

Abstract
This study discusses an analysis of combined airborne and ground observations of particulate nitrate (NO3-(p)) concentrations made during the wintertime DISCOVER-AQ study at one of the most polluted cities in the United States, Fresno, CA in the San Joaquin Valley (SJV) and focuses on development of understanding of the various processes that impact surface nitrate concentrations during pollution events. The results provide an explicit case-study illustration of how nighttime chemistry can influence daytime surface-level NO3-(p) concentrations, complementing previous studies in the SJV. The observations exemplify the critical role that nocturnal chemical production of NO3-(p) aloft in the residual layer (RL) can play in determining daytime surface-level NO3-(p) concentrations. Further, they indicate that nocturnal production of NO3-(p) in the RL, along with daytime photochemical production, can contribute substantially to the build-up and sustaining of severe pollution episodes. The exceptionally shallow nocturnal boundary layer heights characteristic of wintertime pollution events in the SJV intensifies the importance of nocturnal production aloft in the residual layer to daytime surface concentrations. The observations also demonstrate that dynamics within the RL can influence the early-morning vertical distribution of NO3-(p), despite low wintertime wind speeds. This overnight reshaping of the vertical distribution above the city plays an important role in determining the net impact of nocturnal chemical production on local and regional surface-level NO3-(p) concentrations. Entrainment of clean free tropospheric air into the boundary layer in the afternoon is identified as an important process that reduces surface-level NO3-(p) and limits build-up during pollution episodes. The influence of dry deposition of HNO3 gas to the surface on daytime particulate nitrate concentrations is important but limited by an excess of ammonia in the region, which leads to only a small fraction of nitrate existing in the gas-phase even during the warmer daytime. However, in late afternoon, when diminishing solar heating leads to a rapid fall in the mixed boundary layer height, the impact of surface deposition is temporarily enhanced and can lead to a substantial decline in surface-level particulate nitrate concentrations; this enhanced deposition is quickly arrested by a decrease in surface temperature, which drops the gas-phase fraction to near zero. The overall importance of enhanced late afternoon gas-phase loss to the multiday build-up of pollution events is limited by the very shallow nocturnal boundary layer. The case study here demonstrates that mixing down of NO3-(p) from the RL can contribute a majority of the surface-level NO3-(p) in the morning (here, ~80%), and a strong influence can persist into the afternoon even when photochemical production is maximum. The particular day-to-day contribution of aloft nocturnal NO3-(p) production to surface concentrations will depend on prevailing chemical and meteorological conditions. Although specific to the SJV, the observations and conceptual framework further developed here provide general insights into the evolution of pollution episodes in wintertime environments.
1. Introduction
Nocturnal processing of nitrogen oxides, NOx (= NO + NO2) can strongly influence daytime air quality (Dentener and Crutzen, 1993; Brown et al., 2006c). At night, once photochemical reactions shutdown, NOx reacts with ozone (O3) to form nitrate radical (NO3) and dinitrogen pentoxide (N2O5) (Reactions 1 through 3a). N2O5 can react heterogeneously with airborne particles to form either nitric acid (HNO3) (Reaction 4a) or, in the presence of particulate chloride, nitryl chloride (ClNO2) (Reaction 4b, where YClNO2 represents the molar yield of ClNO2 with respect to the N2O5 reacted). In the presence of basic species like ammonia (NH3), HNO3 can be neutralized to form particulate nitrate (NO3-(p)). NO3 radicals can alternatively react with volatile organic compounds (VOCs), which suppresses HNO3 formation (Reaction 3b). Much research has focused on the influence of nocturnal NOx processing on the regional budgets of NOx and O3 and on the oxidative capacity of the atmosphere during subsequent mornings (e.g. Brown et al., 2006b; Thornton et al., 2010; Wild et al., 2016). The corresponding impact of nighttime production of NO3-(g+p), a key nocturnal sink for NOx, on local and regional air quality can be considerable (Lowe et al., 2015; Pusede et al., 2016) but is less often considered in detail.
| (R1) |
| (R2) |
| (R3a) |
| (R3b) |
| (R4a) |
| (R4b) |
The importance of nocturnal NOx chemistry to NO3-(p) production can be especially important in the winter. Relative to summer, nights in winter are longer, colder and more humid, and biogenic VOC emissions tend to be smaller. This allows for a larger fraction of NO2 to be oxidized to HNO3 via the N2O5 hydrolysis pathway (Cabañas et al., 2001; Wagner et al., 2013) and colder temperatures favor partitioning of nitrate to the particle-phase (Stelson and Seinfeld, 1982). In winter, night time HNO3 production can more efficiently compete with daytime photochemically driven production due to the low photolysis rates and hydroxyl radical concentrations (Wagner et al., 2013; Pusede et al., 2016). Multiday pollution events (i.e. periods with elevated particulate matter concentrations) can occur when meteorological conditions inhibit dispersion, as is the case with persistent cold air pool formation often found in valley regions (Whiteman et al., 2014; Baasandorj et al., 2017). During the daytime, sunlight driven convection leads to an evolution of the near-surface temperature profile and causes the atmosphere to be reasonably well mixed up to some height (typically less than 1 km; c.f. Figure S1). Radiative cooling in the late afternoon leads this mixed layer (ML) to decouple and separate into a shallow, near surface-level nocturnal boundary layer (NBL) and a residual layer (RL) aloft, the behavior of which can be further modified by valley flows.
Nocturnal conversion of NOx to NO3-(p) can occur either in the NBL or the RL. Surface NO emissions can substantially limit direct production of NO3-(p) in the NBL by titrating O3, depending on the initial conditions. Nocturnal surface NO emissions do not directly influence the decoupled RL, with chemical production of NO3-(p) dependent on the NOx, O3 and particulate matter in the mixed layer at the time of decoupling. Box and 3D models have been previously used to assess the contribution of nocturnal processes in the RL to the daytime surface concentrations of particulate matter (PM), especially NO3-(p) (Riemer et al., 2003; Curci et al., 2015). Yet, computational models often have difficulty in accurately predicting surface NO3-(p) in many regions, particularly in the winter season, despite good estimations of NOx emissions (Walker et al., 2012; Terrenoire et al., 2015), although this is not always the case (e.g. Schiferl et al., 2014). Here, airborne and ground measurements made over Fresno, CA in the San Joaquin Valley (SJV) during the wintertime 2013 DISCOVER-AQ (Deriving Information on Surface Conditions from COlumn and VERtically resolved observations relevant to Air Quality; Appendix A) (Crawford and Pickering, 2014) study are used to further develop our understanding of the role that different factors play in determining surface-level NO3-(p) concentrations.
Winters in Fresno are characterized by frequent multiday pollution episodes (Chow et al., 1999; Watson and Chow, 2002), when PM2.5 (PM with aerodynamic diameter < 2.5 μm) mass concentrations exceed the 24-hour National Ambient Air Quality Standard (NAAQS) of 35 μg m-3 (Figure 1). Fresno is one of the largest cities in the San Joaquin Valley (SJV), which is largely an agricultural area and suffers from some of the worst air pollution in the United States (American Lung Association, 2014). Shallow daytime mixed layer heights and low wind speeds in winter lead to the accumulation of pollutants across the valley (San Joaquin Valley Air Pollution Control District, 2003). Previous observations in the SJV region have found a build-up of NH4NO3 during pollution episodes (e.g. Chow et al., 2008). Approximately 30 – 80% of the wintertime PM2.5 mass in this region is ammonium nitrate (NH4NO3), with a strong diurnal variability, and most other PM2.5 being organic matter (Chow et al., 2006; Ge, 2012; Young et al., 2016; Parworth et al., 2017). During DISCOVER-AQ specifically, NO3-(p) was found to represent 28% of non-refractory PM1.0 (PM with aerodynamic diameter < 1 μm) mass on average (Young et al., 2016).
Figure 1.
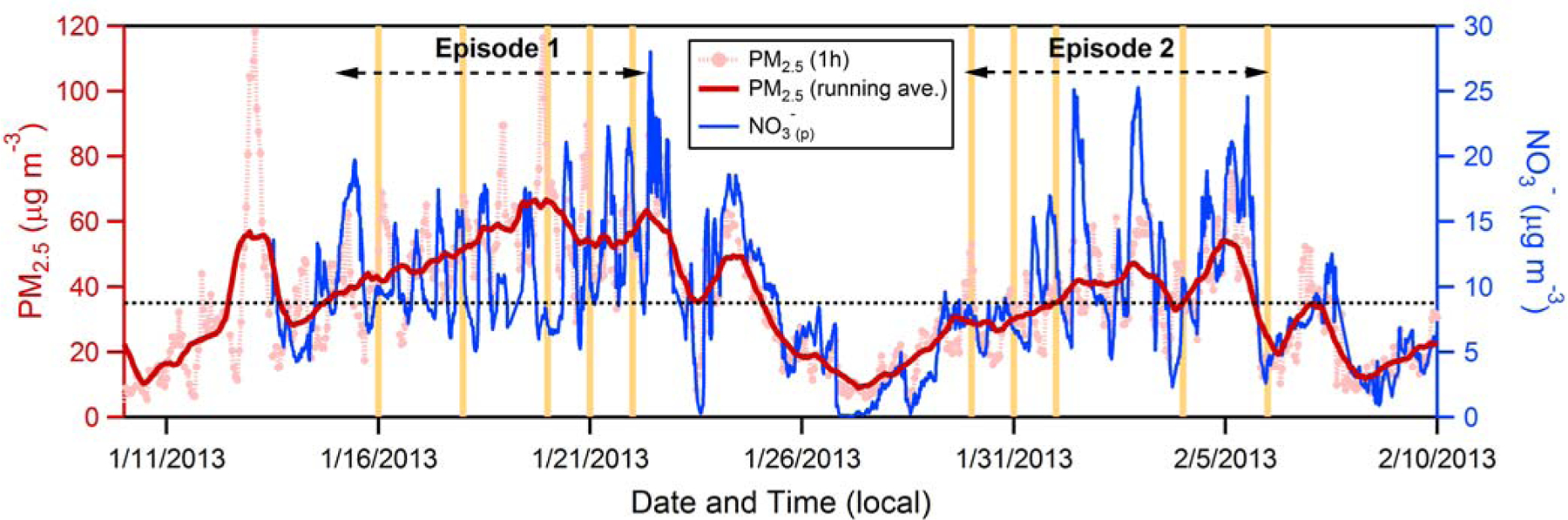
Time series of surface PM2.5 concentration (μg m-3) measured in Fresno during the DISCOVER-AQ campaign for 1 h averages (light red dotted line) and for a running average (red line; smoothed over 24 h), along with the 1 h average NO3-(p) concentration (blue line). The vertical orange lines indicate the days on which airborne measurements were made. The horizontal dashed black line indicates the NAAQS 24 h standard of 35 μg m-3 for PM2.5.
An important role for nocturnal NO3-(p) production in this region has been previously identified based on observations of long-term trends, the spatial and diurnal variability in NO3-(p), and the chemical environment in and around Fresno. For example, Watson and Chow (2002) reported a sharp, early morning (~9 am) increase in surface NO3-(p) concentrations on many days of a severe pollution episode in 2000 and suggested that this behavior was consistent with mixing down of nitrate-rich air from the RL aloft. Young et al. (2016) and Parworth et al. (2017) observed similar behavior more than a decade later during DISCOVER-AQ in 2013. Pusede et al. (2016) characterized the relationship between long-term (multi-year) surface measurements of wintertime NO3-(p) and NO2 in Fresno and Bakersfield and showed that the decline in NO3-(p) in SJV over time (2001–2012) was predominately driven by reduced nocturnal NO3-(p) production in the residual layer. The balance between production, especially night time production, and daytime losses was identified by them as critical to understanding the multiday build-up during pollution events. Further, they concluded from DISCOVER-AQ aircraft measurements that much of the NO3-(p) production was localized over the cities given the sharp urban-rural gradients in NO3-(p); the spatial gradients in 2013 (from (Pusede et al., 2016)) seem to be sharper than gradients in 2000 (from (Chow et al., 2006)), likely reflecting the increasing localization of the NO3-(p) pollution to the urban centers as overall NO3-(p) concentrations in the region have decreased. Brown et al. (2006a) observed that the number concentration of accumulation mode particles (0.32–1.07 μm) often increased above the surface at 90 m AGL compared to surface (7 m AGL) measurements during night, and suggested that this was due to growth of smaller particles into the accumulation mode via NO3-(p) formation. They also observed that the concentration of NO3-(p) at 90 m AGL often increased at night, suggestive of in situ production.
The present study builds on this literature by examining the role that aloft nocturnal nitrate production, in concert with other processes, has in determining surface NO3-(p) concentrations during the DISCOVER-AQ campaign that took place in January and February 2013 in the SJV. Our study combines aircraft and surface observations from DISCOVER-AQ (Fig. S1). During DISCOVER-AQ, two pollution episodes were observed during which PM2.5 concentrations were elevated (Young et al., 2016). The analysis here focuses on quantitative assessment of NO3-(p) concentrations during this first episode (14 – 22 January) in terms of the processes that govern the NO3-(p) diurnal behavior; the observed behavior during this first episode is qualitatively compared with that during the second episode (30 January – 6 February) to examine the factors that contribute to episode-to-episode variability. On flight days, in situ measurements of the vertical profiles of particulate and gas concentrations above Fresno (and other SJV cities) were made three times: in the mid-morning (~9:30 am), around noon and in the mid-afternoon (~2 pm). These measurements allow for assessment of the daytime evolution of the vertical distribution of PM and gases as well as characterization of the time-varying boundary layer height. They also allow for determination of the overnight evolution of the PM vertical distribution, which can be used to characterize the factors that control NO3-(p) concentrations in the RL. The influence of processes occurring aloft on the temporal evolution of NO3-(p) surface concentrations is quantitatively evaluated for this case-study using an observationally constrained 1D box model. The box model accounts for both vertical mixing (entrainment) of air to the surface and for photochemical NO3-(p) production, as well as NO3-(p) loss processes. Ultimately, the observations and analysis further illustrate how daytime surface-level NO3-(p) concentrations depend on a combination of both nocturnal and daytime production of NO3-(p), vertical mixing, altitude-dependent advection in the RL overnight, daytime entrainment of clean air from the free troposphere (FT) and evaporation-driven dry deposition. The model and observations are used to examine the relative importance of these different pathways during the case-study episode considered. This work adds to the existing literature by providing an observationally based, case-study demonstration of how nocturnal processes occurring aloft—in concert with other processes—exert a major control over the evolution of pollution episodes within the SJV specifically, and likely in other regions as well.
2. Materials and Method
Airborne in-situ measurements (such as particle scattering, gas-phase concentrations, RH and temperature) during the DISCOVER-AQ campaign were made by a suite of instruments on board the P3-B NASA aircraft. The flight path flown during each of the three legs for each flight day is shown in Figure S2. The aircraft measurements were complemented by a network of ground measurement sites, of which Fresno was one. At Fresno, continuous, in situ measurements of the chemical composition and physical properties of particulate matter were performed along with measurement of NAAQS regulated pollutants (Young et al., 2016; Zhang et al., 2016; Parworth et al., 2017). Local conditions during DISCOVER-AQ were relatively cool (Tavg = 7.9 °C) and dry (RHavg = 69%) with frequent sunshine and no substantial fog. All data are archived at the DISCOVER-AQ website (NASA Atmospheric Science Data Center). Details of all measurements made are provided in Appendix A and summarized in Table A1.
Table A1.
Summary of instruments deployed and measurements on ground and on aircraft made during the DISCOVER-AQ campaign.
| Platform | Measurement | Instrument | Uncertainty | Response time |
|---|---|---|---|---|
| NASA P3-B aircraft + Ground | Total and submicron scattering at 450, 550 and 700 nm | Integrating Nephelometer (TSI 3563) | 5% | 1 s |
| NASA P3-B Aircraft | Nitrate (gas+particle) | Thermal Dissociation – Laser Induced Fluorescence (TD-LIF) | 15% | 1 s |
| NASA P3-B Aircraft | NASA P3-B Carbon monoxide (CO), Methane (CH4) | Differential Absorption CO Measurement (DACOM) | < 2% | 1 s |
| NASA P3-B Aircraft | Nitrogen monoxide (NO), Nitrogen dioxide (NO2), and Ozone (O3) | 4-channel Chemiluminiscence | 10% for NO, 15% for NO2, and 5% for O3 | 1 to 3 s |
| NASA P3-B Aircraft | Ammonia (NH3) | Picarro G2103 | 35% | 10 s |
| NASA P3-B Aircraft | Aerosol size distribution (0.06 – 1 μm) | Ultra-High Sensitivity Aerosol Spectrometer (UHSAS) | 20% | 1 s |
| NASA P3-B Aircraft | Meteorological and navigational measurements onboard | P3-B Project Data System (PDS) | - | 1 s |
| Ground | PM2.5 mass concentration | Beta-Attenuation Mass (BAM) Monitor | 16% | 1 h |
| Ground | NO, NO2 | Chemiluminiscence | 20% | 1 h |
| Ground | O3 | NIST Standard Reference Photometer (SRP) | 2% | 1 h |
| Ground | Speciated non-refractory PM1.0 | High Resolution Time-of-Flight Aerosol Mass Spectrometer (HR-ToF-AMS) | 25% | 5 min |
| Ground | Water-soluble components of PM2.5 | Particle-Into-Liquid Sampler (PILS) coupled with two Ion chromatography systems | 10 – 20% | 20 min |
| Ground | Aerosol Particle Size | Scanning Mobility Particle Sizer (SMPS) | 10% | 1 min |
| Ground | Aerosol Particle Size | Aerodynamic Particle Sizer (APS) | 20% | 1 s |
| Ground | Refractive black carbon mass concentration | DMT Single Particle Soot Photometer (SP2) | 30% | 5 min |
| Ground | Relative humidity and temperature | Temperature: ± 0.1 K RH: ± 2% |
1 h |
3. Results and Discussion
3.1. Vertical distribution of NO3-(p)
The concentration and vertical distribution of NO3-(p) in the RL ([NO3-(p)]RL) in the morning serves as the initial condition constraint on what is mixed down to the surface as the day advances and the ML rises. Thus, knowledge of the vertical distribution of NO3-(p) in the RL near sunrise is needed to predict the temporal evolution of surface-level NO3-(p) during the daytime, as will be done below. Night time flights were not made during DISCOVER-AQ to allow for characterization of the overnight evolution of the RL. However, the early morning (~09:30 local time) vertical profiles over Fresno allow for characterization of the vertical structure of most of the RL near sunrise (~07:10 local time), as the surface boundary layer height at this point is still quite shallow (~50 m; see Appendix B for a description of the mixed boundary layer height determination method, Figures B1–B2). Fast measurements of total NO3- (gas + particle, NO3-(g+p)) were only available for a subset of flights (Pusede et al., 2016), and particulate-only NO3- measurements were not made with sufficient time resolution, less than about a minute, to allow for robust characterization of the NO3-(p) vertical profile. Therefore, NO3-(p) vertical profiles for each flight during Episode 1 are estimated from in situ measurements of dry particle scattering and the influence of water uptake on scattering, i.e. from the particle hygroscopicity, and calibrated against the slower PILS measurements (Appendix A, Figure A1). The derived, observationally constrained NO3-(p) profiles based on the estimated NO3-(p) exhibit generally good correspondence with the sparser direct measurements of NO3-(g+p), although on one of the two days available for comparison the total NO3- somewhat exceeds the estimated NO3-(p) below ~75 m (Figure 2). This indicates that the estimation method is reasonable, especially since most nitrate is expected to be in the particle-phase (Parworth et al., 2017) given the high relative total ammonium (NH3 + NH4+) concentrations (Figure 3). Only four out of five flight days during Episode 1 have been included in this analysis due to insufficient data on 16 January.
Figure B1.
