

ABSTRACT
Lymphangiogenesis plays a crucial role during development, in cancer metastasis and in inflammation. Activation of VEGFR-3 (also known as FLT4) by VEGF-C is one of the main drivers of lymphangiogenesis, but the transcriptional events downstream of VEGFR-3 activation are largely unknown. Recently, we identified a wave of immediate early transcription factors that are upregulated in human lymphatic endothelial cells (LECs) within the first 30 to 80 min after VEGFR-3 activation. Expression of these transcription factors must be regulated by additional pre-existing transcription factors that are rapidly activated by VEGFR-3 signaling. Using transcription factor activity analysis, we identified the homeobox transcription factor HOXD10 to be specifically activated at early time points after VEGFR-3 stimulation, and to regulate expression of immediate early transcription factors, including NR4A1. Gain- and loss-of-function studies revealed that HOXD10 is involved in LECs migration and formation of cord-like structures. Furthermore, HOXD10 regulates expression of VE-cadherin, claudin-5 and NOS3 (also known as e-NOS), and promotes lymphatic endothelial permeability. Taken together, these results reveal an important and unanticipated role of HOXD10 in the regulation of VEGFR-3 signaling in lymphatic endothelial cells, and in the control of lymphangiogenesis and permeability.
KEY WORDS: HOXD10, Transcription factor, Lymphatic endothelium, Lymphangiogenesis, VEGFR-3, Immediate early gene
Summary: The transcription factor HOXD10 regulates induction of immediate early transcription factors in lymphatic endothelial cells stimulated with the growth factor VEGF-C, and regulates lymphatic endothelial permeability.
INTRODUCTION
The lymphatic system serves to maintain tissue fluid homeostasis, to mediate immune cell trafficking to lymph nodes and to take up dietary fats (Hirakawa et al., 2014). Lymphatic vessels also facilitate the metastatic spread of tumor cells, which is the major cause of cancer-related deaths (Karaman and Detmar, 2014). Induction of lymphatic vessel growth (lymphangiogenesis) is commonly observed in and around tumors, which contributes to tumor dissemination (Mandriota et al., 2001; Skobe et al., 2001). Lymphangiogenesis has also been observed in the draining lymph node and correlates with metastatic spread (Hirakawa et al., 2007, 2005). One of the most prominent inducers of lymphangiogenesis is vascular endothelial growth factor-C (VEGF-C). VEGF-C induces lymphatic vessel sprouting and enlargement (Zheng et al., 2014), and specific blockage of the VEGF-C signaling pathway has been reported to inhibit cancer metastasis to lymph nodes in mice (Burton et al., 2008; He et al., 2002; Roberts et al., 2006; Yang et al., 2011). VEGF-C is also involved in the dynamic response of lymphatic vessels to inflammation. The lymphatic network is known to expand in various inflammatory conditions, both in mice and in humans, for example in chronic skin inflammation (Huggenberger and Detmar, 2011; Kunstfeld et al., 2004). Of note, activation of lymphatic vessels by VEGF-C decreases inflammation, at least in part, by increasing the drainage function of lymphatic vessels (Huggenberger et al., 2011, 2010; Zhou et al., 2011). Taken together, these studies indicate that modulating lymphangiogenesis by application or inhibition of VEGF-C represents a new strategy to treat a variety of pathologic conditions such as cancer and inflammatory diseases.
The main pathway regulating lymphangiogenesis is VEGF-C signaling through its receptor VEGFR-3 (also known as FLT4) on lymphatic endothelial cells (LECs). VEGF-C-knockout mice fail to develop a lymphatic system and are not viable (Karkkainen et al., 2004). Similarly, VEGFR-3-knockout mice die early during embryonic development as they fail to develop a cardiovascular system (Dumont, 1998). Fully mature VEGF-C has affinity for VEGFR-2 (also known as KDR) (Joukov et al., 1997), which is expressed by LECs, but also by blood vessel endothelial cells (BECs). A mutated version of VEGF-C, VEGF-C156S, in which cysteine 156 is replaced by a serine residue, specifically activates VEGFR-3 but not VEGFR-2 (Joukov et al., 1998). Specific activation of VEGFR-3 by VEGF-C156S is sufficient to induce lymphangiogenesis, as demonstrated in K14-VEGF-C156S mice (Veikkola et al., 2001). Upon stimulation, the receptor is phosphorylated at several intracellular tyrosine residues. Phosphorylation of these sites recruits several adaptor molecules, such as CRKI and CRKII, SHC proteins and GRB2, which in turn activate downstream mediators including JNK, ERK, phosphoinositide 3-kinase (PI3K) and AKT (also known as PKB) ultimately leading to proliferation and migration of LECs (Koch et al., 2011; Mäkinen et al., 2001). However, the effects of this signaling cascade on the transcriptional level are only poorly understood.
In a previous study, we investigated the transcriptional changes in LECs mediated by VEGF-C156S-induced VEGFR-3 activation in detail, with a focus on transcription factors involved in this pathway (Dieterich et al., 2015). By treating LECs with VEGF-C156S and analyzing subsequent changes in gene expression, we identified several ‘immediate early’ transcription factors that showed a rapid transient upregulation at the mRNA level after VEGFR-3 stimulation. This first wave of transcription factor induction must be mediated by yet another set of pre-existing ‘front line’ transcription factors that are directly activated by VEGFR-3 signaling at the protein level, independently of transcriptional induction. Here, we identified HOXD10 to be such a front line transcription factor in LECs. Whereas HOXD10 itself is not transcriptionally induced by VEGFR-3 signaling, it is rapidly activated, and is required for induction of the immediate early transcription factor NR4A1. Furthermore, we found that HOXD10 is involved in various key processes in LECs, including migration, tube formation and permeability.
