

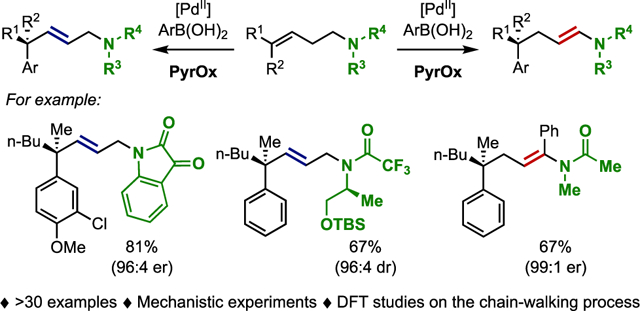
Abstract
The formation of alkyl–palladium complexes via the nucleopalladation of alkenes is the entry point for a wide range of diverse reactions. One possibility is that the intermediate alkyl–Pd complexes can undergo a “chain-walking” event, to allow for remote functionalization through various termination processes. However, there are few methods to selectively interrupt the chain-walking process at a prescribed location. Herein we demonstrate that a variety of homoallylic protected amines undergo an interrupted enantioselective relay Heck reaction to give enantioenriched allylic amine products. The selectivity of this process can be diverted to exclusively yield the ene-amide products by virtue of changing the nature of the amine protecting group. To rationalize this observation, we combine experiment and computation to investigate the mechanism of the chain-walking process and termination events. Isotopic labeling experiments and the computed reaction pathways suggest that the system is likely under thermodynamic control, with the selectivity being driven by the relative stability of intermediates encountered during chain walking. These results illustrate that the chain-walking of alkyl–palladium complexes can be controlled through the alteration of thermodynamic processes and provides a roadmap for exploiting these processes in future reaction development.
Graphical Abstract
Introduction:
Alkene functionalization reactions using π-Lewis acidic metals, such as Pd(II), are often initiated by the addition of a nucleophile to the ligated alkene to form a metal-alkyl intermediate.1 This intermediate can either undergo β-hydride elimination to reform an alkene (e.g., Heck or Wacker type reactions), undergo a subsequent organometallic reaction to ultimately be classified as an alkene difunctionalization reaction,1–4 or the metal can migrate away from the newly formed bond through iterative β-hydride elimination/migratory insertion processes5–10 (often referred to as “chain-walking”).11, 12 Chain-walking strategies allow for either the regeneration of the unsaturation remotely, or incorporation of an external reagent by a terminating organometallic transformation at a site distant from the initial bond forming event.13–15
The majority of these termination processes are directed by the innate nature of the substrate.16–21 As an example, our group,22–36 as well as others,37–45 have used redox acceptors to promote the terminating step. In particular, alkenyl alcohols A have been widely used wherein the alcohol is ultimately converted into the corresponding carbonyl product B through a formal oxidation event (I to B, Figure 1a). Contrastingly, Martin,46–48 Marek,49, 50 Mazet,51, 52 Zhu,53–55 and others,56–63 have used an organometallic step, which often occurs at the termini of the alkyl chain, to trap the chain walking process. As a specific example, the remote functionalization of alkene C is initiated by migratory insertion into a Ni-H followed by chain-walking and ultimate trapping of II with CO2 to yield carboxylic acid D (Figure 1b).47 Clearly, a significant expansion in the scope of remote functionalization would be the ability to intercept the chain walking event at a prescribed position along the alkyl chain.
Figure 1.
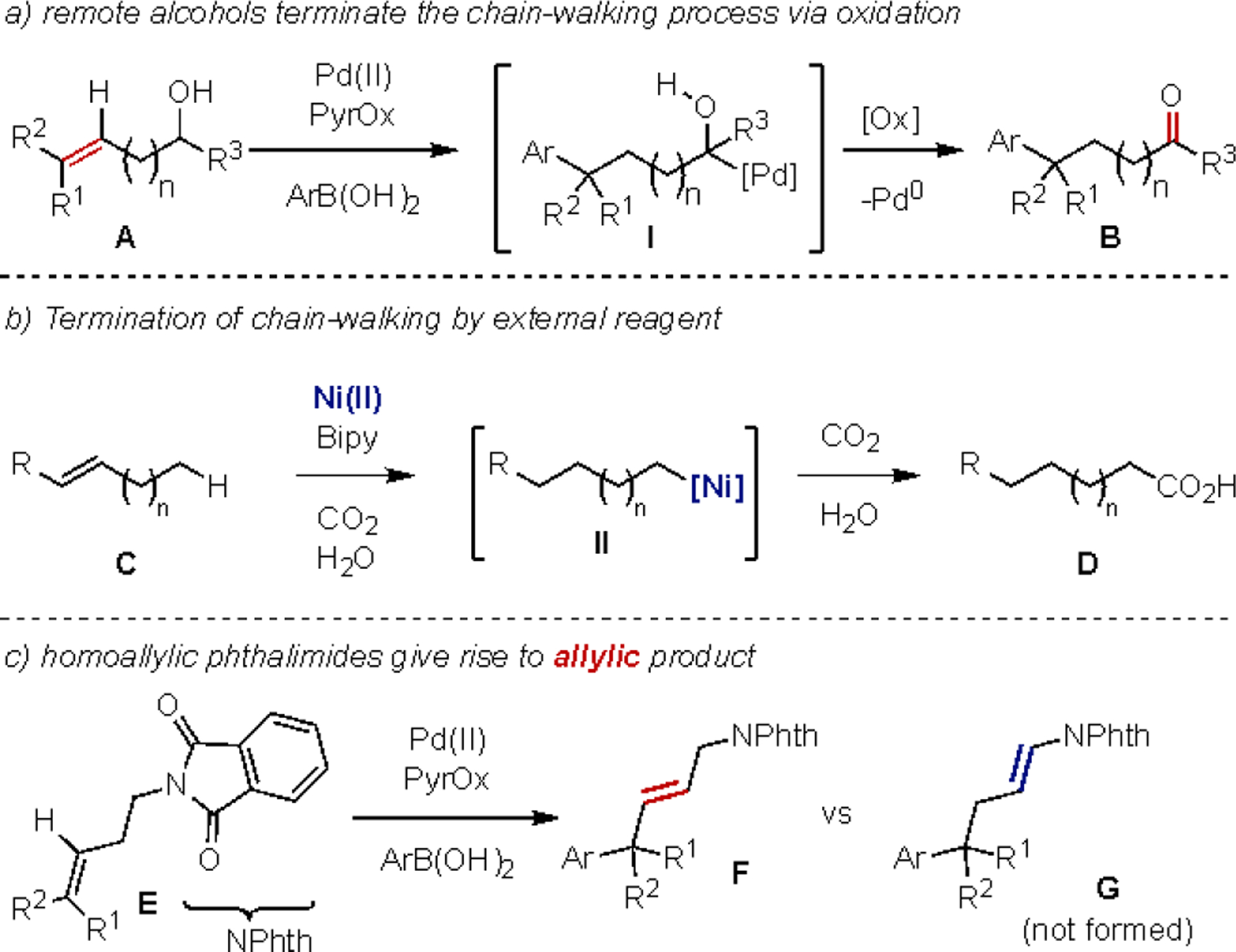
a) redox acceptors such as alcohols terminate the chain-walking process by oxidation, b) termination of the chain-walking process by external reagent, c) relay Heck reaction to give allylic products using homoallylic phthalimide substrates
In this context, we have discovered that a class of alkenyl substrates, namely homoallylic trisubstituted protected amines (for example, phthalimide E), undergo an enantioselective Heck reaction to yield product F (Figure 1c). Instead of relaying the unsaturation to form ene amide products of type G, akin to the corresponding homoallylic alcohols that form carbonyls, this chain-walking process is interrupted to yield allylic phthalimides in a highly selective manner. This surprising result leads to a range of synthetic and mechanistic questions including: 1) what is the synthetic scope of the process? 2) how does the structure of the protected amine impact product selectivity (ene amide vs interrupted chain-walking), and 3) what is the mechanistic origin for the interrupted Heck process; is it a consequence of thermodynamics or kinetics? Herein, we investigate these questions in detail revealing that the reaction has broad scope in terms of the aryl boronic acid, functional group tolerance, and amine protecting group. Additionally, through a combination of experimental and computational techniques, we have explored the basis for the interruption of the chain-walking process.
Results and Discussion:
The optimal conditions for the interrupted enantioselective Heck arylation of trisubstituted homoallylic allylic amine derivatives were found to be similar to those reported for the arylation of homoallylic trisubstituted alcohols24 with minor modifications (see supporting information (SI) for details). The product of this reaction contains a quaternary carbon center with an appended allylic protected amine. Using these optimized conditions, the scope of the arylboronic acid was explored using homoallylic phthalimides 1a and 1b (Table 1). The reaction tolerates the use of unfunctionalized aromatics (2aa–2ab), electron donating and withdrawing substituents at the 3- and 4- positions (2ac–g, 2ba–b, 2be–f), and multiply substituted aryl boronic acids (2bc–d, 2bg–h). These results reveal several general trends. First, it was found that the conversion of phthalimide 1b was higher than 1a (compare 1ag and 1bf) presumably due to the smaller methyl group. Second, the conversion (and subsequently the yield) of the transformation is highly dependent on the electronic nature of the aryl boronic acid used. For example, electron-deficient boronic acids require the reaction to be performed at 40 °C in order to achieve desirable levels of conversion. This is presumably due to the need to overcome the slower migratory insertion as a function of using more electron deficient aryl boronic acids. Fortunately, performing the reaction at elevated temperatures does not cause an observable decrease in enantioselectivity. Alternatively, electron-rich aryl boronic acids were found to add at the newly formed disubstituted alkene to give the 2:1 adduct 3 in low yield (see SI). The absolute configuration of products 2a and 2b were established by comparison of the hydrogenated analogue of 2bg prepared by this route, and via the redox-relay Heck reaction of the alcohol analogue of 1b (see SI). It should be noted that extending the chain length between the alkene and the phthalimide resulted in an inseparable mixture of allylic and homoallylic phthalimides in a 1.3:1 mixture (see SI). Lastly, 1,2-disubstitued alkenes were found to give mixtures arising from poor regioselectivity for arylation of the alkene (see SI).
Table 1.
Scope of aryl boronic acid with homoallylic phthalimides 1a and 1b.