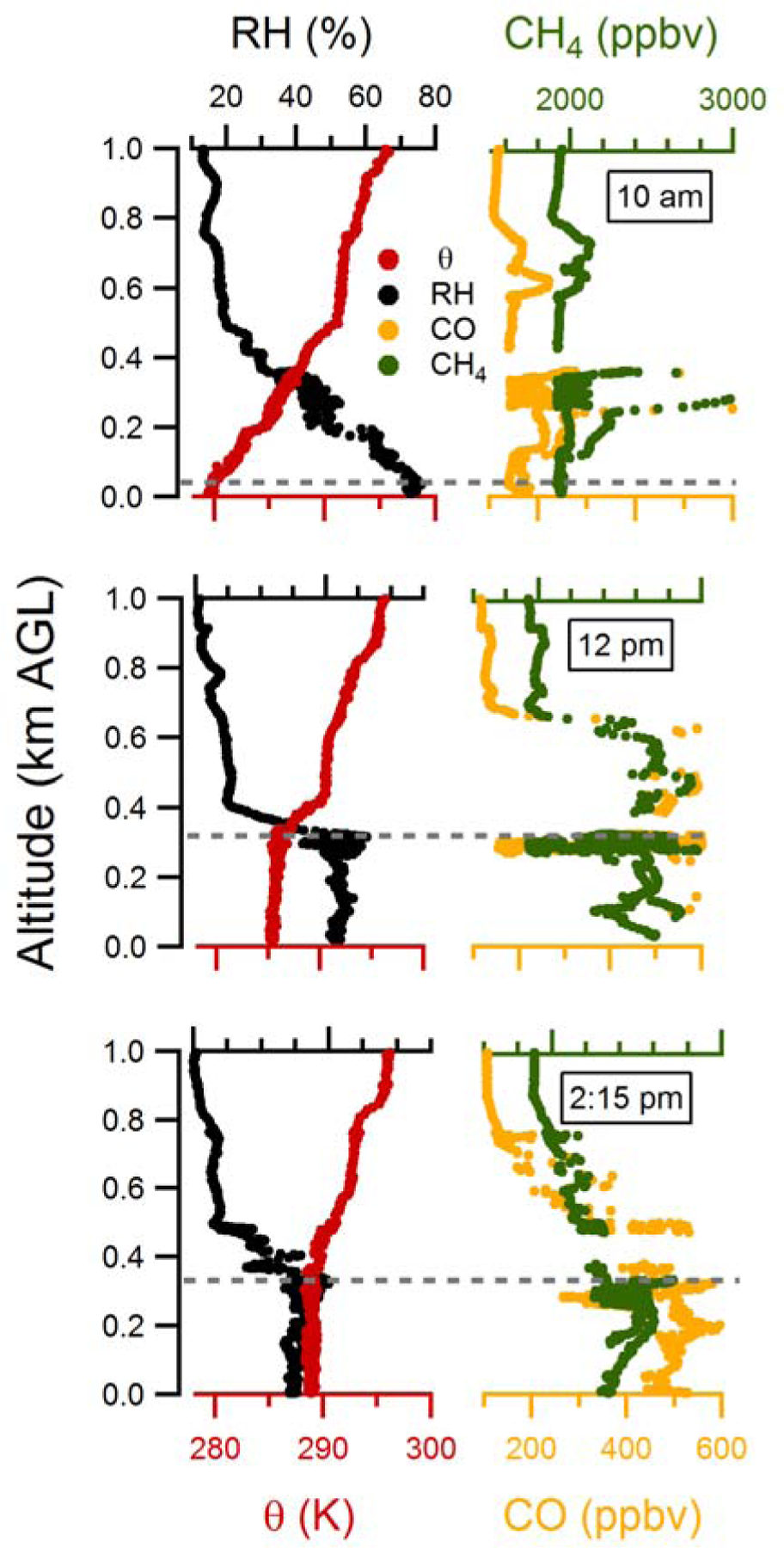
Vertical profiles of potential temperature, θ (K), relative humidity, RH (%), mixing ratios of carbon monoxide, CO (ppbv), and methane, CH4 (ppbv) measured from the P3-B aircraft over Fresno on 18th January, 2013. The horizontal dashed grey line indicates the mixed boundary layer heights.
Figure B2.
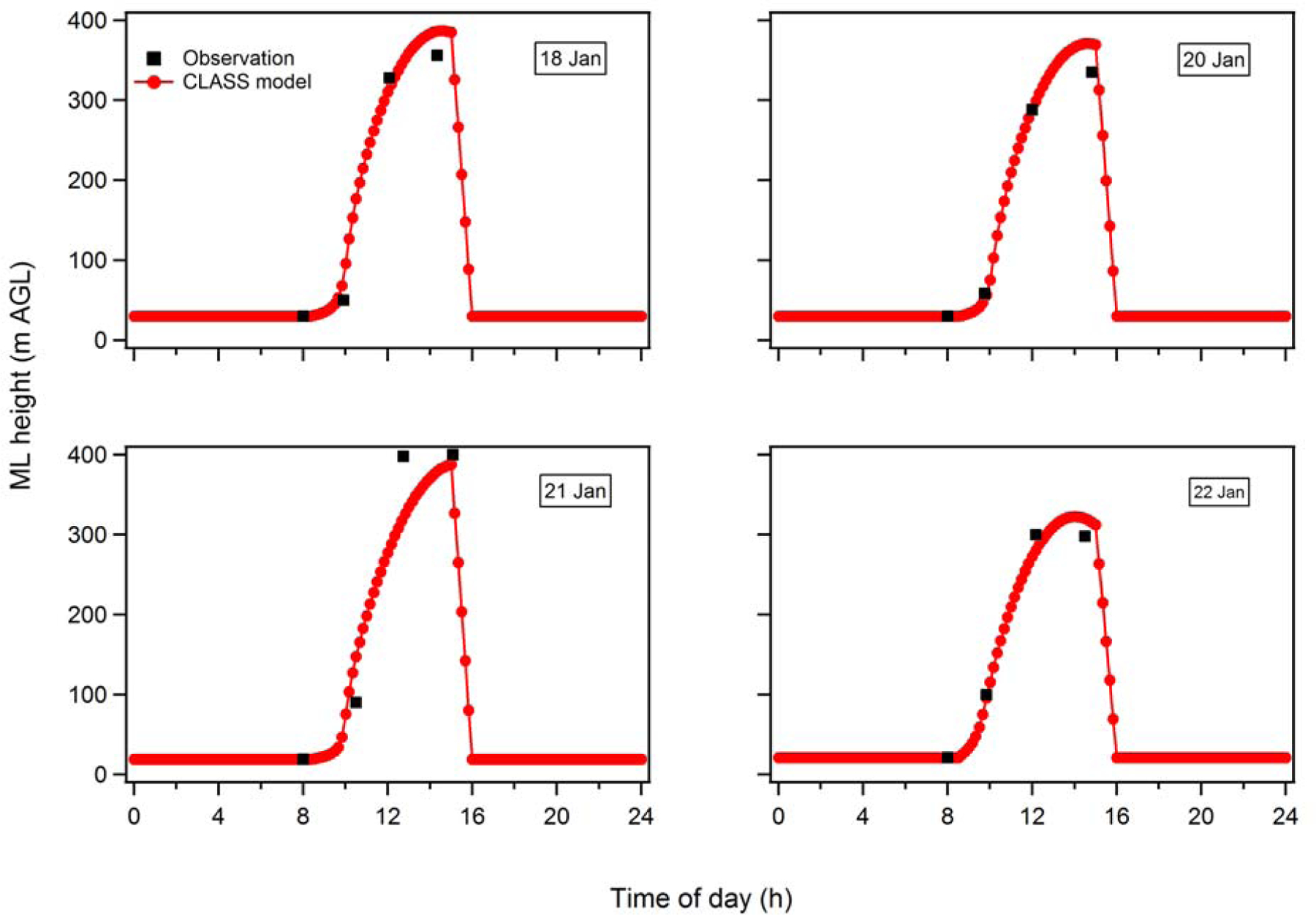
Evolution of the ML height with time (starting at 8 am) on the four flight days in Episode 1. The observational constraints are shown as black circles, where the first point comes from nearby balloon sonde measurements and the last three from the P3-B vertical profiles.
Figure A1.
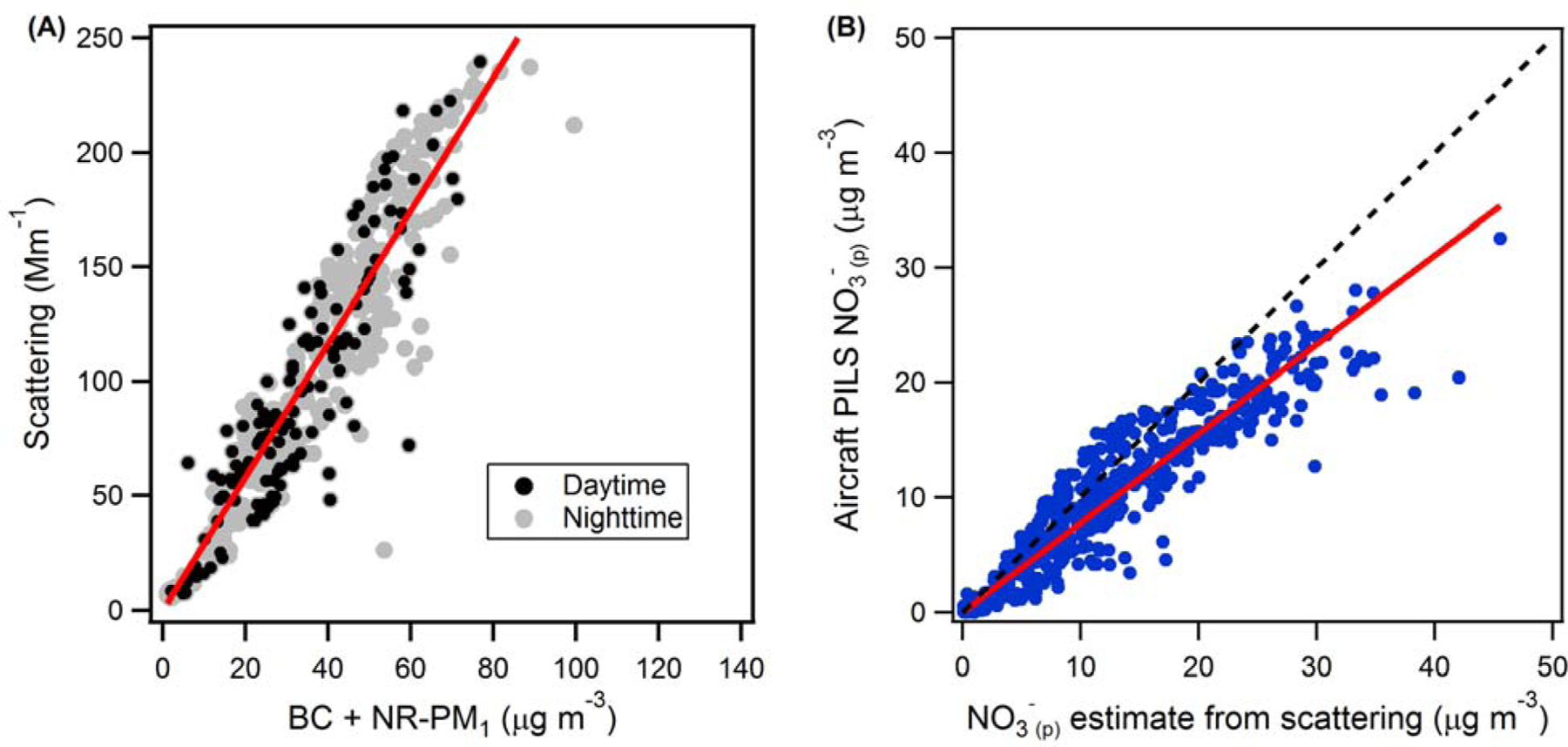
(A) Total particle scattering at 550 nm (Mm-1) versus PM1.0 mass (submicron black carbon, BC + non-refractory PM1.0, NR-PM1) concentration (μg m-3) observed at ground-level in Fresno. The solid red line is the orthogonal distance regression fit including data only during the daytime (black circles) between 8 am and 4 pm; slope = 2.83 Mm2μg-1. (B) NO3-(p) concentration measured by PILS on P3-B aircraft versus that estimated from scattering using the relation NO3-(p) = γσsca,dry/2.83. The solid red line is the linear fit to the data, with slope = 0.78. The dashed black line is the 1:1 line.
Figure 2.
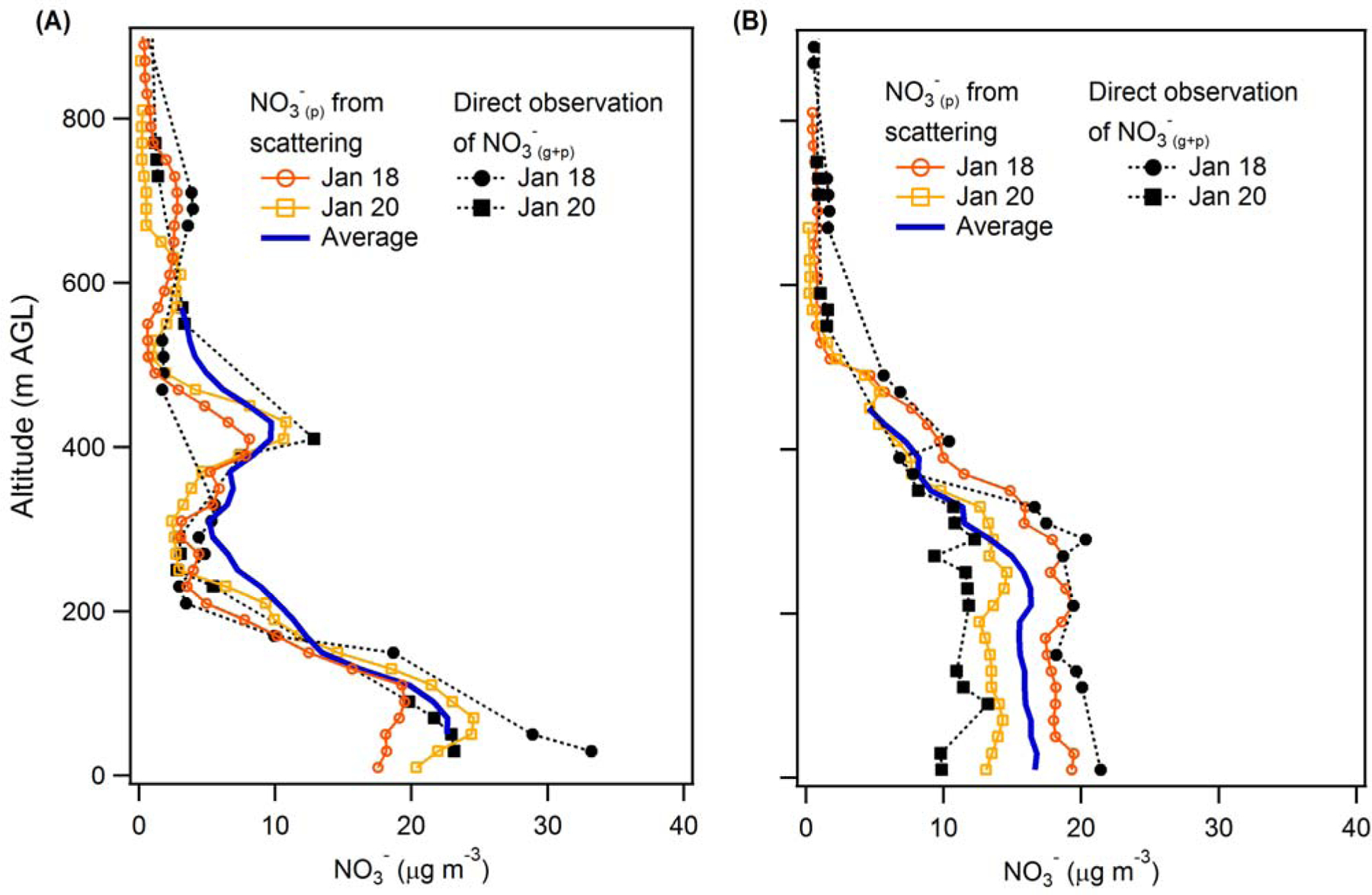
Vertical profiles for two individual flight days of particulate nitrate concentrations estimated from in situ total particle scattering measurements (open markers) and total nitrate (gas + particle) concentrations measured by the TD-LIF (solid black markers) for (A) the morning (~9:30 am) and (B) the afternoon ~2:30 pm. The solid blue lines indicate the average NO3-(p) vertical profiles for all four flight days of Episode 1 (Jan 18, 20, 21 and 22).
Figure 3.
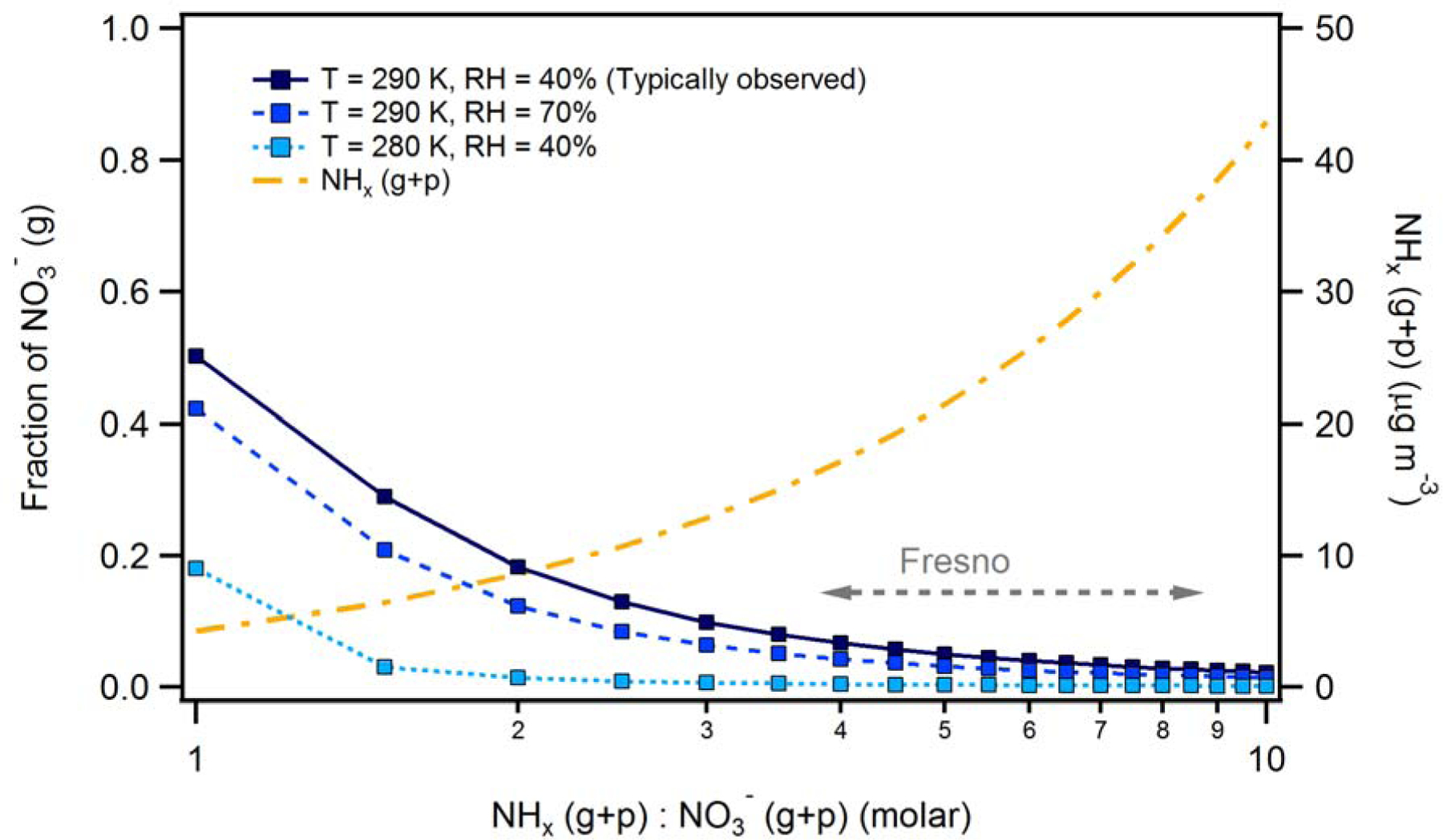
The gaseous fraction of total nitrate versus the molar ratio of total ammonia to total nitrate (ppb) under different environmental conditions (blue lines). The total ammonia is the sum of NH3(g) measured on P3-B close to ground (< 20 m AGL) and NH4+(p) at ground-level measured by PILS at approximately same time. The total nitrate is the NO3-(g+p) measured by TD-LIF close to ground (< 20 m AGL). The grey dashed arrow indicates the observed range of molar ratio values during the campaign period. The total (gas + particle) ammonia is shown for reference (orange line).