RESULTS
HOXD10 is an early mediator of the VEGR3-induced transcriptional response in LECs
Previously, using cap analysis of gene expression (CAGE) RNA sequencing (Kanamori-Katayama et al., 2011; Shiraki et al., 2003), we found upregulation of several immediate early transcription factors, including FOSB, and NR4A1, in LECs stimulated with VEGF-C156S over a time period from 0 h to 8 h (Dieterich et al., 2015). In order to identify transcription factors that might regulate this first wave of transcription factor induction, we used the same data set and screened specifically for transcription factors activated immediately downstream of VEGFR-3 signaling without induction of de novo gene expression. Using the oPOSSUM3 tool (Kwon et al., 2012), we identified eight transcription factors (HOXD10, EGR1, NKX3-1, ELK1, E2F1, FOXD1, NKX2-5, SRY) whose transcription-factor-binding sites (TFBS) were significantly overrepresented in the core promoters (1500 bases upstream to 500 bases downstream of the transcription start sites, Fig. 1A) of differentially expressed transcripts at the earliest time point (15 min) of stimulation (Fig. 1B; Table S1). The frequency of promoters with at least one TFBS for HOXD10, EGR1, NKX3-1, E2F1, or ELK1 was significantly enriched at this time point (Fig. 1B; Table S1), suggesting that the corresponding transcription factors could be active. As most of those transcription factors were either not expressed in LECs (NKX3-1), belong to the previously defined group of immediate early transcription factors induced by VEGF-C156S (EGR1) (Dieterich et al., 2015) or have otherwise well-known, broad functions in various cell types (E2F1 and ELK1), we decided to focus on HOXD10, a transcription factor not previously associated with lymphatic endothelial biology, and to investigate its role in LECs in greater detail. According to the oPOSSUM3 analysis, HOXD10 also showed activity at 30 min, 80 min, 300 min and 360 min after stimulation, indicating that this factor could represent an important, and hitherto unknown, regulator of lymphatic function in response to VEGF-C (Fig. 1C–F; Table S1).
Fig. 1.
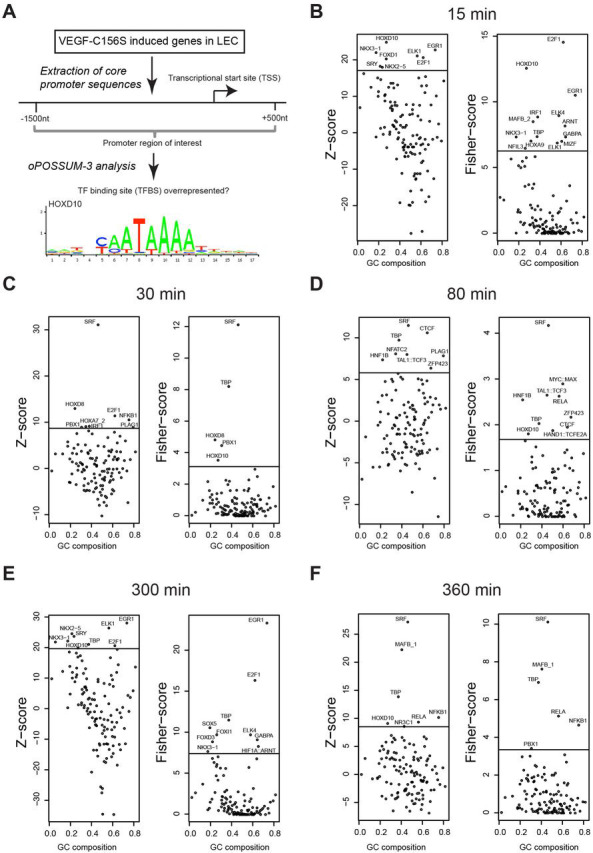
HOXD10 is a downstream mediator of VEGFR-3 induced gene expression in LECs. Transcription factor (TF) activity analysis using oPOSSUM3 on VEGF-C156S-stimulated LECs was performed based on the presence of TFBS in the promoters of differentially expressed transcripts. (A) Schematic overview of the analysis workflow and HOXD10 TFBS derived from JASPAR (jaspar.genereg.net). Data shown represent the 15-min (B), 30-min (C), 80-min (D), 300-min (E) and 360-min time points (F) after VEGF-C156S stimulation. HOXD10 TFBS overrepresentation is indicated by the Z-score (left panels), and overrepresentation of promoters with at least one HOXD10 TFBS is indicated by the Fisher score (right panels). The horizontal line indicates the significance cutoff (mean+1.5×s.d.). Transcription factors are plotted on the x-axis according to the guanine and cytosine (GC) content of their corresponding transcription-factor-binding profiles from JASPAR.
HOXD10 is consistently expressed by LECs and is controlled by the lymphatic transcription factor PROX1
Based on CAGE RNA sequencing, the expression of HOXD10 was not induced over a period of 8 h after VEGF-C156S stimulation (Dieterich et al., 2015). To confirm this data, we performed both quantitative real-time PCR (qPCR) and western blotting. As predicted, we found no induction, but rather a slight decrease of the HOXD10 transcript at ∼80–120 min after stimulation, with no major changes in protein expression levels (Fig. 2A,B; Fig. S1A,B). Next, we compared the expression of HOXD10 in LECs with that in closely related blood vascular endothelial cells (BECs) derived from the same donor. HOXD10 mRNA expression in cultured human LECs was about sixfold higher than in BECs, which was also reflected at the protein level (Fig. 2C,D). In line with this, we also found a significantly higher expression of HOXD10 in LECs than in BECs that were freshly isolated from mouse ears by FACS sorting (Fig. 2E,F). Thus, HOXD10 might exert specific functions in LECs. Interestingly, small interfering RNA (siRNA)-mediated knockdown of the master regulator of lymphatic identity, prospero-related homeobox 1 (PROX1), led to a more than twofold reduction of HOXD10 expression (Fig. 2G), indicating that the selective expression of HOXD10 in LECs is mediated by PROX1.
Fig. 2.

