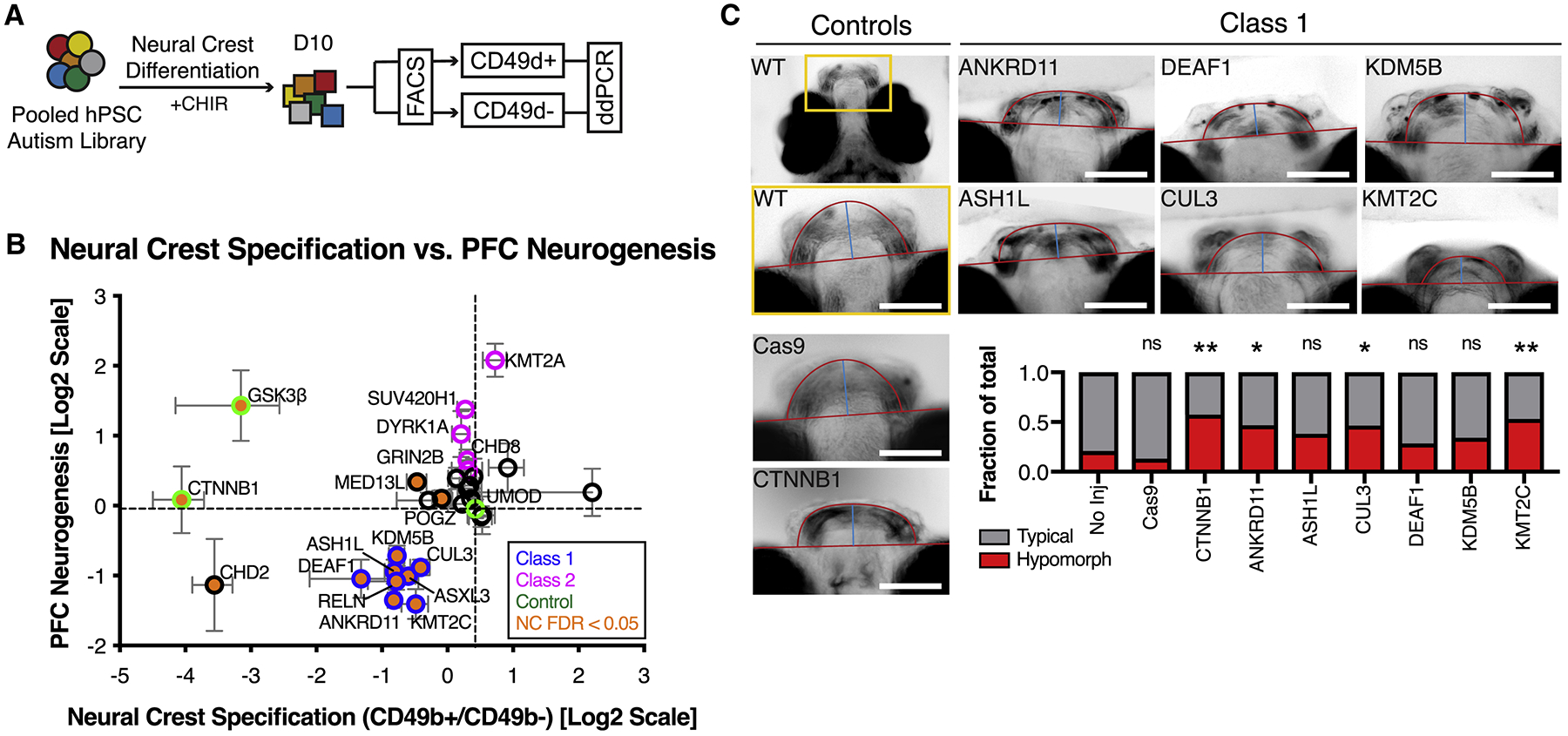
(A) Schematic illustration of multiplex strategy to test autism mutations for WNT/βcatenin response during neural crest development. MIX30 hPSC pools are differentiated toward neural crest for 10 days using an established WNT-dependent protocol. Allele frequencies are then compared between cranial neural crest positive (CD49d+) and negative (CD49d-) sorted fractions. (B) Correlation of neural crest specification with PFC neurogenesis phenotypes reveals that 8/8 Class 1 mutations (blue edge) impair cranial neural crest specification (orange, FDR < 0.05). All error bars are mean ± S.E.M. FDR calculated using Welch ANOVA with post-hoc comparisons to UMOD, p-values corrected for multiple comparisons using Benjamini, Krieger, Yekutieli method. Internal standards are colored green. (C) Analysis of zebrafish jaw development in F0 mosaic loss-of-function animals imaged ventrally at 7 dpf. Top left image depicts area of high magnification. Cas9 alone and CTNNB1 gRNA injections serve as controls. Class 1 mutations exhibit hypomorphic jaw phenotypes that resemble those of CTNNB1 mutants (CTNNB1 p = 0.0064, ANKRD11 p = 0.0488, CUL3 = 0.0488, KMT2C = 0.0088). p values calculated using Fisher’s exact test corrected for multiple comparisons using Benjamini-Hochberg method. No injection n = 43, Cas9 alone n = 23, CTNNB1 n = 40, ANKRD11 n = 32, ASH1L n = 68, CUL3 n = 43, DEAF1 n = 28, KDM5B n = 41, KMT2C n = 47 fish. All injections performed on at least 2 clutches. Dpf, days post fertilization. Scale bars, 100μm. See also Figure S6 and Table S3.
The observed WNT-dependent defects in CNC development could explain the high rate of facial dysmorphism in some autism patients (Cordero et al., 2011; Miles et al., 2008). In fact, facial dysmorphism has been reported in patients for 7 out of 8 Class 1 genes (Balasubramanian et al., 2017; Faundes et al., 2018; Koemans et al., 2017; Ockeloen et al., 2015; Okamoto et al., 2017; Redin et al., 2017; Vulto-van Silfhout et al., 2014). To explore these clinical observations and to further validate our in vitro multiplex data, we generated mosaic F0 loss-of-function zebrafish of Class 1 genes and assessed lower jaw development, a parameter known to critically rely on WNT-dependent CNC function (Curtin et al., 2011; Dougherty et al., 2013; Kamel et al., 2013; Rochard et al., 2016). ANKRD11, CUL3, and KMT2C mutants significantly increased the fraction of jaw hypomorphs, while ASH1L, DEAF1, and KDM5B mutants showed similar but statistically non-significant trends (Figure 6C, S6D). Together these data suggest that Class 1 genes exhibit dysregulated WNT signaling across multiple developmental lineages (Figure S6E).
Clinical correlates of distinct functional subgroups of autism-associated mutations
We next asked whether functional classes of autism-associated mutations defined by our multiplex platform could define cohorts of autism patients with distinct clinical profiles. To define cohorts in an unbiased manner that most accurately represented overall multiplex data, we performed unsupervised hierarchical clustering of all lines across six phenotypic assays related to PFC development and WNT signaling. This analysis revealed two major functional groups (Figure 7A). Cluster A contained all Class 1 mutations and the CTNNB1 control, further supporting a WNT-hyporesponsive phenotype. Cluster B included all Class 2 mutations and the GSK3β control, perhaps suggesting a contrasting relationship to WNT signaling. Based on this analysis, we organized probands from the Simons Simplex Collection (SSC) into 4 cohorts: (1) Cluster A (A, n = 17), (2) Cluster B (B, n = 55), (3) de novo control (DN, n = 263), (4) idiopathic control (Idiopathic, n = 482) (Figure 7B). Cohorts were similar in their demographic profiles (Figure S7A–D), autism severity (Figure S7E) and average head circumference (Figure S7F). Control groups were IQ matched to Cohorts A and B (Figure S7G).
Figure 7. Distinct functional subgroups of autism mutations correlate with differing clinical profiles of language development.