Over Fresno, the observed afternoon (~2:30 pm) NO3-(p) concentrations are nearly constant with altitude up to ~400 m (the daytime boundary layer height) (Figure 2B) whereas the early-morning NO3-(p) concentrations decrease steeply with altitude up to ~350 m (Figure 2A). Corresponding vertical profiles for NO, NO2, O3, relative humidity, temperature and total particle scattering are shown in Figures S2 (early morning) and S3 (afternoon). Like NO3-(p), all indicate substantial differences between the early morning and afternoon profile shapes. This provides a strong indication that altitude-specific processes occur overnight that lead to a reshaping of the NO3-(p) vertical profile. At some altitudes the NO3-(p) in the early-morning RL is greater than the NO3-(p) measured in the previous afternoon, indicating net production, while at other altitudes the early-morning RL NO3-(p) is less than the previous afternoon, indicating net loss (Figure 2). As noted by Pusede et al. (2016), there tend to be sharp concentration gradients in NO3-(p) and NOx between the city and surrounding areas, with lower concentrations outside the city. Thus, whether NO3-(p) at a given altitude increases or decreases overnight results from the competing effects of chemical production versus horizontal advection bringing in this (typically) cleaner air from outside the city. (In the absence of a strong jet aloft and no convective mixing, night time entrainment of cleaner FT air into the RL is expected to be considerably slower than horizontal advection.) Like NO3-(p), the boundary layer is reasonably well mixed with respect to NOx, O3 and particles at the time when decoupling of the RL occurs, around 3 pm the previous day (Figure S4). Box model calculations indicate that the expected local nocturnal chemical production of nitrate in the RL should exhibit relatively minor vertical variation due to variations in temperature and RH alone (Figure S5). In other words, without advective loss or dilution processes of either NO3-(p) or the precursor gases it is expected that the NO3-(p) concentration would increase to a similar extent at all RL altitudes.
The substantial changes observed in the shape of the vertical profile overnight indicate that night time differential advection in the RL is a major factor in determining the shape of the morning NO3-(p) vertical profile during this pollution episode. Differential horizontal advection serves to directly export NO3-(p) from the urban area and import cleaner air from surrounding areas. Secondarily, as NOx concentrations are also lower outside of the Fresno urban area (Pusede et al., 2014), this differential advection will also influence the over-city concentrations of precursors gases (NOx and O3; Figure S3–S4) and consequently the altitude-specific nitrate production, with decreases likely. This is supported by surface-level measurements of NOx and O3 made in Fresno and in the nearby and much more rural cities of Parlier (located 35 km SE of Fresno) and Madera (located 40 km NW of Fresno). The NOx and NO2 concentrations are higher and the O3 lower in Fresno compared to the surrounding cities throughout the day, and the instantaneous nitrate production rate ([NO2][O3]) is substantially higher in Fresno in the late afternoon, when decoupling occurs (Figure S6). The important implication is that overnight advection both directly and indirectly alters the vertical NO3-(p) profile and decreases the over-city NO3-(p) concentrations in the morning, which will consequently serve to limit the extent of localized pollution build-up during events. The impact of overnight differential advection on reshaping the vertical distribution of NO3-(p) has likely increased over the last 15 years as the sharpness of the urban-rural concentration gradients has increased (Chow et al., 2006; Pusede et al., 2016). Nonetheless, the NO3-(p) advected from urban areas in the RL will contribute to the regional SJV background and serve to sustain NO3-(p) levels across the valley during pollution episodes.
In the summer, transport and dispersion of pollutants has been attributed to low-level winds (less than 500 m AGL) in the SJV (Bao et al., 2008). We suggest that a similar, but weaker, circulation may exist even in the winter, just at much slower wind speeds, and that this advection overnight is what leads to differential wash out and the establishment of the particular vertical NO3-(p) concentration profiles in the RL. The concentration of NO3-(p) will likely be lowest in the early-morning RL at altitudes where horizontal advection has the greatest impact. Wind profiler measurements made in nearby Visalia, CA (65 km SE of Fresno) indicate that during the night (19:00 – 07:00) there was local maximum in the mean wind speed at ~250 m, which is around the altitude at which the early-morning NO3-(p) concentration is minimum (a). Below 250 m there was a monotonic increase in the night time mean wind speed with altitude, with very slow speeds observed at the surface. Above 250 m the mean wind speed was relatively constant to ~450 m, above which it increased with altitude. Explicit comparison between the vertical profiles of night time mean wind speed and the estimated early-morning NO3-(p) concentration indicates an inverse relationship (r = −0.98) between the two (Figure S7). This is consistent with the idea that differential advection as a function of altitude overnight serves to shape the early-morning concentration profiles. The wind direction at lower altitudes (~150 m) was generally more variable than those at higher altitudes (285 m or 450 m), and with a general shift from more westerly at lower altitudes (but above the surface) to more northerly near the top of the RL (b). (Note: vector average wind speeds for each individual night were calculated and then a scalar average of these night-specific vector averages was calculated to give the episode-average mean wind speeds. This averaging process emphasizes directional consistency of the winds on a given night, but not between nights.) The increase in NO3-(p) concentration at ~400 m AGL in the early-morning profile, especially noticeable on Jan 21 (Figure S8), could result from a slowing of the winds near the top of the RL or from enhanced recirculation of pollutants at higher altitudes. Regardless of reason, this work indicates that the gradient between the local (above city) and regional NO3-(p) and precursor gases, evident in Pusede et al. (2016), is an important factor in determining the night time evolution of the RL vertical profile. Explicit characterization of the temporal evolution of the vertical structure of NO3-(p) within the night time RL would provide further insights into the altitude-specific processes that control the shape of the early-morning profile (and thus the concentration of NO3-(p) aloft that can be mixed to the surface in daytime).
The difference between the concentration of NO3-(p) at each altitude of the early morning vertical profile and that at 3 pm on the preceding afternoon (∆[NO3-(p)]RL) yields the net overnight NO3- production or loss in the RL. If it is assumed that the layer with the highest NO3-(p) is not influenced by advection, then the ∆[NO3-(p)]RL in this layer provides an estimate of the maximum chemical production (PNO3-). This estimate of PNO3- is certainly a lower bound on actual nitrate formation given the assumption of no influence of horizontal advection, and this also does not account for produced nitrate that remains in the gas-phase (although this is likely to be small). On average, the observations indicate that chemical production overnight in the RL leads to an approximate doubling over the initial NO3-(p) concentration, or 10–25 μg m-3 of NO3-(p) produced over the course of the night for this episode (Table S1). Observed day-to-day variability in PNO3- likely results from day-to-day variations in precursor (NOx and O3) concentrations and N2O5 reactivity, as well as limitations of the assumption of no advection in this layer. To assess the reasonableness of this estimate of PNO3 as a maximum production rate, values of the night-specific average rate coefficients for N2O5 heterogeneous hydrolysis (kN2O5) and associated uptake coefficients (γN2O5) needed to reproduce the observed PNO3- are back-calculated based on the initial NOx, O3, and wet particle surface area and assuming ClNO2 formation is negligible (see Appendix C and Table S1). The derived kN2O5 values range from 1.3 – 5.1 x 10-5 s-1 with corresponding γN2O5 from 2.5 x 10-4 to 4.8 x 10-4. These are smaller than values observed under water-limited conditions in other field studies (Brown et al., 2006c; Bertram et al., 2009) and lower than expected based on lab experiments (Bertram et al., 2009). γN2O5 values separately calculated from the particle composition measurements, following Bertram et al. (2009), are larger than the above back-calculated values, with γN2O5 ~ 10-3, and more consistent with the literature. This suggests that the PNO3- is, in fact, a lower estimate and that the NO3-(p) concentration in even the lower layers of the RL is influenced by advection. Box model calculations using the (too low) back-calculated kN2O5 and γN2O5 yield ~15–42% NOx conversion to HNO3 overnight during this episode. If instead γN2O5 = 10-3 is used, the calculated overnight conversion is somewhat larger, ~52%. Also, if kN2O5 and γN2O5 were assumed sufficiently large such that they are not rate limiting the overnight conversion increases further to ~63%. It should be noted that during this episode the surface O3 overnight is essentially completely titrated away by 6 pm (Figure 5). The reaction between NO2 and O3 (R1) is thus very slow and night time chemical production of NO3-(p) at the surface in the NBL is comparably small.
Figure 5.
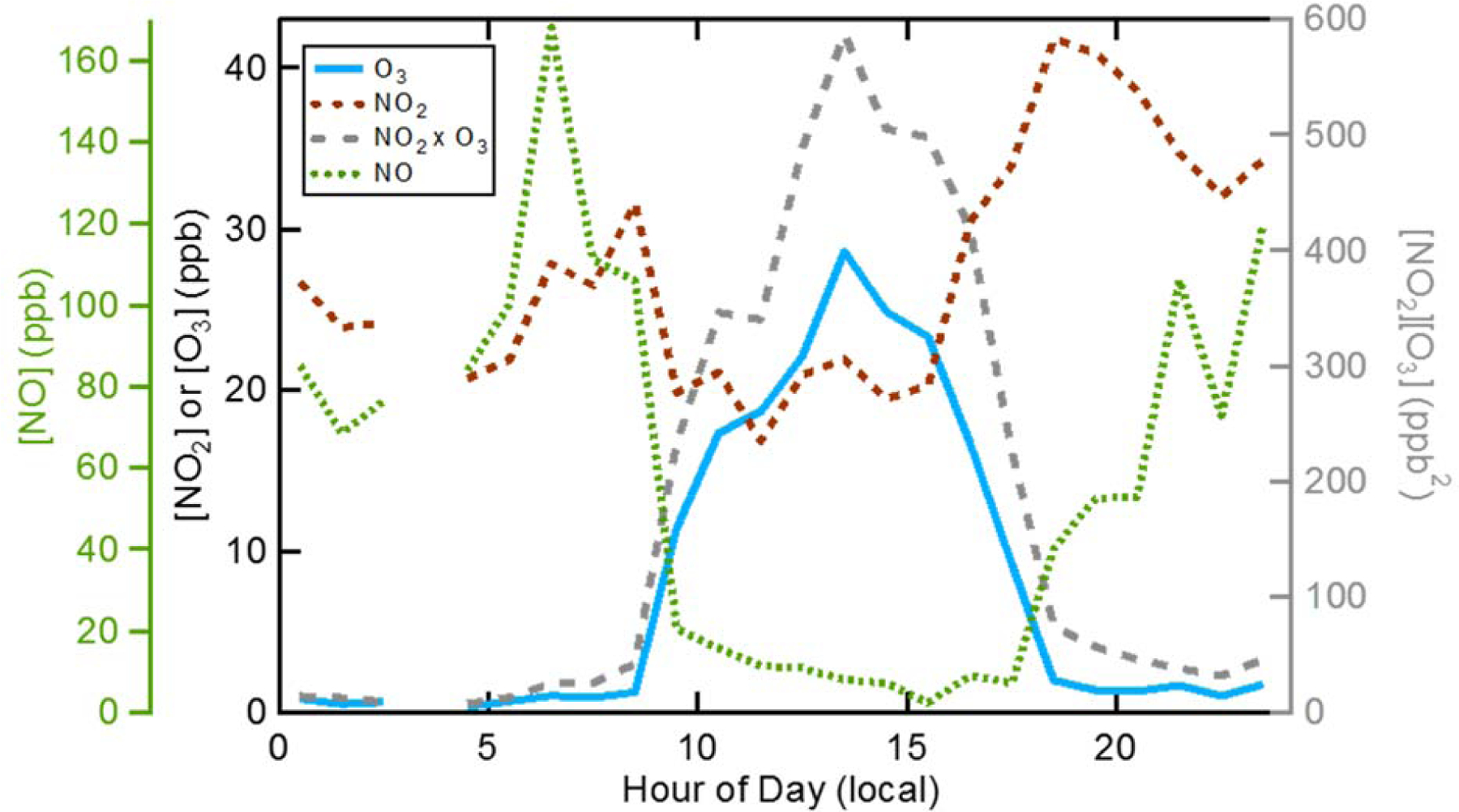
Diurnal profiles for ozone (blue), NO2 (brown), NO (green) and the product of O3 and NO2 (gray) for the first pollution episode.
3.2. Vertical mixing, photochemical production and NO3-(p) sinks
The observed episode average surface-level NO3-(p) concentration exhibits a distinct, rapid increase starting at ~ 8 am, then peaks around 10 – 11 am local time (LT) and decreases fairly continuously after the peak, especially between 1 – 4 pm (Figure 6A). For reference, time series of NO3-(p) during the pollution episode, along with CO, NO, NO2, O3, temperature, surface radiation, and PM1 are shown in Figure S9. Both Young et al. (2016) and Pusede et al. (2016) noted this increase, arguing it is a signature of nocturnal nitrate production. Here, we provide a more detailed examination of the specific influence of vertical mixing and nocturnal NO3-(p) production in the RL on the observed daytime variability in surface-level NO3-(p) using an observationally constrained one dimensional box model (see Appendix D for details). In brief, the model accounts for time-dependent mixing between air in the mixed boundary layer and the RL, daytime photochemical production of nitrate, gas-particle partitioning of nitrate, entrainment of clean air from the free troposphere into the ML and loss of nitrate via dry deposition to calculate the time-dependent evolution of the surface-level NO3-(p) concentration. The observed vertical profiles of NO3-(p) concentrations in the RL (referred to as [NO3-(p)]RL and taken as the observed early-morning and noon profiles) provide a unique constraint for understanding and quantifying the influence of vertical mixing specifically, allowing us to expand on previous studies. The model is additionally constrained by the surface-level concentrations of NO2 and O3, and temporally varying ML height. The evolution of the daytime ML height and rate of entrainment are determined using the Chemistry Land-surface Atmosphere Soil Slab (CLASS) model (https://classmodel.github.io/; Ouwersloot and Vilà-Guerau de Arellano, 2013). The CLASS model is constrained by observations of the time-dependent vertical profile measurements of temperature, RH and other gas-phase species over Fresno and by T and RH profiles and surface sensible heat flux measurements at nearby Huron, CA (~83 km SSW of Fresno) (Appendix B). Starting at around 8 am, the ML begins to grow vertically by entraining air from the RL. It is assumed that air within the ML is instantaneously mixed throughout the volume. Within the (shrinking) RL the NO3-(p) is assumed to retain the initial profile shape until it reaches the maximum ML height observed in the afternoon (~12:30 pm). After this point entrainment of free tropospheric air (FT) begins. The concentration of NO3-(p) in FT air is determined from the vertical profile observed around noon. While entrainment of FT air also alters the NO2 and O3 concentrations in the mixed layer, since these are constrained by the surface observations (within the mixed layer) this is accounted for. Photochemical production of HNO3 is calculated based on the oxidation of NO2 by hydroxyl radicals, with wintertime concentrations estimated to peak around [OH] = 106 molecules cm-3 at noon in the region, with contributions from O(1D) + H2O (from NO2 photolysis), HONO photolysis and CH2O photolysis (Pusede et al., 2016). The OH concentration is assumed to scale linearly with the observed solar radiation (Figure S10).
Figure 6.
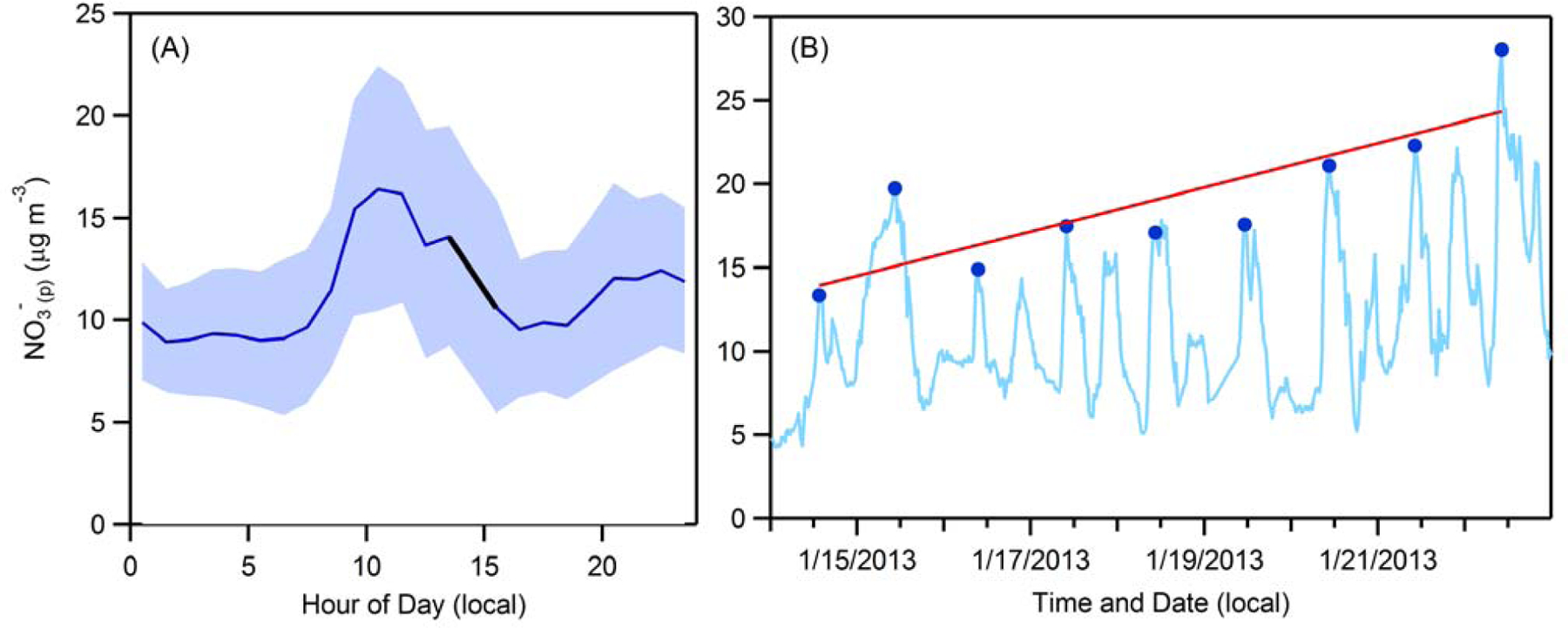
(A) Average diurnal profile (solid line) of surface NO3-(p) for all days of Episode 1. The shaded area indicates the 1σ standard deviation. The solid black line is a linear fit (r2 = 0.99) to the data between 1:30 pm and 3:30 pm. (B) Time series (solid blue line) of surface-level NO3-(p) during Episode 1. The circles indicate the daytime peak values. The linear fit (red line) to the daytime NO3-(p) peaks suggest an increase of 1.32 μg m-3 day-1.
The average calculated daytime temporal evolution of surface NO3-(p) from the observationally constrained box model agrees reasonably well with the average of the surface observations from the four Episode 1 flight days considered (Figure 7A). (The observed diurnal average in Figure 6 uses all of the days from Episode 1 whereas in Figure 7 only four flight days are included. This is because the initial early-morning NO3-(p) vertical profile is required as input to the model.) The model predictions for the individual flight days also exhibit generally good agreement with the NO3-(p) observations except in the late evening, discussed further below (Figure S8). Specifically, the observationally constrained model also shows a rapid increase in NO3-(p) beginning at 8 am, a peak around 10–11 am and a gradual, time-varying decrease through the afternoon.
Figure 7.