HOXD10 is expressed at a higher level in LECs compared to BECs. (A) qPCR showing the expression levels of HOXD10 after VEGF-C156S stimulation of LECs. (B) Western blot analysis for HOXD10 protein and β-actin as loading control in the same samples used in A. (C) HOXD10 expression was higher in LECs than in BECs as measured by qPCR. Bars represent means±s.d. (pooled data from three independent experiments). ***P<0.001 (Student's t-test). (D) Western blot showing HOXD10 protein levels in LECs and BECs. (E) Representative FACS plot to illustrate the gating strategy for sorting of primary LECs and BECs from mouse ears. (F) qPCR showing increased HOXD10 expression in freshly isolated mouse LECs compared to BECs. Results are mean±s.d. (n=4). *P<0.05 (Student's t-test). (G) qPCR of PROX1 and HOXD10 expression levels in LECs. After siRNA-mediated PROX1 knockdown, HOXD10 expression levels are similarly decreased. Bars represent mean±s.d. (a representative experiment with three technical replicates out of four total experiments shown).
HOXD10 regulates expression of FOSB and NR4A1
To investigate the role of HOXD10 during VEGFR-3 signaling in LECs, we used adenoviral vectors to deplete or overexpress HOXD10 (Ad-shHOXD10 and Ad-HOXD10, respectively). Non-targeting short hairpin RNA (shRNA) or GFP expression vectors served as controls (Ad-NT and Ad-GFP, respectively). Transduction of LECs with Ad-HOXD10 resulted in efficient overexpression of HOXD10 (Fig. 3A,B) at 24 h, 48 h and 72 h after viral infection. Downregulation of HOXD10 mRNA after adenoviral delivery of specific shRNA was between 50–70%, depending on the timepoint (24 h, 48 h and 72 h) (Fig. 3C). However, a reduction in the protein level of HOXD10 was only detectable if Ad-shHOXD10 infected cells were treated with VEGF-C156S for 24 h, but not under resting conditions (Fig. 3D). This suggests that the HOXD10 protein is very stable in unstimulated LECs, but is activated and subsequently degraded after activation of the VEGFR-3 pathway. Next, we selected two immediate early transcription factors (Dieterich et al., 2015) with a predicted HOXD10-binding site in their core promoter as identified by oPOSSUM3, namely FOSB and NR4A1 (Table S2), and analyzed their expression in LECs infected with Ad-HOXD10 or Ad-shHOXD10 for 24 h, 48 h and 72 h. As expected, both TFs were strongly upregulated upon HOXD10 overexpression in comparison to control cells (Fig. 3E,F), whereas no major changes in the expression of these genes were detected after depletion of HOXD10 in unstimulated LECs.
Fig. 3.

HOXD10 regulates immediate early transcription factors and is required for VEGF-C156S-induced NR4A1 upregulation. Adenoviral delivery of HOXD10 cDNA (Ad-HOXD10) to LECs resulted in efficient overexpression compared to Ad-GFP control cells, as tested by qPCR (A) and at the protein level by western blotting (B). (C) Knockdown of HOXD10 mRNA in LECs by shRNA (Ad-shHOXD10) in comparison to Ad-NT control cells, as determined by qPCR. (D) HOXD10 protein was not affected by Ad-shHOXD10 in resting LECs (left panel). Upon treatment with VEGF-C156S (1.5 µg/ml) for 24 h, the HOXD10 protein was substantially lower in Ad-shHOXD10-infected cells than in Ad-NT control cells (right panel). (E,F) qPCR analyses showing upregulation of FOSB (E) and NR4A1 (F) in LECs after adenovirus infection. No significant changes of FOSB and NR4A1 were detected after HOXD10 depletion, whereas HOXD10 overexpression resulted in marked upregulation. (G) Schematic overview of the experimental procedure to assess HOXD10-dependent VEGF-C156S-mediated target gene upregulation. LECs were transduced with control virus (Ad-NT) or shHOXD10 (Ad-shHOXD10), starved overnight and treated with VEGF-C156S (1.5 µg/ml) for 60 min or 120 min. RNA was isolated and the transcripts of HOXD10 target genes FOSB and NR4A1 were analyzed by qPCR. Expression of FOSB (H) and NR4A1 (I) after 60 min and 120 min of VEGF-C156S stimulation is shown. NR4A1 upregulation after VEGF-C156S stimulation was significantly reduced in Ad-shHOXD10 cells (I). Bars represent mean±s.d. (pooled data from three independent experiments). *P<0.05; **P<0.01; ***P<0.001; n.s., not significant (Student's t-test).
HOXD10 is necessary for the induction of NR4A1 downstream of VEGFR-3 signaling
As FOSB and NR4A1 are induced upon activation of the VEGFR-3 pathway, we wondered whether HOXD10 is involved in the gene regulation of these genes upon VEGF-C156S stimulation. Therefore, we stimulated HOXD10-depleted LECs with VEGF-C156S for either 1 h, a time point at which the mRNA of FOSB and NR4A1 are upregulated, or 2 h, when the expression has returned to baseline (Fig. 3G) (Dieterich et al., 2015). In control cells (Ad-NT), expression of FOSB and NR4A1 was significantly increased (on average fivefold and 25-fold, respectively) after 1 h stimulation with VEGF-C156S, and returned towards the baseline at the 2 h timepoint, as expected (Fig. 3H,I). In Ad-shHOXD10-transduced cells, induction of FOSB was comparable to that in control cells (threefold after 1 h stimulation, Fig. 3H). In contrast, NR4A1 mRNA upregulation was significantly reduced in cells depleted of HOXD10 (only sevenfold upregulation compared to 25-fold in control cells) after 1 h VEGF-C156S stimulation (Fig. 3I). These results clearly show that HOXD10 is necessary for the VEGF-C156S-mediated upregulation of NR4A1, whereas it is dispensable for the induction of FOSB. No further induction of FOSB nor NR4A1 by VEGF-C156S could be observed in HOXD10-overexpressing cells (data not shown).