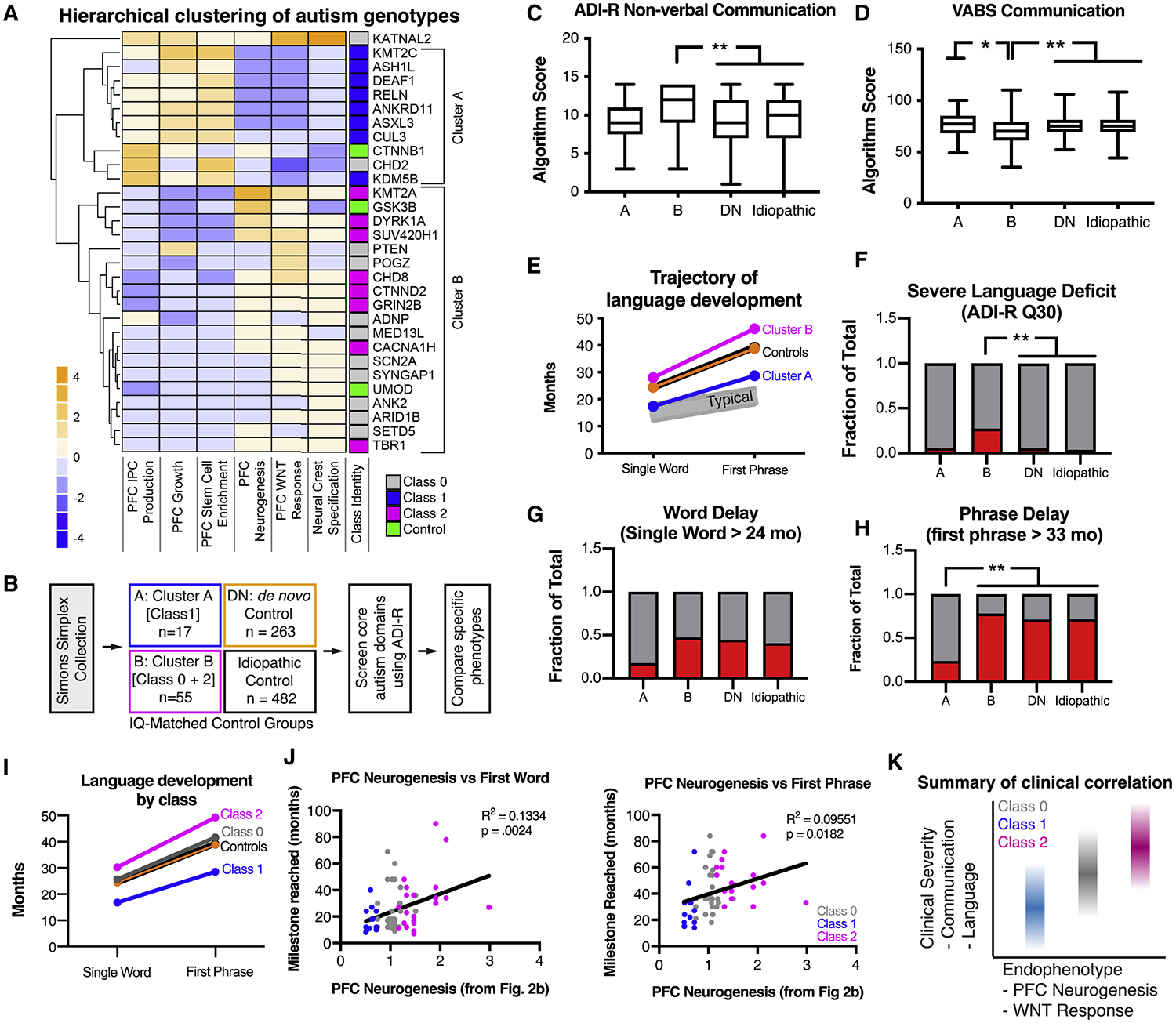
(A) Unbiased hierarchical clustering of phenotypic data from six multiplex assays reveals two major functional groups of autism mutations from the MIX30 library. Class 1 mutations fall into cluster A, while Class 2 mutations fall into cluster B. (B) Strategy for clinical phenotype analysis. Probands from the Simons Simplex Collection (SSC) were segregated into cohorts based on the presence of a de novo mutation in genes from Cluster A (17 patients) or Cluster B (55 patients). Control groups were generated by defining IQ-matched patient cohorts with any other de novo mutation not in the MIX30 library (de novo control, 263 patients) or patients without any known de novo mutation (idiopathic control, 482 patients). First, the ADI-R was used to screen for differences in core autism behavioral domains (communication, social behavior, restricted and repetitive behaviors). Specific behavioral phenotypes were further investigated based on the initial screening. (C), Cluster B patients exhibit increased severity in ADI-R non-verbal communication scores (Kruskal-Wallis p = 0.0072, corrected for multiple comparison of each behavioral domain using Holm-Sidak method; post-hoc Dunn’s multiple comparison test, B vs. DN p = 0.0025; B vs. Idiopathic corrected p = 0.0008). (D) Cluster B patients are on average reduced in the communication domain of the Vineland adaptive behavior scale (ANOVA p = 0.0004; Tukey’s multiple comparison’s test, A vs. B p = 0.037, B vs. DN p = 0.0027, B vs. Idiopathic p = 0.0002). (E) Average trajectories of language development. Control cohorts speak single words at ~24 months (DN=24.4 mo; idiopathic=24.4 mo) and speak their first phrases at ~39 months (other=38.8 mo; idiopathic=39.3 mo). Cluster B patients speak words at 28.02 months and first phrases at 46.1 months. Cluster A patients speak single words at 17.4 months and first phrases at 28.7 months. Typical language development is depicted in gray. (F) Cluster B cohort contains an increased fraction of patients with severe language deficit when compared to control cohorts (chi-squared p < 0.0001; Fisher’s exact tests with Holm-Sidak correction for multiple comparison B vs. DN p = 0.0018, B vs. Idiopathic p = 0.0018). (G) No significant difference in word delay across groups (chi-squared p = 0.114). (H) Cluster A contains a decreased fraction of patients with phrase delay compared to other cohorts (Chi-squared p = 0.0002; Fisher’s exact tests with Holm-Sidak correction for multiple comparison A vs. B p = 0.0018, A vs. DN p = 0.0018, A vs. idiopathic p = 0.0018). (I) Class 0 exhibits an intermediate phenotype of average language development, between Class 1 and 2. (J) Correlation of language development with in vitro PFC neurogenesis. PFC neurogenesis values (Fig. 2b, DCX/SOX2 ratio) were assigned to probands using proband genotypes. Both first word (R2 = 0.1334, p = 0.0024) and first phrases milestone (R2 = 0.09551, p = 0.0182) showed significant positive correlations with the extent of PFC neurogenesis. Each dot represents one proband. (K) Summary of phenotypic segregation of autism patients defined using hPSC-based multiplex analysis platform. ASD, autism spectrum disorder. ADOS, Autism Diagnostic Observation Schedule. ADI-R, Autism Diagnostic Interview-Revised. See also Figure S7.
Autism Diagnostic Interview-Revised (ADI-R) scores were used to first assess major autism behavioral domains and revealed that Cluster B exhibited an increased severity in communication deficits (Figure 7C, Figure S7H–J), and was corroborated by the Vineland adaptive behavioral scale (Figure 7D). We next assessed language development, a major dimension of communication behavior. Interestingly, Cluster A patients on average reached language milestones earlier than control patients, while Cluster B patients were further delayed than controls (Figure 7E–H). These findings are corroborated by assessments of large CHD8 (Bernier et al., 2014) and DYRK1A (Earl et al., 2017) cohorts (Cluster B, Class 2), which demonstrated frequent language abnormalities.
When we further subdivided cluster B by Class (i.e. by PFC neurogenesis phenotype), we noticed that Class 0 patients tended to have an intermediate language phenotype between that of Class 1 and 2 (Figure 7I), mirroring the pattern of PFC neurogenesis phenotypes. Correlating language data with PFC neurogenesis (Figure 3B), using patient genotypes, revealed a positive association with the extent of neurogenesis and the severity of language acquisition phenotype (Figure 7J). Surprisingly, delayed PFC neurogenesis may have a protective effect among autism patients. While we do not imply a causal link between PFC neurogenesis and language acquisition, it will be interesting to explore whether these phenotypes extend to other hPSC-derived lineages potentially involved in language acquisition, and whether those findings replicate in other autism populations.
DISCUSSION
Here we demonstrate that a pooled approach to studying hPSCs is feasible, reproducible, and allows for phenotypic screening of complex genetic disorders. This approach allows for the hypothesis-driven testing of multiple disease phenotypes across a large number of disease-associated mutations, and thus can help to organize the genetic heterogeneity of complex disorders into biological and clinically meaningful subgroups.
