

Abstract
Objective:
To determine the prevalence of cerebrospinal fluid (CSF) markers associated with inflammation (i.e., elevated white blood cell count, protein concentration, and CSF-specific oligoclonal bands) in patients with early active autoimmune encephalitis (AE).
Methods:
CSF characteristics, including WBC count, protein concentration, and oligoclonal banding, were analyzed in patients diagnosed with AE at two tertiary care centers.
Results:
Ninety-five patients were included in the study. CSF white blood cell counts and protein levels were within normal limits for 27% (CI95%: 19–37) of patients with AE. When results of oligoclonal banding were added, 14% (CI95%: 6–16) of patients with AE had “normal” CSF. The median CSF white blood cell count was 8 cells/mm3 (range: 0–544) and the median CSF protein concentration was 0.42 g/L (range: 0.15–3.92).
Conclusions:
White blood cell counts and protein levels were within normal limits in the CSF of a substantial proportion of patients with early active AE. Inclusion of CSF oligoclonal banding identified a higher proportion of patients with an inflammatory CSF profile, especially when CSF was sampled early in the disease process.
Keywords: Autoimmune Encephalitis, Autoimmune Diseases, Encephalitis, diagnostic studies, cerebrospinal fluid
1. Introduction
The number of antibodies associated with autoimmune encephalitis (AE) has markedly increased over the last decade.(Bradshaw and Linnoila, 2018) Despite this expansion, treatment decisions must often be made before results of antibody testing are available, as early treatment is associated with better outcomes.(Balu et al., 2018; Dalmau et al., 2008; Finke et al., 2017, 2012; Hébert et al., 2018; Irani et al., 2013, 2010) To address this issue, diagnostic criteria for AE emphasize early detection of clinical, neuroimaging, and cerebrospinal fluid (CSF) features suggestive of central nervous system inflammation.(Graus et al., 2016) However, cases of definite AE without CSF pleocytosis are increasingly recognized in studies of “paraneoplastic AE”,(Gultekin et al., 2000) epidemiological studies of encephalitis,(Dubey et al., 2018) patients above 60 years-of-age with neuronal cell-surface antibodies,(Escudero et al., 2017) and patients with specific autoantibodies.(Blinder and Lewerenz, 2019; Irani et al., 2013; Warren et al., 2018) The high degree of variability in CSF findings in patients with AE (Blinder and Lewerenz, 2019; Dalmau et al., 2008; Honorat et al., 2017; Petit-Pedrol, 2014; Wang et al., 2016) exemplifies the need to comprehensively evaluate CSF findings in larger cohorts of patients with AE.
To address this clinical gap, we evaluated the period prevalence of commonly-available markers of CSF inflammation in patients presenting with AE at two tertiary care centers. Specifically, we determined the frequency of detection of CSF pleocytosis, elevated protein, and increased CSF-specific oligoclonal bands (OCB) in patients with early active AE. Although protein concentration and CSF-specific OCBs are not part of the current diagnostic criteria for “definite AE”,(Graus et al., 2016) elevated protein may serve as a non-specific marker of central nervous system damage,(Reiber and Peter, 2001) while the detection of OCB may support a diagnosis of “antibody-negative but probable AE”,(Graus et al., 2016) with the potential to identify patients with AE.
2. Material and Methods
2.1. Patients and clinical characteristics
The University Health Network (UHN) in Toronto (Ontario, Canada) and Washington University School of Medicine (WUSM)-affiliated Barnes Jewish Hospital in Saint Louis (Missouri, USA) are tertiary centers that care for patients with AE. Information on patients meeting clinical criteria for AE (Graus et al., 2016)was collected from consecutively-encountered patients between January 1st, 2012 and December 31st, 2019. Patients were included if they met diagnostic criteria for “definite AE” or “antibody-negative but probable AE”,(Graus et al., 2016) and underwent a lumbar puncture (LP) during the early active phase of AE. Patients were excluded from the study if data were missing on either CSF WBC or protein concentration. We defined “early active” disease as ongoing symptoms of a first presentation of AE.
At UHN, the sources of clinical data included paper chart documentation maintained in the clinic and electronic patient record. At WUSM, patients were enrolled in a prospective study of AE, permitting banking of CSF and collection of clinically-relevant data. Data were collected at both sites concerning demographic features (age, sex, enrollment center), time from symptom onset to the first LP, results of the first LP (WBC count, protein and glucose concentration, percentage of lymphocytes, results of OCB), diabetic status, whether the patient was receiving immunosuppression at the time of the LP (“immunosuppressive status”), neuroimaging and electroencephalographic (EEG) features, and results of AE autoantibody panel testing. At UHN, autoantibody testing was performed by Mitogen Labs (Calgary, Canada). At WUSM, autoantibody testing was performed via the Mayo Clinic Neuroimmunology Laboratory (Rochester, Minnesota) using the proprietary Autoimmune or Paraneoplastic Autoantibody Evaluation panels. Both panels included indirect immunofluorescence assays, and were performed by applying specimen to frozen mouse composite tissue, washed and treated with fluorescein-conjugated IgG. Subsequent testing for specific disease-associated antibodies was performed when indicated (e.g., cell binding assays, Western blot, radioimmune assays).(Brooks et al., 2019; Lee and Lee, 2016) Investigations were ordered by treating clinicians at either center, in accordance with clinical indications. A minima, CSF HSV/VZV serologies, inflammatory markers, and neuroimaging with MRI were performed on every patient to exclude alternative etiologies of encephalitis.