HOXD10 regulates LEC migration and formation of cord-like structures
In order to investigate whether HOXD10 expression might be relevant for lymphangiogenesis, we analyzed its role in important functional processes in LECs, namely proliferation, migration and formation of cord-like structures. Treatment with VEGF-A, wild-type (wt) VEGF-C or VEGF-C156S resulted in comparably increased LEC proliferation in vitro as assessed by 4-methylumbelliferyl heptanoate (MUH) fluorescence (Fig. 4A,B). Interestingly, the response towards wt VEGF-C and VEGF-C156S, but not towards VEGF-A, was slightly increased in LECs depleted of HOXD10 (Fig. 4A). In contrast, HOXD10 overexpression resulted in consistently decreased growth-factor-induced proliferation (Fig. 4B), which correlated with a trend towards a lower ratio of living to dead cells (Fig. S2A,B).
Fig. 4.
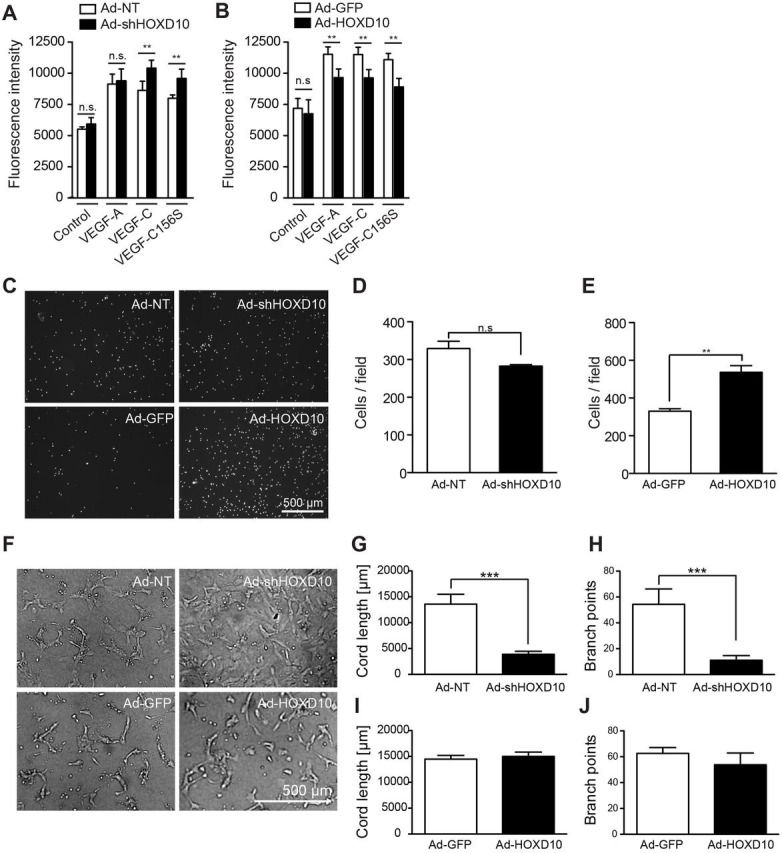
HOXD10 affects proliferation, migration and tubular morphogenesis of LECs. Proliferation of HOXD10-depleted LECs (A) and HOXD10-overexpressing LECs (B) in the absence (medium) or presence of VEGF-A, VEGF-C156S or VEGF-C. A significant increase in proliferation was observed in knockdown cells compared to control cells after treatment with VEGF-C or VEGF-C156S, whereas proliferation of HOXD10-overexpressing cells was slightly reduced. (C) Representative images of the modified Boyden chamber migration assay after 4 h. (D) After HOXD10 knockdown, LEC migration was slightly reduced (not significant). (E) HOXD10-overexpressing cells showed significantly increased migration compared to control cells. (F) Representative images of tubular morphogenesis by transduced LECs. Formation of cord-like structures in HOXD10-depleted cells was severely impaired. Instead, these cells continued to grow as a monolayer. Quantification of total cord length (G) and branch points (H) showing a strong reduction in tube formation in Ad-shHOXD10 cells compared to control (Ad-NT) cells. Cord length (I) and branch points (J) were not significantly affected by HOXD10 overexpression compared to control (Ad-GFP). Bars represent mean±s.d. (n=5 in A,B; n=3 in D,E; n=4 in G–J). **P<0.01; ***P<0.001; n.s., not significant [two-way ANOVA with Bonferroni post-test (A,B) or Student's t-test (all other panels)].
Knockdown of HOXD10 resulted in a trend towards reduced haptotactic LEC migration in a modified Boyden chamber assay, which, however, did not reach statistical significance (Fig. 4C,D). By contrast, migration of HOXD10 overexpressing cells almost doubled in this assay (Fig. 4C,E). Next, we tested whether HOXD10 expression would affect the ability of LECs to form cord-like structures in the presence of a collagen gel. Strikingly, after HOXD10 knockdown, we consistently observed that LECs continued to grow as a monolayer, showing virtually no formation of endothelial cords at all (Fig. 4F–H). HOXD10 overexpression had no major effects on cord-like structure formation (Fig. 4F,I,J). However, we did observe increased numbers of single, rounded cells after HOXD10 overexpression, which might be due to the negative effect of HOXD10 on cell viability, which we observed previously (Fig. 4F). Taken together, these data suggest that HOXD10 is a negative regulator of LEC proliferation and stimulates migration and tube formation.
HOXD10 regulates LEC permeability
Another important aspect of the VEGF-C response in LECs is the regulation of permeability. Previous reports have demonstrated that VEGF-C induces lymphatic permeability both in vitro and in vivo, and facilitates cancer dissemination through the lymphatic system (Breslin et al., 2007; Tacconi et al., 2015). We therefore decided to investigate the role of HOXD10 in lymphatic permeability using two established in vitro methods, transwell LEC permeability for a macromolecular fluorescent tracer (70-kDa FITC–dextran) and transendothelial electric resistance. Of note, HOXD10 depletion strongly reduced the permeability of LEC monolayers towards 70-kDa FITC–dextran, whereas HOXD10 overexpression had the opposite effect (Fig. 5A,B). This effect was also partly reflected by the electric resistance of LEC monolayers. Independently of HOXD10 expression, LECs showed increasing electric resistance during the first 1–2 h after seeding as the monolayers formed (Fig. 5C,D). Whereas HOXD10 knockdown had no significant effects on the resistance over the entire observation period, overexpression resulted in reduced electric resistance after monolayer formation, indicating a loosening of endothelial junctions (Fig. 5C,D). These data suggest that HOXD10 regulates VEGF-C-induced permeability changes in LECs.