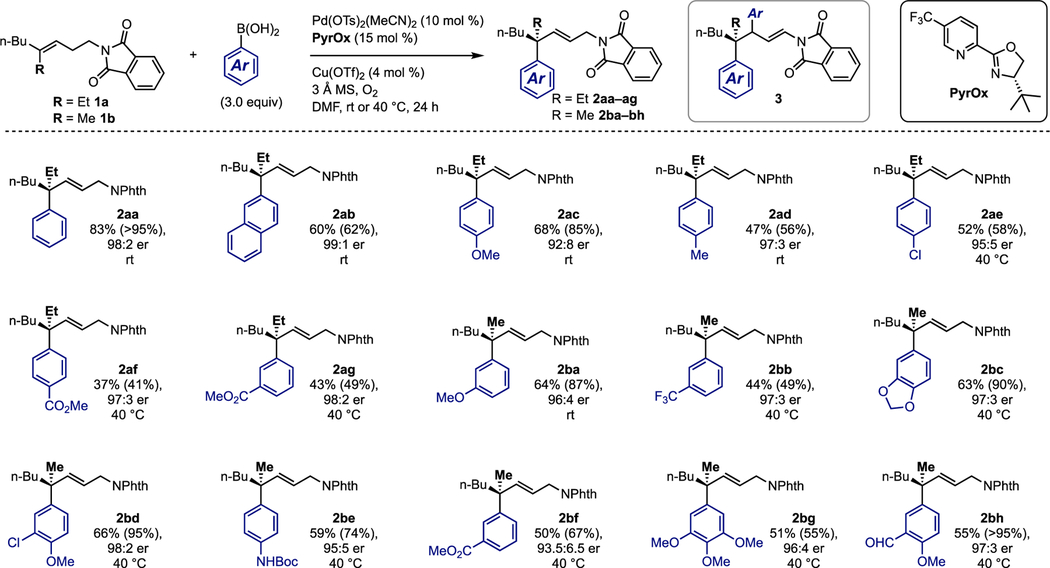
|
For each entry the following notation is used: yield % (conversion %), enantiomeric ratio, reaction temperature. Aryl boronic acid was added batch-wise in either two (room temperature reactions) or three (40 °C reactions) portions.
Scope of functional group on the alkene.
We next turned our attention to probing the effect of functional groups attached to the alkene on the reaction outcome (Table 2). It was found that azides (2c), nitriles (2d), halides (2h), tosylates (2f), malonates (2g), protected alcohols (2e, 2j), and protected amines (2i) are all compatible with the reaction. In general, the yields are modestly diminished when compared to unfunctionalized substrates, but the enantioselectivity remains high. The ability to use electrophiles such as 2e and 2h allows for the introduction of a wide scope of additional functionality. Additionally, the use of malonate 2g highlights the compatibility of this reaction with competing intramolecular nucleophiles. It should be noted that a coeluting isomeric product arising from addition of the aryl group to the internal carbon of the alkene was formed in low yields (up to 15%, see table 2).
Table 2.
Scope of non-participating functional groups.
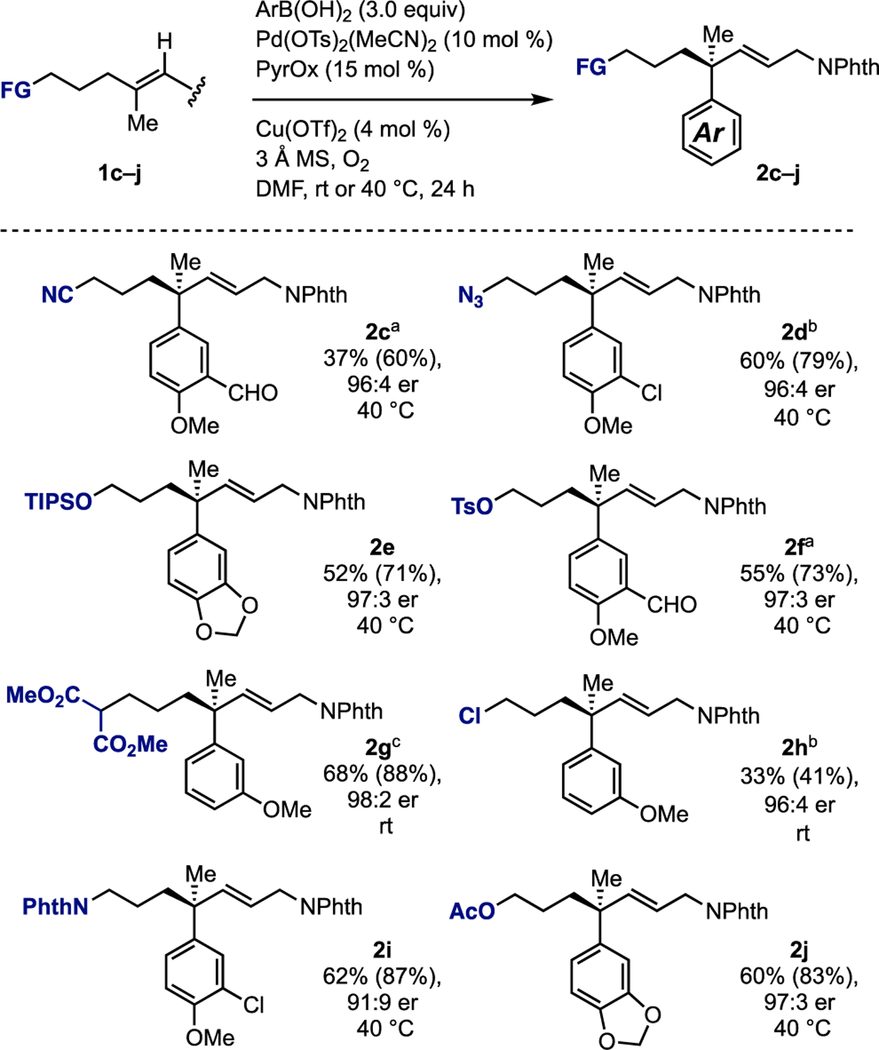
|
For each entry the following notation is used: yield % (conversion %), enantiomeric ratio, reaction temperature. Aryl boronic acid was added batch-wise in either two (room temperature reactions) or three (40 °C reactions) portions. a isolated in 90% purity, b isolated in 95% purity, c isolated in 85% purity.
Scope of “acceptor” groups.
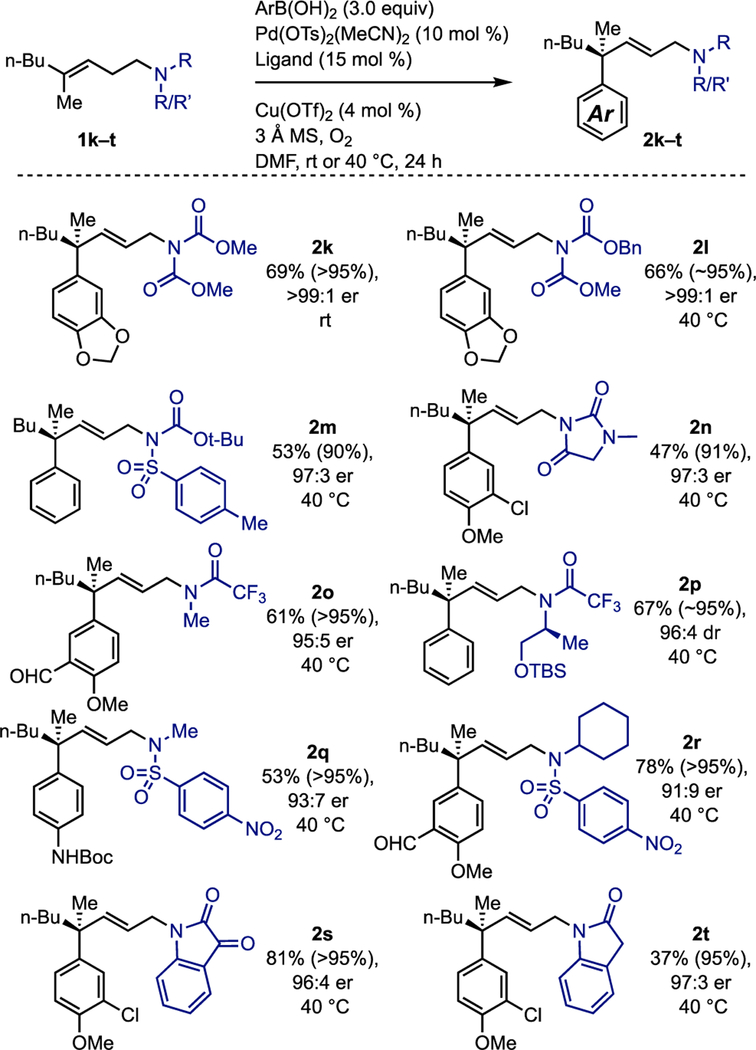
Having established the scope of the reaction in terms of aryl boronic acids (Table 1) and attached functional groups on the alkene (Table 2), we next explored what other amine derivatives at the termini were capable of selectively forming the allylic product (Table 3). We focused our attention on groups that contained useful N-protecting groups, or desirable heterocycles. For example, bisprotected amines (2k–m), monoprotected amines (2o–2r), hydantoins (2n), isatin (2s), and indolinone (2t) are compatible. This greatly increases the scope of this transformation to include protected primary amines with or without orthogonal protecting groups, as well as protected secondary amines. Alkylated amines with trifluoroacetamide (2o, 2p) and sulfonamide protecting groups (2q, 2r) are compatible, which can be readily removed to give rise to allylic secondary amines. Additionally, the N–alkyl substituent tolerates both primary (2o, 2q) and tertiary (2p, 2r) substituents. Notably, the remote stereocenter in 2p does not influence the enantioselectivity of the transformation. Finally, the enantioselectivity of these transformations remains high regardless of the nature of the group at the amine.
Table 3.
Scope of compatible functional groups at the termini.
|
For each entry the following notation is used: yield % (conversion %), enantiomeric ratio, reaction temperature. Aryl boronic acid was added batch-wise in either two (room temperature reactions) or three (40 °C reactions) portions.
Formation of ene products.
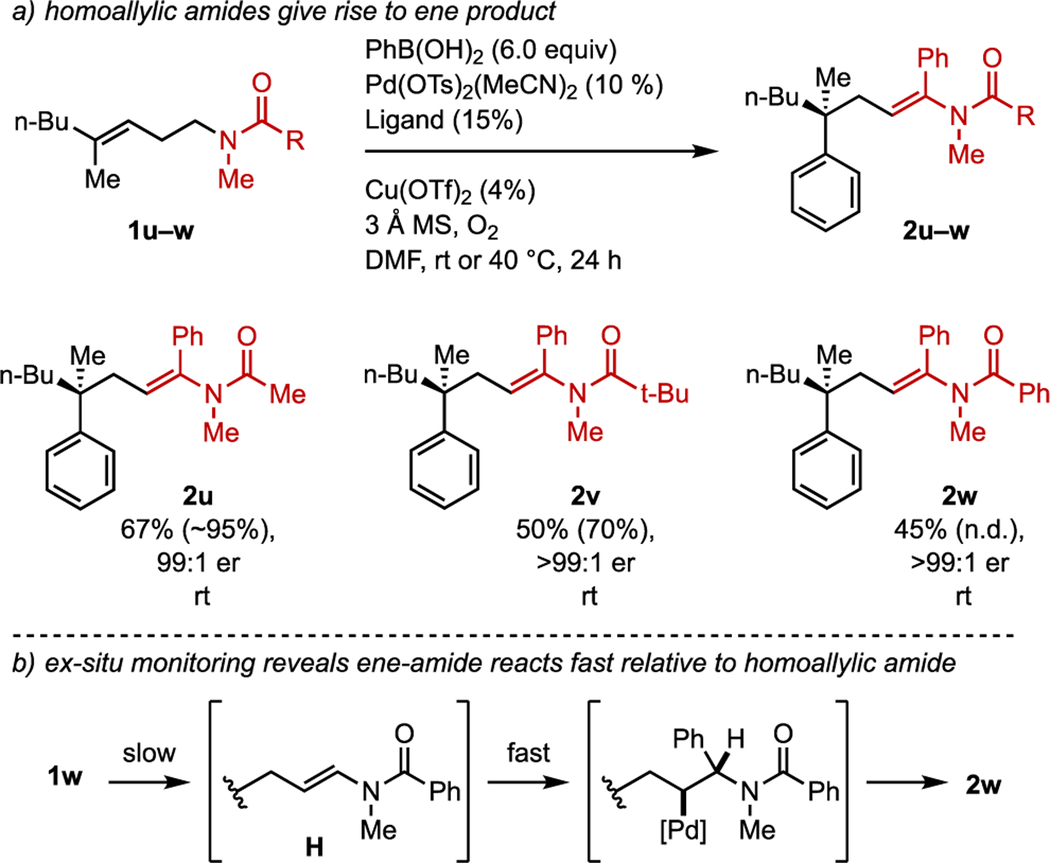
In each example reported in tables 1–3, there was no detectable amount of the isomeric ene products. However, when N-methyl acetamide 1u was subjected to the standard reaction conditions, the bisarylated ene-amide 2u was unexpectedly formed in moderate yield (44%), with none of the isomeric allyl product. Doubling the amount of boronic acid to six equivalents in the reaction resulted in a significantly improved yield (67%, Table 4a). It was reasoned that the cause for this switch in selectivity for the formation of the ene amide type product compared to the structurally similar trifluoroacetamide could either be due to the significantly smaller size of the methyl group relative to the trifluoromethyl group, or due to the more electron rich carbonyl. To test these hypotheses, the pivaloyl amide 1v and benzamide 1w were subjected to the reaction conditions. In both instances, the ene-amides 1v and 2v were formed exclusively in comparable yields and high enantioselectivity. These results suggest that the difference in the electron density of the amide carbonyl is responsible for this observed change in selectivity. To further probe the origin of the addition of two aryl groups, the formation of 2w was investigated at early time points using ex-situ monitoring (Table 4b). If the initially formed ene-amide H reacts at a comparable rate with 1w, then at early time points only a small amount of 2w should be present. However, at early conversions (<10%) only a trace amount of a 1:1 adduct presumed to be H was evident, with the primary product being 2w. This result suggests that the ene-amide H reacts significantly faster than 1w.