2.2. Outcome variables
The outcome variables for this study were the prevalence of CSF pleocytosis (defined as >5 cells/mm3) (Graus et al., 2016), elevated protein concentration (defined as >0.45 g/L), and presence of three or more CSF-specific OCB at the first LP in patients with early active AE. At WUSM, CSF cell count was performed on the IQ200 IRIS automated analyzer (Beckman Coulter). At UHN, cell count was performed manually. At both centers, results were reported with correction for dilution factors. The interaction between results of the CSF analysis were considered when two parameters were included (CSF WBC and protein concentration), and following the addition of a third parameter (CSF OCB).
2.3. Exposure variables
The exposure variables for this study were age (categorized by decade), sex, diabetic status, immunosuppressive status, delay from symptom onset to LP (in weeks), presence of features of AE on neuroimaging, abnormal EEG, and antibody subtypes (i.e., cell-surface, intracellular, or negative).
2.4. Statistical analysis
Descriptive statistics were calculated for clinical data. Differences between the study samples from the two sites were assessed using the Mann-Whitney U test (continuous variables) or Fisher’s exact test (categorical variables). The prevalence of outcome variables in our sample was calculated with 95% confidence intervals. The predictive value of the aforementioned exposure variables—with and without ancillary studies (i.e., EEG and MRI) as predictive variables—on the outcome variables was analysed using multivariable logistic regression analyses. Statistical analysis was completed using R (R Core Team, Vienna, 2018). Statistical significance was defined as p-value <0.05 unless otherwise specified.
2.5. Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by research ethics boards at each institution. All procedures performed in human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all participants or their delegates.
2.6. Data Availability
Anonymyzed data will be shared upon request addressed to the corresponding author by qualified investigators.
3. Results
One-hundred-and-two patients with AE were enrolled between January 1st, 2012 and December 31st, 2019. Seven patients were excluded due to incomplete information concerning the first LP obtained during the early active phase of AE: their median age was 15 years (range 10–29); five were female; CSF NMDAR IgG autoantibodies were detected in 6 patients; no AE-associated antibodies were detected in 1 patient. Of the 95 patients meeting inclusion criteria, 41 were recruited at WUSM and 54 at UHN. Demographic details and clinical features are summarized in Table 1. The majority of patients were female (62%). The median age-at-symptomatic onset was 51 years (range 14–83). Patients were more likely to present with subacute seizures and working memory impairment at UHN than WUSM; there were no other significant differences in the study samples’ characteristics between the two sites. Cell-surface antibodies associated with AE were detected in 59 (62%) patients, most commonly NMDAR and LGI1 autoantibodies. Antibodies were detected in the CSF, except for LGI1 which was detected in serum only in 10/20 (50%) of affected individuals. Antibodies against intracellular antigens were identified in 15 patients (16%); 19 patients (20%) met criteria for antibody-negative but probable AE (Table 2). Sixty-three patients (66%) had their first LP performed within 8 weeks from symptom onset (median = 32 days; range 0–2034)—87% within 6 months and 96% within one year. OCB were measured in 51 (54%) patients.
Table 1:
Description of study sample
| Participant Characteristics | UHN | WUSM | Combined | p-valuea |
|---|---|---|---|---|
| Demographics | ||||
| Patients included, n (%) | 54 (57) | 41 (43) | 95 (100) | -- |
| Median age, year (range) | 40 (14–83) | 57 (18–83) | 51 (14–83) | 0.13 |
| Female, n (%) | 37 (69) | 22 (54) | 59 (62) | 0.20 |
| Median delay from symptom onset to first LP, days (range) | 37 (1–2034) | 21 (0–376) | 32 (0–2034) | 0.07 |
| Clinical featuresb | ||||
| Subacute onset working memory deficits, n (%) | 51 (94) | 31 (76) | 82 (86) | 0.01* |
| Subacute onset of seizure, n (%) | 34 (63) | 16 (39) | 50 (53) | 0.03* |
| Subacute onset of psychiatric symptoms, n (%) | 40 (74) | 32 (78) | 71 (76) | 0.81 |
| MRI findings suggestive of AE, n (%)c | 15 (28) | 18 (46) | 33 (36) | 0.08 |
| EEG abnormalities suggestive of AE, n (%)d | 37 (79) | 31 (79) | 68 (79) | 0.94 |
| CSF Pleocytosis, n (%)e | 26 (48) | 27 (66) | 53 (56) | 0.10 |
| Patients admitted to ICU, n (%) | 21 (39) | 17 (42) | 38 (40) | 0.83 |
| Status epilepticus at presentation, n (%) | 9 (17) | 1 (2) | 10 (11) | 0.04* |
p-value of the difference between the UHN and WUSM sites using Mann-Whitney U test for continuous variables and Fisher’s exact test for categorical variables.
p<0.05
Subacute-onset is defined as a “rapid progression of less than 3 months”, based on clinical criteria from Graus et al. 2016.
Temporal FLAIR signal changes highly restricted to the medial temporal lobes.
Epileptiform or slow-wave activity involving the temporal lobes.
>5cells/mm3.
AE: autoimmune encephalitis; ICU: intensive care unit; LP: lumbar puncture; UHN: University Health Network; WUSM: Washington University School of Medicine.