In this study, 27 isogenic hPSC lines harboring de novo autism associated mutations were pooled and screened for early neurodevelopmental phenotypes. Dysregulation of PFC neural stem cells emerged as a convergence point among the tested lines; class 1 mutations led to a decrease in PFC neurogenesis while class 2 mutations enhanced PFC neurogenesis. In addition, class 1 mutations were uniformly associated with an abnormal cellular response to stimulation of the WNT/βcatenin pathway. Finally, the observed in vitro phenotypes correlated with the clinical severity of language delay in a large cohort of genetically defined patients with mutations in the corresponding genes (Figure 7K). Together, these data support the notion that diverse genetic mutations associated with autism converge on a smaller number of molecular and cellular pathways.
In the future, it will be interesting to explore the mechanistic basis of the phenotypic convergence within genotype classes. Class 1 genes were hyporesponsive to CHIR99021-induced neural stem cell proliferation and neural crest specification, suggesting dysregulation of WNT/βcatenin signaling. One possibility is that Class 1 mutations globally dysregulate βcatenin activity and affect canonical targets of the pathway. However, another possibility is that Class 1 mutations perturb WNT signaling in a locus specific manner. The polycomb pathway regulates WNT-dependent transcriptional responses to proneural gene loci (Hirabayashi et al., 2009). Interestingly, at least 5/8 Class 1 genes are known regulators of Polycomb activity (Figure 3I). Thus, one model arising from our data is that Class 1 genes regulate WNT/βcatenin transcriptional activity specifically at polycomb-dependent proneural gene loci. Furthermore, at least 2/5 Class 2 genes, KMT2A and CHD8, have been shown to also affect Polycomb signaling. However, out data do not rule out the possibility that class 1 mutations affect a much broader set of developmental pathways beyond WNT/βcatenin.
In addition to studying isogenic hPSCs, the multiplex platform could be adapted to patient-specific autism iPSCs (using natural genetic variation as readout for each pooled genotype) to explore polygenic risk or to assess the impact of genetic background, as even highly penetrant autism mutations can lead to distinct phenotypes in different patients (Bernier et al., 2014). Similarly, this approach can be easily adapted to test the impact of autism-related genes in other hPSC-derived lineages of potential relevance to the study of autism such as striatal lineages, cortical interneurons, cerebellar neurons, amygdala or in non-neuronal lineages such as astrocytes or microglia. More broadly, our technology bridges a widening gap between the rapid accumulation of genetic information and the limited ability to assess functional impact in disease-relevant human cell types, thus elucidating the pathogenesis and potentially discovering new therapeutic targets of complex human disease.
Limitations of Study
There are several limitations to the pooling approach and its use in this study. First, interpretation of phenotypic readouts may be complicated by non-autonomous processes. For example, a single hyperproliferating line in the pool may outcompete other lines for nutrients, and thus suppress the proliferation of lines with lower fitness. Such competition for resources could exaggerate the phenotypes observed in pooled cultures. Validation of phenotypes with independent clones and in single line assays is therefore critical. Second, differences in the proliferation rate of individual lines at the hPSC stage could lead to over or under-representation within the library. To safeguard against this, we successfully tested a cryopreservation strategy that allows expansion and storage of pools at low passage numbers. On the other hand, differences in baseline proliferation and cell density impact important aspects of neuronal biology including neurogenesis and synaptic maturation, and thus represent a significant source of experimental variability across hPSC-based disease models. In fact, the multiplex approach controls for equal cell density across lines and other environmental variables, allowing for more precise phenotypic comparisons. Third, an important challenge for the multiplex hPSC platform is to generate mature neuronal lineages and models of network connectivity between neuronal subtypes, a biology that lies at the core of autism.
With regard to insights into autism biology, this study identifies PFC neurogenesis and perhaps dysregulated WNT/βcatenin signaling as a convergence point among autism mutations. The ability to discern such convergence relied on the selection of de novo early-developmental autism genes, and the ability to generate specific hypotheses. It remains unclear whether other classes of autism mutations, such as copy number variants, non-coding variants, structural variants, and common variants, might also converge on PFC neurogenesis. To identify convergence among the broader set of autism variants, whose expression and function are less well defined, would likely necessitate screening a significantly larger number of cell lines and phenotypes. Finally, due to limitations in CRISPR technology, many of the lines harbored biallelic mutations, though patients harbor monoallelic mutations. While biallelic mutations did not exhibit more pronounced phenotypes than monoallelic mutations, the degree to which biallelic mutations phenocopy disease phenotypes normally caused by monoallelic mutations remains to be further explored.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for reagents may be directed to the Lead Contact, Lorenz Studer (studerl@mskcc.org).
MATERIALS AVAILABILITY
Cell lines generated and used in this study are available upon reasonable request from the Lead contact
DATA AND SOFTWARE AVAILABILITY
The RNA sequencing data generated in this paper is uploaded to GEO with accession number GSE146760. This dataset includes the counts-table output from HTSeq (Anders et al., 2015) and quantification of fold changes of each cell type compared to the starting hESCs.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
MEL1 (46XY, p38–50) and derivatives were maintained with Essential 8 medium or Essential 8 flex (E8) in feeder-free conditions on vitronectin (VTN-N) substrate. hPSCs were passaged as clumps with an EDTA dissociation solution (0.5 μM EDTA/PBS). Pooling was performed by dissociating lines to single cell with the EDTA dissociation solution and adding cells at desired frequency. Pools were initially established in the presence 10 μM ROCK inhibitor for 1 day. Pooled hPSCs were frozen in E8 supplemented with 10% DMSO media and thawed in the presence of 10 μM ROCK inhibitor. The MIX30 pool contains 30 hPSC lines derived from a MEL1 founder. Each of the lines contains an indel in a separate gene (see Table 1). In addition, a MIX32 pool containing 32 lines was generated for validation experiments. The MIX32 pool contains 14 autism clones from the original MIX30 library as well as the 13 independently generated clones with indels in the same genes (1 line was not paired with an independent clone). Nine of the independent clones were generated with a distinct gRNA, while 4 clones were generated with the same gRNA. Three control lines (UMOD, GSK3β, and CTNNB1) from the original library were also included in the MIX32 library, plus 2 additional UMOD clones. All cells were cultured at 37°C with 5% CO2. Media was changed every day unless using E8-flex. The MEL1 founder was tested for karyotype abnormalities. Cell lines are periodically authenticated using STR analysis and routinely checked for mycoplasma.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-PAX6 | Biolegend | 901301, RRID: AB_256503 |
| Rabbit Anti-FOXG1 | Clontech | M227, RRID: AB_2827749 |
| Goat Anti-SP8 | Santa-Cruz | sc-104661, RRID: AB_2194626 |