Table 4.
Scope of amide substrates
|
For each entry the following notation is used: yield % (conversion %), enantiomeric ratio, reaction temperature. Aryl boronic acid was added batch-wise in two portions.
Mechanistic investigations.
Above it is demonstrated that a wide variety of alkenyl amine derivatives selectively undergo the redox relay Heck reaction to form allylic products (Tables 1–3), while alkyl and aryl amides selectively form ene products (Table 4). This observed disparity led us to explore the mechanism by which the terminal group imparts selectivity in the chain walking process. Importantly, a detailed mechanistic understanding could guide the future development of site-selective termination of chain-walking processes. Therefore, we employed a combination of experimental and computational approaches to interrogate these observations.
We reasoned that the selectivity of these processes could be a result of either the thermodynamics or kinetics of the chain-walking process (Figure 2a). If the system is under thermodynamic control, then the alkene ligated Pd–H species III, alkyl–Pd complex IV, and alkene ligated Pd–H species V could establish an equilibrium (Figure 2a). If this were the case, then the selective formation of the allylic products would be a result of the relative stability of III compared to V. Alternatively, the selectivity could be driven by the kinetics of the migratory insertion and β-hydride elimination processes. For example, if the barrier for migratory insertion of the Pd–H to form IV, or the β-hydride elimination of Pd–He to form V were prohibitively high, then the selective formation of the allylic product would be under kinetic control. Differentiating between these two scenarios could allow for insight into how to design future systems.
Figure 2.
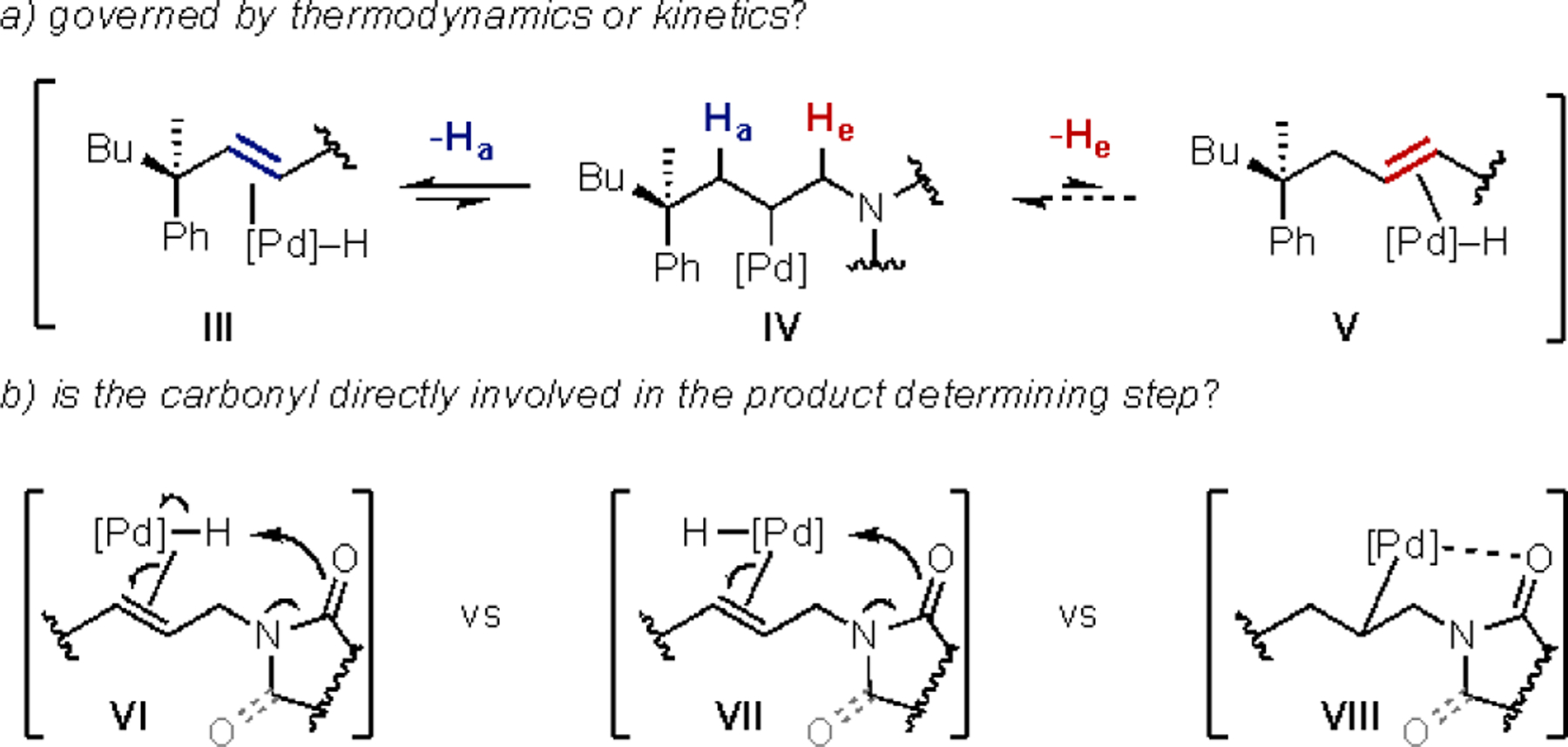
Mechanistic hypotheses for the interrupted chain-walking: a) kinetic and/or thermodynamic driven, b) direct involvement of the carbonyl
Alternatively, the selective formation of the allyl product could arise from direct involvement of the carbonyl (Figure 3b). For example, the carbonyl could deprotonate the alkene bound Pd–H intermediate VI, displace the alkene through ligand exchange (VII), or stabilize the intermediate Pd–alkyl species (VIII) (Figure 2b). In each case, the selective formation of the allylic product could arise from the ability of each of these processes to occur through a proposed six-membered palladacycle transition state (VI and VII), or intermediate (VIII).
Figure 3.
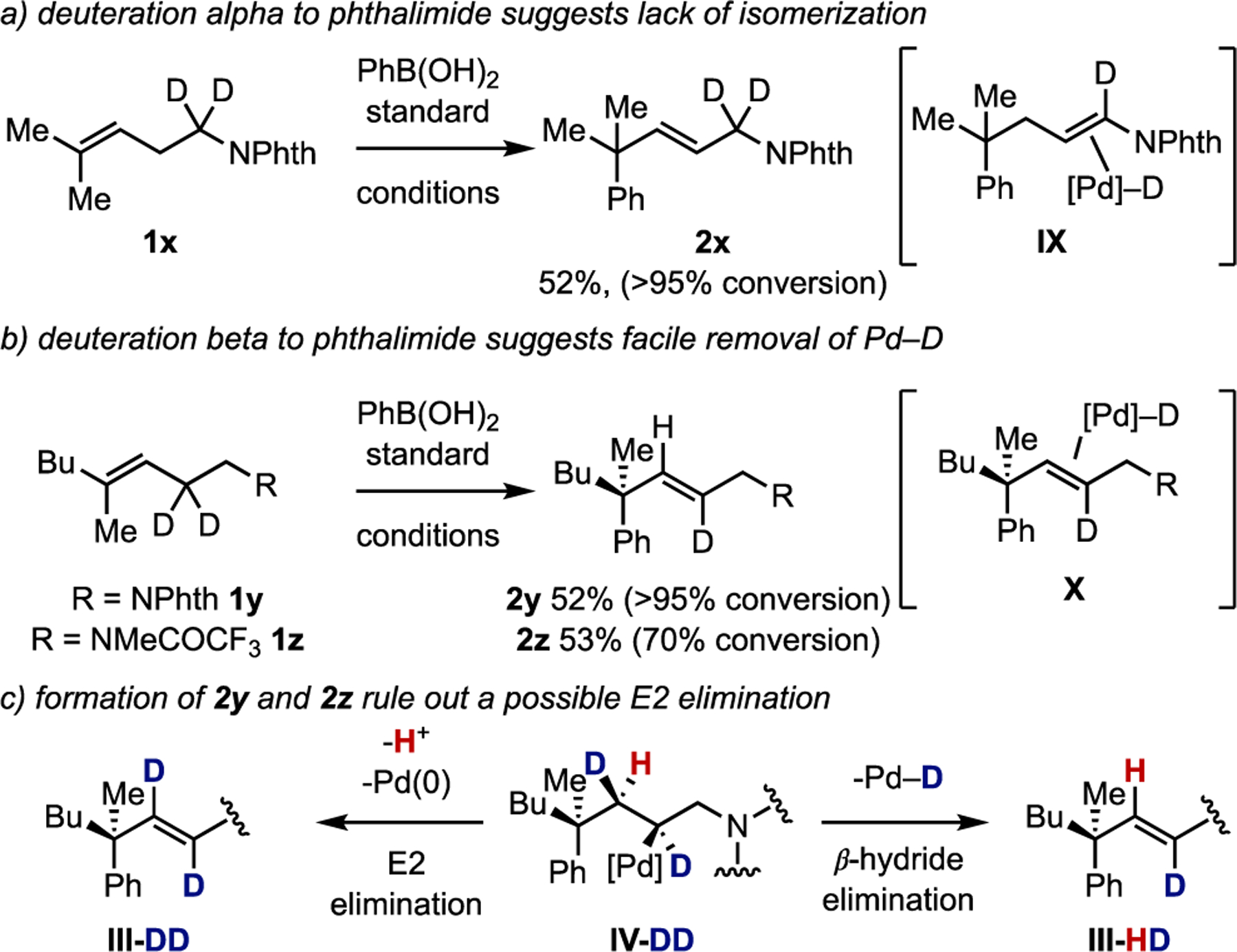
Deuterium labeling studies: a) deuteration α to phthalimide, b) deuteration β to phthalimide, and c) formation of monodeuterated product rules out an E2 elimination.