Table 2:
Autoantbodies detected in combined sample
| Autoantibodies, n (%) | |
|---|---|
| Cell-surface (total) | 59 (62) |
| AMPAR | 2 (2) |
| CASPR2 | 1 (1) |
| GABAb | 3 (3) |
| GlyR | 1 (1) |
| LGI1 | 20 (21) |
| NMDAR | 31 (33) |
| VGCC | 1 (1) |
| Intracellular (total) | 15 (16) |
| GAD | 4 (4) |
| GFAP | 1 (1) |
| Hu | 2 (2) |
| Ma2 | 4 (4) |
| SOX1 | 1 (1) |
| Yo | 3 (3) |
| Others (total) | 2 (2) |
| ADEM | 1 (1) |
| SREAT | 1 (1) |
| Antibody-negative AE | 19 (20) |
| Total | 95 (100) |
AE: autoimmune encephalitis; AMPA: alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor; ADEM: acute disseminated encephalomyelitis; CASPR2: contacting-associated protein-like 2; GABAb: gamma aminobutyric acid; GFAP: glial fibrillary acidic protein; GlyR: glycine Receptor; LGI1: leucine-rich glioma inactivated 1; NMDAR: N-methyl-D-aspartate receptor; SREAT: steroid-responsive encephalopathy associated with autoimmune thyroiditis; VGCC: voltage-gated calcium channel.
Fifty-three cases (53/95, 56%, CI95%: 45–66) met criteria for pleocytosis (Table 1). The median WBC count was 8 cells/mm3 (range: 0–544), with median CSF lymphocyte proportion 90% (range: 3–100) (Figure 1A). Protein concentration was elevated in 45/95 (47%, CI95%: 37–59; Figure 1B), with a median CSF protein concentration of 0.42 g/L (range: 0.15–3.92). The median CSF glucose concentration was 66.6 mg/dL (range: 37.8–136.8).
Fig.1. Distribution of CSF findings in patients with early active autoimmune encephalitis.
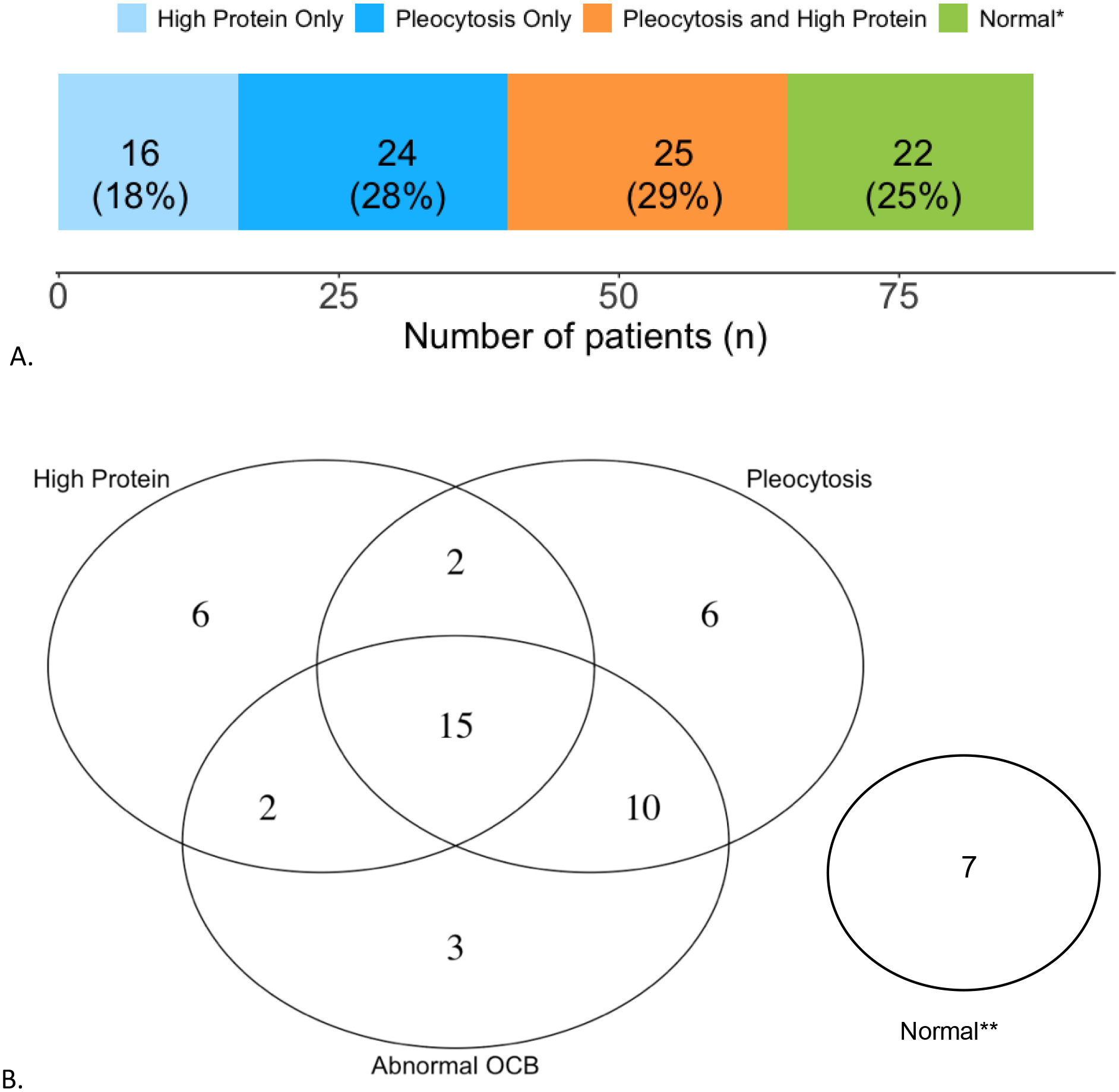
A. CSF white blood cell count density for patients with early active autoimmune encephalitis. Pleocytosis threshold is marked by the dotted red line and the median CSF white blood cell count for our sample with a blue dashed line. B. CSF protein concentration density for patients with early active autoimmune encephalitis. Elevated protein threshold is marked by the dotted red line and the median CSF protein concentration for our sample with a blue dashed line. Graph created with R (R Core Team, Vienna, 2018).