| Mouse Anti-COUPTF-1 [H8132] | Abcam | ab41858 (discontinued) |
| Rabbit Anti-NKX2.1 | Abcam | ab76013, RRID: AB_10861200 |
| Rabbit Anti-SOX2 | Biolegend | 630802, RRID: AB_2195784 |
| Mouse Anti-PAX6 | BD Biosciences | 561462, RRID: AB_10715442 |
| Mouse Anti-ZO-1 | BD Biosciences | 610966, RRID: AB_398279 |
| Rabbit Anti-GSH2 | Millipore | ABN162, RRID: AB_11203296 |
| Mouse Anti-TBR2 | eBioscience | 14-4877-82, RRID: AB_2572882 |
| Mouse Anti-TUJ1 | Covance | MMS-435P, RRID: AB_2315514 |
| Mouse anti-Ki67 | DAKO | M724001 −2, RRID: AB_2631211 |
| Rabbit anti-NeuN | Cell Signaling Tech | 24307, RRID:AB_2651140 |
| Rat anti-CTIP2 | Abcam | Ab18465, RRID_2064130 |
| Rabbit anti-TBR1 | Abcam | Ab31940, RRID_AB2200219 |
| Mouse anti-SATB2 | Abcam | Ab51502, RRID_AB_882455 |
| Mouse anti-ISL1/2 | DSHB | 39.4D5, RRID_AB2314683 |
| Rabbit Anti-GABA | Sigma | A2052, RRID: AB_477652 |
| Mouse Anti-TBR2-FITC | eBioscience | 11-4877-42, RRID: AB_2572499 |
| Mouse Anti-DCX-PE | BD Biosciences | 561505, RRID: AB_10643766 |
| Mouse Anti-SOX2-A647 | BD Biosciences | 560302, RRID: AB_1645323 |
| Mouse Anti-CD49d-PE/Cy7 | Biolegend | 304314, RRID: AB_10643278 |
| Mouse anti-PAX6-A647 | BD Biosciences | 562249, RRID: AB_11152956 |
| Mouse anti-SSEA4-A647 | BD Biosciences | 560796, RRID: AB_2033991 |
| Mouse anti-OCT4-PE | BD Biosciences | 560186, RRID: AB_1645331 |
| AlexaFluor Donkey Anti-Goat 488 | Thermo Fisher Scientific | A-11055, RRID: AB_2534102 |
| AlexaFluor Donkey Anti-Goat 568 | Thermo Fisher Scientific | A-11057, RRID: AB_142581 |
| AlexaFluor Donkey Anti-Goat 647 | Thermo Fisher Scientific | A-21447, RRID: AB_141844 |
| AlexaFluor Donkey Anti-Rabbit 488 | Thermo Fisher Scientific | A-21206, RRID: AB_141708 |
| AlexaFluor Donkey Anti-Rabbit 555 | Thermo Fisher Scientific | A-31572, RRID: AB_162543 |
| AlexaFluor Donkey Anti-Rabbit 647 | Thermo Fisher Scientific | A-31573, RRID: AB_2536183 |
| AlexaFluor Donkey Anti-Mouse 488 | Thermo Fisher Scientific | R37114, RRID: AB_2556542 |
| AlexaFluor Donkey Anti-Mouse 555 | Thermo Fisher Scientific | A-31570, RRID: AB_2536180 |
| AlexaFluor Donkey Anti-Mouse 647 | Thermo Fisher Scientific | A-21235, RRID: AB_141693 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEME:F12 w/ high glucose | Thermo Fisher Scientific | 11320–033 |
| L-Glutamine (100X) | Thermo Fisher Scientific | 25030–081 |
| Non-essential Amino Acids (100X) | Thermo Fisher Scientific | 11140–050 |
| Penicillin Streptomycin | Thermo Fisher Scientific | 15140–122 |
| 2-mercaptoethanol | Thermo Fisher Scientific | 21985–023 |
| Ascorbic Acid (AA) | Sigma Aldrich | 4034–100g |
| Recombinant human SHH (C24II) | R&D | 1845-SH |
| CHIR99021 (CHIR) | R&D | 4423 |
| Y-27632 (ROCKi) | R&D | 1254 |
| SB431542 (SB) | R&D | 1614 |
| LDN193189 (LDN) | Stem Cell Technologies | 72142 |
| XAV-939 (XAV) | R&D | 3746 |
| Recombinant human FGF8 | R&D | 423-F8 |
| Poly-L-Ornithine (PO) | Sigma Aldrich | P3655 |
| Mouse Laminin I (LAM) | R&D | 3400-010-1 |
| Fibronectin (FN) | Thermo Fisher Scientific | 356008 |
| Essential 8 (E8) | Thermo Fisher Scientific | A1517001 |
| Essential 6 (E6) | Thermo Fisher Scientific | A1516401 |
| 0.5M EDTA, pH 8.0 | Thermo Fisher Scientific | 15575020 |
| Accutase | Innovative Cell Technologies | AT104–500 |
| Vitronectin | Thermo Fisher Scientific | A14700 |
| Knockout Serum Replacement | Thermo Fisher Scientific | 10828–028 |
| Essential 8 FLEX | Thermo Fisher Scientific | A28558501 |
| Matrigel | BD Biosciences | 356234 |
| 50X B27 supplement w/o Vit A | Thermo Fisher Scientific | 17504044 |
| N2 supplement B | Stem Cell Technologies | 07156 |
| Recombinant human BMP4 | R&D | 314-BP |
| T rizol | Thermo Fisher Scientific | 15596026 |
| Dispase | Stemcell Technologies | 07913 |
| Progesterone | Sigma Aldrich | P8783 |
| Recombinant human FGF2 | R&D | 233-FB-001MG/CF |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | Qiagen | 74104 |
| SsoFast™ EvaGreen® Supermix | Bio-Rad | 172–5202 |
| iScript™ Reverse Transcription Supermix | Bio-Rad | 170–8841 |
| EdU Click-iT | Thermo Fisher Scientific | C10640 |
| Cytofix/Cytoperm Kit | BD Biosciences | 554722 |
| BD Perm/Wash Buffer | BD Biosciences | 554723 |
| ddPCR Supermix for probes no dUTP | Bio-Rad | 1863024 |
| Deposited Data | ||
| RNA-Seq | This study | NCBI GEO: GSE146760 |
| Experimental Models: Cell Lines | ||
| MEL-1 hESC line | Stem Cells Ltd | NIHhESC-11–0139 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish | ab | ZDB-GENO-960809–7 |
| Oligonucleotides | ||
| See Supplemental Table 4 | N/A | N/A |
| Recombinant DNA | ||
| PX458 Cas9-GFP | Addgene | Addgene: 48138 |
| Software and Algorithms | ||
| Poly Peak Parser | http://yosttools.genetics.utah.edu/PolyPeakParser/ | N/A |
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| FlowJo 9 | http://www.flojo.com | N/A |
| DESeq2 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html | (Love et al., 2014) |
| STAR 2.5.0 | https://github.com/alexdobin/STAR | N/A |
| R | https://cran.r-project.org/ | N/A |
| CHOPCHOP | http://chopchop.cbu.uib.no/ | N/A |
| Quantasoft (Bio-Rad) | https://www.bio-rad.com/en-us/sku/1864011-quantasoft-software-regulatory-edition?ID=1864011 | N/A |
Zebrafish husbandry
Zebrafish work was approved by the Institutional Animal Care and Use Committee (IACUC) at MSKCC. Zebrafish were bred and maintained in the Zuckerman fish facility, in temperature (28°C), pH (7.4), and salinity-controlled conditions. All fish were maintained on a 14hr on/10hr off light cycle. Zebrafish used were of the ab strain.
METHOD DETAILS
Experimental Design
To ensure the reproducibility of the differentiation strategies provided, several members of the Studer lab independently performed particular aspects of the prefrontal and occipital cortical differentiation and the protocol is consistently used in the laboratory. No specific methods were used for randomization and investigators were not blinded to the methods of differentiation. No statistical methods were utilized to determine sample size.
Gene selection for autism library
Gene selection for the MIX30 library was performed in the Spring of 2015 using the SFARI gene database. First, all genes with a score of 1 or 2 (high confidence) were selected. Second, genes were filtered for early developmental expression using the BrainSpan human fetal brain transcriptional atlas (https://brainspan.org) expressed at PCW8) and a hPSC-derived cortical neuron transcriptional atlas (http://cortecon.neuralsci.org, expressed on or before day 50). This resulted in a candidate list of 28 high-confidence, early-developmental autism genes.
Generation of the hPSC-multiplex library
CRISPR/Cas9 was used to introduce frameshift mutations into high-confidence autism genes. Guide RNAs (gRNAs) were designed to target exons in which indels or single nucleotide variant (SNV) mutations have been found in patients. If no suitable target sequence was found, then an upstream site was chosen. gRNAs were cloned into the bicistronic PX458 Cas9-GFP vector and introduced into MEL1 hPSCs by nucleofection. After 24 hours post nucleofection, cells were FACS sorted for GFP, and individual clones were collected and cultured on a mouse embryonic fibroblast feeder layer in the presence of 10 μM ROCK inhibitor in knockout serum replacement stem cell media as previously described (Fattahi et al., 2016) for two weeks. ROCK-inhibitor was removed from culture media after 4 days. Clones were picked onto a vitronectin substrate and further maintained in E8 and clones were screened for indels using sanger sequencing. True homozygous or heterozygous clones were preferred over compound heterozygotes. Heterozygous clones were inferred bioinformatically using Poly Peak Parser. All frozen stocks were re-sequenced for validation to confirm the presences of an indel. Since patient mutations could be gain-of-function or loss-of-function, DNA sequencing rather than protein expression was used for validation.