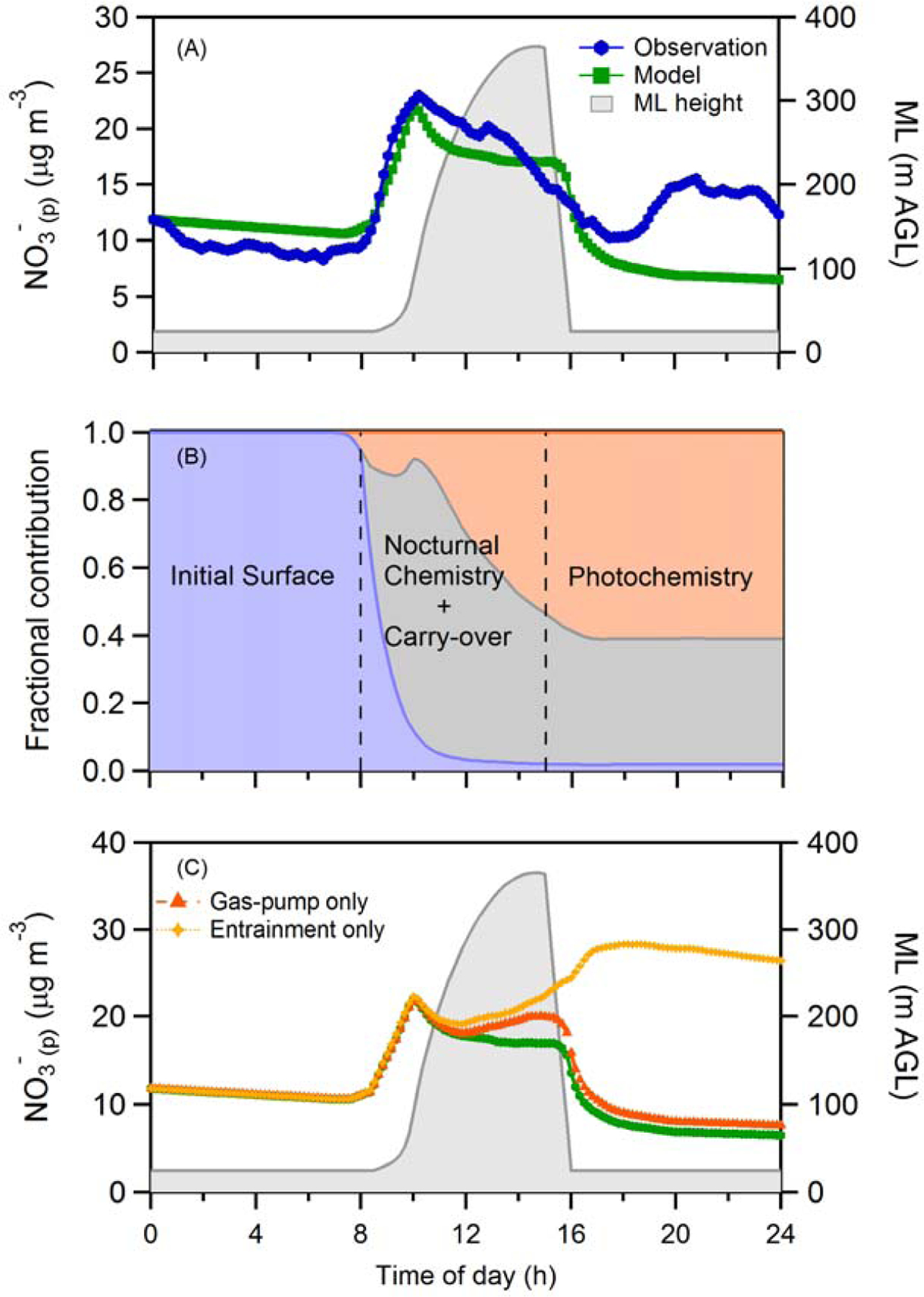
(A) Comparison between the observed (blue circles) and observationally constrained model predicted (green squares) diurnal profile of the surface NO3-(p) concentration (μg m-3) for the four flight days (18th, 20th, 21st and 22nd January, 2013) during Episode 1. Also shown is the diurnal variation in the boundary layer height (gray), as constrained by daytime measurements. (B) The diurnal variation in the simulated fraction of the total surface-level NO3-(p) contributed by the initial surface-level NO3-(p) (i.e. that at surface-level at 12:00 am), the NO3-(p) mixed down from the RL, and NO3-(p) produced from daytime photochemical reactions. (C) Comparison between the simulated diurnal profile when all processes are included (green squares, same as Panel A) and when only one NO3-(p) sink at a time is considered. The individual sinks considered are only entrainment of free troposphere air (yellow crosses) or only dry deposition of HNO3 via the gas-phase pump (orange triangles).
Consideration of the individual processes occurring in the model demonstrates that vertical mixing down of [NO3-(p)]RL and the shape of the [NO3-(p)]RL vertical profile predominately control the morning-time evolution of the surface NO3-(p) during this episode (Figure 7 and Figure 8). The particularly steep rise in the surface-level NO3-(p) in the morning results from the combination of the NBL height being exceptionally shallow (only ~20 m) and the NO3-(p) in the low-altitude region of the RL being greater than the NO3-(p) in the early-morning NBL. The peak and turnover in surface-level NO3-(p) occurs when even higher RL layers, where [NO3-(p)]RL < [NO3-(p)]ML, are entrained. In other words, the temporal evolution of the surface-level NO3-(p) is linked to the shape of the early-morning vertical NO3- profile. Further, it should be noted that the exact model behavior is dependent on the timing of the CLASS-predicted boundary layer height increase, with the initial increase and timing of the surface-level NO3-(p) peak being particularly sensitive to the shape of the rise between 8 and 10 am. Nonetheless, because the NBL is so shallow here, only ~3–12% of the daytime ML height, the surface concentration is strongly impacted by the concentrations in the RL and the initial (pre-8 am) surface-level nitrate has control over daytime concentrations. Thus, the model results demonstrate that the observation of the large 10 am peak in NO3-(p) is a clear indication of the strong influence of nocturnal processes occurring aloft—both chemical production and advection-driven local loss—on daytime surface concentrations.
Figure 8.
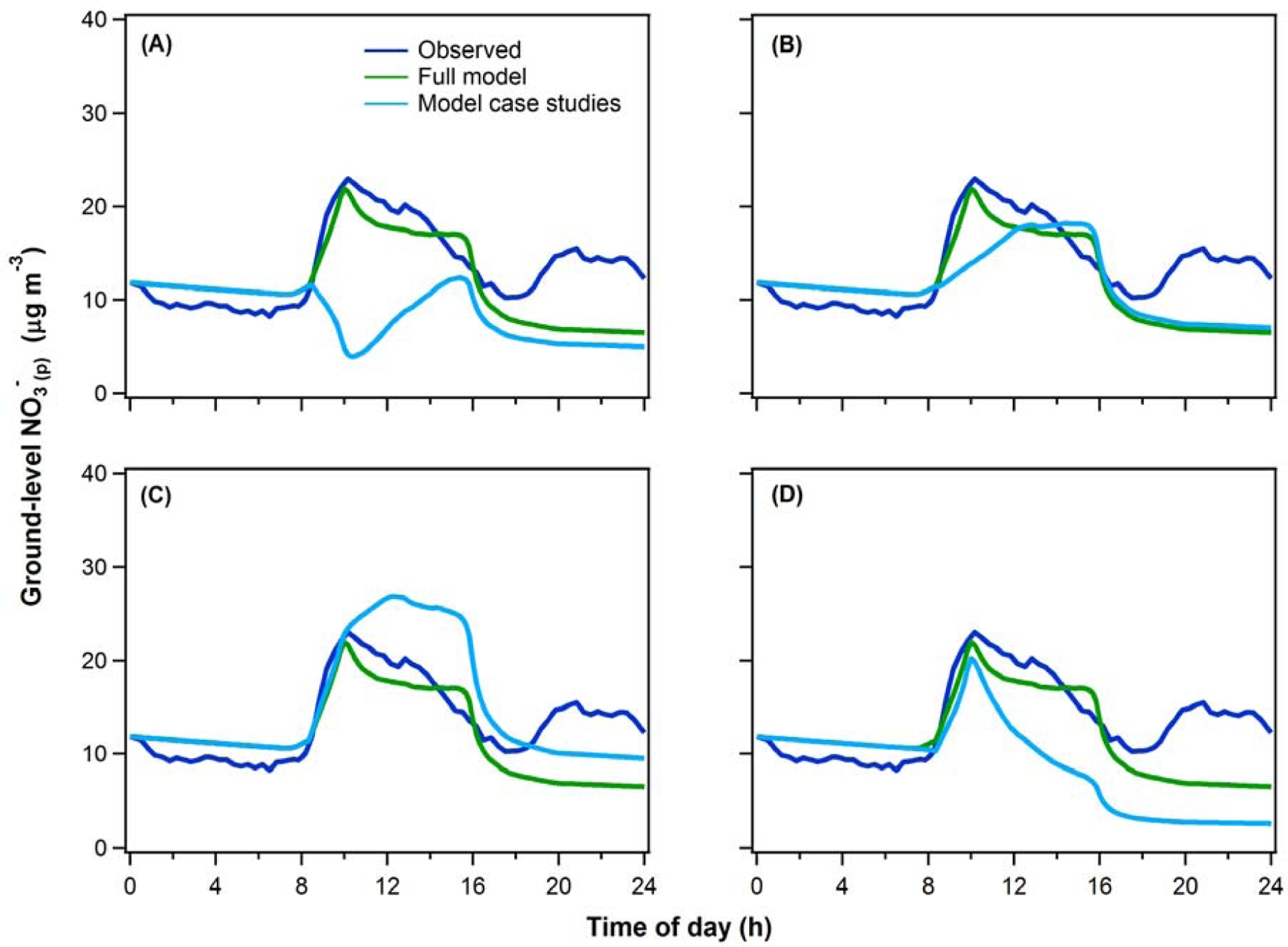
Model predictions of the diurnal variation in surface-level NO3-(p) under (A-C) different assumptions regarding the NO3-(p) concentration and vertical variability in the early-morning RL, or (D) without daytime photochemical production of NO3-(p). In all panels the blue curve shows the observations and the green curve shows the full observationally constrained model results (identical to Figure 6) for the average of the four flight days in Episode 1. For (A-C), the assumptions were: (A) The [NO3-(p)]RL is equal to zero; (B) The [NO3-(p)]RL is constant with altitude and equal to the NO3-(g+p) at 3 pm previous afternoon, corresponding to a case of zero net production or loss; (C) the [NO3-(p)]RL is constant with altitude and equal to the maximum observed [NO3-(p)] in the early-morning RL profile.
As an extreme counter-example, if there were no NO3-(p) in the RL, mixing would have led to an initial decline in the early morning surface NO3-(p) (Figure 8A). Alternatively, if the aloft NO3-(p) concentration were assumed to be equal to that from the previous day at 3 pm (and with no vertical variability), there would not have been a sharp increase in the morning surface NO3-(p) (Figure 8B). Instead, there would have been a more gradual increase from the morning into the afternoon due largely to the increasing influence of photochemical production. This is representative of a case in which there was neither aloft production of NO3-(p) nor losses from advection, such that the early-morning RL concentration was determined entirely by carry-over from the prior day; in this case the difference between the early-morning surface concentration and that in the RL is small compared to the observations. If, instead, the RL NO3-(p) concentration at all altitudes had been equal to the maximum NO3-(p) observed in the RL (no vertical gradient in the RL), then the morning peak in surface-level NO3-(p) would have occurred later and the NO3-(p) concentration would be substantially higher throughout a greater fraction of the day (Figure 8C). This is representative of a case in which nocturnal production in the RL occurred, but where advection did not serve to reshape the NO3-(p) vertical profile in the RL. Clearly, export of pollution from the relatively compact Fresno urban area to the broader region (and import of cleaner air) plays an important role in determining the daytime surface-level concentration of NO3-(p), multi-day build up and the population exposure in this urban area. While it has previously been suggested that the morning increase in surface-level NO3-(p) is indicative of mixing down of NO3-(p) in the RL (Watson and Chow, 2002; Pusede et al., 2016; Young et al., 2016), the current study provides an explicit, observationally constrained demonstration of this effect and highlights the dual roles of chemical production and advective loss in the RL.
The time-evolving relative contributions of surface-level NO3-(p) from the NBL, the RL and photochemical production are individually quantifiable from the model for this episode (Figure 7B). As the ML rises, the relative contribution of NO3-(p) from the RL rapidly increases reaching ~80% at the 10–11 am peak. After this point, the relative contribution of NO3-(p) from photochemical production increases continuously. By the time that decoupling of the NBL occurs (~3 pm), photochemically produced NO3-(p) comprises 58% of surface-level NO3-(p) while NO3-(p) from the previous nights’ RL still comprises 40%; the contribution of NO3-(p) that was in the NBL is negligible (<2%). Pusede et al. (2016) showed that future decreases in NOx emissions are more likely to decrease night time than daytime NO3-(p) production. The results here therefore suggest that decreases in NO3-(p) may be more apparent, on average, in the morning than the afternoon since the fractional contributions of night time-produced versus daytime-produced NO3-(p) shift throughout the day. However, care must be taken when interpreting observations from individual days since the meteorological conditions that favor observation of an early morning increase will not always occur (discussed further below). Since it is assumed here that OH scales with solar radiation, the potential for enhanced production of OH (and subsequently NO3-(p)) in the early morning via e.g. HONO photolysis is not accounted for in the model (Pusede et al., 2016). If this process were included, the increase in morning surface-level NO3-(p) would be even greater than is already calculated from mixing down of NO3-(p) in the RL. Since the observationally constrained model already predicts a somewhat larger peak at 10 am for surface-level NO3-(p) concentrations compared to the observations, early-morning photochemical production appears to have had a relatively limited influence on the morning surface-level NO3-(p) compared to mixing down of nocturnal NO3-(p) during this episode.
While vertical mixing and the shape of the NO3-(p) vertical profile are what predominately drive the morning temporal evolution in the surface-level NO3-(p) (especially the peak) for this episode, the afternoon behavior, especially between ~1 pm and 4 pm, is shaped by the balance between photochemical production and loss via (i) dilution by entrainment of FT air and (ii) evaporation of NO3-(p) and subsequent dry deposition of HNO3 gas, i.e. a gas-phase pump for NO3-(p) loss. Here, the relative importance of these loss pathways is considered. The latter process (gas-phase pump) has been previously considered by Pusede et al. (2016) while the former (FT entrainment) was not. Loss through dry deposition of NO3-(p) is negligible since deposition velocities for HNO3 (vd = 1 – 10 cm s-1) are much larger than for particles (vd = 0.001 – 0.1 cm s-1) (Meyers et al., 1989; Horii et al., 2005; Farmer et al., 2013; Pusede et al., 2016). These loss mechanisms ultimately limit the extent of the pollution episode build up. Once the daytime model ML reaches maximum height entrainment into the ML of typically cleaner air from just above the ML (i.e. from the FT) occurs. The time-evolving entrainment rates are estimated from the CLASS model (Appendix C).
Considering the gas-phase pump, the warm (typically 290 K) and dry (RH = 40% or less during the campaign) afternoon conditions enhance evaporation of NO3-(p) relative to night time and early morning conditions, thereby increasing loss through dry deposition of HNO3 gas in the afternoon (Pusede et al., 2016). However, total ammonia is in substantial excess (3.8 – 8.9 times NO3-(g+p) on a molar basis), with thermodynamic calculations indicating that the gas-phase fraction of NO3- is <0.15 during the daytime and near zero at night when it is colder and RH is higher (Figure 3). These estimates of the gas-phase fraction of NO3- are similar to the observational measurements of Parworth et al. (2017), who determined the daytime and night time averages during the first episode were 0.08 ± 0.03 (1σ) and 0.04 ± 0.05 (1σ), respectively. Importantly, the gas-phase fraction here is substantially smaller than that estimated in Pusede et al. (2016) who found a daytime gas-phase fraction of 0.4 (median) and a 24-h average of 0.15. Consequently, loss of nitrate via the gas-phase pump is less than in their analysis and suggests that the role of this pathway was likely overestimated. The general influence of the gas-phase fraction on loss via dry deposition is shown in Figure S11. In general, the results indicate that the gas-phase fraction has a strong influence on the loss of NO3-(p) due to HNO3 deposition.
Including both FT entrainment and dry deposition, the box model reasonably reproduces the observed afternoon decrease in surface-level NO3-(p). This allows assessment of the relative importance of these two loss processes by turning them off one at a time (Figure 7C). The calculations indicate that entrainment of clean FT air plays an important role in the afternoon surface concentration decline. Without entrainment, the model predicts that the afternoon NO3-(p) would be ~18% higher, leading to a double-humped daytime profile. Despite the relatively low gas-phase fraction, the gas-phase pump also contributes to the afternoon decline. The model results indicate that these two loss processes contribute approximately equally to the afternoon decline. There are, however, a few hours when the gas-phase pump is potentially of extreme importance. When the RL decouples and the surface mixed layer becomes quite shallow the rate of loss due to dry deposition is enhanced. This leads to a rapid decrease in surface-level NO3-(p). Yet, the concurrent decrease in the NBL temperature and increase in RH and NH3 enhances partitioning of nitrate to the particle-phase, thereby limiting the impact of this rapid decline over time. (In the model here, the decoupling is assumed to occur very rapidly while the temperature and RH changes are from observations and occur more gradually. If the decoupling was actually slower the influence of the gas-phase pump at this point in time would be reduced and the modelled decrease in NO3-(p) that occurs around 3–5 pm would be less than shown.)
The model predicts that after decoupling and cooling occur the surface-level NO3-(p) will continue to decrease at ~2% h-1 overnight via the gas-phase pump, which is similar to the loss rate observed between midnight and 7 am (Figure 7A). If the gas-phase pump is turned off completely (i.e. the nitric acid deposition velocity is set to zero) there is an increase in the modelled NO3-(p) that begins at ~3 pm (when decoupling occurs) and continues until 6 pm (Figure 7C). This is a result of the continual decrease in temperature and increase in RH enhancing partitioning to the particle-phase. Although not a focus of this study, on some days, there is a sharp increase in surface-level NO3-(p) observed in the evening, starting around 8 pm (LT). While this could theoretically result from enhanced partitioning to the particle-phase at night, the timing does not match the observed temperature and RH variations. Surface-level chemical production of nitrate via N2O5 hydrolysis could alternatively be the source of this increase, but given the near-zero surface-level O3 concentration due to titration by NO the production via this pathway would be insufficient. This evening increase is observed on many days, although with somewhat variable timing and magnitude (Figure 8). Thus, it may be that the evening increase results from advection to the measurement site of air from a not-to-distant location (given low wind speeds) that has higher surface concentrations. Regardless, while the reason for this night time increase in surface NO3-(p) remains unclear, the occurrence does not impact the analysis of the early-morning and daytime NO3-(p) behavior.
The cumulative impact of the nocturnal production in the RL, daytime photochemical production and afternoon loss processes is that the NO3-(p) concentration at ~3 pm, the point when decoupling of the RL occurs, is slightly higher than that at 8 am during the episode. Therefore, there is a gradual net increase (average of 1.32 μg m-3 day-1) in surface-level NO3-(p) as the episode progresses, albeit with day-to-day variability (Figure 6B). For comparison, the 24-h average surface-level NO3-(p) increases by 0.66 μg m-3 day-1. While decreasing NOx emissions and NO3-(p) production, especially nocturnal production (Pusede et al., 2016), is the most direct and reliable route towards decreasing surface NO3-(p) concentrations (Kleeman et al., 2005), decreases in NH3 could theoretically also have some influence on NO3-(p) by increasing the efficiency of the gas-phase pump. However, this will only be the case if NH3 decreases exceed decreases in NOx by at least a factor of five such that the ratio between the two is changed substantially and the gas-phase fraction is increased (Figure 3). Such preferential targeting of NH3 sources is therefore highly unlikely to be an efficient control strategy, at least for the SJV where the total ammonia-to-nitrate ratio is large. In regions where the NH4+(g+p):NO3-(g+p) molar ratio is closer to unity, the nitrate partitioning is more sensitive to changes in this ratio and thus ammonia control could potentially prove effective.