Fig. 5.

HOXD10 regulates the permeability of LEC monolayers. (A,B) Macromolecular permeability was determined using a transwell assay, in which the fluorescence intensity in the outer well was measured 5 min, 30 min and 60 min after addition of 70-kDa FITC–dextran to the inner well. LECs depleted of HOXD10 (Ad-shHOXD10) showed significantly decreased permeability compared to control cells (Ad-NT) (A), whereas the permeability of HOXD10-overexpressing cells was strongly increased (B). Results represent mean+s.d. (a representative experiment with three technical replicates of three individual experiments is shown). **P<0.01, ***P<0.001 (two-way ANOVA with Bonferroni post-test). (C,D) Trans-endothelial electrical resistance was measured using the ECIS system. Resistance was not affected by HOXD10 knockdown (C), whereas resistance was reduced in HOXD10-overexpressing cells (D) (one representative experiment with four technical replicates of two experiments is shown). The dotted vertical line indicates the time point of cell seeding.
Identification of HOXD10 target genes in LECs
In order to shed light upon the mechanisms behind the functional effects of HOXD10 on LECs in vitro, we performed CAGE RNA sequencing 24 h after infection of LECs with Ad-HOXD10 or Ad-shHOXD10. The efficiency of overexpression and knockdown was validated separately by qPCR in those samples (Fig. S3A,B). In HOXD10-overexpressing LECs, we identified differentially expressed transcripts [false discovery rate (FDR)<0.05, log2 fold change>1], corresponding to 1599 transcriptional start sites (TSS), 1046 of which were upregulated and 553 were downregulated (Fig. 6A; Table S3). Gene ontology analysis indicated that the upregulated transcripts upon HOXD10 overexpression were significantly enriched for genes involved in nuclear factor κB (NFκB) signaling and in (negative) regulation of cell death and apoptosis, respectively (Fig. 6B). Of note, the genes associated with negative apoptosis regulation (according to the gene ontology annotation) again included several genes related to the NFκB pathway (e.g. NFKB1, IL1B and FAS) (Table S4). The NFκB pathway in general, and proteins such as FAS in particular, are, however, known to exert both pro- and anti-apoptotic activity, depending on the context (Brint et al., 2013). Thus, upregulation of these genes might also lead to an induction of apoptosis. Genes downregulated by HOXD10 were frequently associated with cell migration and blood vessel development (Fig. 6C). Thus, the global effects of HOXD10 overexpression in gene regulation are well in line with the phenotypic effects observed in proliferation, migration and tube formation described above.
Fig. 6.

RNA sequencing of LECs after knockdown or overexpression of HOXD10. LECs were infected with adenovirus to knockdown or overexpress HOXD10, and RNA was extracted and subjected to CAGE RNA sequencing 24 h later. (A) Summary of the results: HOXD10 overexpression resulted in the significant (FDR<0.05, log2FC>1) upregulation of 1046 TSS and downregulation of 553 TSS. Knockdown of HOXD10 had only minor effects on unstimulated LECs (upregulation of 2 TSS). (B,C) Gene ontology analysis of genes associated with upregulated (B) and downregulated (C) TSS in HOXD10-overexpressing LECs. The ten most significantly enriched ‘biologic process’ (GO_BP) terms are shown. (D) Heat map of selected TSS differentially expressed in HOXD10-overexpressing cells, whose associated genes have been linked to endothelial biology previously. A number after a p indicates different TSS within the same genes.
By contrast, depletion of HOXD10 in resting LECs only led to minimal changes in gene expression (Fig. 6A; Table S3), which is not surprising given the stability of HOXD10 in unstimulated cells (Fig. 3D). Of note, among the transcripts affected by HOXD10 overexpression, we found several corresponding to genes with well-known functions in the regulation of (lymph-) angiogenesis and endothelial permeability, such as NOS3, IL7, VEGFC, NRP2, ENG, ROBO4, PTPRB, TEK, CDH5, CLDN5, ROBO4 and DLL4 (Fig. 6D).
HOXD10 inhibits expression of CDH5, CLDN5, TEK and DLL4, whereas it increases expression of NOS3
In order to validate and extend the data obtained by CAGE RNA sequencing, we analyzed expression of selected genes involved in permeability and lymphangiogenesis (CDH5, CLDN5, TEK, NOS3 and DLL4) by qPCR in LECs 24 h, 48 h and 72 h after infection with Ad-HOXD10 and Ad-shHOXD10, respectively. Expression of the permeability-associated genes CDH5, CLDN5 and TEK was strongly reduced in LECs overexpressing HOXD10 24 h after infection, and remained repressed to variable extents up to 72 h after infection (Fig. 7A–C). NOS3, which promotes endothelial permeability, was not significantly induced at the earliest timepoint, but expression increased over time and reached a twofold increase at 72 h after infection (Fig. 7D). Expression of DLL4, a gene involved in endothelial sprouting and tubular morphogenesis, was decreased by HOXD10 expression (Fig. 7E). Given that VE-cadherin, the protein encoded by CDH5, is an important component of endothelial junctions and is needed for junctional integrity, we investigated whether the HOXD10-mediated repression of CDH5 would also result in a reduction at the protein level. Staining of LEC monolayers indeed demonstrated that VE-cadherin was largely absent from endothelial junctions in HOXD10-overexpressing cells, whereas overall cellular density was not affected (Fig. 7F). In addition, total cellular VE-cadherin protein levels were significantly reduced, as demonstrated by western blotting (Fig. 7G,H). Apart from a small reduction of DLL4 and CDH5 mRNA (but not protein) levels at the 72 h timepoint, possibly due to the increased viability and, thus, faster monolayer formation, knockdown of HOXD10 had no major effects on the expression of CLDN5, TEK and NOS3, and did not affect junctional integrity, as expected (Fig. S4A–H). These data suggest that HOXD10 regulates expression of several genes known to be involved in key aspects of (lymphatic) endothelial biology, and thereby controls lymphangiogenesis and lymphatic permeability.