When considering antibody subtypes, the proportion of patients with CSF pleocytosis was 59% (CI95%:42–69) among those with AE associated with neuronal cell-surface autoantibodies (antibody-mediated encephalitis), 60% (CI95%: 32–84) among those with intracellular antibodies, and 47% (CI95%:24–71) among those with negative antibody testing. The prevalence of CSF pleocytosis was lower in patients with AE associated with LGI1 30% (CI95%: 12–54) than NMDAR (77%; CI95%: 59–90; p=0.04). On the multivariable logistic regression (AUC=0.65), increasing age correlated with a decreased odds of having CSF pleocytosis (OR 0.97; CI95%: 0.95–0.99; p=0.03). Findings of AE on MRI (OR 0.78; CI95%: 0.30–2.22; p=0.65) or an abnormal EEG (OR: 1.99; CI95%: 0.60–6.70; p=0.22) were not associated with CSF pleocytosis. Similarly, when considering antibody subtypes, the proportion of patients with elevated protein concentration was 37% (CI95%: 25–50) among patients with neuronal cell-surface autoantibodies, 53% (CI95%: 27–79) among those with intracellular antibodies, and 68% (CI95%: 43–87) in patients without AE-associated antibodies. Protein was elevated in 32% (CI95%: 17–51) of patients with NMDAR autoantibodies, and 40% (CI95%: 19–64) of patients with LGI1 autoantibodies (p=0.42). On the multivariable logistic regression (AUC=0.75), increasing age (OR 1.03; CI95%: 1.00–1.05, p=0.03) was associated with high protein concentration when adjusting for female sex, delay from symptom onset to LP, immunomodulatory treatment status at time of LP, and autoantibody subtype. Findings of AE on MRI (OR 0.40; CI95%: 0.12–1.36; p=0.14) or an abnormal EEG (OR: 0.22; CI95%: 0.60–9.30; p=0.22) did not correlate with elevated protein.
Of the 51 patients with OCB measurements, an abnormal number of CSF-specific bands (≥3) were detected in 59% (CI95%: 44–72), including 42% (CI95%: 25–61) of patients with cell-surface autoantibodies, 38% (CI95%: 9–76) of patients with intracellular autoantibodies, and 50% (CI95%:16–84) of patients with negative antibody-testing. Eighty-seven percent (CI95%: 60–98) of patients with NMDAR autoantibodies and 69% (CI95%: 39–91) of patients with LGI1 autoantibodies had OCB in the CSF (p=0.39). In multivariate logistic regression adjusting for the aforementioned exposure variables (AUC=0.79), increasing age (OR 0.96; CI95%: 0.93–0.99, p=0.05) and delay from symptom onset to LP of more than 8 weeks (OR 0.19; CI95%: 0.03–0.99, p=0.05) were associated with a lower odds of having ≥3 CSF-specific OCB. Abnormal EEG (OR 0.36; CI95%: 0.05–2.42; p=0.28) and MRI findings of AE (OR 2.09; CI95%: 0.40–1.34; p=0.39) were not correlated with abnormal OCB.
The interactions among different CSF parameters were systemically considered. The interaction between CSF pleocytosis and protein concentration—parameters that are commonly reported on the same day for hospitalized patients—are summarized in table 3 and figure 2A. When considering CSF WBC and protein only, 27% (CI95%: 19–37; 26/95) of patients with early active AE had CSF WBC and protein concentration within normal limits. On multivariable logistic regression, none of the exposure variables, including antibody subtype, correlated with the presence of either (AUC=0.65) or a combination (AUC=0.70) of CSF pleocytosis and elevated protein. Although findings did not achieve statistical significance when controlling for other variables, it is notable that CSF WBC and protein levels were within normal limits in almost one-third of patients with antibodies against cell-surface antigens, including LGI1 (55%, CI95%: 32–77; Table 3).
Table 3:
Distribution of CSF pleocytosis and high protein in patients with early active autoimmune encephalitis
| CSF Parameters | ||||
|---|---|---|---|---|
| High proteina only | Pleocytosisb only | Pleocytosis and high protein | Normal WBC and protein | |
| Antibody Subtypes | ||||
| Overall, n (%, CI95%) | 16 (17, 10–26) | 24 (25, 17–35) | 29 (31, 21–40) | 26 (27, 19–37) |
| LGI1, n (%, CI95%) | 3 (15, 3–38) | 1 (5, 0–25) | 5 (25, 9–49) | 11 (55 32–77) |
| Intracellular, n (%, CI95%) | 2 (13, 2–40) | 3 (20, 4–48) | 6 (40, 16–68) | 4 (27, 55–78) |
| Negative, n (%, CI95%) | 6 (32, 13–57) | 2 (11, 1–33) | 7 (37, 16–62) | 4 (21, 6–46) |
| Others, n (%, CI95%) | 0 (0, 0–84) | 0 (0, 0–84) | 2 (100, 16–100) | 0 (0, 0–84) |
The percentage distribution of the the different combinations of CSF parameters are given by antibody subtype and should be read “horizontally”.
High protein: >0.45 g/L.
Pleocytosis: >5cells/mm3.