Measurement of allele frequencies using droplet digital PCR (ddPCR)
ddPCR was used to deconvolute allele frequencies from pooled cultures. ddPCR is able to measure the allele frequency of any DNA variant within a population of DNA. It does so using the same principle as traditional PCR, except the reaction mixture is partitioned into thousands of droplets that each contain approximately one molecule of DNA. In addition, the ddPCR reaction contains a fluorescent probe of one color (e.g. FAM) to the DNA variant of interest, and fluorescent probe of another color (e.g. HEX) to the corresponding wild-type allele for that variant. All droplets containing the DNA variant sequence will fluoresce with FAM, while all droplets containing the wild-type allele with fluoresce with HEX. Allele frequency can then be determined by measuring the number of FAM and HEX droplets. To deconvolute the allele frequencies for all lines in the autism pool, pairs of allele-specific probes were designed for each line in the autism library. A separate ddPCR reaction was run for each probe pair. Thus, to ascertain allele frequencies for all lines in the MIX30 autism pool, 30 separate reactions were run.
ddPCR probes were generated using the manufacturer’s design engine (BioRad, see Supplementary Table S4), and incorporated a 5’ fluorescently labeled HEX or FAM probe for wild-type and mutant alleles respectively, and a 3’ ZEN quencher. ddPCR was performed according to the manufacturer protocol. Briefly, a bulk PCR reaction (10–50ng of genomic DNA from pooled culture, 10 units of restriction enzyme (NEB), 900nM forward and reverse primer each, 250nM mutant and wild-type probe each, 1X ddPCR Supermix for probes no dUTP, up to 20ul ddH2O) was partitioned into droplets using the QX200 droplet generator (BioRad, 1864002). DNA was quantified using a fluorometer (Qubit 3.0, Thermo Q33216). PCR reactions were run with a standard thermocycler (C1000 Touch, BioRad) with annealing temperatures optimized for each probe pair. PCR reactions were allowed to incubate at 4°C for at least 2 hours prior to droplet reading. Droplets were read using the QX200 Droplet Reader (BioRad, 1864003) and analyzed using QuantaSoft Software (BioRad), which estimates the absolute number of DNA copies of wild-type and mutant alleles in a reaction by assuming a Poisson distribution of the fluorescence reads and converting this to fractional abundance estimates. Mutant allele frequency is then calculated as: total mutant alleles / (total mutant + wild-type alleles). Growth and cell-state phenotypes were determined by calculating changes in relative allele frequency across phenotypic fractions and normalizing each line to the internal negative standard, UMOD, for each replicate. WNT response phenotypes from day 45 PFC cultures were determined by comparing changes in relative allele frequency between treated and untreated conditions.
Prefrontal (PFC) and occipital (OCC) cortex differentiation
hPSCs were dissociated to single cells and plated on matrigel substrate in E8 at a density of 250,000 cells/cm2 in the presence of 10 μM ROCK inhibitor (Day −1). From Day 0 to 6–8, cells were cultured in Essential 6 medium in the presence of 100 nM LDN193189 (TGFβi) and 10 μM SB431542 (BMPi). To enrich for forebrain patterning, 2uM XAV939 (WNTi) was also included from days 0–2. On day 6–8, monolayer cultures were dissociated with Accutase and gently triturated and passed through a 45-micron cell strainer. Cells were spun down and replated as high-density droplets on poly-ornithine/laminin/fibronectin coated dishes and cultured in N2 media with B27 (1:1000, without Vitamin A), FGF8 (50ng/ml), and SHH (25ng/ml) for 4 days, until neuroepithelial rosettes were visible. Droplets were then passaged 1:2 with trypsin onto poly-ornithine/laminin/fibronectin coated plates and cultured in the same media. At day 20, cultures were passaged using Accutase or dispase to a density of 200,000 cells/cm2 to 400,000 cells/cm2 and cultured in N2 media with B27 (1:50, without Vitamin A), FGF8 (50ng/ml), for up to an additional 20 days. FGF8 was removed from the culture media after day 40. Cultures in which excessive flat morphology cells arose were discarded. OCC cultures were generated in the same manner as PFC cultures, except FGF8 was removed from all culture media as it is known to specify PFC identity. Low concentration SHH (25ng/ml) was included in the culture media for three reasons. First, SHH is found at low concentrations in the dorsal telencephalon and regulates cortical progenitor proliferation (Komada et al., 2008; Wang et al., 2016). The concentration of SHH used here is not sufficient to induce cortical interneuron identity (Maroof et al., 2013).Second, SHH helps to temporally synchronize the PFC and OCC protocols so they can be compared. If SHH was not included then only the PFC protocol would contain a mitogenic factor, making it difficult to determine whether differential gene expression between OCC and PFC cultures is due to temporal or regional differences. Third, rosette formation is inefficient without FGF8 and SHH, and therefore SHH was required to form rosettes in the OCC culture protocol. However, SHH is not required for the PFC differentiation protocol (data not shown).
Neural crest differentiation
hPSCs were dissociated to single cells and plated on matrigel substrate in E8 at a density of 200,000 cells/cm2 in the presence of 10 μM ROCK inhibitor (Day −1). From day 0–2, cells were cultured in E6 with BMP4 (1ng/mL), 0.6 μM CHIR99021, and 10 μM SB431542. From day 3–10, cells were cultured in 1.5 μM CHIR99021 and 10μM SB431542. Cells were dissociated with Accutase for FACS.
Cell-cycle exit analysis
Day 20 PFC cultures were treated with CHIR99021 (0.6 μM) for 2 days or left untreated. At day 22, cultures were pulsed with EdU using the EdU Click-iT system according to manufacturer protocol. Briefly, cells were treated with EdU for 1 hour, dissociated with Accutase for 30 minutes at 37C, and passaged onto poly-ornithine/laminin/fibronectin coated plates at a density of 200,000–400,000 cells/cm2 in the presence of 10 μM ROCK inhibitor. Cells were fixed 18 hours later and fixed for immunocytochemistry. ImageJ was used for cell counting. Images were thresholded to define individual cells (particles) and cells were counted using the analyze particle function.
Immunocytochemistry
Cells were fixed in 4% PFA for 15 minutes at room temperature and washed three times with PBS. Cells were blocked for 30 minutes in 10% FBS, 1% BSA, 0.3% Triton-X diluted in PBS, and incubated with primary antibody overnight. The next day, sections were washed with PBS then incubated with secondary antibody for 1 hour at room temperature. Microscopy was performed using a standard inverted epifluorescence microscope (Olympus IX71 or Zeiss Axio Observer). Images were acquired using Cell Sens (Olympus) or Zen Pro (Zeiss) Software. Min, max and gamma (midtone) adjustments were applied uniformly to images during processing with Adobe Photoshop Creative Cloud.
Flow cytometry and genomic DNA extraction from fixed cells.
Cultures were dissociated with Accutase and fixed and permeabilized with BD Cytofix/Cytoperm for 45 minutes on ice. Fixed cells were washed with BD Perm/Wash Buffer and stained with primary antibody for 1 hour on ice and secondary antibody for 30 minutes on ice and sorted using a FACSAria III flow cytometer (BD Bioscience) and analyzed using FlowJo Software. Isotype controls were used for gating. The DCX-PE antibody (BD 561505) was validated by analyzing a time-course of DCX expression during neural differentiation, which demonstrated, as expected, that the fraction of DCX positive cells increased over time and was largely negative for PAX6 expression. Sorted fixed cells were centrifuged for 5 minutes at 20,000 rcf, resuspended in 500 μl lysis buffer (10mM Tris-HCL pH 8.0, 100 mM NaCl, 10mM EDTA, 0.5% SDS, 40mg/mL proteinase K) and incubated at 65°C, shaking, overnight. The next day, 300 μl NaCl was added to lysis and incubated on ice for 10 minutes. Samples were centrifuged at 20,000 rcf for 10 minutes and aqueous phase DNA was precipitated in 650 μl of isopropanol, washed with 70% ethanol, and resuspended in ddH2O.