Fig. 7.

HOXD10 regulates expression of CDH5, CLDN5, TEK, NOS3 and DLL4. (A–E) LECs were infected with adenovirus to overexpress HOXD10 (Ad-HOXD10) or control virus (Ad-GFP), and the expression of the target genes CDH5 (A), CLDN5 (B), TEK (C), NOS3 (D) and DLL4 (E) was determined by qPCR after 24 h, 48 h and 72 h. Bars represent mean±s.d. (pooled data from five individual experiments). *P<0.05, **P<0.01, ***P<0.001 (two-way ANOVA with Bonferroni post-test). (F) Representative microscopic images of VE-cadherin staining in LEC monolayers 72 h after infection with Ad-GFP and Ad-HOXD10. (G,H) Western blot analysis of total cellular VE-cadherin in LECs 24 h, 48 h and 72 h after infection with Ad-GFP and Ad-HOXD10. (G) Representative blot showing reduced levels of VE-cadherin in HOXD10-overexpressing cells (one representative experiment out of three experiments is shown). (H) Densitometric analysis of relative VE-cadherin signal intensity in western blots, normalized to β-actin (pooled data of three individual experiments). *P<0.05, **P<0.01 (two-way ANOVA with Bonferroni post-test).
DISCUSSION
The VEGF-C–VEGFR-3 signaling axis likely represents the most prominent pathway driving lymphangiogenesis, both during development and in pathologic conditions. Activation of VEGFR-3 by VEGF-C leads to receptor dimerization and phosphorylation at several tyrosine residues (Salameh et al., 2005), which then activate downstream kinases, including MAPKs, PI3K and PKC, which ultimately induce proliferation, migration and sprouting of lymphatic endothelial cells (Koch et al., 2011). However, despite the central importance of this pathway, it is currently not completely understood how its activation is translated into a transcriptional response. Recently, using CAGE RNA sequencing of LECs stimulated with the VEGFR-3-specific ligand VEGF-C156S (Joukov et al., 1998), we have described a wave of immediate early transcription factors, which are upregulated at the mRNA level within minutes to a few hours after stimulation (Dieterich et al., 2015). Apart from many well-known immediate early transcription factors induced by MAPK signaling in various cell types (e.g. FOS, ATF3 and EGR proteins), we also identified a transcription factor whose induction and activation was rather specific for LECs, namely MAFB. Together, these immediate early transcription factors are thought to be responsible for the subsequent changes in cellular behavior, such as growth and proliferation, by the induction of further downstream target genes. But how is the transcriptional induction of these immediate early transcription factors regulated? It is conceivable that this requires yet another class of transcription factors, which we call here ‘front line’ transcription factors. These transcription factors must already be present in unstimulated cells, as they are activated very rapidly in case of an appropriate stimulus, and independently of de novo gene expression. Furthermore, their activation is transient, meaning that the transcription factors either get ‘consumed’ (i.e. degraded) or otherwise shut off after induction of their immediate early targets.
Here, we identified and characterized HOXD10 as one such front line transcription factor in lymphatic endothelial cells stimulated with VEGF-C156S. Combining gene expression data and transcription factor activity prediction based on the presence of known TFBS in the promoters of differentially expressed genes, we found that HOXD10 is not induced by VEGFR-3 stimulation on the mRNA level, but is activated as early as 15 min after the stimulus. The mechanism by which activated VEGFR-3 triggers HOXD10 activity is entirely unknown, but it has been reported that the transcription factors of the HOX cluster 10, to which HOXD10 belongs, contain several putative tyrosine phosphorylation sites in the homeodomain flanking regions (Guerreiro et al., 2012). Thus, the trigger of HOXD10 activity downstream of VEGFR-3 stimulation might well be a phosphorylation event. Furthermore, consistent with the model of a front line transcription factor described above, the HOXD10 protein was present and fairly stable in unstimulated cells, even after knockdown of HOXD10 mRNA. Reductions at the protein level could only be observed after stimulation, indicating that activated HOXD10 is rapidly shut off by degradation.
Using oPOSSUM3, we found HOXD10-binding sites in the promoters of several genes upregulated in LECs by VEGF-C156S (Table S2), including the immediate early transcription factors FOSB and NR4A1. HOXD10 overexpression indeed resulted in increased expression of all these transcription factors, but only the upregulation of NR4A1 induced by VEGF-C156S was dependent on HOXD10. This suggests that there is considerable redundancy among transcription factors regulating the immediate early response. A possible candidate to compensate for a lack of HOXD10 in LECs is HOXD8, which has previously been described to be a PROX1 target gene and to regulate ANGPT2 expression in LECs (Harada et al., 2009). The DNA-consensus-binding motifs of HOXD10 (CAATAAA) and HOXD8 (TAATTAAT) are fairly similar, and we observed signs of HOXD8 activity in VEGF-C156S-stimulated LECs, although at much later time points than HOXD10 (Fig. 1; Table S1). Although the expression level of HOXD8 was not affected by HOXD10 overexpression or knockdown (Fig. S4H,I), double knockdown of HOXD10 and HOXD8 might have additional effects on immediate early transcription factor expression in VEGF-C-treated LECs.