LGI1: Leucine-rich glioma inactivated 1; NMDAR: N-methyl-D-aspartate receptor.
Fig.2. Interactions between CSF parameters in early active autoimmune encephalitis.
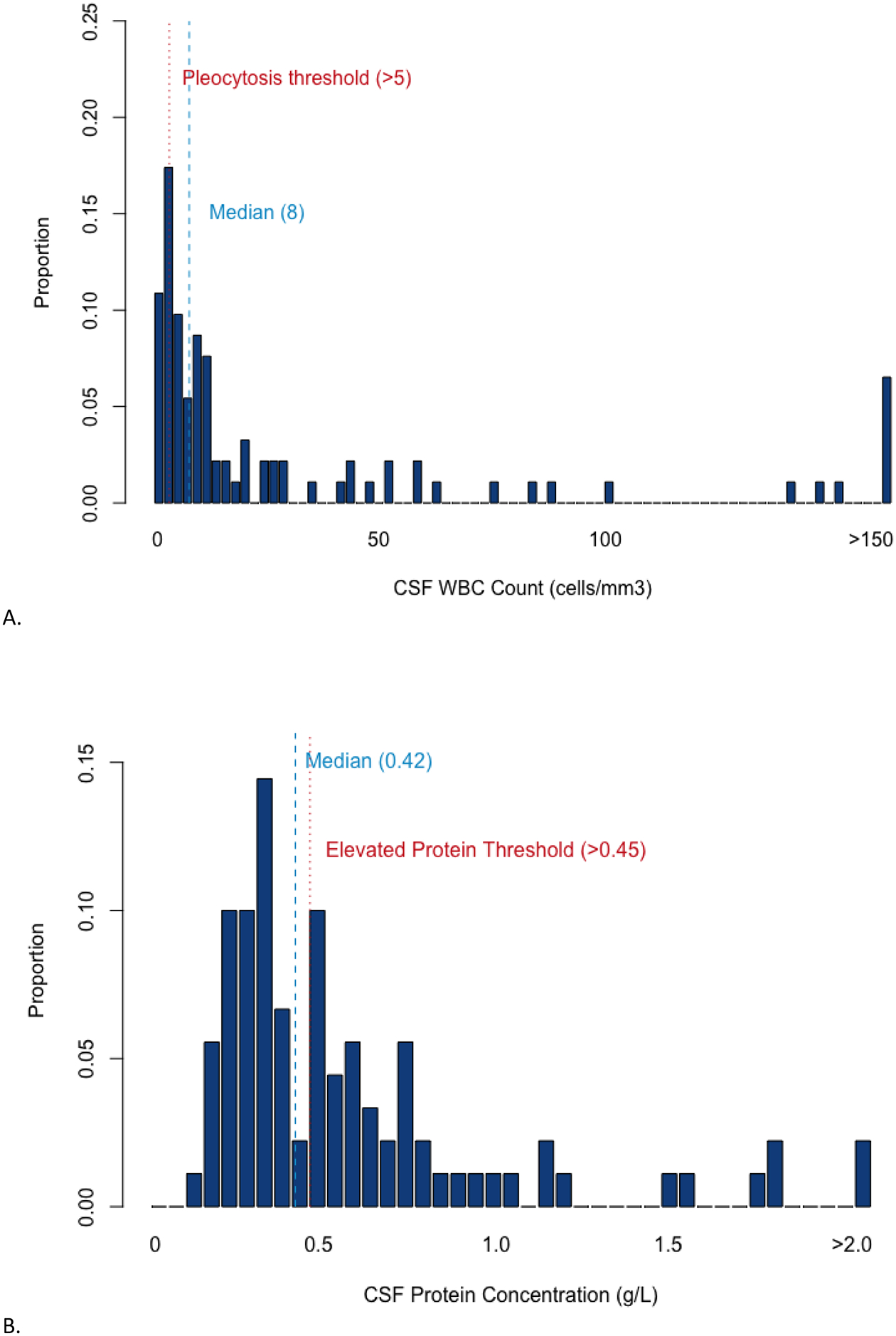
A. CSF analysis of pleocytosis and high protein concentration in 95 patients with early active autoimmune encephalitis. B. CSF analysis of three parameters: pleocytosis, high protein concentration, and abnormal CSF-specific OCB in 51 patients with early active autoimmune encephalitis. Pleocytosis: >5cells/mm3. High protein: >0.45 g/L. Abnormal OCB: ≥3 abnormal CSF bands. *Absence of CSF pleocytosis and high protein.**Absence of CSF pleocytosis, high protein, and abnormal OCB. CSF: cerebrospinal fluid. OCB: oligoclonal bands. Graph created with R (R Core Team, Vienna, 2018) using package ggplot2 (New York, 2016).
The interactions among all three parameters (pleocytosis, elevated protein, and OCB) are summarized in table 4 and figure 2B. Seven patients (7/51; 14%; CI95%: 6–26) had normal CSF WBC, protein, and OCB (“triple negative”). The results of antibody testing for these patients were as follows: negative (1), NMDAR (1), LGI1 (3), GABAb receptor (1) and Ma2 (1). All of these patients had abnormal EEG; four had findings of AE on MRI. None of the exposure variables were statistically associated with “triple negative” CSF findings on multivariable logistic regression (AUC=0.55).