RNA extraction and qRT-PCR
RNA was extracted using Trizol reagent followed by chloroform extraction. RNA was precipitated in isopropanol and resuspended in ddH2O. cDNA synthesis was performed using iScript Reverse Transcriptase with 1 μg of total RNA. RT-PCR was performed with EvaGreen Supermix and analyzed on a CFx96 Real-Time System (BioRad). Occipital versus prefrontal differentially expressed transcripts that were used to assess areal patterning were selected using a multi-step process. A list of candidate transcripts was first identified using the differential gene expression search function from the https://brainspan.org transcriptome atlas. Seven prefrontal enriched and seven occipital enriched transcripts were further selected from the candidate list based on literature search to corroborate cell-type and region-specific expression.
RNA sequencing and gene expression analysis
RNA was isolated from hPSC-derived forebrain neural stem cells (NSCs), PFC and OCC patterned neurons at day 30 (described above). Total RNA was sent to the MSKCC Integrated Genomics Operation for RNA quality control, library preparation and paired-end sequencing (30–40 million reads). Raw FASTQ files were aligned to the ENSEMBL GRCh38 genome build using STAR 2.5.0. Read counts were tabulated using HTSeq and imported to DESeq2 for further analysis using a standardized pipeline. PFC and OCC samples were compared against each other to identify differentially expressed genes between the two cell types (Supplemental Table S2).
Comparison of hPSC-derived cells to the BrainSpan Developmental Transcriptome
To compare the molecular profiles of hPSC-derived PFC and OCC to neurons in vivo, we generated a list of the top 200 differentially expressed genes between PFC (averaging: OFC, DFC, VFC and MFC) and OCC (averaging: PCx, Ocx and ITC) regions at PCW 8 from the Developmental Transcriptome dataset (Hawrylycz et al., 2012) (BrainSpan, RNA-Seq Gencode v10 summarized to genes). The expression of the top 200 PFC and OCC genes was then compared with their differential expression in hPSC derived cultures using a 2×2 contingency table. The resulting list is made available in Supplemental Table S2.
Creation of zebrafish CRISPR F0 mosaic mutants
We designed targeting sgRNAs for the genes of interest for homologous exons that were targeted in hPSC lines, in two zebrafish paralogues if applicable. CHOPCHOP (cite: http://chopchop.cbu.uib.no/). gRNA/Cas9/Tracer complexes were then synthesized using the ALT-R system and prepared according to previously published protocols (https://www.idtdna.com/pages/products/crispr-genome-editing/alt-r-crispr-cas9-system) CRISPR activity was confirmed from a random subset of injected embryos using a surveyor assay (IDT), for at least 1 paralogue of each gene for conditions that showed a significant jaw phenotype.
Zebrafish imaging and image processing
Fish were imaged at 7 dpf using an upright Zeiss Discovery V16 equipped with a motorized stage, brightfield, GFP and tdTomato filter sets. To acquire images, fish were lightly anaesthetized with Tricaine 4 mg ml-1 and placed into agarose molds to properly image the head from a ventral vantage point. Images were acquired with the Zeiss Zen software v1, and the post image processing was done using ImageJ. Zebrafish images were quantified by a blinded observer using ImageJ software. Jaw length was measured as an angle between one line from the top of the eyes and a second line from the top of the right eye to middle of the jaw, depicted in Figure S7D. Jaw angle phenotypes were scored on a binary scale, with a cut-off of < 1 s.d. below WT average.
Functional Clustering of Multiplex Data and Clinical Analysis
Functional classes of autism mutations (see Figure 3) were defined based on a positive PFC neurogenesis phenotype. A positive PFC neurogenesis phenotype was assigned if a genotype exhibited altered PFC neuronal production or altered PFC stem cell enrichment with an FDR < 0.05, or if a genotype exhibited both altered PFC neuronal production and altered PFC stem cell enrichment with an FDR <0.1. Aggregate multiplex data across PFC and WNT-related assays (PFC growth, PFC IPC production, PFC neurogenesis, PFC stem cell enrichment, PFC WNT response, Neural crest induction) were aggregated, weighted equally and normalized to the UMOD control. The matrix was scaled and clustered in R using the pheatmap package with default clustering parameters (Figure 7A).
Proband data was ascertained from the Simons Simplex Collection Clinical Database (SFARIBase). Genotypes were assigned using previously published results from sequencing studies (Iossifov et al., 2014; Krumm et al., 2015; Sanders et al., 2015). Patients in Cluster A and Cluster B were assigned genotypes based on the presence of de novo coding or splice-site variants. Non-splice site intronic and inherited mutations were not considered. Patients with de novo loss-of-function or MIS3 missense mutations that did not fit into Cluster A or Cluster B were included in the de novo Control. All other patients were included in the idiopathic control group. De novo and idiopathic control cohorts were IQ-matched to Cluster A and B. To do this, patients in control groups were sorted from lowest to highest IQ. Starting with patients with an IQ of 54 (lowest IQ found in cluster B), patients were sequentially added with increasingly higher IQs until the cohort average reached the average of cluster B. The ADI-R verbal communication score excludes patients with severe language deficits, and thus the ADI-R non-verbal communication score was used in order to compare all patients regardless of language ability (Figure 7C). Correlations in Figure 7J were performed by assigning a PFC neurogenesis value to each patient based on their genotype. The PFC neurogenesis value is the neuronal production value (DCX/SOX2) for each genotype from Figure 3C.
QUANTIFICATION AND STATISTICAL ANALYSIS
All reported measurements are from distinct samples. At least three independent biological replicates were used for each multiplex experiment in the main text, derived from at least two independent MIX30 pools for multiplex experiments. Specific data on replicates (n) is given in the figure legends. Data are presented as mean ± S.E.M., except where noted in the figure legends. Biological replicates for multiplex experiments were transformed to a Log2 scale as values were typically ratios. Samples were normalized using a z-transformation: (x-μ)/ σ (μ = sample mean, σ = sample standard deviation). When samples contained multiple outliers, a modified z-transformation was used: 0.6745*(x- med)/MAD (med = sample median, MAD = median absolute deviation). Welch ANOVA, which does not assume equal variance between groups, was then used to compare groups to the internal control UMOD. Post-hoc Benjamini, Krieger, Yekutieli False Discovery Rate correction for multiple comparisons was used to generate corrected p values. Comparisons of clinical cohorts were performed using Kruskal-Wallis with Dunn’s test or ANOVA with Tukey test (for normally distributed parameters). For comparison of language phenotypes (Figure 7F–H), exact p-values for Fisher’s test were corrected for multiple testing with Holm-Sidak method. Fisher’s tests were two-sided. Statistical analysis was performed using Prism 7 (Graphpad) or Excel (Microsoft) software. Mean and corrected p values from multiplex assays are included in Supplemental Table S3.
Supplementary Material
Table S1. Description of lines in autism library, related to Figure 1, including function, indel type (monoallelic or biallelic frameshift), indel length, indel sequence, indel location, and expression in hPSC-derived cortical neuron transcriptional atlas (Cortecon), and post-conception week (PCW) 8 human fetal tissue (BrainSpan).
Table S3. Summary of results from MIX30 multiplex assays, related to Figure 3, Figure 5 and Figure 6. Table includes false discover rates (FDR) and phenotypes from 7 phenotypic assays. P values are corrected for multiple comparison using the Benjamini, Krieger, Yekutieli method. Boxes are color coded according to FDR or phenotype magnitude.
Table S2. hPSC-derived PFC RNA sequencing, related to Figure 2 and STAR METHODS. Top 200 differentially expressed genes between day 30 PFC and OCC cultures.
Table S4. Table of reagents, related to STAR METHODS. Includes oligonucleotides and antibodies used in this study.