3.3. Comparison between episodes
The above analysis focuses on observations made during one pollution episode, but there was a second pollution episode observed during DISCOVER-AQ (Jan. 30-Feb. 5, 2013). The episode-averaged diurnal behavior of the surface NO3-(p) concentration for this second episode showed evidence of an early morning increase, but the increase is not as sharp as the first episode (Figure 9). Additionally, the day-to-day variability in the surface NO3-(p) was much greater during the second episode; on some days, there was minimal evidence of an early-morning increase but on others there was a substantial increase. The shapes of the early morning vertical NO3-(p) profiles (around 9:30 am) were notably different during Episode 2 on two of the flight days as well, as was the evolution of the profiles from morning to afternoon (Figure S12). The afternoon mixed layer heights were much higher during Episode 2 than Episode 1, ranging from 600–700 m AGL compared to 300–400 m AGL, respectively. The early-morning mixed layer heights were also higher during Episode 2 (~170 m) compared to Episode 1 (around 70 m). During Episode 1, the surface-level winds exhibit a consistent shift in direction from easterly in the early morning (5–8 am) to southerly in the later morning (9 am-12 pm), and the mean surface-level wind speed increased over this same period, from 0.31 m s-1 to 0.82 m s-1 (Figure 9). In contrast, during Episode 2 there was a lack of day-to-day consistency in the surface wind direction, especially during the early morning (5–8 am), and there was a more substantial change in the mean surface-level wind speed from early morning to later morning, from 0.32 m s-1 to 1.12 m s-1 (Figure 9). The Episode 2 mean night time aloft wind speeds were also overall lower and more constant with altitude, with little variability from 150 m to 400 m, although still with a substantial increase from the surface (Figure S13). The aloft nocturnal winds during Episode 2 were somewhat more variable than Episode 1 winds in terms of the wind direction (Figure 4 versus Figure S13).
Figure 9.
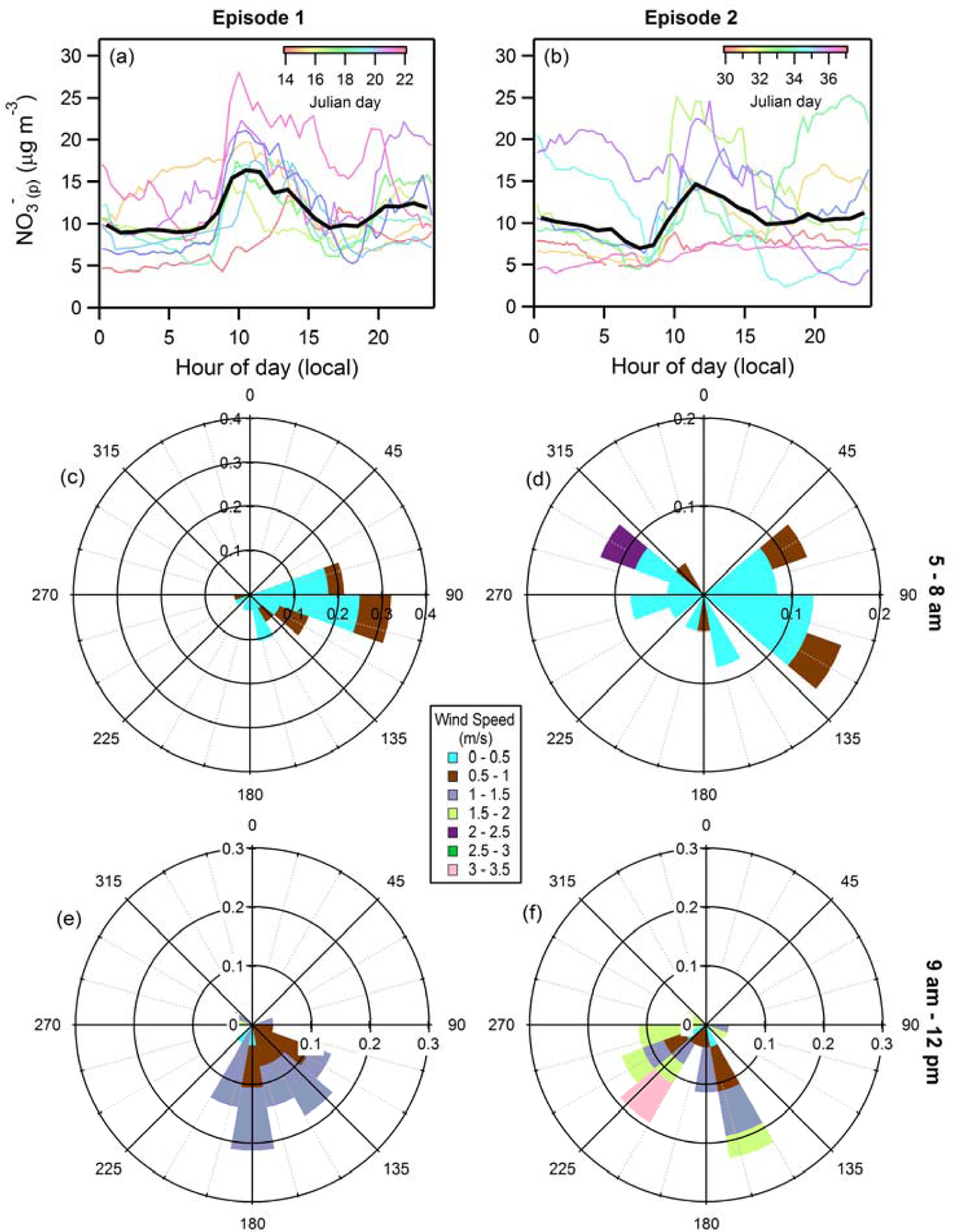
(Top panels) Diurnal variation in the surface-level particulate nitrate concentration during (a) the first episode and (b) the second episode. The solid black lines are the average profile over the episode and the colored lines are for individual days. (Middle/Bottom panels) Wind roses for surface-level (10 m) winds in Fresno for the early morning (5 – 8 am) during (c) episode 1 and (d) episode 2, and for the late morning (9 am – 12 pm) during (e) episode 1 and (f) episode 2.
Figure 4.
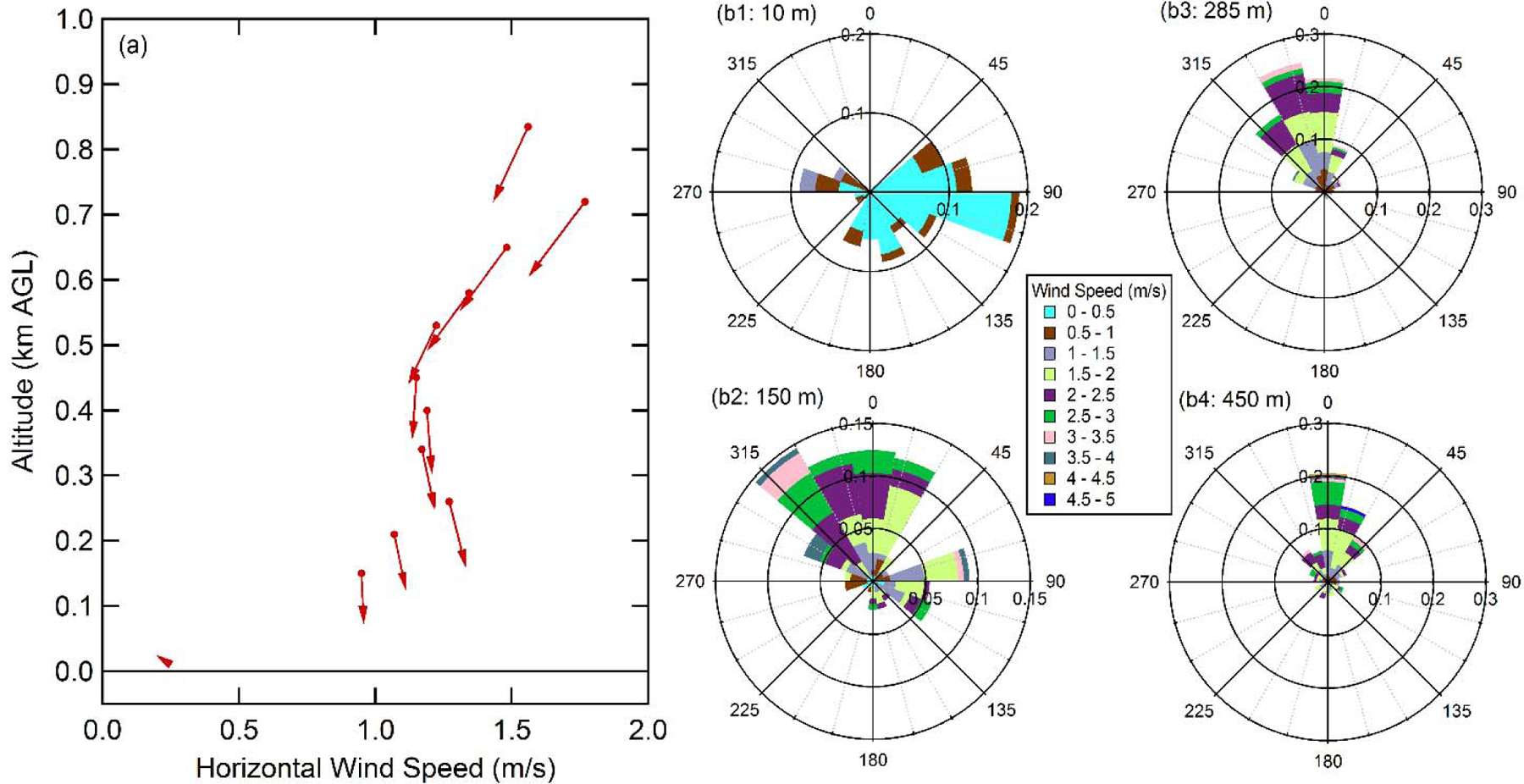
(a) Vertical profile of the average night time (19:00–07:00) horizontal winds over Visalia, CA (65 km SE of Fresno) and the surface (10 m) wind in Fresno for flight days during Episode 1 (Jan. 18, 20, 21, and 22). The length of the arrows corresponds to the wind speed and the direction to the average wind direction, with the measurement height indicated by the small circle on the tail of the arrow. (b) Corresponding wind roses for (b1) the surface, (b2) 125–175 m, (b3) 225–345 m, and (b4) 400–500 m. The length of each arc corresponds to the normalized probability and the colors indicate the wind speed (m/s; see legend). Data are from the National Oceanic and Atmospheric Administration, Earth System Research Laboratory, Physical Sciences Division Data and Image Archive (https://www.esrl.noaa.gov/psd/data/obs/datadisplay/, accessed 3 June 2017).
Overall, this increased day-to-day variability in both the surface NO3-(p) and wind behavior, and a difference in the evolution of the NO3-(p) vertical profiles from early morning to late morning/early afternoon in Episode 2 compared to Episode 1, suggests that the meteorological conditions during the second episode were generally less conducive to simple interpretation using the mixing model discussed above. Instead, it seems that advection and export from the urban area were of increased importance during Episode 2, both overnight and especially in the early-to-mid morning. The contrasting behavior between the two episodes suggests that while the observation of a sharp, early-morning rise and peak in surface-level NO3-(p) (such as during the first episode) might be generally considered a strong indicator of the production of NO3-(p) in the RL, the absence of such a feature does not preclude an important role for nocturnal production aloft.
3.4. Linking to other regions
Production of NO3-(p) in the RL can vary widely based on initial concentrations of its precursor gases, as well as the rate of heterogeneous uptake of N2O5 by particles. It may be that production of NO3-(p) via the N2O5 hydrolysis pathway may be significant in the aloft RL in other regions with similar geographical and meteorological conditions, such as Salt Lake Valley, Utah (Kuprov et al., 2014; Baasandorj et al., 2017). However, in valley regions with lower NOx or O3 the nocturnal PNO3- may be lower, thus limiting the importance of this pathway (Akira et al., 2005; Bigi et al., 2012). Among other factors, the extent to which nocturnal NO3-(p) formation occurs more-so in the surface layer versus in layers aloft will depend importantly on the extent of NOx emissions at the surface (which titrate O3, suppressing particulate nitrate formation), the absolute and relative height of the nocturnal boundary layer (which affects the rate HNO3 deposition and the air volumes in which nitrate production occurs), and gradients in RH, T and NH3 (Kim et al., 2014).
For example, Baasandorj et al. (2017) observe at their valley wall and valley floor sites in wintertime Utah that O3 concentrations near the surface remain well-above zero even during pollution episodes, thus allowing for surface-level NO3-(p) formation overnight, substantiated by direct measurements of N2O5, in addition to formation aloft. Nitrate-specific diurnal profiles were not reported. In Shanghai, China, Wang et al. (2009) observed in fall 2007 that both O3 and NO2 remain elevated at night at the surface, with a concomitant increase in surface NO3-(p). And in wintertime Seoul, Korea, Kim et al. (2017) observed relatively limited diurnal variability in O3 and NO2 concentrations measured at 60 m, with both remaining elevated throughout the night. However, they did not observe any notable build up in NO3-(p) overnight, but did observe NO3-(p) to increase and peak in the morning, as here. In contrast, in Fresno here the night time surface O3 levels during Episode 1 here nearly zero, suppressing surface NO3-(p) formation. This near-zero nocturnal O3 is similar to observations by Kuprov et al. (2014) made a few years before Baasandorj et al. (2017) at one of the same valley floor sites in Utah, reflecting year-to-year differences. Such differences can influence the extent to which a notable increase in NO3-(p) is observed to occur in the early morning as air is entrained from the residual layer to the surface. This is because if surface production and production in the residual layer are similar in magnitude the contrast between the two will be reduced and entrainment will appear to have a less apparent impact on the diurnal profile. However, because the effective volume of the residual layer is typically much larger than the nocturnal boundary layer (as is the case here), even without an observed increase in NO3-(p) at the surface in the morning the NO3-(p) produced in the residual layer can still dominate the overall NO3-(p) burden during the day.
Additionally, comparison between the Baasandorj et al. (2017) observations of late afternoon surface NO2 and O3 (which reflect the initial conditions within the residual layer) with the Fresno observations indicates differences can exist in how nocturnal production in layers aloft influences the build-up and sustaining of PM2.5 in pollution episodes. They observed during a strong PM2.5 episode a slow build-up of PM2.5 followed by a plateau lasting multiple days. During this period, late-afternoon O3 concentrations decreased over time while late-afternoon NO2 was approximately constant (in the daily average). Consequently, the nitrate radical production rate in the residual layer, and thus the N2O5 and NO3-(p) production rates, decreased over time in their study. In contrast, for Episode 1 here, the late-afternoon nitrate radical production rate increased over time across the episode (by 0.25 μg m-3 day-1), with only a moderate decrease in the daytime O3 over time (Figure S9). These differences reflect the different photochemical conditions between the regions, and illustrate the coupling between the daytime photochemical conditions (i.e. O3 production) and night time NO3-(p) formation above the surface.
4. Conclusion
This work combines surface and aircraft observations made during a pollution episode in 2013 to demonstrate that in the San Joaquin Valley (specifically Fresno, CA) production of NO3-(g+p) in the nocturnal residual layer can play a crucial role in determining daytime surface concentrations of particulate NO3- in winter, when photochemical production is relatively slow and morning boundary layers are extremely shallow. The influence of processes occurring in the aloft RL on NO3-(p) surface concentrations is evident in the NO3-(p) diurnal variability, specifically the occurrence of a mid-morning peak in surface-level NO3-(p). While the mid-morning peak has been previously suggested as a signature of nocturnal nitrate production aloft (Watson and Chow, 2002; Brown et al., 2006a; Lurmann et al., 2006; Pusede et al., 2016; Young et al., 2016), the current study makes novel use of vertical profiles of NO3-(p) concentrations measured multiple times on individual days to quantitatively illustrate the importance of nocturnal processes on surface concentrations. The analysis shows that the NO3-(p) concentration in the morning-time mixed boundary layer can be dominated by nocturnally produced NO3-(p); vertical mixing in the early morning, which entrains air from the residual layer into the surface mixed layer, has a particularly large impact on the surface concentrations here due to the nocturnal boundary layer being exceptionally shallow. In the afternoon, photochemically produced nitrate contributes the majority of the total NO3-(p) burden for the episode examined, but still with a substantial contribution from nocturnal production. The case-study here illustrates that nocturnal NO3-(p) production can play a critically important role in the build-up and sustaining of pollution episodes in the SJV, supporting previous suggestions made, in part, on the basis of calculated chemical production values and an assessment of multi-year trends in the relationship between NO3-(p) and NO2 (Pusede et al., 2016).
The current work also demonstrates that a difference exists between the shape of the typical vertical profiles of NO3-(p) in afternoon and early-morning over Fresno. This difference is shown to very likely result from altitude-specific horizontal advection in the nocturnal RL leading to differential wash-out of NO3-(p) and precursor gases, rather than from differences in chemical production rates. Consequently, there is a steep vertical gradient in NO3-(p) in the early-morning RL that, in turn, influences the temporal evolution of surface-level NO3-(p) during the day, especially in early morning. Ultimately, differential advection is shown to have an important role in limiting the maximum surface-level concentration of NO3-(p) observed within the urban area during the day, a result of the urban-rural gradients being particularly steep (Pusede et al., 2016). Absent this overnight export of pollution from the city, nitrate pollution would build up during pollution events to a much greater extent. However, advection likely contributes to the build-up of NO3-(p) throughout the valley, outside of the cities. Daytime loss processes are also shown to help limit the multi-day build-up of surface-level NO3-(p). Afternoon entrainment of air from the cleaner free troposphere into the ML (and export of mixed-layer air to the FT) is shown to be an important loss process for particulate nitrate. Janssen et al. (2012; 2013) have similarly identified afternoon loss via FT entrainment as an important process shaping the diurnal variability of surface-level organic aerosol concentrations in forested areas that are dominated by organic aerosol. Loss of NO3-(p) via dry deposition of HNO3 and subsequent evaporation of NH4NO3 is found to contribute to afternoon particulate nitrate loss, but the effect is limited by the (relatively) high afternoon boundary layer and the small gas-phase fraction of nitrate (<0.15). However, this gas-phase pump may have a substantial influence on the surface concentrations in the few hours just after decoupling of the RL occurs, when the boundary layer height is low and it is still sufficiently warm. Consistent with previous suggestions (Kleeman et al., 2005; Pusede et al., 2016), we conclude that control strategies for the region should focus on reduction of concentrations of NOx and O3 (the latter of which might require VOC controls) in the mid-afternoon, specifically around the time that the RL decouples from the surface layer, as this largely determines the production rate of nitrate in the aloft RL.
Supplementary Material
Figure C1.