Table 4:
Distribution of three CSF markers of inflammation in patients with early active autoimmune encephalitis
| CSF Parameters | ||||||
|---|---|---|---|---|---|---|
| High proteina only | Pleocytosisb only | Abnormal OCBc only | Combination of two CSF abnormalities | Pleocytosis, OCB, and high protein | Normal WBC, protein, and OCB | |
| By antibody subtype | ||||||
| Overall, n (%, CI95%) | 6 (12, 4–24) | 6 (12, 4–24) | 3 (6, 1–16) | 14 (27, 16–42) | 15 (29, 17–44) | 7 (14, 6–26) |
| LGI1, n (%, CI95%) | 3 (2, 5–54) | 1 (8, 0–36) | 2 (15, 2–45) | 2 (15, 2–45) | 2 (15, 2–45) | 3 (23, 5–54) |
| Intracellular, n (%, CI95%) | 0 (0, 0–37) | 2 (25, 3–65) | 0 (0, 0–37) | 2 (25, 3–65) | 3 (38, 9–76) | 1 (10, 0–45) |
| Negative, n (%, CI95%) | 1 (12, 0–53) | 2 (25, 3–65) | 1 (12, 0–53) | 0 (0, 0–37) | 3 (38, 9–76) | 1 (12, 0–53) |
| Others, n (%, CI95%) | 0 (0, 0–84) | 0 (0, 0–84) | 0 (0, 0–84) | 0 (0, 0–84) | 2 (100, 16–100) | 0 (0, 0–84) |
The percentage distribution of the the different combinations of CSF parameters are given by antibody subtype and should be read “horizontally”.
High protein: >0.45 g/L.
Pleocytosis: >5cells/mm3.
Abnormal OCB: ≥3 abnormal CSF bands.
LGI1: Leucine-rich glioma inactivated 1; NMDAR: N-methyl-D-aspartate receptor; OCB: oligoclonal bands.
A combination of abnormal CSF pleocytosis, protein, and CSF-specific OCB—“triple positive”—was seen in 15 patients (15/51; 29%; CI95%: 17–44), including 7 patients with cell-surface receptor autoantibodies. “Triple positive” status was associated with male sex (OR 1.15; CI95%: 1.06–1.97; p=0.04) and delay from symptom onset to lumbar puncture less than 8 weeks (OR 40.2; CI95%: 1.47–1102; p=0.03). Increased OCB were more frequently found in the subtype of patients with pleocytosis (81%, CI95%: 62–94) than in those with a normal CSF WBC count (47%, CI95%: 33–62, p= 0.02). Elevated protein was not correlated with abnormal CSF OCB (p=0.54).
4. Discussion
Forty-four percent of patients with AE in this series did not have CSF pleocytosis, thus lacking a key element of the proposed diagnostic criteria for possible AE.(Graus et al., 2016) When taking into account the presence of CSF pleocytosis and high protein only, a substantial proportion (26/95; 27%) of patients with AE had a “normal” CSF in the early active phase of their disease. This proportion is similar to that reported in an epidemiological study of AE in Olmsted County, USA (10/24; 42%; p=0.22).(Dubey et al., 2018) The addition of a third CSF parameter—OCB—reduced the share of patients in this series with “non-inflammatory” CSF or normal-appearing to 14%. However, caution is warranted when interpreting CSF findings in patients with symptoms persisting beyond 8 weeks, recognizing the strong association between delays in CSF sampling and normal CSF WBC, protein and OCB findings.
These findings emphasize that a LP with neither CSF pleocytosis nor elevated protein should not deter clinicians from considering a diagnosis of AE in patients meeting other diagnostic criteria—namely new focal central nervous system findings, seizures not explained by a previously known seizure disorder, and MRI or EEG features suggestive of encephalitis.(Graus et al., 2016) The prevalence of CSF abnormalities did not differ between the different antibody subtypes when adjusting for sex, age-at-onset, delay from symptom onset to LP, and diabetic and immunosuppressive status. This may, however, have been due to the smaller sample size in this series, recognizing that a prior meta-analysis of 1305 patients found such a difference between antibody subtypes.(Blinder and Lewerenz, 2019) Looking at specific antibodies, our study lends further support to the suggestion that patients with NMDAR encephalitis frequently have abnormal CSF studies,(Blinder and Lewerenz, 2019; Dalmau et al., 2008; Titulaer et al., 2013; Wang et al., 2016; Warren et al., 2018) in contrast to patients with LGI1 antibodies in whom the CSF may be “normal”/non-inflammatory.(Blinder and Lewerenz, 2019; Gadoth et al., 2017; Irani et al., 2011; Shin et al., 2013)
We further explored the relationship between older age and CSF findings, noting prior publications suggesting that over 20% of AE patients >60 years-of-age did not have signs of inflammation in the CSF or on neuroimaging.(Escudero et al., 2017) The finding that older age was inversely associated with the presence of CSF pleocytosis or abnormal OCB affirms these observations. Older age was also associated with elevations in CSF protein in our series, consistent with reports in healthy individuals and patients with chronic inflammatory demyelinating polyneuropathy.(Breiner et al., 2019; Garton et al., 1991)
The main strength of this study was the inclusion of patients in the early active phase of their illness, at which point diagnostic uncertainty was high and the diagnosis of AE in question. Accordingly, our results inform the prevalence of CSF abnormalities in “undifferentiated” patients with AE. This study also considered the association between the prevalence of CSF abnormalities in patients with AE and sex, age, antibody subtype, diabetic status, immunosuppressive status, and delay between symptom onset and LP. Limitations of our study include the relatively small sample size—especially when considering AE subtype in isolation—retrospective data collection, and lack of OCB testing on a substantial proportion of patients. The inclusion of patients from two sites that used different techniques to analyze CSF also presents challenges,(Kleine et al., 2010; Zimmermann et al., 2011) although it is reassuring that CSF cell counts were similar across sites (Table 1). Further studies recruiting patients from other locations and practice environments within and beyond North America are needed to ensure generalizability of findings.