HOXD10 knockdown and overexpression had various functional effects on LECs, the most striking of which was its impact on permeability. High HOXD10 expression resulted in abnormal junctions and increased permeability, whereas HOXD10 depletion resulted in reduced permeability through LEC monolayers. Currently, the role of VEGFR-3 signaling in regulating LEC permeability in vitro is not entirely clear, with some reports showing a reduction in transendothelial electric resistance of LECs after stimulation with VEGF-C (Breslin et al., 2007; Tacconi et al., 2015), whereas others found no effect of VEGF-C156S on macromolecular permeability (Cromer et al., 2014). Therefore, it is possible that wild-type VEGF-C induces permeability by activating VEGFR-2, whereas specific VEGFR-3 activation by VEGF-C156S has no such effects. By contrast, changes in electrical resistance and macromolecular permeability might reflect different cellular processes, which are only partially dependent on VEGFR-3. In our hands, wild-type VEGF-C did not alter macromolecular permeability in LECs (data not shown). Nevertheless, our data suggest that chronic overexpression (and thus, overactivation) of HOXD10, and also lack of HOXD10, do have a clear effect on LEC junctional integrity and permeability. In this context, it is interesting that NR4A1, the immediate early transcription factor dependent on HOXD10, has been found to regulate permeability in blood vascular endothelial cells, by reducing expression of VE-cadherin and claudin-5, and by inducing NOS3 (also known as e-NOS) (Hamers et al., 2014; Zhao et al., 2011). In line with those findings, we found similar changes in gene expression in LECs overexpressing HOXD10, suggesting that HOXD10 could regulate LECs permeability through NR4A1. In conclusion, reduced or exaggerated HOXD10 activity in lymphatic vessels in vivo due to, for example, HOXD10-targeted therapy or therapeutic application of VEGF-C, might be a way to control lymphatic vascular permeability, and thereby, lymphatic fluid uptake and transport.
Another striking biologic effect of HOXD10 was its role in tubular morphogenesis, with LECs depleted of HOXD10 being unable to form any cord-like structures. Instead, these cells continued to grow as monolayers, implying that HOXD10 is required for the morphological changes LECs undergo while forming tube-like structures. Overexpression of HOXD10 resulted in the dysregulation of several genes previously implicated in (lymph-) angiogenesis and vascular morphogenesis, including DLL4 (Niessen et al., 2011; Zheng et al., 2011), IL7 (Iolyeva et al., 2013), AMOTL2 (Hultin et al., 2014), ROBO4 (Yu et al., 2014) and PTPRB (also called VE-PTP) (Hayashi et al., 2013), indicating that these genes are direct or indirect targets of HOXD10. In line with our hypothesis that HOXD10 is only required in acutely stimulated cells, we did not see any changes in the expression of these genes in unstimulated LECs after HOXD10 knockdown. However, it is conceivable that during collagen-induced tubular morphogenesis, lack of HOXD10 could result in aberrant expression of one or several of these genes, leading to the observed phenotype.
Interestingly, various HOX genes have been found to regulate cell adhesion and migration (reviewed in Taniguchi, 2014). For example, in blood vessel endothelial cells, HOXB3 and HOXD3 have been reported to promote angiogenesis by altering adhesion molecule expression and tubular morphogenesis (Boudreau et al., 1997; Myers et al., 2000). HOXD10, by contrast, has been found to exert anti-angiogenic activity, and inhibited migration and sprouting when overexpressed in venous endothelial cells (Myers et al., 2002). In addition, both negative and positive roles for HOXD10 in (tumor) cell proliferation, migration, and invasiveness have been reported, depending on the specific cell line analyzed (Cho et al., 2008; Hakami et al., 2014; Sharpe et al., 2014; Yang et al., 2015; Zha et al., 2012). Taken together, this indicates that HOXD10 exerts different functional roles in different cell types. Clearly, further studies will be needed to elucidate the mechanistic basis for this functional difference, to identify the putative upstream kinase and other interaction partners, and to characterize the role of HOXD10 in other cell types and other signal transduction pathways.
In conclusion, our study demonstrates that HOXD10, a transcription factor which is activated but not induced by VEGF-C–VEGFR-3 signaling, serves as a link between VEGFR-3 stimulation and the upregulation of immediate early transcription factors in LECs. Furthermore, our results implicate a role of HOXD10 in LEC permeability, migration and tubular morphogenesis. These results might help to better understand the dynamic responses of lymphatic vessels to physiological and pathological stimuli in vivo, and to design novel therapeutics for the modulation of lymphatic vessel growth and function.
MATERIALS AND METHODS
CAGE RNA sequencing of primary human lymphatic endothelial cells stimulated with VEGF-C156S
Generation and analysis of the CAGE RNA sequencing data set of primary human LECs has been described previously (Dieterich et al., 2015). In brief, primary human dermal microvascular LECs from foreskin (Hirakawa et al., 2003) were serum-starved overnight in endothelial basal medium (EBM) supplemented with 0.2% bovine serum albumin (BSA), 2 mM L-glutamine and penicillin-streptomycin. Subsequently, cells were stimulated with recombinant human VEGF-C156S (1.5 µg/ml, kind gift of Kari Alitalo, Vihuri Research Center, Helsinki, Finland), and TRIzol lysates were collected at 16 different time points (0 min, 15 min, 30 min, 45 min, 60 min, 80 min, 100 min, 120 min, 150 min, 180 min, 210 min, 240 min, 300 min, 360 min, 420 min, 480 min), with non-stimulated cells serving as a control. RNA samples were sequenced according to the CAGE method at the RIKEN Institute, Yokohama, using the HeliScope CAGE protocol (Kanamori-Katayama et al., 2011; Shiraki et al., 2003), and differential gene expression analysis was performed using edgeR (Robinson et al., 2010). The entire gene expression data are available at http://fantom.gsc.riken.jp/5/datafiles/phase2.0/extra.
oPOSSUM3 transcription factor activity analysis
oPOSSUM3 (Kwon et al., 2012) was performed as described previously (Dieterich et al., 2015). Using the TSS defined by CAGE sequencing, which are associated to known refSeq transcripts, we extracted differentially expressed refSeq transcripts using edgeR (P<0.01) at each timepoint t compared to timepoint t−1. Then, the oPOSSUM3 tool was applied to core promoter sequences (1500 bp upstream to 500 bp downstream of the TSS) of each transcript using transcription-factor-binding profiles from the JASPAR database (jaspar.genereg.net) (Mathelier et al., 2014). Transcription-factor-binding profiles were defined as significantly overrepresented if their corresponding Z-score was >mean+1.5 × s.d. Similarly, overrepresentation of promoters with at least one TFBS was considered significant if the corresponding Fisher score was >mean+1.5 × s.d. HOXD10 target genes predicted by this method are summarized in Table S2.