Deuterium labeling experiments.
On the basis of previous mechanistic investigations from our group using deuterium labeling studies to probe the chain-walking process,64 we investigated the outcome of the reaction when either of the two methylene subunits of the homoallylic chain were deuterated (Figure 3). We began with dideuterosubstrate 1x, which would allow us to probe whether the reaction proceeds through the intermediacy of Pd–D intermediate IX (Figure 3a). Subjecting 1x to the standard reaction conditions resulted in the formation of 2x with no observable deuterium scrambling, as well as a small amount of the double arylation product (see SI). This suggests that if Pd–D alkene intermediate IX is formed, that it exists in equilibrium amounts and no dissociation of the Pd–D occurs from IX.
To confirm the dehydrogenation process occurs from the allylic carbon, substrates 1y and 1z were subjected to the reaction conditions (Figure 3b). This resulted in the formation of monodeuterated products 2y and 2z with no detection of deuterium scrambling, or dideutero-products. This result suggests that the reaction is proceeding through Pd–D alkene complex X, and the final product is formed via dissociation of the ligated Pd–D or deprotonation of the Pd–D. The formation of 2y and 2z also rule out the possibility of an E2-type elimination occurring from the alkyl-Pd complex IV (Figure 3c). Because the migratory insertion and β-hydride elimination processes are stereospecific, the alkyl-Pd complex IV-DD would be formed from 1y or 1z. The exclusive formation of the monodeuterated alkene III-HD rules out an E2 elimination via deprotonation of the hydrogen anti to the Pd to form III-DD.
DFT studies were performed at the IEFPCM(DMF)-M06/6–31+G(d)//M06/SDD-6–311+G(d,p) level of theory. All energies listed are reported in kcal/mol. See the supporting information for details
DFT studies.
In order to further understand the factors that differentiate between the formation of the allyl and ene products, we next turned to DFT calculations. The reaction pathway was calculated for simplified substrate analogues of phthalimide (Phth) 1a and N-methyltrifluoroacetamide (TFA) 1o, both of which showed high selectivity for formation of the allyl product, and N-methylacetamide (Acet) 1u, which showed high selectivity for formation of the ene product. Geometry optimizations were performed with the SDD effective core potential (ECP) for Pd and M06 functional and 6–31+G(d) basis set for all other atoms. Solvent effects were added as energy correction using the IEFPCM (DMF) implicit solvation model at the M06/6–311+G(d,p) level (see SI). Since the selectivity determining step would be determined after the initial migratory insertion of the aryl-Pd complex, all stationary points beyond this step were computed. The results of these calculations are detailed in Schemes 1–5.
Scheme 1.
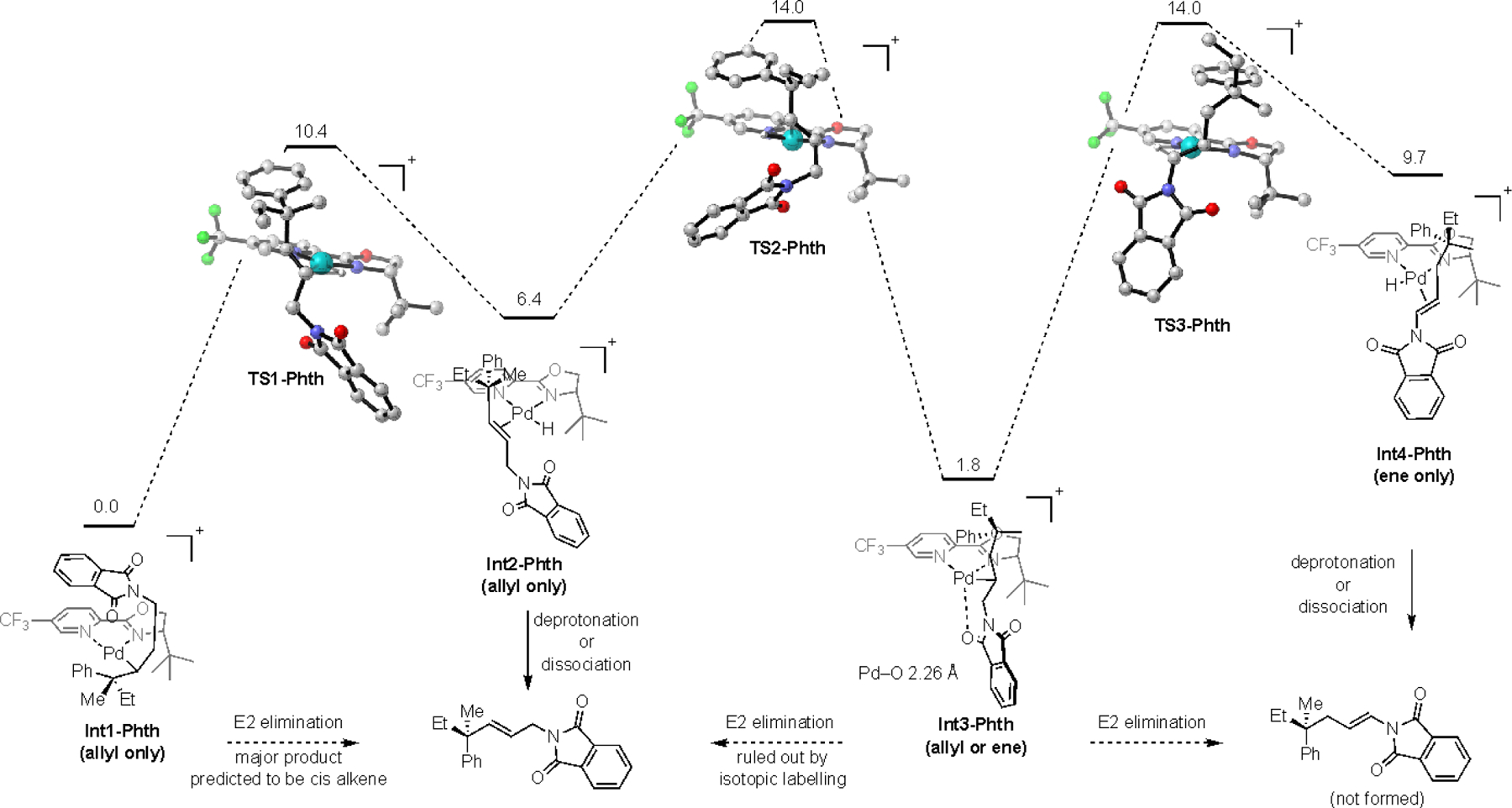
Computed reaction pathway of homoallylic phthalimides
Scheme 5.
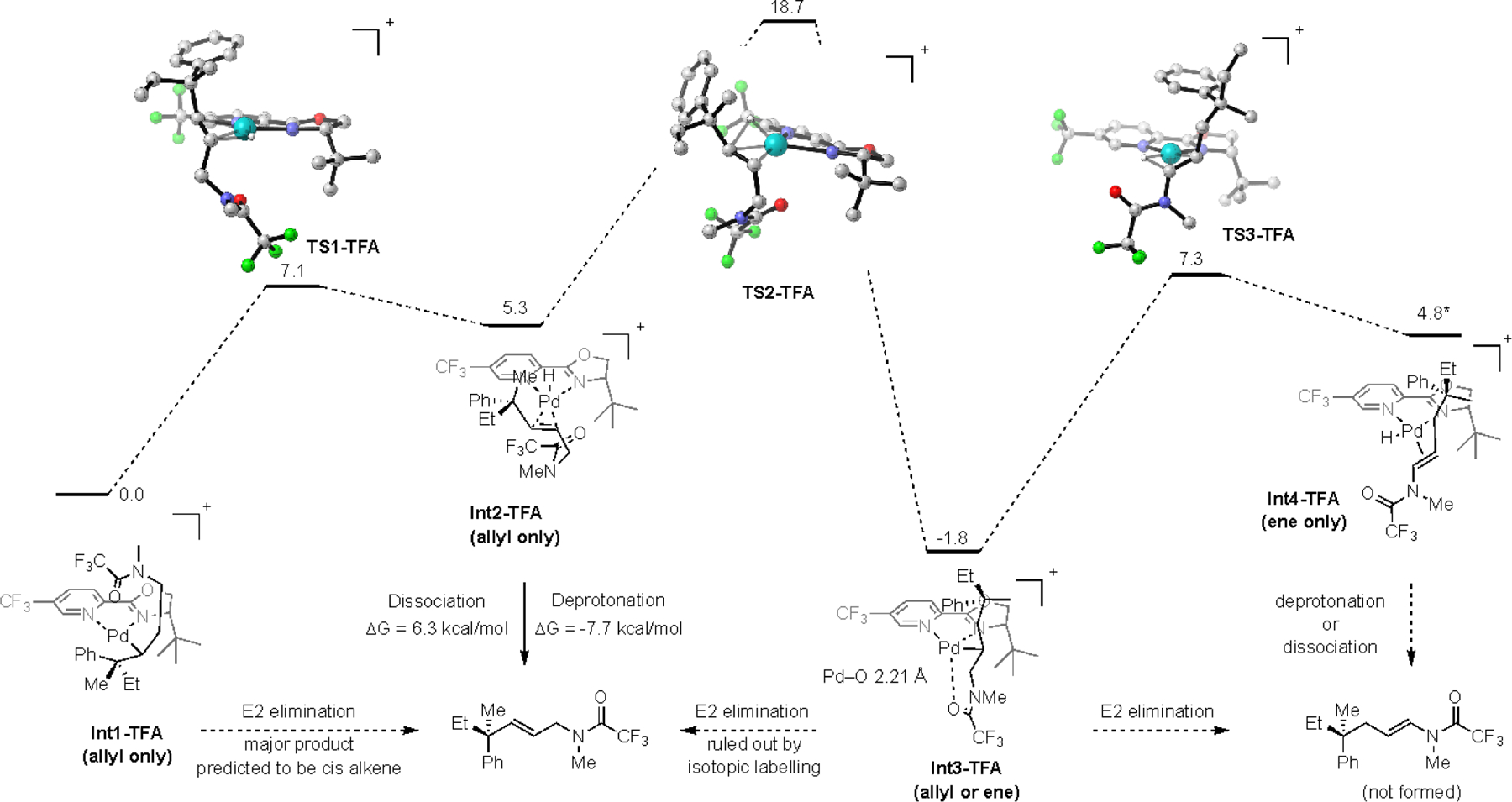
Computed reaction pathway of N-trifluoromethylacetamides
Potential energy surface of the relay Heck reaction with homoallylic phthalimides.