4.1. Conclusions
A substantial proportion of the 95 patients with AE included in this multicenter study did not have elevated WBC or protein in the CSF in the early active phase of their illness. The inclusion of OCB measures identified substantially more inflammatory CSF profiles in this patient population, and should be considered when evaluating patients with clinical presentations concerning for AE. Further studies are required to assess predictors of early changes in non-specific CSF markers of inflammation and the prognostic value of early CSF findings in patients with AE.
Acknowledgement
The Authors thank Dr. Melanie Yarbrough (Department of Pathology and Immunology, Washington University School of Medicine) for clarification concerning laboratory procedures and measures at Washington University School of Medicine.
Study funding:
Funding for work completed at WUSM was provided by the National Institutes of Health (K23AG064029 to GSD), American Academy of Neurology/American Brain Foundation (Clinical Research Training Fellowship to GSD), and via philanthropic contributions from patients and family members to promote research and education into Autoimmune Encephalitis (GSD). JH received funding from the University of Toronto Postgraduate Medical Education bursary program for this work.
Appendix 1: Authors
| Name | Location | Role | Contribution |
|---|---|---|---|
| Julien Hébert | University of Toronto, Canada | Author | Design and conceptualized study; analyzed the data; data acquisition; drafted the manuscript for intellectual content |
| Priti Gros | University of Toronto, Canada | Author | Major role in data acquisition; data interpretation; reviewed manuscript for intellectual content |
| Sarah Lapointe | University of Toronto, Canada; University Health Network, Toronto, Canada | Author | Major role in data acquisition; data interpretation; reviewed manuscript for intellectual content |
| Fatima S. Amtashar | Washington University School of Medicine, Dept of Neurology, MO | Author | Major role in data acquisition; data interpretation; reviewed manuscript for intellectual content |
| Claude Steriade | New York University Langone Comprehensive Epilepsy Center, NY | Author | Major role in data acquisition; data interpretation; reviewed manuscript for intellectual content |
| Catherine Maurice | University of Toronto, Canada; University Health Network, Toronto, Canada | Author | Major role in data acquisition; data interpretation; reviewed manuscript for intellectual content |
| Richard A. Wennberg | University of Toronto, Canada; University Health Network, Toronto, Canada | Author | Data interpretation; reviewed manuscript for intellectual content |
| Gregory S. Day | Mayo Clinic Florida, Department of Neurology; Jacksonville, FL | Author | Design and conceptualized study; data interpretation; reviewed manuscript for intellectual content |
| David F. Tang-Wai; University Health Network, Toronto, Canada | University of Toronto, Canada; University Health Network, Toronto, Canada | Author | Design and conceptualized study; data interpretation; reviewed manuscript for intellectual content |
Footnotes
Ethical standards
All human and animal studies have been approved by the appropriate ethics committee and have been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All persons gave their informed consent prior to their inclusion in the study.
Conflicts of interest:
Dr. Hébert reports no disclosures.
Dr. Gros reports no disclosures.
Dr. Lapointe reports no disclosures.
Mrs. Amtashar reports no disclosures.
Dr. Steriade receives research funding from Finding A Cure for Seizure and Epilepsy (FACES), UCB and the American Epilepsy Society. She serves as a consultant to the non-profit organization The Epilepsy Study Consortium. She has received compensation for serving on an advisory board for UCB.
Dr Maurice reports no disclosures.
Dr Wennberg reports no disclosures.
Dr. Day has served as a topic editor on dementia for DynaMed Plus (EBSCO Industries, Inc) and as clinical director for the Anti-NMDA Receptor Encephalitis Foundation (uncompensated). He receives research/grant support from the National Institutes of Health (K23AG064029) and holds stock in ANI Pharmaceuticals, Inc. GSD has provided record review and expert medical testimony on legal cases pertaining to management of Wernicke encephalopathy.
Dr. Tang-Wai reports no disclosures.