Highlights.
hPSC-based multiplex platform for interrogation of autism-associated mutations
Prefrontal cortex paradigm in hPSCs identifies autism-related neurogenesis defects
Abnormal WNT/βcatenin responses in class of autism genes with neurogenesis defects
Endophenotypes in hPSCs correlate with clinical data in autism patients
ACKNOWLEDGEMENTS
We would like to thank members of the Studer lab for their valuable input on experimental design and feedback on manuscript. This work was supported in part through NYSTEM award C030137, an award from the Melanoma Research Alliance (L.S.) the NYSTEM facility award N13S-011 (LS & MJT) and through the NIH Cancer Center support grant P30 CA008748. G.C. is supported by a Ruth L. Kirschstein F30 M.D./Ph.D. pre-doctoral fellowship (F30 MH113343-01A1) and a training grant from the National Institute of General Medical Sciences (T32GM007739) to the Weill Cornell/Rockefeller/Sloan-Kettering Tri-Institutional MD-PhD Program. R.M.W. is supported by the NIH Director’s New Innovator Award (DP2CA186572), Mentored Clinical Scientist Research Career Development Award (K08AR055368), the Melanoma Research Alliance, The Starr Cancer Consortium, The Pershing Square Sohn Foundation, The Alan and Sandra Gerry Metastasis Research Initiative at the Memorial Sloan Kettering Cancer Center and The Harry J. Lloyd Foundation, Consano). S.J.C is supported by the Kirschstein-NRSA predoctoral fellowship (F31) award as part of the National Cancer Institute of the National Institutes of Health under Award Number F31CA196305 as well as the Joanna M. Nicolay Melanoma Foundation Research Scholar Award 2014 and the Robert B. Catell Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
G.C. and L.S. are listed as inventors of a related patent application filed by the Memorial Sloan Kettering Cancer Center. L.S. is a co-founder and paid consultant of BlueRock Therapeutics. All other authors declare no competing financial interests.
REFERENCES
- Ahn J, Jang J, Choi J, Lee J, Oh SH, Lee J, Yoon K, and Kim S (2014). GSK3beta, but not GSK3alpha, inhibits the neuronal differentiation of neural progenitor cells as a downstream target of mammalian target of rapamycin complex1. Stem Cells Dev 23, 1121–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arranz J, Balducci E, Arato K, Sanchez-Elexpuru G, Najas S, Parras A, Rebollo E, Pijuan I, Erb I, Verde G, et al. (2019). Impaired development of neocortical circuits contributes to the neurological alterations in DYRK1A haploinsufficiency syndrome. Neurobiol Dis 127, 210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian M, Willoughby J, Fry AE, Weber A, Firth HV, Deshpande C, Berg JN, Chandler K, Metcalfe KA, Lam W, et al. (2017). Delineating the phenotypic spectrum of Bainbridge-Ropers syndrome: 12 new patients with de novo, heterozygous, loss-of-function mutations in ASXL3 and review of published literature. J Med Genet 54, 537–543. [DOI] [PubMed] [Google Scholar]
- Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C, Vulto-van Silfhout AT, et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, and Sabatini DM (2014). Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A (2008). Wnt/beta-catenin signaling in cerebral cortical development. Organogenesis 4, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero DR, Brugmann S, Chu Y, Bajpai R, Jame M, and Helms JA (2011). Cranial neural crest cells on the move: their roles in craniofacial development. Am J Med Genet A 155A, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corley M, and Kroll KL (2015). The roles and regulation of Polycomb complexes in neural development. Cell Tissue Res 359, 65–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, Barnes CC, and Pierce K (2011). Neuron number and size in prefrontal cortex of children with autism. JAMA 306, 2001–2010. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, and Morgan J (2007). Mapping early brain development in autism. Neuron 56, 399–413. [DOI] [PubMed] [Google Scholar]
- Curtin E, Hickey G, Kamel G, Davidson AJ, and Liao EC (2011). Zebrafish wnt9a is expressed in pharyngeal ectoderm and is required for palate and lower jaw development. Mech Dev 128, 104–115. [DOI] [PubMed] [Google Scholar]
- De Ferrari GV, and Moon RT (2006). The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene 25, 7545–7553. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguchi K, Inoue K, Avila WE, Lopez-Terrada D, Antalffy BA, Quattrocchi CC, Sheldon M, Mikoshiba K, D’Arcangelo G, and Armstrong DL (2003). Reelin and disabled-1 expression in developing and mature human cortical neurons. J Neuropathol Exp Neurol 62, 676–684. [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Moon RT, and Raible DW (1998). Control of neural crest cell fate by the Wnt signalling pathway. Nature 396, 370–373. [DOI] [PubMed] [Google Scholar]
- Dougherty M, Kamel G, Grimaldi M, Gfrerer L, Shubinets V, Ethier R, Hickey G, Cornell RA, and Liao EC (2013). Distinct requirements for wnt9a and irf6 in extension and integration mechanisms during zebrafish palate morphogenesis. Development 140, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, Liu CY, Watson LA, and Tsai LH (2016). Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci 19, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 39, 25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl RK, Turner TN, Mefford HC, Hudac CM, Gerdts J, Eichler EE, and Bernier RA (2017). Clinical phenotype of ASD-associated DYRK1A haploinsufficiency. Mol Autism 8, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst C (2016). Proliferation and Differentiation Deficits are a Major Convergence Point for Neurodevelopmental Disorders. Trends Neurosci 39, 290–299. [DOI] [PubMed] [Google Scholar]
- Fattahi F, Steinbeck JA, Kriks S, Tchieu J, Zimmer B, Kishinevsky S, Zeltner N, Mica Y, El-Nachef W, Zhao H, et al. (2016). Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature 531, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faundes V, Newman WG, Bernardini L, Canham N, Clayton-Smith J, Dallapiccola B, Davies SJ, Demos MK, Goldman A, Gill H, et al. (2018). Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am J Hum Genet 102, 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi-Shimogori T, and Grove EA (2001). Neocortex patterning by the secreted signaling molecule FGF8. Science 294, 1071–1074. [DOI] [PubMed] [Google Scholar]
- Gallagher D, Voronova A, Zander MA, Cancino GI, Bramall A, Krause MP, Abad C, Tekin M, Neilsen PM, Callen DF, et al. (2015). Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev Cell 32, 31–42. [DOI] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, and Vitkup D (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompers AL, Su-Feher L, Ellegood J, Copping NA, Riyadh MA, Stradleigh TW, Pride MC, Schaffler MD, Wade AA, Catta-Preta R, et al. (2017). Germline Chd8 haploinsufficiency alters brain development in mouse. Nat Neurosci 20, 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory GD, Vakoc CR, Rozovskaia T, Zheng X, Patel S, Nakamura T, Canaani E, and Blobel GA (2007). Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol Cell Biol 27, 8466–8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm K, Sum EY, Fujiwara Y, Lindeman GJ, Visvader JE, and Orkin SH (2004). Defective neural tube closure and anteroposterior patterning in mice lacking the LIM protein LMO4 or its interacting partner Deaf-1. Mol Cell Biol 24, 2074–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond VE, So E, Cate HS, Britto JM, Gunnersen JM, and Tan SS (2010). Cortical layer development and orientation is modulated by relative contributions of reelin-negative and -positive neurons in mouse chimeras. Cereb Cortex 20, 2017–2026. [DOI] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, et al. (2012). An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazlett HC, Gu H, Munsell BC, Kim SH, Styner M, Wolff JJ, Elison JT, Swanson MR, Zhu H, Botteron KN, et al. (2017). Early brain development in infants at high risk for autism spectrum disorder. Nature 542, 348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Munoz I, Lund AH, van der Stoop P, Boutsma E, Muijrers I, Verhoeven E, Nusinow DA, Panning B, Marahrens Y, and van Lohuizen M (2005). Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc Natl Acad Sci U S A 102, 7635–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ, Vessella RL, and Tewari M (2013). Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods 10, 1003–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N, and Gotoh Y (2004). The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 131, 2791–2801. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M, and Gotoh Y (2009). Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63, 600–613. [DOI] [PubMed] [Google Scholar]
- Huang YC, Shih HY, Lin SJ, Chiu CC, Ma TL, Yeh TH, and Cheng YC (2015). The epigenetic factor Kmt2a/Mll1 regulates neural progenitor proliferation and neuronal and glial differentiation. Dev Neurobiol 75, 452–462. [DOI] [PubMed] [Google Scholar]