We began our investigation with the homoallylic phthalimide (Scheme 1). The phthalimide complex Int1-Phth was calculated to be the most stable intermediate along the pathway. This is in contrast to previous studies with an alcohol redox acceptor in which the C–Pd and alkene-Pd–H complexes were found to be more stable as the Pd migrated towards the redox acceptor.64 The calculated energy barriers of the following β-hydride elimination and migratory insertion steps (TS1–3-Phth) are relatively low, which suggests that the system is under thermodynamic control. Notably, the allyl Pd–H complex Int2-Phth and the ene Pd–H complex Int4-Phth are relatively high energy intermediates, with low barriers leading to either Int1-Phth or Int3-Phth. A possible mechanism to account for the formation of the allyl product is an E2 elimination to form Pd(0) from either Int1-Phth or Int3-Phth. The lowest energy conformer of Int1-Phth has a favorable anti-periplanar arrangement for an E2 elimination to occur. However, this geometry would lead to the formation of the unobserved cis alkene product. Alternatively, an E2 elimination from Int3-Phth can be ruled out by the isotopic labelling experiments discussed in Figure 3c. Therefore, we computationally investigated the possible mechanisms for loss of Pd–H from Int2-Phth.
We envisioned that the Pd–H complex Int2-Phth could undergo either deprotonation of the Pd–H to directly generate Pd0, ligand exchange of the alkene with the phthalimide carbonyl, or dissociation of the ligated Pd–H through an associative mechanism with DMF.64 To determine the feasibility of intra- or intermolecular deprotonation of the Pd–H by either the phthalimide carbonyl (Scheme 2) or DMF (Scheme 3), respectively, the reaction pathways for these two processes were calculated. For the phthalimide to deprotonate the Pd–H, Int2-Phth must first adopt a conformation in which the carbonyl is in close contact to the hydride (Pre-TS4-Phth). The intramolecular deprotonation of the Pd–H from this high energy intermediate occurs with a relatively high barrier of 20.7 kcal/mol. The resulting protonated phthalimide Post-TS4-Phth would likely undergo rapid deprotonation by trace water or DMF to yield the corresponding neutral Pd(0) complex. To investigate whether DMF could serve as a competent base to deprotonate the Pd–H of Int2-Phth, the solvate DMF-PdH-Phth and protonated DMF adduct DMFH-Pd-Phth were calculated (Scheme 3). This process was found to be thermodynamically favorable, which suggests that this is a viable pathway for the formation of the allyl product (see SI for details).
Scheme 2.
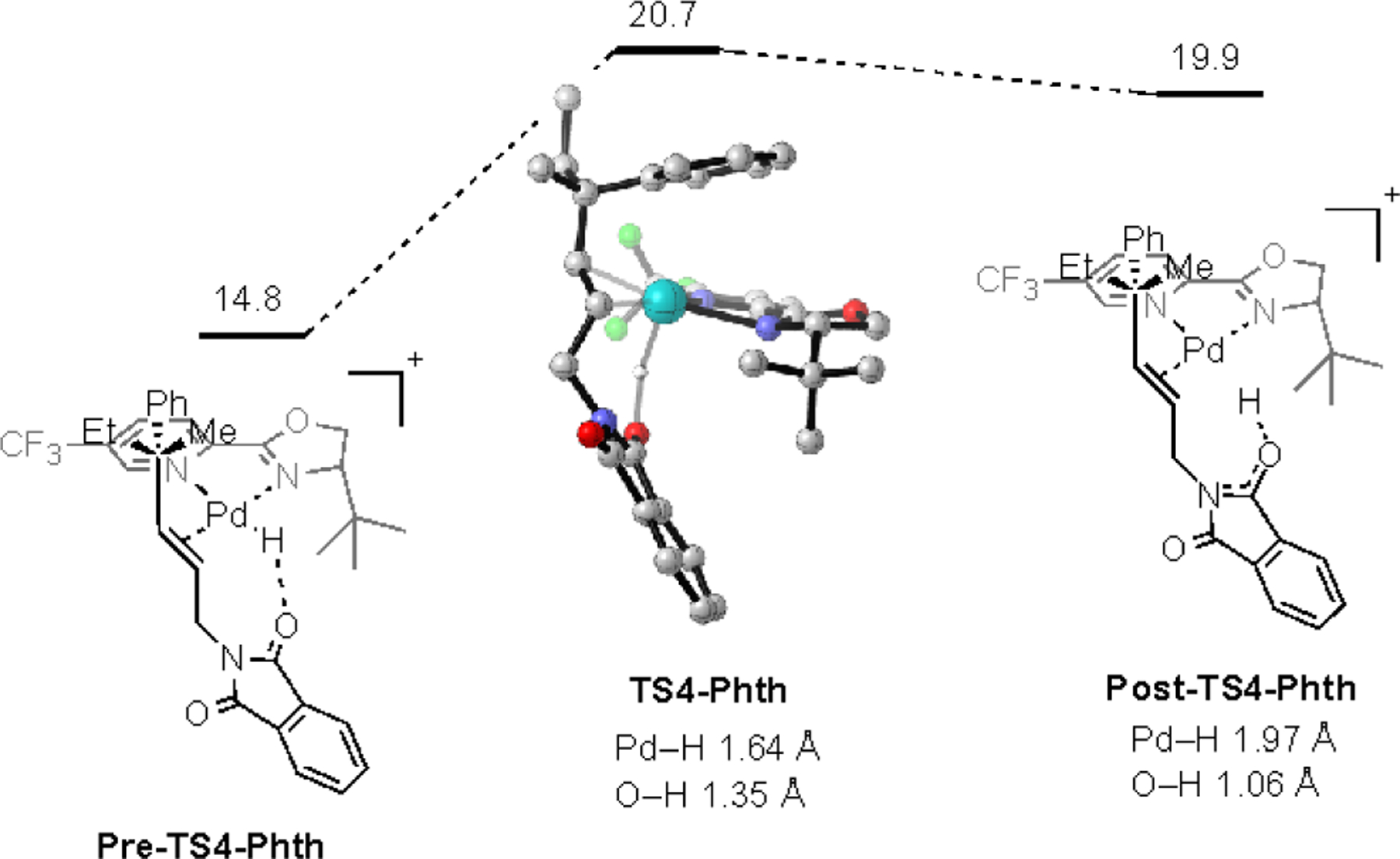
Intramolecular deprotonation of Pd–H
Scheme 3.
Intermolecular deprotonation of Pd–H
However, a reasonable alternative mechanism is direct dissociation of the ligated Pd–H by DMF. To test this hypothesis, the energetics for the displacement of Int2-Phth and Int4-Phth by DMF were calculated (Figure 4). Previous reports have shown that the transition state for similar processes could not be located.65 In line with these, the dissociation process was found to be endergonic for both Int2-Phth and Int4-Phth (Figure 4). Importantly, the process is less endergonic from Int4-Phth. If we assume that the transition states for these two process show the same trend, this data suggests that the selectivity is likely not driven by the relative rates of the dissociation of the ligated Pd–H from Int2-Phth and Int4-Phth by DMF.
Figure 4.
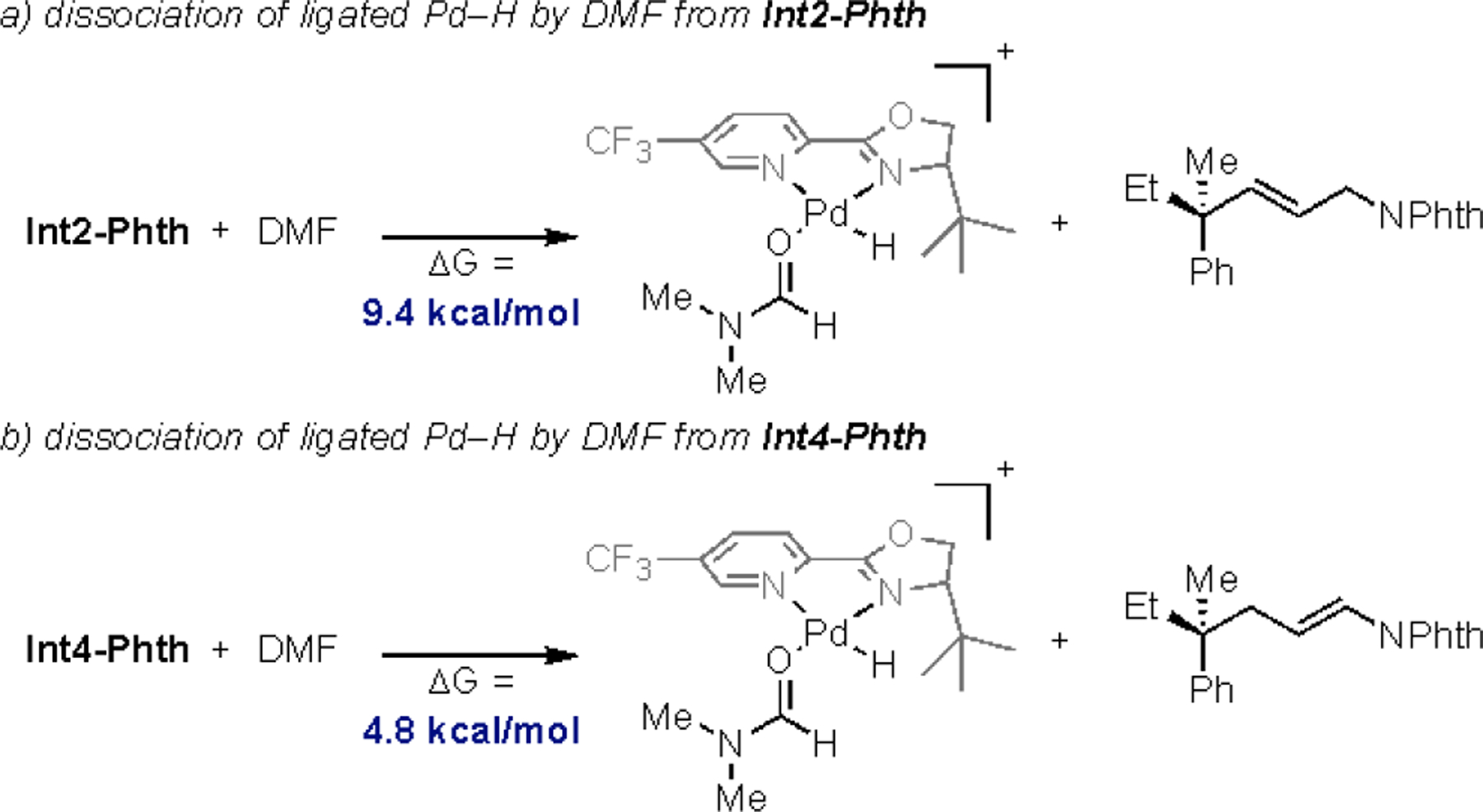
Dissociation of ligated Pd–H by DMF: a) dissociation from Int2-Phth, b) dissociation from Int4-Phth
Overall, these results suggest that the preferential formation of the allylic product is likely a result of the thermodynamics of the system. Additionally, the results presented in Schemes 2–3 suggest that if the final product determining step is deprotonation, it is occurring intermolecularly by DMF or trace water. Alternatively, the results in Figure 4 show that dissociation of the alkene from the ligated Pd–H by DMF is also an energetically viable pathway.
Potential energy surface of the relay Heck reaction with homoallylic acetamides and trifluoroacetamides.
We next carried out a similar analysis for the acetamide (Acet) acceptor group (Scheme 4). In contrast to the phthalimide, the carbonyl of the acetamide shows a significant amount of ligation to the cationic Pd center throughout the reaction pathway consistent with the amides enhanced Lewis basicity. In particular, the degree of ligation of the carbonyl to the Pd center destabilizes TS2-Acet, and stabilizes Int3-Acet and Int4-Acet. This results in the formation of Int3-Acet being essentially irreversible. These two factors combined presumably lead to the preferential formation of the ene-product.