References
- Balu R, McCracken L, Lancaster E, Graus F, Dalmau J, Titulaer MJ, 2018. A score that predicts 1-year functional status in patients with anti-NMDA receptor encephalitis. Neurology. 10.1212/WNL.0000000000006783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinder T, Lewerenz J, 2019. Cerebrospinal Fluid Findings in Patients With Autoimmune Encephalitis-A Systematic Analysis. Front. Neurol 10, 804 10.3389/fneur.2019.00804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw MJ, Linnoila JJ, 2018. An Overview of Autoimmune and Paraneoplastic Encephalitides. Semin. Neurol 38, 330–343. 10.1055/s-0038-1660821 [DOI] [PubMed] [Google Scholar]
- Breiner A, Bourque PR, Allen JA, 2019. Updated cerebrospinal fluid total protein reference values improve chronic inflammatory demyelinating polyneuropathy diagnosis. Muscle Nerve 60, 180–183. 10.1002/mus.26488 [DOI] [PubMed] [Google Scholar]
- Brooks J, Yarbrough ML, Bucelli RC, Day GS, 2019. Testing for N-methyl-D-aspartate receptor autoantibodies in clinical practice. Can. J. Neurol. Sci. J. Can. Sci. Neurol 1–27. 10.1017/cjn.2019.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, Dessain SK, Rosenfeld MR, Balice-Gordon R, Lynch DR, 2008. Anti-NMDA-receptor encephalitis: case series and analysis of the effect of antibodies. Lancet Neurol 7, 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, Gadoth A, Smith CY, Bryant SC, Klein CJ, Aksamit AJ, Toledano M, Boeve BF, Tillema J-M, Flanagan EP, 2018. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann. Neurol 83, 166–177. 10.1002/ana.25131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero D, Guasp M, Ariño H, Gaig C, Martínez-Hernández E, Dalmau J, Graus F, 2017. Antibody-associated CNS syndromes without signs of inflammation in the elderly. Neurology 89, 1471–1475. 10.1212/WNL.0000000000004541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke C, Kopp U, Pruss H, Dalmau J, Wandinger K, Ploner C, 2012. Cognitive deficits following anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry 83, 195–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke C, Prüss H, Heine J, Reuter S, Kopp U, Wegner F, Then Bergh F, Koch S, 2017. Evaluation of Cognitive Deficits and Structural Hippocampal Damage in Encephalitis With Leucine-Rich, Glioma-Inactivated 1 Antibodies. JAMA Neurol 74, 50–59. [DOI] [PubMed] [Google Scholar]
- Gadoth A, Pittock S, Dubey D, McKeon A, Britton J, Schmeling J, Smith A, Kotsenas A, 2017. Expanded phenotypes and outcomes among 256 LGI1/CASPR2-IgG-positive patients. Ann Neurol 82, 79–92. [DOI] [PubMed] [Google Scholar]
- Garton MJ, Keir G, Lakshmi MV, Thompson EJ, 1991. Age-related changes in cerebrospinal fluid protein concentrations. J. Neurol. Sci 104, 74–80. [DOI] [PubMed] [Google Scholar]
- Graus F, Titulaer M, Ramani B, Benseler S, Bien C, Cellucci T, Cortese I, 2016. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 15, 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gultekin SH, Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J, 2000. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain J. Neurol 123 (Pt 7), 1481–1494. 10.1093/brain/123.7.1481 [DOI] [PubMed] [Google Scholar]
- Hébert J, Day GS, Steriade C, Wennberg RA, Tang-Wai DF, 2018. Long-Term Cognitive Outcomes in Patients with Autoimmune Encephalitis. Can. J. Neurol. Sci. J. Can. Sci. Neurol 45, 540–544. 10.1017/cjn.2018.33 [DOI] [PubMed] [Google Scholar]
- Honorat JA, Komorowski L, Josephs KA, Fechner K, St Louis EK, Hinson SR, Lederer S, Kumar N, Gadoth A, Lennon VA, Pittock SJ, McKeon A, 2017. IgLON5 antibody: Neurological accompaniments and outcomes in 20 patients. Neurol. Neuroimmunol. Neuroinflammation 4, e385 10.1212/NXI.0000000000000385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani S, Alexander S, Waters P, Kleopas K, Pettingill P, Zuliani L, Peles E, 2010. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 133, 2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani S, Michelle A, Lang B, Pettingill P, Waters P, Johnson M, Schott J, 2011. Faciobrachial Dystonic Seizures Precede Lgi1 Antibody Limbic Encephalitis. Ann Neurol 69, 892–900. [DOI] [PubMed] [Google Scholar]
- Irani S, Stagg C, Schott J, Rosenthal C, Schneider S, Pettiingill P, 2013. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 136, 3151–3162. [DOI] [PubMed] [Google Scholar]
- Kleine TO, Nebe CT, Löwer C, Lehmitz R, Geilenkeuser W-J, Kruse R, Dorn-Beineke A, 2010. Evaluation of cell counting and leukocyte differentiation in cerebrospinal fluid controls using hematology analyzers by the German Society for Clinical Chemistry and Laboratory Medicine. Clin. Chem. Lab. Med 48, 839–848. 10.1515/CCLM.2010.168 [DOI] [PubMed] [Google Scholar]
- Lee SK, Lee S-T, 2016. The Laboratory Diagnosis of Autoimmune Encephalitis. J. Epilepsy Res 6, 45–52. 10.14581/jer.16010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit-Pedrol M, 2014. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 13, 276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiber H, Peter JB, 2001. Cerebrospinal fluid analysis: disease-related data patterns and evaluation programs. J. Neurol. Sci 184, 101–122. [DOI] [PubMed] [Google Scholar]
- Shin Y, Lee S, Shin J, Moon J, Lim J, Byun J, Kim T, Lee K, Kim Y, 2013. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 165, 75–81. [DOI] [PubMed] [Google Scholar]
- Titulaer M, McCraken L, Inigo J, Armangue T, Glaser C, 2013. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 12, 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li J-M, Hu F-Y, Wang R, Hong Z, He L, Zhou D, 2016. Anti-NMDA receptor encephalitis: clinical characteristics, predictors of outcome and the knowledge gap in southwest China. Eur. J. Neurol 23, 621–629. [DOI] [PubMed] [Google Scholar]
- Warren N, Siskind D, O’Gorman C, 2018. Refining the psychiatric syndrome of anti-N-methyl-d-aspartate receptor encephalitis. Acta Psychiatr. Scand 138, 401–408. 10.1111/acps.12941 [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Ruprecht K, Kainzinger F, Heppner FL, Weimann A, 2011. Automated vs. manual cerebrospinal fluid cell counts: a work and cost analysis comparing the Sysmex XE-5000 and the Fuchs-Rosenthal manual counting chamber. Int. J. Lab. Hematol 33, 629–637. 10.1111/j.1751-553X.2011.01339.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymyzed data will be shared upon request addressed to the corresponding author by qualified investigators.