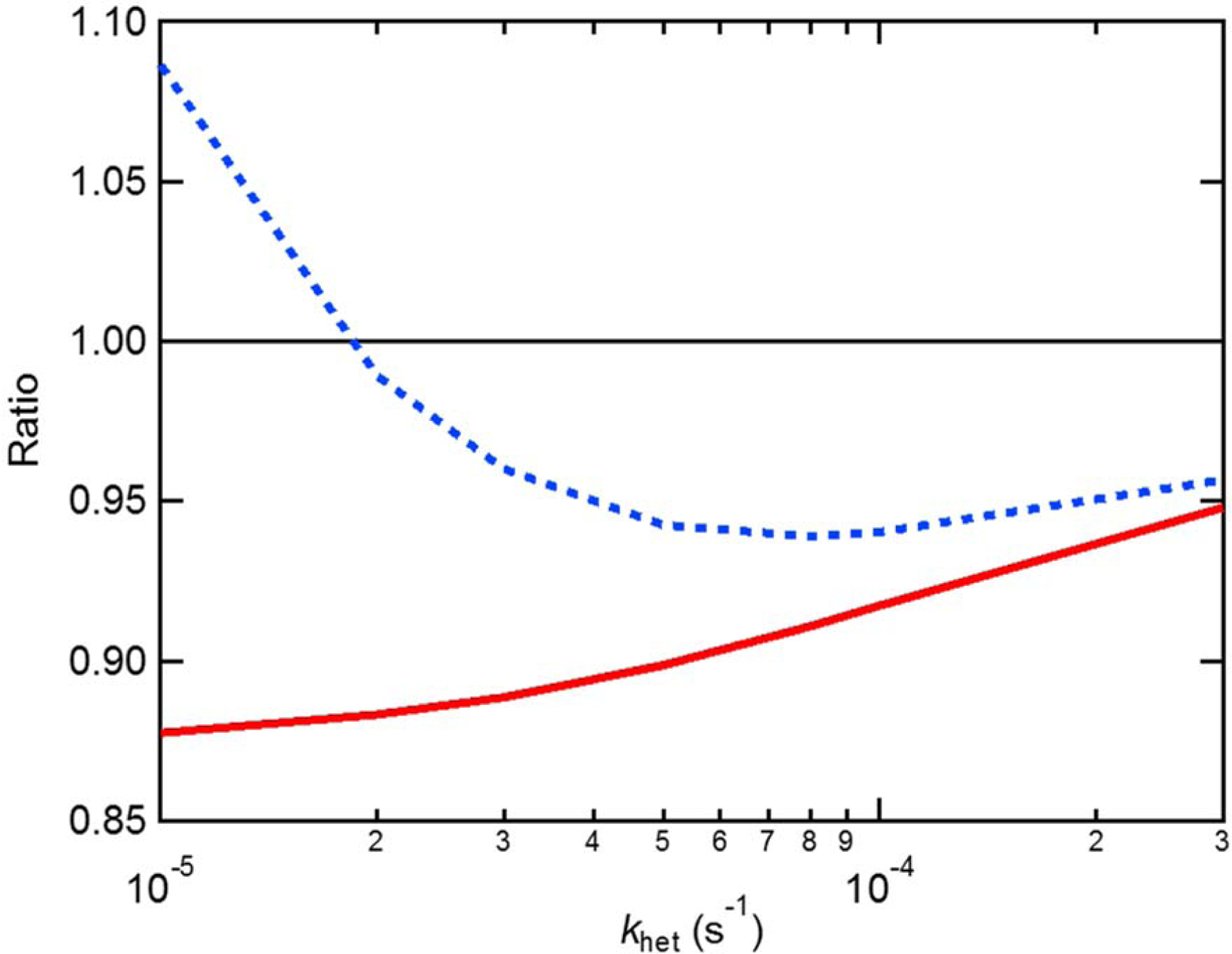
Model results showing the influence of including NO3 + VOC reactions on HNO3 production via the heterogeneous hydrolysis of N2O5, as a function of the heterogeneous oxidation rate. The red line shows the ratio between the HNO3 produced via N2O5 hydrolysis when reactions with VOCs are considered and when they are not. Reaction of NO3 with VOCs reduces the HNO3 formed via hydrolysis. The blue line shows the ratio between the total HNO3 produced from either N2O5 hydrolysis or NO3 + VOC reactions when reactions with VOCs are considered and when they are not.
Acknowledgements
This work was funded by the California Air Resources Board (14–307) and the DISCOVER-AQ campaign was supported by NASA. ICF is supported in part by California Agricultural Experiment Station (Hatch project CA-D-LAW-2229-H). The authors thank the entire DISCOVER-AQ science team for their contributions to the DISCOVER-AQ dataset. Profs. Sally Pusede (Univ. Virginia) and Ron Cohen (UC Berkeley), and Prof. Mike Kleeman (UC Davis) are thanked for their particularly useful comments and discussion.
Appendix A: Measurements
A1. Airborne Measurements
Airborne measurements used in this paper were made from the P3-B NASA aircraft during the DISCOVER-AQ field campaign in January-February, 2013 in San Joaquin Valley (SJV), California. All data are available from the publicly accessible DISCOVER-AQ website (NASA Atmospheric Science Data Center).
The P3-B was equipped with an array of instruments to measure both gas and particle-phase properties. A TSI-3563 nephelometer provided total scattering from dry particles at 450, 550 and 700 nm and scattering at 550 nm by particles at 80% RH (Beyersdorf et al., 2016). Gas-phase NH3 was measured using a cavity ringdown spectroscopy with a Picarro G2103 (von Bobrutzki et al., 2010), using the NOAA aircraft NH3 inlet and calibration scheme as in Nowak et al. (2010). Measurements of NO, NO2, NOx, and O3 were obtained through a 4-channel chemiluminiscence instrument (Brent et al., 2015). CO and CH4 were measured with a differential absorption CO measurement spectrometer (Sachse et al., 1987). Total gas (HNO3) + particle (NH4NO3) nitrate were measured using thermal dissociation – laser induced fluorescence (TD-LIF), where HNO3 and volatilizable particulate nitrate are converted into NO2 for detection (Day et al., 2002). While the TD-LIF instrument is not optimized for particle sampling, most of the particulate mass was in the submicron size range and thus inertial losses will likely only lead to a small (if any) negative bias in the measured particulate nitrate (Pusede et al., 2016). Aerosol size distributions for 0.06 – 1.0 μm diameter particles were measured with an ultra-high sensitivity aerosol spectrometer (UHSAS). The UHSAS uses an optical sizing method, but is calibrated relative to mobility diameter. The P3-B flew throughout the SJV on 10 days and performed vertical spirals over six sites across the valley, including Fresno. The location of these sites and the flight path are shown in Figure S2. This same flight path was repeated three times every day between approximately 8:30 am and 3:00 pm, with vertical profiles over Fresno at approximately 9:30–10:00 am, 12–12:30 pm and 2:30–3:00 pm. This enables assessment of the evolution of the species-specific vertical profile during the day across the valley. Out of the ten research flights during the campaign, only eight of them have been used here due to gaps in the dataset. Four of these days are during the first pollution episode (Jan. 18, 20, 21 and 22) and four are during the second pollution episode (Jan. 30 and 31, and Feb. 1 and 4).
Observations of the light scattering coefficient at 550 nm (σsca) for dry and humidified particles (no size cut-off) made from the P3-B (Beyersdorf et al., 2016) have been used to estimate the vertical distribution of PM mass and NO3-(p) concentrations. Scattering is linearly related to the total mass concentration of PM. The observed hygroscopicity is dependent on particle composition, with higher hygroscopicity indicative of a higher particulate inorganic fraction and lower hygroscopicity indicative of a higher particulate organic fraction; the relationship between hygroscopicity and the inorganic fraction (or the organic fraction) is reasonably linear when the inorganic species are primarily ammonium sulfate and ammonium nitrate (Zhang et al., 2014), as these have similar hygroscopicities (Petters and Kreidenweis, 2007). The particulate nitrate concentration is much larger than the particulate sulfate concentration, as determined from both the surface and aircraft measurements (< 600 m AGL), with nitrate-to-sulfate mass ratios of 8 and 16, respectively (both determined from PILS measurements). Thus, the observed hygroscopicity is primarily reflective of the particulate nitrate fraction (Parworth et al., 2017). More specifically, a linear relationship was observed between surface-level measurements of dry σscat and PM1.0 (= black carbon (BC) + non-refractory PM1.0, (NR-PM1)) mass concentrations in Fresno (slope = 2.83 m2 g-1 with intercept forced through zero; Figure A1a). Only data points between 8 am and 4 pm were included in determining this relationship to reflect the time period during which the airborne measurements were obtained. The observed relationship for dry, surface-level σsca and NR-PM1 is used to estimate the NR-PM1 concentration during the vertical profiles from the aircraft dry σscat measurements. The hygroscopicity (water uptake) of a particle depends on its chemical composition. Inorganic components, predominantly NO3- and ammonium in the wintertime SJV region (Young et al., 2016), are highly hygroscopic while organic components of PM1 tend to have much lower hygroscopicity (Petters and Kreidenweis, 2007). Thus, measurements of the particle hygroscopicity can be used to estimate the ratio of inorganic to organic mass in the sampled PM (Massoli et al., 2009; Parworth et al., 2017). The average particle hygroscopicity was characterized by the optical hygroscopicity parameter, γ defined by Equation A1.
| (A1) |
where σscat,wet and σscat,dry are the scattering coefficients (in Mm-1) measured under wet (RHwet = 80%) and dry (RHdry = 20%) conditions respectively. The parameter γ varies reasonably linearly with the particle inorganic mass fraction (Massoli et al., 2009). Therefore, an initial estimate of NO3-(p) concentrations at high time resolution, and thus as a function of altitude, is obtained from the equation [NO3-(p)] = γσsca,dry/2.83. The factor of 2.83 has units of m2 g-1 and comes from the σscat versus NR-PM1 relationship determined above. However, previous studies show some variability in the linear relationship between γ and inorganic mass fraction and, importantly, typically have slopes somewhat less than unity and non-zero intercepts, as is assumed in the above conversion (e.g. Massoli et al., 2009). Therefore, the low-time-resolution aircraft PILS NO3-(p) measurements (which are not appropriate for vertical profiles) were used to calibrate the above high-time-resolution NO3-(p) estimates. There was a strong, linear correlation between the NO3-(p) observed by the PILS and the initially estimated NO3-(p) (Figure A1b). This demonstrates the general validity of the estimation approach. However, the PILS NO3-(p) concentrations were, on average, 22% lower than the initially estimated NO3-(p). Therefore, the initially estimated NO3-(p) concentrations were adjusted downwards by 22%, and the final expression relating σsca,dry (in Mm-1) and γ to NO3-(p) concentrations (in μg m-3) is:
| (A2) |
The uncertainty in the estimated [NO3-(p)] is approximately 20%, based on the scatter around the best-fit line in Figure A1.
A2. Ground Measurements
Fresno (36.745 °N, 119.77 °W) was a “supersite” where comprehensive, continuous measurements of the chemical and physical properties of particulate matter were made. Chemical composition of non-refractory PM1.0 was measured by a High Resolution Time-of-Flight-Aerosol Mass Spectrometer (HR-ToF-AMS) (Young et al., 2016). The soluble fraction of PM3.0 was characterized using a Particle-Into-Liquid Sampler (PILS) coupled to an ion chromatograph (Parworth et al., 2017). Gas-phase water-soluble species were collected at 5 – 7 hr time resolution using an automatic-switching annular denuder system placed in front of the PILS and were analyzed offline with ion chromatography after extraction (Parworth et al., 2017). The combination of the denuder measurements and the particle measurements allowed for determination of the gas-phase fraction of nitrate. Light extinction and light absorption coefficients were measured using the UC Davis cavity ringdown-photoacoustic spectrometer, and scattering coefficients were determined by difference (Cappa et al., 2012; Lack et al., 2012). Refractory black carbon concentrations were measured using a single particle soot photometer (Schwarz et al., 2006). In situ gas-phase measurements of NO, NO2 and O3, along with environmental factors (T and RH) were made by the California Air Resources Board. Particle size distributions were measured using a Scanning Mobility Particle Sizer (SMPS; size range: 10 – 800 nm), and an Aerodynamic Particle Sizer (APS; 700 nm – 6 μm). Measurements included in the current study are listed in Table A.
Additionally, a radiosonde was used to obtain vertical profiles of pressure, temperature and humidity over nearby Huron (36.203 °N, 120.103 °W) twice a day, once in the morning around 8 AM and again in the evening 4 PM. Diurnal measurements of the surface heat flux and friction velocity were determined from measurements made with a sonic anemometer at Huron. Measurements of wind speed and wind direction as a function of altitude at nearby Visalia, CA are from the National Oceanic and Atmospheric Administration (NOAA) Profiler Network (https://www.esrl.noaa.gov/psd/data/obs/instruments/WindProfilerDescription.html).
Appendix B: Determining Mixed Boundary Layer Height
The mixed layer (ML) heights have been determined from each of the vertical profiles of potential temperature (θ), relative humidity (RH), CO and CH4 measured from the P3-B aircraft. Example profiles for each of the three flight legs on 18 January 2013 are shown in Fig. B1. The altitude at which there is a strong change in the slope, from approximately altitude-independent to having a steep gradient, is determined to be the top of the ML. The vertical profile measurements allow for determination of the ML height over Fresno around 10:00 am, 12:30 pm and 2:30 pm. The ML height at 8 am is separately determined from the radiosonde measurements at nearby Huron (located 83 km SSW), as the flight data do not allow for characterization of ML height this early. It is assumed that the 8 am ML measurements at Huron are representative of the ML heights in Fresno. The observed ML height increases with time from 8 am until approximately noon or 1 pm, after which it is approximately constant. The rise in ML height with time is modelled using the Chemistry Land-surface Atmosphere Soil Slab (CLASS) model (Vilà-Guerau De Arellano, 2015). The CLASS model allows for estimation of ML heights with finer time resolution than the observations (i.e. in between flights; shown as black dots in Figure B2) and of the corresponding time-dependent entrainment velocities. The model input parameters are constrained by observations from nearby Huron of the nocturnal boundary layer height, the morning inversion strength (~ 8 am), the sensible surface heat flux, the friction velocity, and the lapse rate through the residual layer, as well as by an estimate of the subsidence rate based on Trousdell et al. (2016). The model inputs are adjusted to ensure that the modelled ML growth agrees reasonably well with the observations from the P3-B over Fresno (Figure B2). The resulting average entrainment velocities in the afternoon (1 – 4 pm) from the CLASS model agree well with independently determined entrainment rates based on afternoon decline in SO42-(p) for the Episode 1 days. Since SO42-(p) is effectively non-volatile and since photochemical production via oxidation of SO2 is relatively slow, the decline in SO4-(p) in the afternoon can be attributed solely to dilution from entrainment of “clean” FT air since the influence of the gas-phase pump is small. After 3 pm the boundary layer is assumed to linearly drop over a 1-hour period to the NBL height observed at 8 am the same day. A relatively rapid (~1 h) decline in the mixed layer height is consistent with wintertime observations of diurnal BLH profiles (Bianco et al., 2011).
The sensitivity of the box model to the boundary layer growth predicted by the (observationally constrained) CLASS model has been examined. An alternative boundary layer growth profile was estimated by fitting the observed P3-B ML heights using a sigmoidal function (Figure B3). The general shapes of the CLASS and sigmoidal profiles are similar, although the sigmoidal profile exhibits a somewhat faster rise. Entrainment of FT air in the afternoon for the sigmoidal growth profile was accounted for using the average entrainment rates estimated from the observed SO42-(p) loss rates and assuming that entrainment begins at noon, when the BL height was near the maximum. The same linear decrease in the BL height starting at 3 pm was assumed. The use of this alternative model yields a diurnal NO3-(p) profile for Episode 1 that is very similar to that obtained using the CLASS model (Figure B3). This indicates that the general behavior of the diurnal surface NO3-(p) profile is not particularly sensitive to the treatment of the BL rise and that the results obtained here are robust.
Figure B3.
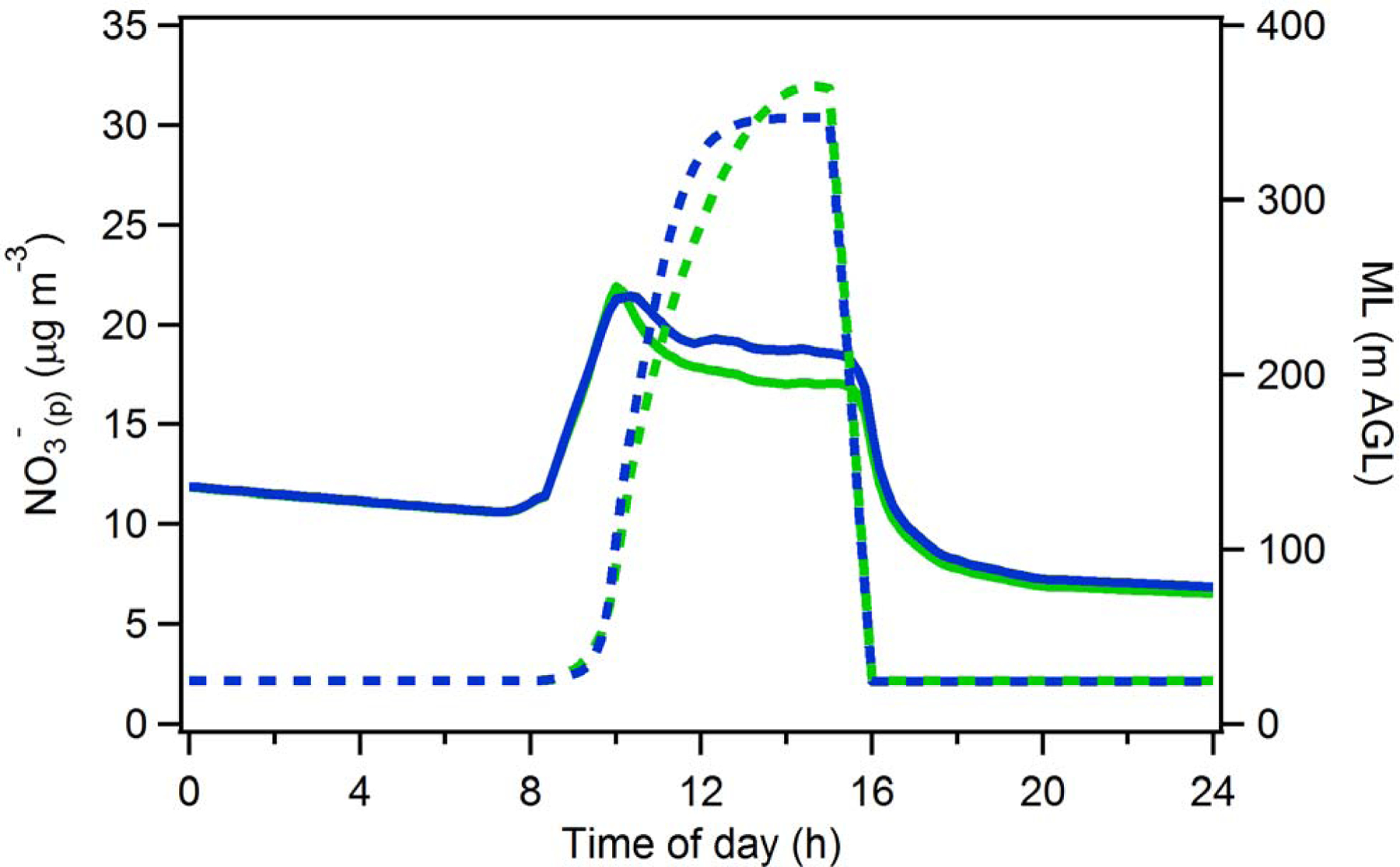
Average modelled surface NO3-(p) (solid lines) using the CLASS model output (green) and a sigmoid fit to the observed ML heights (blue). The ML heights used in the model are shown in dashed lines.
Appendix C: Nocturnal Reactions in the RL
C1. N2O5 production and heterogeneous reactivity
The gas-phase and heterogeneous chemistry occurring in the RL was assumed to follow the reaction scheme indicated by Reactions 1–4. Focusing first on the heterogeneous hydrolysis of N2O5, one estimate of the night-specific average rate coefficients for N2O5 heterogeneous hydrolysis (kN2O5) is obtained through consideration of the initial concentrations of precursor gases and the observed maximum overnight increase in the RL NO3-(p), PNO3-. More specifically, a 1D box model including nocturnal gas-phase chemistry and heterogeneous reaction of N2O5 with particles was run iteratively to determine an average kN2O5 for the night (19:00–08:00; 13 hours) such that it reproduced the observed PNO3-. The observed chloride at Fresno was small (1% of PM1.0) during the episode, and thus formation of nitryl chloride (ClNO2) can be reasonably neglected (Young et al., 2016). Since, the boundary layer is fairly well-mixed in the afternoon, surface-level observations of NOx, O3, NO3-(p), particle wet surface area and temperature at 3 pm on the preceding day were used as initial conditions. Based on back-calculated kN2O5 values, night-specific values of heterogeneous N2O5 uptake coefficient (γN2O5) were determined from:
| (C1) |
where ω is the mean molecular speed of N2O5 (256 m s-1), Sa is wet particle surface area, and γ is the N2O5 heterogeneous uptake coefficient (Brown et al., 2006c). The wet particle surface area was calculated from the observed dry particle size distributions, particle hygroscopicity and RH. The resulting back-calculated kN2O5 values from Eqn. C1 were in the range 1.3 – 5.1 x 10-5 s-1. The corresponding back-calculated γN2O5 were in the range 2.5 x 10-4 to 4.8 x 10-4 (Table S1), which as noted in the main text are somewhat smaller than values observed under water-limited conditions in other field studies and lower than expected based on lab experiments (Bertram et al., 2009).