Scheme 4.
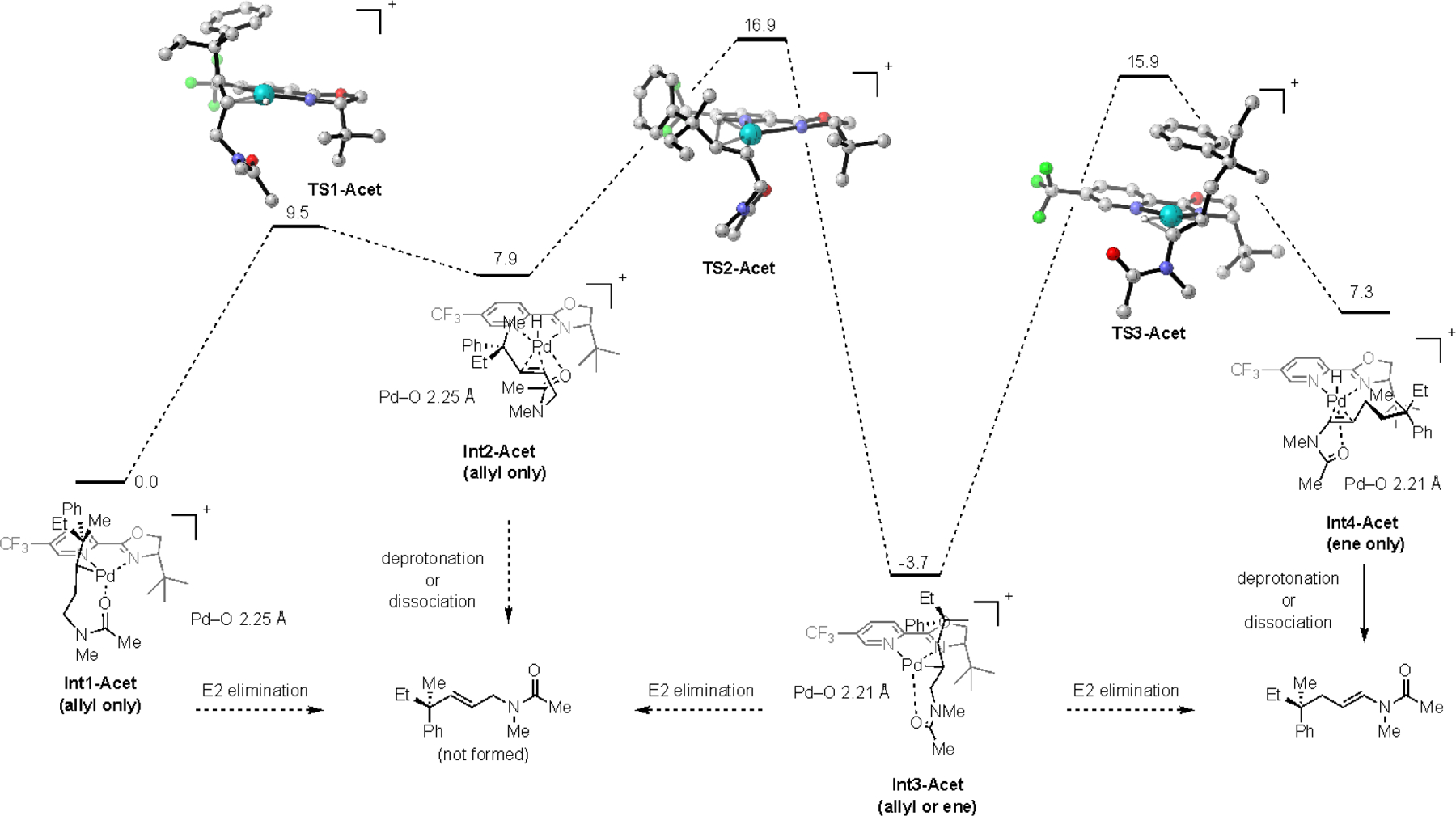
Computed reaction pathway of N-methylacetamides
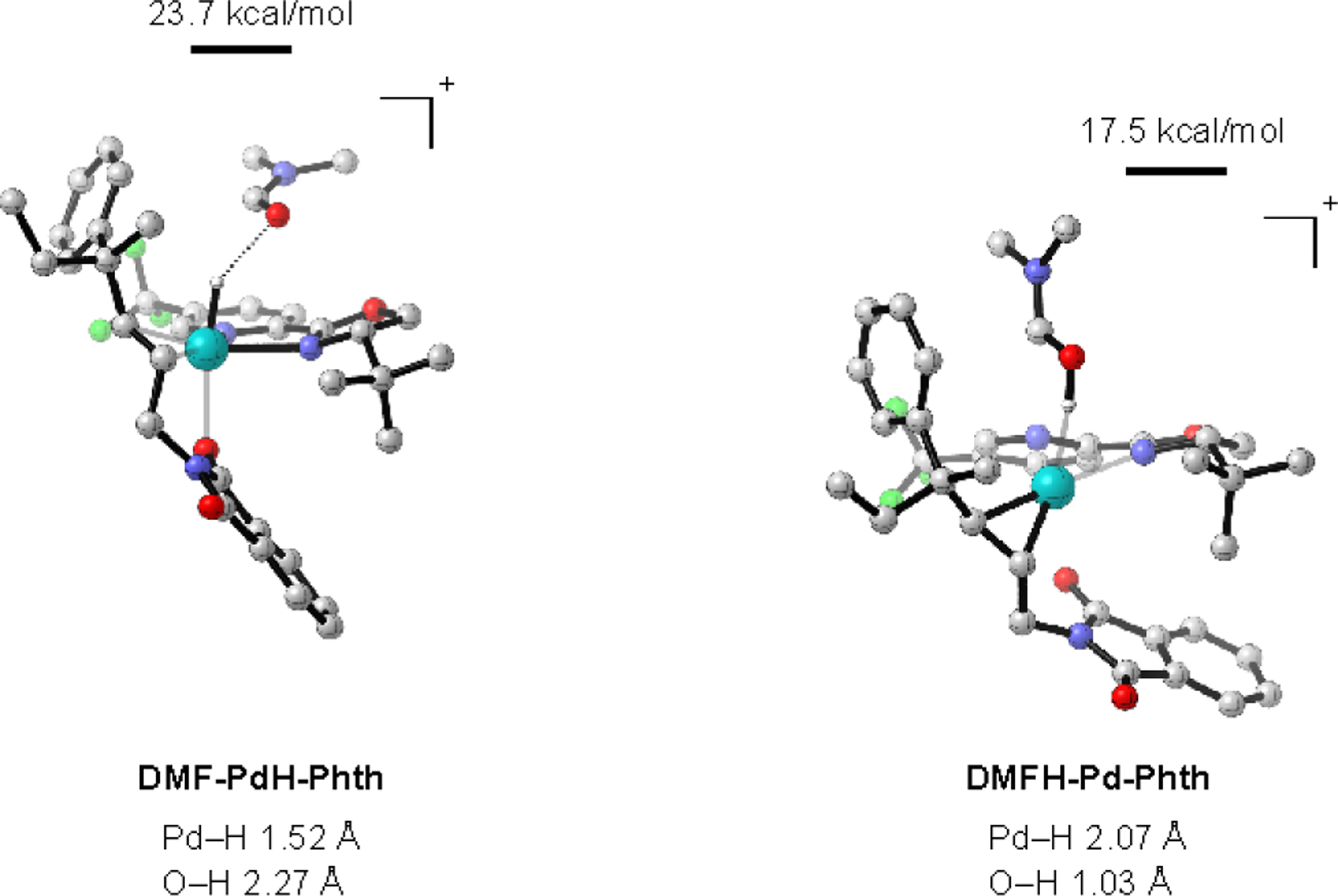
Conversely, the potential energy surface of the trifluoroacetamide (TFA, Scheme 5) is similar to that of the phthalimide (Phth). One notable difference is Int3-TFA is the lowest energy species along the pathway. However, the relative stabilities of Int2-TFA relative to reversion back to Int1-TFA combined with the relatively high barrier of TS2-TFA suggests that this system is potentially under kinetic control. This would require that the barrier for either deprotonation of Int2-TFA or dissociation of Int2-TFA by DMF be lower than conversion to Int3-TFA. The DMF adduct DMF-PdH-TFA was found to be significantly lower in energy compared to the phthalimide analogue (15 kcal/mol vs. 24 kcal/mol, Scheme 5) and the deprotonation of the Pd–H by DMF was found to be energetically favorable (see SI for details). Additionally, the dissociation by DMF was found to be more favorable in the case of Int2-TFA (6 kcal/mol vs. 9 kcal/mol, Scheme 5). Ultimately, these results suggest that the decomposition of Int2-TFA to product may preferentially occur via a dissociation or deprotonation pathway that has a lower barrier than the formation of Int3-TFA. Importantly, this is consistent with the experimentally observed allyl selectivity of the trifluoroacetamide substrates.
DFT studies were performed at the IEFPCM(DMF)-M06/6–31+G(d)//M06/SDD-6–311+G(d,p) level of theory. All energies listed are reported in kcal/mol. See the supporting information for details
DFT studies were performed at the IEFPCM(DMF)-M06/6–31+G(d)//M06/SDD-6–311+G(d,p) level of theory. All energies listed are reported in kcal/mol. *Minimum using loose convergence criteria. See the supporting information for details
Summary of mechanistic studies.
The mechanistic studies presented here have demonstrated that the selective formation of the allyl and ene products is driven primarily by thermodynamics (Figure 5). Additionally, deuterium labeling studies showed that an E2 elimination from Int3-D2 does not occur based on the exclusive formation of 2-HD without any evidence for the formation of 2-DD (Figure 5a). The computational studies revealed that the nature of the carbonyl has a direct impact on the relative stabilities of Int2, Int3, and Int4, with the electron rich amide carbonyl serving to stabilize Int3 and Int4 (Figure 5b). Importantly, the stabilization of these intermediates likely results in the preferential formation of the ene product. Lastly, the computations revealed that the allyl selective pathways greatly favor the formation of Int1 and Int3 through the intermediacy of Int2 (Figure 5c). Overall, the results suggest that the selective formation of the allyl product is a result of deprotonation of the Pd–H of Int2 or direct dissociation of Pd–H from Int2.
Figure 5.