- Iossifov I, Levy D, Allen J, Ye K, Ronemus M, Lee YH, Yamrom B, and Wigler M (2015). Low load for disruptive mutations in autism genes and their biased transmission. Proc Natl Acad Sci U S A 112, E5600–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 34, 27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Wang PP, Atabay KD, Murphy EA, Doan RN, Hecht JL, and Walsh CA (2015). Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat Neurosci 18, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung EM, Moffat JJ, Liu J, Dravid SM, Gurumurthy CB, and Kim WY (2017). Arid1b haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat Neurosci 20, 1694–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkman HO (2012). A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol Autism 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel G, Hoyos T, Rochard L, Dougherty M, Kong Y, Tse W, Shubinets V, Grimaldi M, and Liao EC (2013). Requirement for frzb and fzd7a in cranial neural crest convergence and extension mechanisms during zebrafish palate and jaw morphogenesis. Dev Biol 381, 423–433. [DOI] [PubMed] [Google Scholar]
- Kim WY, Wang X, Wu Y, Doble BW, Patel S, Woodgett JR, and Snider WD (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci 12, 1390–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Khoshkhoo S, Frankowski JC, Zhu B, Abbasi S, Lee S, Wu YE, and Hunt RF (2018). Chd2 Is Necessary for Neural Circuit Development and Long-Term Memory. Neuron 100, 1180–1193 e1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koemans TS, Kleefstra T, Chubak MC, Stone MH, Reijnders MRF, de Munnik S, Willemsen MH, Fenckova M, Stumpel C, Bok LA, et al. (2017). Functional convergence of histone methyltransferases EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. PLoS Genet 13, e1006864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada M, Saitsu H, Kinboshi M, Miura T, Shiota K, and Ishibashi M (2008). Hedgehog signaling is involved in development of the neocortex. Development 135, 2717–2727. [DOI] [PubMed] [Google Scholar]
- Krumm N, O’Roak BJ, Shendure J, and Eichler EE (2014). A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci 37, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX, et al. (2015). Excess of rare, inherited truncating mutations in autism. Nat Genet 47, 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurabayashi N, and Sanada K (2013). Increased dosage of DYRK1A and DSCR1 delays neuronal differentiation in neocortical progenitor cells. Genes Dev 27, 2708–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakoma J, Garcia-Alonso L, and Luque JM (2011). Reelin sets the pace of neocortical neurogenesis. Development 138, 5223–5234. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus EA, Kintner C, and Harris W (1998). The role of GSK3beta in regulating neuronal differentiation in Xenopus laevis. Mol Cell Neurosci 12, 269–280. [DOI] [PubMed] [Google Scholar]
- Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, Amenduni M, Szekely A, Palejev D, Wilson M, et al. (2015). FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 162, 375–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying SW, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG, et al. (2013). Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan J, and King MC (2010). Genetic heterogeneity in human disease. Cell 141, 210–217. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, et al. (2017). Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles JH, Takahashi TN, Hong J, Munden N, Flournoy N, Braddock SR, Martin RA, Bocian ME, Spence MA, Hillman RE, et al. (2008). Development and validation of a measure of dysmorphology: useful for autism subgroup classification. Am J Med Genet A 146A, 1101–1116. [DOI] [PubMed] [Google Scholar]
- Munji RN, Choe Y, Li G, Siegenthaler JA, and Pleasure SJ (2011). Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci 31, 1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary DD, Chou SJ, and Sahara S (2007). Area patterning of the mammalian cortex. Neuron 56, 252–269. [DOI] [PubMed] [Google Scholar]
- Ockeloen CW, Willemsen MH, de Munnik S, van Bon BW, de Leeuw N, Verrips A, Kant SG, Jones EA, Brunner HG, van Loon RL, et al. (2015). Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Eur J Hum Genet 23, 1176–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto N, Miya F, Tsunoda T, Kato M, Saitoh S, Yamasaki M, Kanemura Y, and Kosaki K (2017). Novel MCA/ID syndrome with ASH1L mutation. Am J Med Genet A 173, 1644–1648. [DOI] [PubMed] [Google Scholar]
- Packer A (2016a). Enrichment of factors regulating canonical Wnt signaling among autism risk genes. Mol Psychiatry. [DOI] [PubMed] [Google Scholar]
- Packer A (2016b). Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci Biobehav Rev 64, 185–195. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, and Geschwind DH (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, and Goldstein DB (2013). Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9, e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, and Shilatifard A (2016). Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352, aad9780. [DOI] [PubMed] [Google Scholar]
- Redin C, Brand H, Collins RL, Kammin T, Mitchell E, Hodge JC, Hanscom C, Pillalamarri V, Seabra CM, Abbott MA, et al. (2017). The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet 49, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochard L, Monica SD, Ling IT, Kong Y, Roberson S, Harland R, Halpern M, and Liao EC (2016). Roles of Wnt pathway genes wls, wnt9a, wnt5b, frzb and gpc4 in regulating convergent-extension during zebrafish palate morphogenesis. Development 143, 2541–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronemus M, Iossifov I, Levy D, and Wigler M (2014). The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet 15, 133–141. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz SU, Albert M, Malatesta M, Morey L, Johansen JV, Bak M, Tommerup N, Abarrategui I, and Helin K (2011). Jarid1b targets genes regulating development and is involved in neural differentiation. EMBO J 30, 4586–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T, Ji F, Yuan Z, and Jiao J (2015). CHD2 is Required for Embryonic Neurogenesis in the Developing Cerebral Cortex. Stem Cells 33, 1794–1806. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Ritesh KC, Tsan YC, Liao R, Su F, Cao X, Hannibal MC, Keegan CE, Chinnaiyan AM, Martin DM, et al. (2016). De novo dominant ASXL3 mutations alter H2A deubiquitination and transcription in Bainbridge-Ropers syndrome. Hum Mol Genet 25, 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterneckert JL, Reinhardt P, and Scholer HR (2014). Investigating human disease using stem cell models. Nat Rev Genet 15, 625–639. [DOI] [PubMed] [Google Scholar]
- Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, Wynshaw-Boris A, Colamarino SA, Lein ES, and Courchesne E (2014). Patches of disorganization in the neocortex of children with autism. N Engl J Med 370, 1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet DJ (2017). iPSCs Meet GWAS: The NextGen Consortium. Cell Stem Cell 20, 417–418. [DOI] [PubMed] [Google Scholar]
- Testa G (2011). The time of timing: how Polycomb proteins regulate neurogenesis. Bioessays 33, 519–528. [DOI] [PubMed] [Google Scholar]
- van de Leemput J, Boles NC, Kiehl TR, Corneo B, Lederman P, Menon V, Lee C, Martinez RA, Levi BP, Thompson CL, et al. (2014). CORTECON: a temporal transcriptome analysis of in vitro human cerebral cortex development from human embryonic stem cells. Neuron 83, 51–68. [DOI] [PubMed] [Google Scholar]
- Vulto-van Silfhout AT, Rajamanickam S, Jensik PJ, Vergult S, de Rocker N, Newhall KJ, Raghavan R, Reardon SN, Jarrett K, McIntyre T, et al. (2014). Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. Am J Hum Genet 94, 649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Hou S, and Han YG (2016). Hedgehog signaling promotes basal progenitor expansion and the growth and folding of the neocortex. Nat Neurosci 19, 888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz A, Peretz M, Aharony A, Sagi I, and Benvenisty N (2018). Defining essential genes for human pluripotent stem cells by CRISPR-Cas9 screening in haploid cells. Nat Cell Biol 20, 610–619. [DOI] [PubMed] [Google Scholar]
- Yu C, Mannan AM, Yvone GM, Ross KN, Zhang YL, Marton MA, Taylor BR, Crenshaw A, Gould JZ, Tamayo P, et al. (2016). High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol 34, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Description of lines in autism library, related to Figure 1, including function, indel type (monoallelic or biallelic frameshift), indel length, indel sequence, indel location, and expression in hPSC-derived cortical neuron transcriptional atlas (Cortecon), and post-conception week (PCW) 8 human fetal tissue (BrainSpan).
Table S3. Summary of results from MIX30 multiplex assays, related to Figure 3, Figure 5 and Figure 6. Table includes false discover rates (FDR) and phenotypes from 7 phenotypic assays. P values are corrected for multiple comparison using the Benjamini, Krieger, Yekutieli method. Boxes are color coded according to FDR or phenotype magnitude.
Table S2. hPSC-derived PFC RNA sequencing, related to Figure 2 and STAR METHODS. Top 200 differentially expressed genes between day 30 PFC and OCC cultures.
Table S4. Table of reagents, related to STAR METHODS. Includes oligonucleotides and antibodies used in this study.
Data Availability Statement
The RNA sequencing data generated in this paper is uploaded to GEO with accession number GSE146760. This dataset includes the counts-table output from HTSeq (Anders et al., 2015) and quantification of fold changes of each cell type compared to the starting hESCs.