A second estimate of the γN2O5 values is calculated from the particle composition following Bertram et al. (2009). The calculated γN2O5 depend on the particle water content (specifically, the [H2O]/[NO3-(p)] and thus RH) and the chloride fraction. The composition-calculated γN2O5 (~ 10-3) are larger than the above back-calculated values and more consistent with the literature although on the lower side of previous measurements (Brown et al., 2006c; Bertram et al., 2009). That the back-calculated γN2O5 are smaller than the γN2O5 calculated from the composition is likely a consequence of the PNO3- being an under-estimate relative to the true overnight production in the RL. This is because the observed PNO3- is taken as the difference between the previous afternoon and early morning NO3-(p) concentration in the aloft RL layer having the maximum morning concentration. This does not account for the influence of advection, which is most likely going to reduce the morning NO3-(p) relative to if there were no advection.
C2. Reactions with VOCs
Not considered in the above is the reaction of the NO3 radical with VOCs. NO3 radicals react rapidly with alkenes and more slowly with alkanes and other species. NO3 reaction with VOCs can lead to hydrogen abstraction and direct formation of HNO3, especially for reactions with alkanes. For alkenes and aromatics, NO3 reaction typically proceeds via NO3 addition and formation of organic nitrates. The latter would suppress formation of particulate inorganic nitrate but can serve as an important source of particulate organic nitrate (Kiendler-Scharr et al., 2016). Organic nitrate formation has been observed as an important source of summertime organic aerosol in Bakersfield, CA (also in the SJV) (Rollins et al., 2012). VOC concentrations and reactivity are likely much lower during the colder winter compared to the warmer summer (Pusede et al., 2014), and thus reaction of VOCs with NO3 radicals is likely much suppressed.
Concentrations of a broad suite of VOCs were measured via whole air canister sampling during DISCOVER-AQ. These measurements can be used to assess the potential influence of NO3 reaction with VOCs on HNO3 and NO3-(p) formation. The nitrate reactivity towards each VOC is calculated as kVOC+NO3.[VOC], where the kVOC+NO3 is the VOC-specific rate coefficient and [VOC] is the VOC concentration (Ng et al., 2017). Average afternoon VOC concentrations are used, which should be representative of the initial concentrations in the RL. The VOCs are ranked according to their reactivity with NO3. The top 20 VOCs are considered explicitly, and all other VOCs are lumped into a common VOC species with the average concentration and krxn of these species (Table S2). Reactions between NO3 and alkenes and aromatics are assumed to form (unreactive) organic nitrates while reactions between NO3 and all other species are assumed to form HNO3 and an organic product species. The influence of NO3 reaction with VOCs on NO3-(p) is assessed by calculating the overnight production of HNO3 both with and without VOCs using typical afternoon NO (3 ppb), NO2 (20 ppb) and O3 (27 ppb) concentrations and for kN2O5 ranging from 1 x 10-5 s-1 to 3 x 10-4 s-1. HNO3 produced from N2O5 hydrolysis is tracked separately from HNO3 produced from NO3 reaction with VOCs. The HNO3 production via N2O5 hydrolysis decreases marginally when VOC reactions are included. The HNO3 suppression ranges from ~12% for kN2O5 = 10-5 s-1 to 5% for kN2O5 = 10-4 s-1 (Figure D1). However, the calculations indicate that much of this HNO3 suppression is potentially offset by HNO3 production from reaction of NO3 with non-alkene or aromatic compounds. For larger kN2O5 the net suppression is only 5%, with the suppression decreasing as kN2O5 decreases. At the lowest kN2O5 (10-5 s-1) the calculations indicate that the inclusion of the NO3 + VOC reaction actually leads to an increase in the net HNO3 production (Figure D1). Overall, these calculations suggest that reaction of NO3 with VOCs has a relatively minor influence on the overnight local production of HNO3 in the RL.
Figure D1.
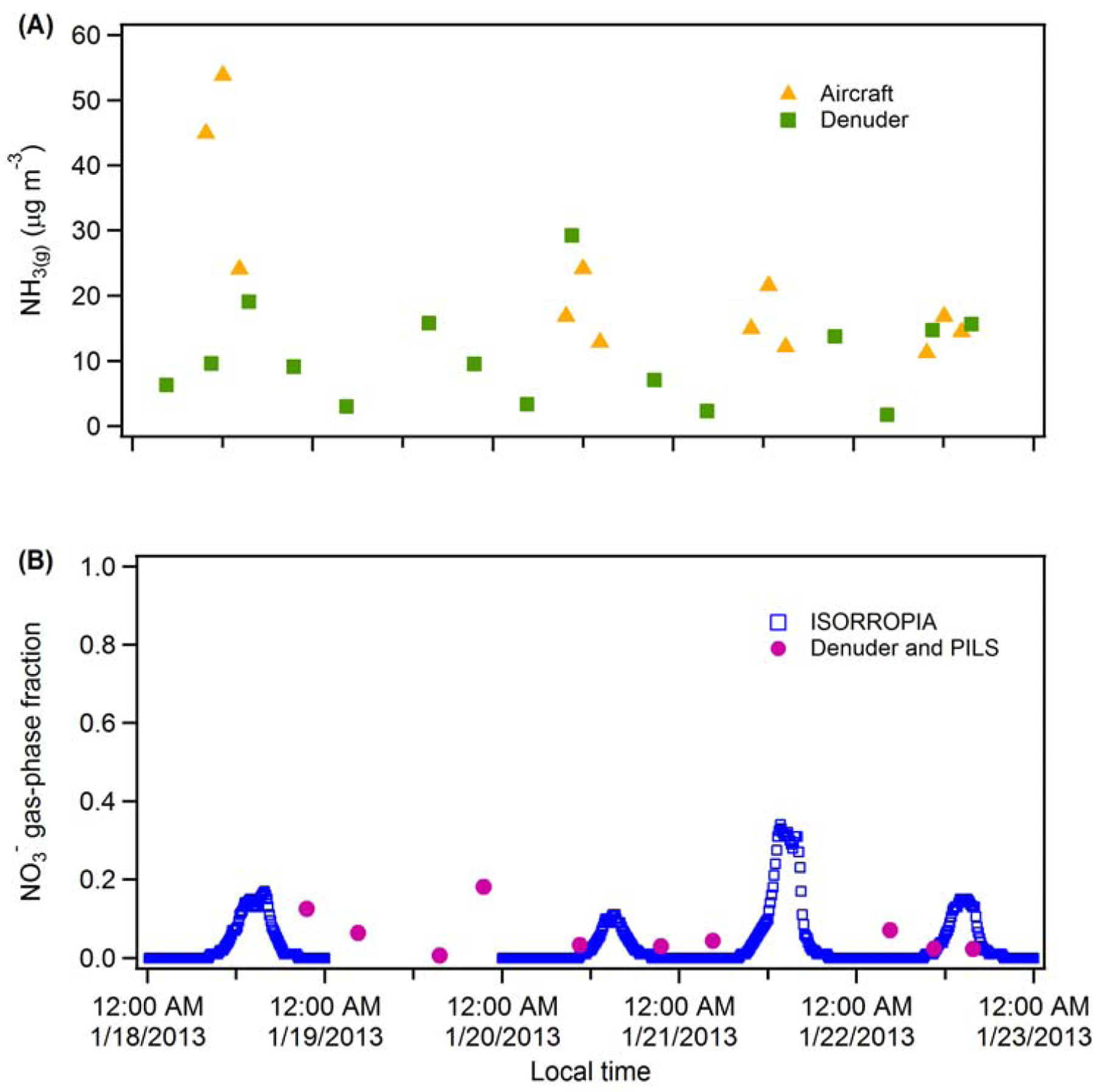
(A) Time series of NH3(g) (μg m-3) measured with the denuder at the surface (green squares) and at the lowest altitudes by CIMS onboard P3-B aircraft (yellow triangles). (B) The nitrate gas-phase fraction estimated by ISORROPIA (blue squares) and the observed fraction determined from the denuder HNO3(g) and PILS NO3-(p) measurements (pink circles) (Parworth et al., 2017).
Appendix D: Box Model Details
The box model for calculating the time-varying surface concentrations of NO3-(p) accounts for: (i) mixing of air in the aloft RL with the surface air, including the time-dependent rise and fall of the boundary layer; (ii) daytime photochemical production of HNO3 from the OH + NO2 reaction; (iii) T-and RH-dependent gas-particle partitioning of ammonium nitrate; (iv) afternoon entrainment of air from the free troposphere; (iv) competition between condensation of HNO3 onto existing suspended particles versus loss via dry deposition; (v) dry deposition of particulate NO3-(p). The kinetic equations were solved in the data analysis program Igor (Wavemetrics) and set up using the kinsim Igor package, developed by Harold Stark (http://www.igorexchange.com/node/1333). The model was initialized with the observed NO3-(p) measured by the AMS at surface-level at 12 am and run in 10 minute steps. For each time step, the photochemical production equations used the instantaneous observed NO2 and temperature, and estimated OH concentration and ML height. The fraction of NO3- in gas-phase, f, for each time step was determined based on the instantaneous conditions using the chemical thermodynamic model, ISORROPIA II in the forward mode, with the phase state set as metastable (Fountoukis and Nenes, 2007). ISORROPIA was initialized with the observed particulate composition, specifically NO3- and SO42- (AMS), and Na+, K+ and Cl- (PILS). (The PILS and AMS sampled particles of somewhat different size, with the PILS sampling PM3 and the AMS PM1. The AMS observations are available at higher time-resolution, and thus preferable to use here. However, there are challenges in quantitative characterization of Na+, K+ and Cl- using the AMS, and thus the PILS was used instead for these species. Comparison of the AMS and PILS NO3- and SO42- indicates that the AMS-measured concentrations are ~10% lower than for the PILS, attributable to mass between 1 and 3 μm (Parworth et al., 2017). The Na+, K+ and Cl- ions are minor components of the total PM3, and thus the AMS-PILS difference has minor influence on the calculations here.) Since the PILS was not functioning on 18th January, 2013, the ionic compositions of K+ and Cl- were estimated from the linear relationship between PILS and AMS composition (Equations C1–C2). Since Na+ measured by PILS was generally constant during the episode it was assumed to be the same on 18th. The diurnally varying concentrations of total NH3(g+p) for ISORROPIA were calculated as the sum of NH4+(p) measured by AMS and NH3(g) measured by the denuder at the surface in Fresno; since the denuder measurements were averages over 6–7 hours, the NH3(g) concentration was linearly interpolated between the individual measurements to allow for estimation with higher time resolution. The 6–7 hour average denuder-based NH3(g) measurements compare reasonably with the point NH3(g) measurements made on board the P3-b at the lowest altitude over Fresno. The fraction of NO3- predicted to be in the gas-phase was also found to be in generally good agreement with the observations (Figure D1; (Parworth et al., 2017).
| (D1) |
| (D2) |
As the boundary layer rises, starting around 8 am, and air from the RL is mixed into the surface air, the instantaneous NO3-(p) concentration at the surface ([NO3-(p)]surf) is calculated as:
| (D3) |
where t and t-1 represent the current and previous time steps respectively, BLH is the boundary layer height (m AGL), we is the entrainment velocity and [NO3-(p)]RL,t is the concentration of NO3-(p) in the layer of air that is entrained. Between 8 am and (approximately) noon, the vertical NO3-(p) profile within the remaining RL (above the instantaneous BLH) is assumed to remain unchanged from the early-morning observed profile. The vertical NO3-(p) profile is updated to that observed during the second flight once the BLH (from the CLASS model) reaches the ML height observed around noon.
The daytime photochemical production of HNO3 was calculated from Reaction DR1 (Burkholder et al., 2015, http://jpldataeval.jpl.nasa.gov).
| (DR1) |
The OH concentration at a given time step was assumed to scale with the solar radiation flux (SR) as:
| (D4) |
where the maximum daytime OH concentration is assumed to be [OH]max = 1 x 106 molecules cm-3, after (Pusede et al., 2016). The rate coefficient for condensation of HNO3(g) onto suspended particulates, kcond, was calculated based on collision theory (Seinfeld and Pandis, 2006) as:
| (D5) |
where the summation is over particle size, Dp,i is the mean particle diameter in the size bin i (m), and dNi is the number concentration in the size bin i (m-3). The term βi is the size-dependent Fuchs correction in the continuum regime, given by:
| (D6) |
where Kn = λ/Dp,i, and λ is the gas mean free path (65 nm). The parameter D is the diffusion coefficient of HNO3 gas in air (cm2 s-1) given by (De Andrade et al., 1992):
| (D7) |
where T is ambient temperature (K). The corresponding evaporation rate coefficient (kevap) is determined as:
| (C8) |
where Keqm is the instantaneous (temperature and RH-dependent) equilibrium partitioning coefficient for ammonium nitrate. The rate coefficient for loss of gas-phase HNO3 or NO3-(p) from dry deposition, kdep (s-1) is:
| (C9) |
where vd is the deposition velocity (cm s-1) and BLH is the time-dependent boundary layer height. The HNO3(g) deposition velocity has been shown to vary linearly with wind speed (Ma and Daggupaty, 2000). Here, it was assumed that:
| (C10) |
where ws is the observed wind speed, and wsmin and wsmax are the minimum and maximum values observed. The bounds of Eqn. C10 (lower limit vd = 1 cm s-1 and upper limit 10 cm s-1) were chosen to span previously observed ranges. While the accuracy of the empirical Eqn. C10 is not known, we note that use of a constant vd of 0.07 cm s-1 does not change the box model output substantially (Figure S14). Of course, if the actual vd is lower than estimated here the influence of dry deposition on NO3-(p) concentrations would be decreased. The NO3-(p) deposition velocity was assumed to be 0.01 cm s-1, consistent with much slower deposition of particles than soluble gases such as HNO3. Dry deposition occurred both during the daytime and nighttime.
Footnotes
Author Contributions
The authors declare that they have no conflict of interest.
References
- Akira K, Akikazu K, Kiyoshi I, Yoshio I, Masahiko S, Lal SM, and Balkrishan S: Investigation of air pollution concentration in Kalthmandu valley during winter season, Journal of Environmental Sciences, 17, 1008–1013, doi:1001-0742(2005)06-1008-06, 2005. [PubMed] [Google Scholar]
- American Lung Association: State of the air: 2014. report, http://www.stateoftheair.org/2014/assets/ALA-SOTA-2014-Full.pdf, last access: 3 April 2017, 2014.
- Baasandorj M, Hoch SW, Bares R, Lin JC, Brown SS, Millet DB, Martin R, Kelly K, Zarzana KJ, Whiteman CD, Dube WP, Tonnesen G, Jaramillo IC, and Sohl J: Coupling between Chemical and Meteorological Processes under Persistent Cold-Air Pool Conditions: Evolution of Wintertime PM2.5 Pollution Events and N2O5 Observations in Utah’s Salt Lake Valley, Environ. Sci. Technol, doi: 10.1021/acs.est.6b06603, 2017. doi: 10.1021/acs.est.6b06603, 2017. [DOI] [PubMed] [Google Scholar]
- Bao J-W, Michelson SA, Persson POG, Djalalova IV, and Wilczak JM: Observed and WRF-Simulated Low-Level Winds in a High-Ozone Episode during the Central California Ozone Study, J. Appl. Meteor. Clim, 47, 2372–2394, doi:doi: 10.1175/2008JAMC1822.1, 2008. [DOI] [Google Scholar]
- Bertram TH, Thornton JA, Riedel TP, Middlebrook AM, Bahreini R, Bates TS, Quinn PK, and Coffman DJ: Direct observations of N2O5 reactivity on ambient aerosol particles, Geophysical Research Letters, 36, L19803, doi: 10.1029/2009gl040248, 2009. [DOI] [Google Scholar]
- Beyersdorf AJ, Ziemba LD, Chen G, Corr CA, Crawford JH, Diskin GS, Moore RH, Thornhill KL, Winstead EL, and Anderson BE: The impacts of aerosol loading, composition, and water uptake on aerosol extinction variability in the Baltimore–Washington, D.C. region, Atmos. Chem. Phys, 16, 1003–1015, doi: 10.5194/acp-16-1003-2016, 2016. [DOI] [Google Scholar]
- Bianco L, Djalalova IV, King CW, and Wilczak JM: Diurnal Evolution and Annual Variability of Boundary-Layer Height and Its Correlation to Other Meteorological Variables in California’s Central Valley, Boundary-Layer Meteorology, 140, 491–511, doi: 10.1007/s10546-011-9622-4, 2011. [DOI] [Google Scholar]
- Bigi A, Ghermandi G, and Harrison RM: Analysis of the air pollution climate at a background site in the Po valley, J. Environ. Monitoring, 14, 552–563, doi: 10.1039/C1EM10728C, 2012. [DOI] [PubMed] [Google Scholar]
- Brent LC, Thorn WJ, Gupta M, Leen B, Stehr JW, He H, Arkinson HL, Weinheimer A, Garland C, Pusede SE, Wooldridge PJ, Cohen RC, and Dickerson RR: Evaluation of the use of a commercially available cavity ringdown absorption spectrometer for measuring NO2 in flight, and observations over the Mid-Atlantic States, during DISCOVER-AQ, J. Atmos. Chem, 72, 503–521, doi: 10.1007/s10874-013-9265-6, 2015. [DOI] [Google Scholar]
- Brown SG, Hyslop NP, Roberts PT, McCarthy MC, and Lurmann FW: Wintertime Vertical Variations in Particulate Matter (PM) and Precursor Concentrations in the San Joaquin Valley during the California Regional Coarse PM/Fine PM Air Quality Study, J. Air & Waste Manage. Assoc, 56, 1267–1277, doi: 10.1080/10473289.2006.10464583, 2006a. [DOI] [PubMed] [Google Scholar]
- Brown SS, Neuman JA, Ryerson TB, Trainer M, Dubé WP, Holloway JS, Warneke C, de Gouw JA, Donnelly SG, Atlas E, Matthew B, Middlebrook AM, Peltier R, Weber RJ, Stohl A, Meagher JF, Fehsenfeld FC, and Ravishankara AR: Nocturnal odd-oxygen budget and its implications for ozone loss in the lower troposphere, Geophysical Research Letters, 33, L08801, doi: 10.1029/2006GL025900, 2006b. [DOI] [Google Scholar]
- Brown SS, Ryerson TB, Wollny AG, Brock CA, Peltier R, Sullivan AP, Weber RJ, Dube WP, Trainer M, Meagher JF, Fehsenfeld FC, and Ravishankara AR: Variability in nocturnal nitrogen oxide processing and its role in regional air quality, Science, 311, 67–70, doi: 10.1126/science.1120120, 2006c. [DOI] [PubMed] [Google Scholar]
- Burkholder JB, Sander SP, Abbatt J, Barker JR, Huie RE, Kolb CE, Kurylo MJ, Orkin VL, Wilmouth DM, and Wine PH: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, Jet Propulsion Laboratory, Pasadena, 2015, http://jpldataeval.jpl.nasa.gov.
- Cabañas B, Salgado S, Martín P, Baeza MT, and Martínez E: Night-time Atmospheric Loss Process for Unsaturated Aldehydes: Reaction with NO3 Radicals, J. Phys. Chem. A, 105, 4440–4445, doi: 10.1021/jp0029459, 2001. [DOI] [Google Scholar]
- Cappa CD, Onasch TB, Massoli P, Worsnop DR, Bates TS, Cross ES, Davidovits P, Hakala J, Hayden KL, Jobson BT, Kolesar KR, Lack DA, Lerner BM, Li S-M, Mellon D, Nuaaman I, Olfert JS, Petäjä T, Quinn PK, Song C, Subramanian R, Williams EJ, and Zaveri RA: Radiative Absorption Enhancements Due to the Mixing State of Atmospheric Black Carbon, Science, 337, 1078–1081, doi: 10.1126/science.1223447, 2012. [DOI] [PubMed] [Google Scholar]
- Chow JC, Chen LWA, Watson JG, Lowenthal DH, Magliano KA, Turkiewicz K, and Lehrman DE: PM2. 5 chemical composition and spatiotemporal variability during the California Regional PM10/PM2. 5 Air Quality Study (CRPAQS), Journal of Geophysical Research-Atmospheres, 111, D10S04, doi: 10.1029/2005JD006457, 2006. [DOI] [Google Scholar]
- Chow JC, Watson JG, Lowenthal DH, Hackney R, Magliano K, Lehrman D, and Smith T: Temporal variations of PM2. 5, PM10, and gaseous precursors during the 1995 integrated monitoring study in central California, J. Air Waste Manage. Assoc, 49, 16–24, 1999. [DOI] [PubMed] [Google Scholar]
- Chow JC, Watson JG, Lowenthal DH, and Magliano KL: Size-resolved aerosol chemical concentrations at rural and urban sites in Central California, USA, Atmos. Res, 90, 243–252, doi: 10.1016/j.atmosres.2008.03.017, 2008. [DOI] [Google Scholar]
- Crawford JH and Pickering KE: DISCOVER-AQ: Advancing strategies for air quality observations in the next decade. In: EM, Air and Waste Management Association, 2014.