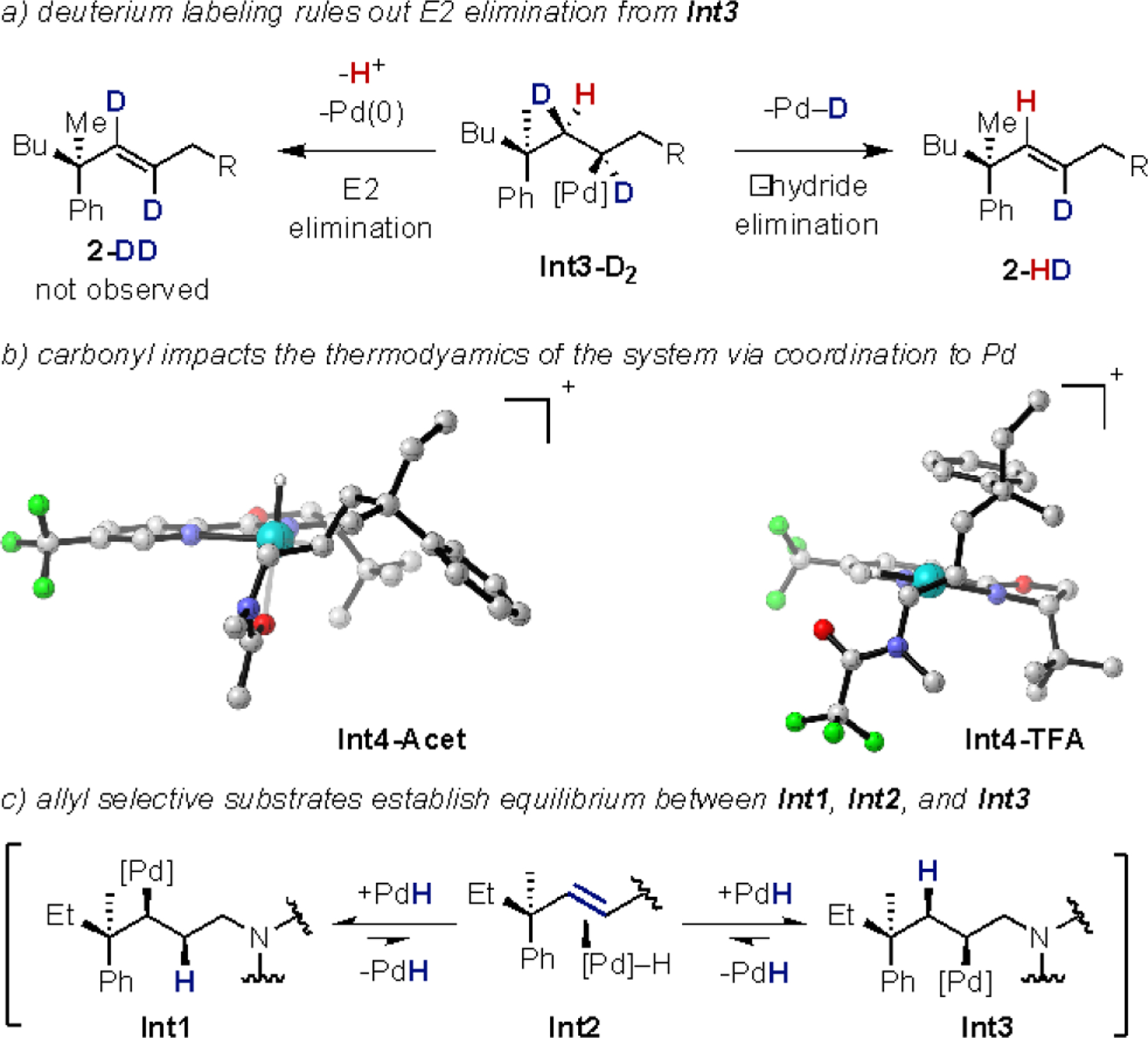
Summary of mechanistic studies: a) Int3 does not undergo E2 elimination, b) ligation of the carbonyl to the Pd effects the selectivity, and c) system is under thermodynamic control
Conclusion
In summary, we have demonstrated that a variety of homoallylic imides, biscarbamates, sulfonamides, and trifluoroacetamides are capable of engaging in the relay Heck reaction to form enantiomerically enriched allylic amines containing a quaternary center. The scope of this process is broad with modest to good yields and high enantioselectivity for all examples. Alternatively, we have found that homoallylic amides give rise to related ene-amides products. To explain the observed difference in selectivity between these two substrate classes, isotopic labeling experiments were combined with DFT calculations. These studies demonstrated that the preferential formation of the allylic products is governed by a combination of the reversible nature of the chain-walking process and the instability of intermediates that would lead to ene amide type products. In contrast, the formation of the ene-amide is governed by the stability imparted by the general Lewis basicity of the amide carbonyl. The underlying strategies of using modest changes to substrate structure to control chain-walking termination events should impact the discovery and development of new synthetically attractive remote functionalization reactions.
Supplementary Material
ACKNOWLEDGMENT
M.S.S. thanks the National Institute of Health (NIGMS R01GM063540), S.P.R thanks the National Institute of Health for financial support through a F32 Ruth L. Kirschtein NRSA fellowship (F32 GM128354-01A1). The computational portion of this work was supported by the National Science Foundation’s Extreme Science and Engineering Discovery Environment (XSEDE, CHE190028) and the Center for High Performance Computing (CHPC) at the University of Utah. NMR results included in this report were recorded at the David M. Grant NMR Center, a University of Utah Core Facility. Funds for construction of the Center and the helium recovery system were obtained from the University of Utah and the National Institutes of Health awards 1C06RR017539-01A1 and 3R01GM063540-17W1 respectively. NMR instruments were purchased with support of the University of Utah and the National Institutes of Health award 1S10OD25241-01.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures for all reactions; spectroscopic characterization data for all new compounds; detailed computational methods; copies of 1H and 13C NMR spectra; copies of SFC chromatograms. (PDF)
REFERENCES
- (1).McDonald RI; Liu G; Stahl SS, Palladium(II)-catalyzed alkene functionalization via nucleopalladation: stereochemical pathways and enantioselective catalytic applications. Chem. Rev 2011, 111, 2981–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zeng T; Liu Z; Schmidt MA; Eastgate MD; Engle KM, Directed, Palladium(II)-Catalyzed Intermolecular Aminohydroxylation of Alkenes Using a Mild Oxidation System. Org. Lett 2018, 20, 3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Liu Z; Gao Y; Zeng T; Engle KM, Transition-Metal-Catalyzed 1,2-Carboboration of Alkenes: Strategies, Mechanisms, and Stereocontrol. Is. J. Chem 2019, doi: 10.1002/ijch.201900087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Liu Z; Wang Y; Wang Z; Zeng T; Liu P; Engle KM, Catalytic Intermolecular Carboamination of Unactivated Alkenes via Directed Aminopalladation. J. Am. Chem. Soc 2017, 139, 11261–11270. [DOI] [PubMed] [Google Scholar]
- (5).Melpolder JB; Heck RF, Palladium-catalyzed arylation of allylic alcohols with aryl halides. J. Org. Chem 1976, 41, 265–272. [Google Scholar]
- (6).Chalk AJ; Magennis SA, Palladium-catalyzed vinyl substitution reactions. I. New synthesis of 2- and 3-phenyl-substituted allylic alcohols, aldehydes, and ketones from allylic alcohols. J. Org. Chem 1976, 41, 273–278. [Google Scholar]
- (7).Chalk AJ; Magennis SA, Palladium-catalyzed vinyl substitution reactions. II. Synthesis of aryl substituted allylic alcohols, aldehydes, and ketones from aryl halides and unsaturated alcohols. J. Org. Chem 1976, 41, 1206–1209. [Google Scholar]
- (8).Larock RC; Leung W-Y; Stolz-Dunn S, Synthesis of aryl-substituted aldehydes and ketones via palladium-catalyzed coupling of aryl halides and non-allylic unsaturated alcohols. Tet. Lett 1989, 30, 6629–6632. [Google Scholar]
- (9).Kochi T; Kanno S; Kakiuchi F, Nondissociative chain walking as a strategy in catalytic organic synthesis. Tet. Lett 2019, 60, 150938–150948. [Google Scholar]
- (10).Gauthier D; Lindhardt AT; Olsen EPK; Overgaard J; Skrydstrup T, In Situ Generated Bulky Palladium Hydride Complexes as Catalysts for the Efficient Isomerization of Olefins. Selective Transformation of Terminal Alkenes to 2-Alkenes. J. Am. Chem. Soc 2010, 132, 7998–8009. [DOI] [PubMed] [Google Scholar]
- (11).Sommer H; Julia-Hernandez F; Martin R; Marek I, Walking Metals for Remote Functionalization. ACS Cent. Sci 2018, 4, 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Larionov E; Li H; Mazet C, Well-defined transition metal hydrides in catalytic isomerizations. Chem. Comm 2014, 50, 9816–9826. [DOI] [PubMed] [Google Scholar]
- (13).Molloy JJ; Morack T; Gilmour R, Positional and Geometrical Isomerisation of Alkenes: The Pinnacle of Atom Economy. Angew. Chem., Int. Ed. Engl 2019, 58, 13654–13664. [DOI] [PubMed] [Google Scholar]
- (14).Ren W; Sun F; Chu J; Shi Y, A Pd-Catalyzed Site-Controlled Isomerization of Terminal Olefins. Org. Lett 2020, 22, 1868–1873. [DOI] [PubMed] [Google Scholar]
- (15).Vasseur A; Bruffaerts J; Marek I, Remote functionalization through alkene isomerization. Nat. Chem 2016, 8, 209–219. [DOI] [PubMed] [Google Scholar]
- (16).Moriya T; Suzuki A; Miyaura N, A stereoselective preparation of γ-alkoxyallylboronates via catalytic isomerization of pinacol [(E)-3-alkoxy-1-propenyl]boronates. Tet. Lett 1995, 36, 1887–1888. [Google Scholar]
- (17).Yamamoto Y; Miyairi T; Ohmura T; Miyaura N, Synthesis of Chiral Esters of (E)-3-(Silyloxy)-2-propenylboronic Acid via the Iridium-Catalyzed Isomerization of the Double Bond. J. Org. Chem 1999, 64, 296–298. [DOI] [PubMed] [Google Scholar]
- (18).Yamamoto Y; Kurihara K; Yamada A; Takahashi M; Takahashi Y; Miyaura N, Intramolecular allylboration of γ-(ω-formylalkoxy)allylboronates for syntheses of trans- or cis-2-(ethenyl)tetrahydropyran-3-ol and 2-(ethenyl)oxepan-3-ol. Tet 2003, 59, 537–542. [Google Scholar]
- (19).Shimizu H; Igarashi T; Miura T; Murakami M, Rhodium-Catalyzed Reaction of 1-Alkenylboronates with Aldehydes Leading to Allylation Products. Angew. Chem., Int. Ed. Engl 2011, 50, 11465–11469. [DOI] [PubMed] [Google Scholar]
- (20).Miura T; Nishida Y; Morimoto M; Murakami M, Enantioselective Synthesis of Anti Homoallylic Alcohols from Terminal Alkynes and Aldehydes Based on Concomitant Use of a Cationic Iridium Complex and a Chiral Phosphoric Acid. J. Amer. Chem. Soc 2013, 135, 11497–11500. [DOI] [PubMed] [Google Scholar]
- (21).Miura T; Nishida Y; Murakami M, Construction of Homoallylic Alcohols from Terminal Alkynes and Aldehydes with Installation of syn-Stereochemistry. J. Amer. Chem. Soc 2014, 136, 6223–6226. [DOI] [PubMed] [Google Scholar]
- (22).Mei TS; Werner EW; Burckle AJ; Sigman MS, Enantioselective redox-relay oxidative heck arylations of acyclic alkenyl alcohols using boronic acids. J. Am. Chem. Soc 2013, 135, 6830–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Werner EW; Mei TS; Burckle AJ; Sigman MS, Enantioselective Heck Arylations of Acyclic Alkenyl Alcohols Using a Redox-Relay Strategy. Science 2012, 338, 1455–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mei TS; Patel HH; Sigman MS, Enantioselective construction of remote quaternary stereocentres. Nature 2014, 508, 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Patel HH; Sigman MS, Palladium-Catalyzed Enantioselective Heck Alkenylation of Acyclic Alkenols Using a Redox-Relay Strategy. J. Am. Chem. Soc 2015, 137, 3462–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang C; Santiago CB; Crawford JM; Sigman MS, Enantioselective Dehydrogenative Heck Arylations of Trisubstituted Alkenes with Indoles to Construct Quaternary Stereocenters. J. Am. Chem. Soc 2015, 137, 15668–15671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chen ZM; Hilton MJ; Sigman MS, Palladium-Catalyzed Enantioselective Redox-Relay Heck Arylation of 1,1-Disubstituted Homoallylic Alcohols. J. Am. Chem. Soc 2016, 138, 11461–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Patel HH; Sigman MS, Enantioselective Palladium-Catalyzed Alkenylation of Trisubstituted Alkenols To Form Allylic Quaternary Centers. J. Am. Chem. Soc 2016, 138, 14226–14229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Race NJ; Schwalm CS; Nakamuro T; Sigman MS, Palladium-Catalyzed Enantioselective Intermolecular Coupling of Phenols and Allylic Alcohols. J. Am. Chem. Soc 2016, 138, 15881–15884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chen ZM; Nervig CS; DeLuca RJ; Sigman MS, Palladium-Catalyzed Enantioselective Redox-Relay Heck Alkynylation of Alkenols To Access Propargylic Stereocenters. Angew. Chem., Int. Ed. Engl 2017, 56, 6651–6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Patel HH; Prater MB; Squire SO; Sigman MS, Formation of Chiral Allylic Ethers via an Enantioselective Palladium-Catalyzed Alkenylation of Acyclic Enol Ethers. J. Am. Chem. Soc 2018, 140, 5895–5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Race NJ; Yuan Q; Sigman MS, Enantioselective C2-Alkylation of Indoles through a Redox-Relay Heck Reaction of 2-Indole Triflates. Chem. Eur. J 2019, 25, 512–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bahamonde A; Al Rifaie B; Martín-Heras V; Allen JR; Sigman MS, Enantioselective Markovnikov Addition of Carbamates to Allylic Alcohols for the Construction of α-Secondary and α-Tertiary Amines. J. Amer. Chem. Soc 2019, 141, 8708–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Allen JR; Bahamonde A; Furukawa Y; Sigman MS, Enantioselective N-Alkylation of Indoles via an Intermolecular Aza-Wacker-Type Reaction. J. Am. Chem. Soc 2019, 141, 8670–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Liu J; Yuan Q; Toste FD; Sigman MS, Enantioselective construction of remote tertiary carbon–fluorine bonds. Nat. Chem 2019, 11, 710–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Prater MB; Sigman MS, Enantioselective Synthesis of Alkyl Allyl Ethers via Palladium-Catalyzed Redox-Relay Heck Alkenylation of O-Alkyl Enol Ethers. Isr. J. Chem doi: 10.1002/ijch.201900077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Carmona RC; Correia CRD, Stereoselective Synthesis of 3-Hydroxy-4-arylcyclopentanones and 4-Arylcyclopentenones through a Heck-Matsuda Desymmetrization ofmeso cis-4-Cyclopentene-1,3-diol. Adv. Synth. Cat 2015, 357, 2639–2643. [Google Scholar]
- (38).Oliveira CC; Angnes RA; Correia CRD, Intermolecular Enantioselective Heck–Matsuda Arylations of Acyclic Olefins: Application to the Synthesis of β-Aryl-γ-lactones and β-Aryl Aldehydes. J. Org. Chem 2013, 78, 4373–4385. [DOI] [PubMed] [Google Scholar]
- (39).Liu Z-S; Qian G; Gao Q; Wang P; Cheng H-G; Wei Q; Liu Q; Zhou Q, Palladium/Norbornene Cooperative Catalysis To Access Tetrahydronaphthalenes and Indanes with a Quaternary Center. ACS Cat. 2018, 8, 4783–4788. [Google Scholar]
- (40).Larionov E; Lin L; Guénée L; Mazet C, Scope and Mechanism in Palladium-Catalyzed Isomerizations of Highly Substituted Allylic, Homoallylic, and Alkenyl Alcohols. J. Amer. Chem. Soc 2014, 136, 16882–16894. [DOI] [PubMed] [Google Scholar]
- (41).Lin L; Romano C; Mazet C, Palladium-Catalyzed Long-Range Deconjugative Isomerization of Highly Substituted α,β-Unsaturated Carbonyl Compounds. J. Amer. Chem. Soc 2016, 138, 10344–10350. [DOI] [PubMed] [Google Scholar]
- (42).Singh S; Bruffaerts J; Vasseur A; Marek I, A unique Pd-catalysed Heck arylation as a remote trigger for cyclopropane selective ring-opening. Nat. Comm 2017, 8, 14200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hou X-L; Xu B; Ding C-H; Li H; Gao A; Liu X-Y, Kinetic Resolution of 5-Substituted Cyclohexenols by Palladium-Catalyzed Asymmetric Redox-Relay Heck Reaction. Synthesis 2016, 49, 159–166. [Google Scholar]
- (44).Singh S; Simaan M; Marek I, Pd-Catalyzed Selective Remote Ring Opening of Polysubstituted Cyclopropanols. Chem. Eur. J 2018, 24, 8553–8557. [DOI] [PubMed] [Google Scholar]
- (45).Grotjahn DB; Larsen CR; Gustafson JL; Nair R; Sharma A, Extensive Isomerization of Alkenes Using a Bifunctional Catalyst: An Alkene Zipper. J. Am. Chem. Soc 2007, 129, 9592–9593. [DOI] [PubMed] [Google Scholar]
- (46).Juliá-Hernández F; Moragas T; Cornella J; Martin R, Remote carboxylation of halogenated aliphatic hydrocarbons with carbon dioxide. Nature 2017, 545, 84–88. [DOI] [PubMed] [Google Scholar]
- (47).Gaydou M; Moragas T; Julia-Hernandez F; Martin R, Site-Selective Catalytic Carboxylation of Unsaturated Hydrocarbons with CO2 and Water. J. Am. Chem. Soc 2017, 139, 12161–12164. [DOI] [PubMed] [Google Scholar]
- (48).Sun SZ; Börjesson M; Martin-Montero R; Martin R, Site-Selective Ni-Catalyzed Reductive Coupling of α-Haloboranes with Unactivated Olefins. J. Am. Chem. Soc 2018, 140, 12765–12769. [DOI] [PubMed] [Google Scholar]
- (49).Chinkov N; Majumdar S; Marek I, New Approach to the Stereoselective Synthesis of Metalated Dienes via an Isomerization-Elimination Sequence. J. Am. Chem. Soc 2002, 124, 10282–10283. [DOI] [PubMed] [Google Scholar]
- (50).Ho GM; Judkele L; Bruffaerts J; Marek I, Metal-Catalyzed Remote Functionalization of ω-Ene Unsaturated Ethers: Towards Functionalized Vinyl Species. Angew. Chem., Int. Ed 2018, 57, 8012–8016. [DOI] [PubMed] [Google Scholar]
- (51).Romano C; Mazet C, Multicatalytic Stereoselective Synthesis of Highly Substituted Alkenes by Sequential Isomerization/Cross-Coupling Reactions. J. Am. Chem. Soc 2018, 140, 4743–4750. [DOI] [PubMed] [Google Scholar]
- (52).Romano C; Fiorito D; Mazet C, Remote Functionalization of α,β-Unsaturated Carbonyls by Multimetallic Sequential Catalysis. J. Amer. Chem. Soc 2019, 141, 16983–16990. [DOI] [PubMed] [Google Scholar]
- (53).He Y; Cai Y; Zhu S, Mild and Regioselective Benzylic C–H Functionalization: Ni-Catalyzed Reductive Arylation of Remote and Proximal Olefins. J. Am. Chem. Soc 2017, 139, 1061–1064. [DOI] [PubMed] [Google Scholar]
- (54).Chen F; Chen K; Zhang Y; He Y; Wang YM; Zhu S, Remote Migratory Cross-Electrophile Coupling and Olefin Hydroarylation Reactions Enabled by in Situ Generation of NiH. J. Am. Chem. Soc 2017, 139, 13929–13935. [DOI] [PubMed] [Google Scholar]
- (55).Xiao J; He Y; Ye F; Zhu S, Remote sp3 C-H Amination of Alkenes with Nitroarenes. Chem. 2018, 4, 1645–1657. [Google Scholar]
- (56).Larock RC; Lu YD; Bain AC; Russell CE, Palladium-catalyzed coupling of aryl iodides, nonconjugated dienes and carbon nucleophiles by palladium migration. J. Org. Chem 1991, 56, 4589–4590. [Google Scholar]
- (57).Larock RC; Wang Y; Lu Y; Russell CA, Synthesis of Aryl-Substituted Allylic Amines via Palladium-Catalyzed Coupling of Aryl Iodides, Nonconjugated Dienes, and Amines. J. Org. Chem 1994, 59, 8107–8114. [Google Scholar]
- (58).Dupuy S; Zhang KF; Goutierre AS; Baudoin O, Terminal-Selective Functionalization of Alkyl Chains by Regioconvergent Cross-Coupling. Angew. Chem., Int. Ed 2016, 55, 14793–14797. [DOI] [PubMed] [Google Scholar]
- (59).Borah AJ; Shi Z, Rhodium-Catalyzed, Remote Terminal Hydroarylation of Activated Olefins through a Long-Range Deconjugative Isomerization. J. Am. Chem. Soc 2018, 140, 6062–6066. [DOI] [PubMed] [Google Scholar]
- (60).Aspin S; Goutierre AS; Larini P; Jazzar R; Baudoin O, Synthesis of aromatic alpha-aminoesters: palladium-catalyzed long-range arylation of primary C sp 3-H bonds. Angew. Chem., Int. Ed 2012, 51, 10808–10811. [DOI] [PubMed] [Google Scholar]
- (61).Peng L; Li Y; Li Y; Wang W; Pang H; Yin G, Ligand-Controlled Nickel-Catalyzed Reductive Relay Cross-Coupling of Alkyl Bromides and Aryl Bromides. ACS Catal. 2018, 8, 310–313. [Google Scholar]
- (62).Peng L; Li Z; Yin G, Photochemical Nickel-Catalyzed Reductive Migratory Cross-Coupling of Alkyl Bromides with Aryl Bromides. Org. Lett 2018, 20, 1880–1883. [DOI] [PubMed] [Google Scholar]
- (63).Han C; Fu Z; Guo S; Fang X; Lin A; Yao H, Palladium-Catalyzed Remote 1,n-Arylamination of Unactivated Terminal Alkenes. ACS Cat. 2019, 9, 4196–4202. [Google Scholar]
- (64).Hilton MJ; Xu LP; Norrby PO; Wu YD; Wiest O; Sigman MS, Investigating the nature of palladium chain-walking in the enantioselective redox-relay Heck reaction of alkenyl alcohols. J. Org. Chem 2014, 79, 11841–11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Xu L; Hilton MJ; Zhang X; Norrby PO; Wu YD; Sigman MS; Wiest O, Mechanism, reactivity, and selectivity in palladium-catalyzed redox-relay Heck arylations of alkenyl alcohols. J. Am. Chem. Soc 2014, 136, 1960–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.