- Curci G, Ferrero L, Tuccella P, Barnaba F, Angelini F, Bolzacchini E, Carbone C, Denier van der Gon HAC, Facchini MC, Gobbi GP, Kuenen JPP, Landi TC, Perrino C, Perrone MG, Sangiorgi G, and Stocchi P: How much is particulate matter near the ground influenced by upper-level processes within and above the PBL? A summertime case study in Milan (Italy) evidences the distinctive role of nitrate, Atmos. Chem. Phys, 15, 2629–2649, doi: 10.5194/acp-15-2629-2015, 2015. [DOI] [Google Scholar]
- Day DA, Wooldridge PJ, Dillon MB, Thornton JA, and Cohen RC: A thermal dissociation laser-induced fluorescence instrument for in situ detection of NO2, peroxy nitrates, alkyl nitrates, and HNO3, Journal of Geophysical Research-Atmospheres, 107, D64046, doi: 10.1029/2001JD000779, 2002. [DOI] [Google Scholar]
- De Andrade JB, De Aragão NM, and Araújo FRJ: Nitric Acid-Air Diffusion Coefficient: Experimental Determination Using a Diffusion Cell, Int. J. Environ. Anal. Chem, 49, 103–109, doi: 10.1080/03067319208028130, 1992. [DOI] [Google Scholar]
- Dentener FJ and Crutzen PJ: Reaction of N2O5 on tropospheric aerosols - Impact on the global distributions of NOX, O3, and OH, Journal of Geophysical Research-Atmospheres, 98, 7149–7163, doi: 10.1029/92jd02979, 1993. [DOI] [Google Scholar]
- Farmer DK, Chen Q, Kimmel JR, Docherty KS, Nemitz E, Artaxo PA, Cappa CD, Martin ST, and Jimenez JL: Chemically Resolved Particle Fluxes Over Tropical and Temperate Forests, Aerosol Sci. Technol, 47, 818–830, doi: 10.1080/02786826.2013.791022, 2013. [DOI] [Google Scholar]
- Fountoukis C and Nenes A: ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+, Ca2+, Mg2+,NH4+, Na+, SO42-, NO3-, Cl-, H2O aerosols, Atmos. Chem. Phys, 7, 4639–4659, doi: 10.5194/acp-7-4639-2007, 2007. [DOI] [Google Scholar]
- Ge X, Setyan A, Sun Y, and Zhang Q: Primary and secondary organic aerosols in Fresno, California during wintertime: Results from high resolution aerosol mass spectrometry, Journal of Geophysical Research-Atmospheres, doi: 10.1029/2012JD018026, 2012. 10.1029/2012JD018026, 2012. [DOI] [Google Scholar]
- Horii CV, Munger JW, Wofsy SC, Zahniser M, Nelson D, and McManus JB: Atmospheric reactive nitrogen concentration and flux budgets at a Northeastern US forest site, Ag. Forest Meteor, 133, 210–225, doi: 10.1016/j.agrformet.2004.08.009, 2005. [DOI] [Google Scholar]
- Janssen RHH, Vilà-Guerau de Arellano J, Ganzeveld LN, Kabat P, Jimenez JL, Farmer DK, van Heerwaarden CC, and Mammarella I: Combined effects of surface conditions, boundary layer dynamics and chemistry on diurnal SOA evolution, Atmos. Chem. Phys, 12, 6827–6843, doi: 10.5194/acp-12-6827-2012, 2012. [DOI] [Google Scholar]
- Janssen RHH, Vilà-Guerau de Arellano J, Jimenez JL, Ganzeveld LN, Robinson NH, Allan JD, Coe H, and Pugh TAM: Influence of boundary layer dynamics and isoprene chemistry on the organic aerosol budget in a tropical forest, Journal of Geophysical Research-Atmospheres, 118, 9351–9366, doi: 10.1002/jgrd.50672, 2013. [DOI] [Google Scholar]
- Kiendler-Scharr A, Mensah AA, Friese E, Topping D, Nemitz E, Prevot ASH, Äijälä M, Allan J, Canonaco F, Canagaratna M, Carbone S, Crippa M, Dall Osto M, Day DA, De Carlo P, Di Marco CF, Elbern H, Eriksson A, Freney E, Hao L, Herrmann H, Hildebrandt L, Hillamo R, Jimenez JL, Laaksonen A, McFiggans G, Mohr C, O’Dowd C, Otjes R, Ovadnevaite J, Pandis SN, Poulain L, Schlag P, Sellegri K, Swietlicki E, Tiitta P, Vermeulen A, Wahner A, Worsnop D, and Wu HC: Organic nitrates from night-time chemistry are ubiquitous in the European submicron aerosol, Geophysical Research Letters, 43, 7735–7744, doi: 10.1002/2016GL069239, 2016. [DOI] [Google Scholar]
- Kim H, Zhang Q, Bae GN, Kim JY, and Lee SB: Sources and atmospheric processing of winter aerosols in Seoul, Korea: insights from real-time measurements using a high-resolution aerosol mass spectrometer, Atmos. Chem. Phys, 17, 2009–2033, doi: 10.5194/acp-17-2009-2017, 2017. [DOI] [Google Scholar]
- Kim YJ, Spak SN, Carmichael GR, Riemer N, and Stanier CO: Modeled aerosol nitrate formation pathways during wintertime in the Great Lakes region of North America, Journal of Geophysical Research-Atmospheres, 119, 12420–12445, doi: 10.1002/2014jd022320, 2014. [DOI] [Google Scholar]
- Kleeman MJ, Ying Q, and Kaduwela A: Control strategies for the reduction of airborne particulate nitrate in California’s San Joaquin Valley, Atmos. Environ, 39, 5325–5341, doi: 10.1016/j.atmosenv.2005.05.044, 2005. [DOI] [Google Scholar]
- Kuprov R, Eatough DJ, Cruickshank T, Olson N, Cropper PM, and Hansen JC: Composition and secondary formation of fine particulate matter in the Salt Lake Valley: Winter 2009, J. Air & Waste Manage. Assoc, 64, 957–969, doi: 10.1080/10962247.2014.903878, 2014. [DOI] [PubMed] [Google Scholar]
- Lack DA, Richardson MS, Law D, Langridge JM, Cappa CD, McLaughlin RJ, and Murphy DM: Aircraft instrument for comprehensive characterization of aerosol optical properties, Part 2: black and brown carbon absorption and absorption enhancement measured with photo acoustic spectroscopy, Aerosol Sci. Technol, 46, 555–568, doi: 10.1080/02786826.2011.645955, 2012. [DOI] [Google Scholar]
- Lowe D, Archer-Nicholls S, Morgan W, Allan J, Utembe S, Ouyang B, Aruffo E, Le Breton M, Zaveri RA, and Di Carlo P: WRF-Chem model predictions of the regional impacts of N2O5 heterogeneous processes on night-time chemistry over north-western Europe, Atmos. Chem. Phys, 15, 1385–1409, doi: 10.5194/acp-15-1385-2015, 2015. [DOI] [Google Scholar]
- Lurmann FW, Brown SG, McCarthy MC, and Roberts PT: Processes Influencing Secondary Aerosol Formation in the San Joaquin Valley during Winter, J. Air Waste Manag. Assoc, 56, 1679–1693, doi: 10.1080/10473289.2006.10464573, 2006. [DOI] [PubMed] [Google Scholar]
- Ma J and Daggupaty SM: Effective Dry Deposition Velocities for Gases and Particles over Heterogeneous Terrain, J. Appl. Meteor, 39, 1379–1390, doi:, 2000. [DOI] [Google Scholar]
- Massoli P, Bates TS, Quinn PK, Lack DA, Baynard T, Lerner BM, Tucker SC, Brioude J, Stohl A, and Williams EJ: Aerosol optical and hygroscopic properties during TexAQS-GoMACCS 2006 and their impact on aerosol direct radiative forcing, Journal of Geophysical Research-Atmospheres, 114, D00F07, doi: 10.1029/2008JD011604, 2009. [DOI] [Google Scholar]
- Meyers T, Huebert B, and Hicks B: HNO3 deposition to a deciduous forest, Boundary Layer Meteor, 49, 395–410, doi: 10.1007/BF00123651, 1989. [DOI] [Google Scholar]
- NASA Atmospheric Science Data Center: DISCOVER-AQ data and information, last access: 01/26/2017, doi: doi: 10.5067/Aircraft/DISCOVER-AQ/Aerosol-TraceGas. [DOI] [Google Scholar]
- Ng NL, Brown SS, Archibald AT, Atlas E, Cohen RC, Crowley JN, Day DA, Donahue NM, Fry JL, Fuchs H, Griffin RJ, Guzman MI, Herrmann H, Hodzic A, Iinuma Y, Jimenez JL, Kiendler-Scharr A, Lee BH, Luecken DJ, Mao J, McLaren R, Mutzel A, Osthoff HD, Ouyang B, Picquet-Varrault B, Platt U, Pye HOT, Rudich Y, Schwantes RH, Shiraiwa M, Stutz J, Thornton JA, Tilgner A, Williams BJ, and Zaveri RA: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys, 17, 2103–2162, doi: 10.5194/acp-17-2103-2017, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak JB, Neuman JA, Bahreini R, Brock CA, Middlebrook AM, Wollny AG, Holloway JS, Peischl J, Ryerson TB, and Fehsenfeld FC: Airborne observations of ammonia and ammonium nitrate formation over Houston, Texas, Journal of Geophysical Research-Atmospheres, 115, D22304, doi: 10.1029/2010JD014195, 2010. [DOI] [Google Scholar]
- Ouwersloot HG and Vilà-Guerau de Arellano J: Analytical Solution for the Convectively-Mixed Atmospheric Boundary Layer, Boundary-Layer Meteor, 148, 557–583, doi: 10.1007/s10546-013-9816-z, 2013. [DOI] [Google Scholar]
- Parworth CL, Young DE, Kim H, Zhang X, Cappa CD, Collier S, and Zhang Q: Wintertime water-soluble aerosol composition and particle water content in Fresno, California: Results from DISCOVER-AQ 2013, Journal of Geophysical Research-Atmospheres, 122, 3155–3170, doi: 10.1002/2016JD026173, 2017. [DOI] [Google Scholar]
- Petters MD and Kreidenweis SM: A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys, 7, 1961–1971, doi: 10.5194/acp-7-1961-2007, 2007. [DOI] [Google Scholar]
- Pusede SE, Duffey KC, Shusterman AA, Saleh A, Laughner JL, Wooldridge PJ, Zhang Q, Parworth CL, Kim H, Capps SL, Valin LC, Cappa CD, Fried A, Walega J, Nowak JB, Weinheimer AJ, Hoff RM, Berkoff TA, Beyersdorf AJ, Olson J, Crawford JH, and Cohen RC: On the effectiveness of nitrogen oxide reductions as a control over ammonium nitrate aerosol, Atmos. Chem. Phys, 16, 2575–2596, doi: 10.5194/acp-16-2575-2016, 2016. [DOI] [Google Scholar]
- Pusede SE, Gentner DR, Wooldridge PJ, Browne EC, Rollins AW, Min KE, Russell AR, Thomas J, Zhang L, Brune WH, Henry SB, DiGangi JP, Keutsch FN, Harrold SA, Thornton JA, Beaver MR, St. Clair, J. M., Wennberg, P. O., Sanders, J., Ren, X., VandenBoer, T. C., Markovic, M. Z., Guha, A., Weber, R., Goldstein, A. H., and Cohen, R. C.: On the temperature dependence of organic reactivity, nitrogen oxides, ozone production, and the impact of emission controls in San Joaquin Valley, California, Atmos. Chem. Phys, 14, 3373–3395, doi: 10.5194/acp-14-3373-2014, 2014. [DOI] [Google Scholar]
- Riemer N, Vogel H, Vogel B, Schell B, Ackermann I, Kessler C, and Hass H: Impact of the heterogeneous hydrolysis of N2O5 on chemistry and nitrate aerosol formation in the lower troposphere under photosmog conditions, Journal of Geophysical Research-Atmospheres, 108, D44144, doi: 10.1029/2002jd002436, 2003. [DOI] [Google Scholar]
- Rollins AW, Browne EC, Min K-E, Pusede SE, Wooldridge PJ, Gentner DR, Goldstein AH, Liu S, Day DA, Russell LM, and Cohen RC: Evidence for NOx Control over Nighttime SOA Formation, Science, 337, 1210–1212, doi: 10.1126/science.1221520, 2012. [DOI] [PubMed] [Google Scholar]
- Sachse GW, Hill GF, Wade LO, and Perry MG: Fast-response, high-precision carbon monoxide sensor using a tunable diode laser absorption technique, Journal of Geophysical Research-Atmospheres, 92, 2071–2081, doi: 10.1029/JD092iD02p02071, 1987. [DOI] [Google Scholar]
- San Joaquin Valley Air Pollution Control District: San Joaquin Airshed 2003. PM10 Plan https://www.valleyair.org/Air_Quality_Plans/docs/2003%20PM10%20Plan/PDF%202003%20PM10%20Plan%20adpt%20chapters/Ch%202-Final.pdf, last access: 6/20/2016, 2003.
- Schiferl LD, Heald CL, Nowak JB, Holloway JS, Neuman JA, Bahreini R, Pollack IB, Ryerson TB, Wiedinmyer C, and Murphy JG: An investigation of ammonia and inorganic particulate matter in California during the CalNex campaign, Journal of Geophysical Research-Atmospheres, 119, 1883–1902, doi: 10.1002/2013JD020765, 2014. [DOI] [Google Scholar]
- Schwarz JP, Gao RS, Fahey DW, Thomson DS, Watts LA, Wilson JC, Reeves JM, Darbeheshti M, Baumgardner DG, Kok GL, Chung SH, Schulz M, Hendricks J, Lauer A, Kärcher B, Slowik JG, Rosenlof KH, Thompson TL, Langford AO, Loewenstein M, and Aikin KC: Single-particle measurements of midlatitude black carbon and light-scattering aerosols from the boundary layer to the lower stratosphere, Journal of Geophysical Research-Atmospheres, 111, doi: 10.1029/2006JD007076, 2006. [DOI] [Google Scholar]
- Seinfeld JH and Pandis SN: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley and sons, 2006. [Google Scholar]
- Stelson A and Seinfeld JH: Relative humidity and temperature dependence of the ammonium nitrate dissociation constant, Atmos. Environ, 16, 983–992, doi: 10.1016/0004-6981(82)90184-6, 1982. [DOI] [Google Scholar]
- Terrenoire E, Bessagnet B, Rouïl L, Tognet F, Pirovano G, Létinois L, Beauchamp M, Colette A, Thunis P, Amann M, and Menut L: High-resolution air quality simulation over Europe with the chemistry transport model CHIMERE, Geosci. Model Dev, 8, 21–42, doi: 10.5194/gmd-8-21-2015, 2015. [DOI] [Google Scholar]
- Thornton JA, Kercher JP, Riedel TP, Wagner NL, Cozic J, Holloway JS, Dubé WP, Wolfe GM, Quinn PK, Middlebrook AM, Alexander B, and Brown SS: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, doi: 10.1038/nature08905, 2010. [DOI] [PubMed] [Google Scholar]
- Vilà-Guerau De Arellano J, van Heerwaarden CC, van Stratum BJH & van den Dries K: Atmospheric Boundary Layer, Integrating Air Chemistry and Land Interactions, Cambridge University Press, New York, 2015. [Google Scholar]
- von Bobrutzki K, Braban CF, Famulari D, Jones SK, Blackall T, Smith TEL, Blom M, Coe H, Gallagher M, Ghalaieny M, McGillen MR, Percival CJ, Whitehead JD, Ellis R, Murphy J, Mohacsi A, Pogany A, Junninen H, Rantanen S, Sutton MA, and Nemitz E: Field inter-comparison of eleven atmospheric ammonia measurement techniques, Atmos. Meas. Tech, 3, 91–112, doi: 10.5194/amt-3-91-2010, 2010. [DOI] [Google Scholar]
- Wagner NL, Riedel TP, Young CJ, Bahreini R, Brock CA, Dubé WP, Kim S, Middlebrook AM, Öztürk F, Roberts JM, Russo R, Sive B, Swarthout R, Thornton JA, VandenBoer TC, Zhou Y, and Brown SS: N2O5 uptake coefficients and nocturnal NO2 removal rates determined from ambient wintertime measurements, J. Geophys. Res.-Atmos, 118, 9331–9350, doi: 10.1002/jgrd.50653, 2013. [DOI] [Google Scholar]
- Walker JM, Philip S, Martin RV, and Seinfeld JH: Simulation of nitrate, sulfate, and ammonium aerosols over the United States, Atmos. Chem. Phys, 12, 11213–11227, doi: 10.5194/acp-12-11213-2012, 2012. [DOI] [Google Scholar]
- Wang X, Zhang Y, Chen H, Yang X, Chen J, and Geng F: Particulate Nitrate Formation in a Highly Polluted Urban Area: A Case Study by Single-Particle Mass Spectrometry in Shanghai, Environ. Sci. Technol, 43, 3061–3066, doi: 10.1021/es8020155, 2009. [DOI] [PubMed] [Google Scholar]
- Watson JG and Chow JC: A wintertime PM 2.5 episode at the Fresno, CA, supersite, Atmos. Environ, 36, 465–475, doi: 10.1016/S1352-2310(01)00309-0, 2002. [DOI] [Google Scholar]
- Whiteman CD, Hoch SW, Horel JD, and Charland A: Relationship between particulate air pollution and meteorological variables in Utah’s Salt Lake Valley, Atmos. Environ, 94, 742–753, doi: 10.1016/j.atmosenv.2014.06.012, 2014. [DOI] [Google Scholar]
- Wild RJ, Edwards PM, Bates TS, Cohen RC, de Gouw JA, Dubé WP, Gilman JB, Holloway J, Kercher J, Koss AR, Lee L, Lerner BM, McLaren R, Quinn PK, Roberts JM, Stutz J, Thornton JA, Veres PR, Warneke C, Williams E, Young CJ, Yuan B, Zarzana KJ, and Brown SS: Reactive nitrogen partitioning and its relationship to winter ozone events in Utah, Atmos. Chem. Phys, 16, 573–583, doi: 10.5194/acp-16-573-2016, 2016. [DOI] [Google Scholar]
- Young DE, Kim H, Parworth C, Zhou S, Zhang X, Cappa CD, Seco R, Kim S, and Zhang Q: Influences of emission sources and meteorology on aerosol chemistry in a polluted urban environment: results from DISCOVER-AQ California, Atmos. Chem. Phys, 16, 5427–5451, doi: 10.5194/acp-16-5427-2016, 2016. [DOI] [Google Scholar]
- Zhang X, Kim H, Parworth CL, Young DE, Zhang Q, Metcalf AR, and Cappa CD: Optical Properties of Wintertime Aerosols from Residential Wood Burning in Fresno, CA: Results from DISCOVER-AQ 2013, Environ. Sci. Technol, 50, 1681–1690, doi: 10.1021/acs.est.5b04134, 2016. [DOI] [PubMed] [Google Scholar]
- Zhang X, Massoli P, Quinn PK, Bates TS, and Cappa CD: Hygroscopic growth of submicron and supermicron aerosols in the marine boundary layer, Journal of Geophysical Research-Atmospheres, 119, 8384–8399, doi: 10.1002/2013JD021213, 2014. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.