Isolation of primary mouse endothelial cells
For isolation of primary mouse endothelial cells, ears of FVB mice were split, minced, and digested with collagenase IV (Life Technologies). Cells were depleted of erythrocytes using PharmLyse buffer (BD Bioscience), and stained with primary antibodies [rat anti-mouse-CD45 antibody conjugated to APC–Cy7 (BioLegend 103115, 1:200); rat anti-mouse-CD31 antibody conjugated to APC (BD 551262, 1:300); hamster anti-mouse-podoplanin antibody conjugated to PE (eBioscience 12-5381, 1:200)]. 7AAD was used for discrimination between living and dead cells. All animal experiments were performed according to approved guidelines.
cDNA synthesis and qPCR
RNA from cultured or FACS-sorted LECs and BECs was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. cDNA was generated from RNA samples using the high-capacity cDNA reverse transcriptase kit (Life Technologies). Gene expression was analyzed by SYBR-Green real-time PCR with the AB7900 HT Fast real-time PCR System (Life Technologies), using RPLP0 as internal housekeeping control to normalize the Ct values. The complete list of primer sequences is provided in Table S5.
siRNA-mediated knockdown of PROX1
siRNA silencing of PROX1 in LECs was performed as previously described (Lee et al., 2009). Sequences for PROX1 shRNAs were 5′-GCAAAGAUGUUGAUCCUUCTT-3′ and 5′-GAAGGAUCAACAUCUUUGCTT-3′. Briefly, LECs were electroporated with shRNAs using the Nucleofactor II kit (Amaxa Biosystems). Four individual experiments were performed and the expression of PROX1 and HOXD10 was analyzed after 48 h by qPCR.
Knockdown and overexpression of HOXD10 in LECs in vitro
Knockdown and overexpression of HOXD10 was achieved using adenoviral vectors (Sirion Biotech) with either a pre-validated shRNA targeting HOXD10 under the U6 promoter (Ad-shHOXD10) or HOXD10 cDNA under control of the CMV promoter (Ad-HOXD10). A non-targeting shRNA construct (Ad-NT) and a CMV-GFP construct (Ad-GFP) served as controls. For transduction, subconfluent LECs were infected with adenovirus at a multiplicity of infection (MOI) of 25. Medium was replaced after 4 h.
CAGE RNA sequencing of HOXD10-depleted or -overexpressing LECs
LECs were infected with adenovirus, and RNA was extracted and subjected to CAGE RNA sequencing according to the nAnT-iCAGE protocol (Murata et al., 2014). Differential expression was analyzed using DESeq2 (Love et al., 2014). The results are summarized in Table S3. Gene ontology (GO) analysis was performed using DAVID (https://david.ncifcrf.gov/home.jsp) (Huang et al., 2009a,b), considering ‘biologic process’ terms only. Raw data can be downloaded at http://fantom.gsc.riken.jp/5/suppl/Klein_et_al_2015/
Immunofluorescence staining
Cells were cultured on collagen-coated chamber slides for 72 h before fixing in 4% paraformaldehyde (PFA). Blocking and permeabilization was performed with PBS plus 0.3% Triton X-100, 5% donkey serum, 0.2% BSA and 0.05% NaN3 for 30 min before staining with a goat anti-VE-cadherin antibody (sc-6458, Santa Cruz Biotechnology, 1:200) for 1 h at room temperature. Cells were washed in PBS and incubated with an Alexa-Fluor-594-conjugated donkey anti-goat-IgG antibody (1:200, Life Technologies) for 30 min, followed by nuclear staining with Hoechst 33342 (2 µg/ml). Images were taken with an LSM800 confocal microscope (Zeiss), using a 20× objective.
Western blot analyses
Protein was extracted from TRIzol lysates or directly from cells using a modified RIPA buffer [50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% NP-40, 5 mM EDTA, 1% Triton X-100, complete protease inhibitors (Roche)]. Proteins were separated by SDS-PAGE on 10% polyacrylamide gels and transferred onto PVDF membranes (Immobilon-P, Millipore). The membrane was blocked and incubated with primary antibodies [rabbit anti-HOXD10 (sc-66926, Santa Cruz Biotechnology, 1:200), goat anti-VE-cadherin (AF938, R&D Systems, 1:1000), rabbit anti-β-actin (ab8227, Abcam, 1:1000), mouse anti-β-catenin (MAB2081, Chemicon, 1:1000) and mouse anti-β-tubulin (T9026, Sigma-Aldrich, 1:500)] in 5% milk in TBS plus 0.1% Tween 20, followed by washes and incubation with secondary antibodies [donkey anti-rabbit-IgG (NA9340V, GE Healthcare, 1:10,000), sheep anti-mouse-IgG (GE-Healthcare NA931V, 1:10,000) and donkey anti-goat-IgG (ABIN101172, antibodies online, 1:10,000)] conjugated to horseradish peroxidase (HRP). Signal was developed with ECL Prime (GE Healthcare) or Pierce ECL substrate (Thermo Scientific). Signal density analysis on scanned films was performed using ImageJ.
Cell proliferation, viability, migration and tube formation assays
For proliferation assays, 2500 LECs per well were seeded on collagen-coated 96-well plates. Cells were incubated over night in EBM containing 1% fetal bovine serum (FBS) before 20 ng/ml VEGF-A, 200 ng/ml VEGF-C156S or 200 ng/ml recombinant VEGF-C (kind gift of Kari Alitalo) were added. After 72 h, cell proliferation was analyzed by adding 100 µg/ml 4-methylumbelliferyl heptanoate (MUH, Sigma-Aldrich) in PBS as previously described (Stadler et al., 1989). Fluorescence (corresponding to the proliferation rate) was measured on a SpectraMax Reader (Molecular Devices) at 355 nm excitation and 460 nm emission. Quintuplicates were analyzed for each condition.
To measure LEC viability, 100,000 cells were seeded on collagen-coated six-well plates. Cells were infected with adenovirus as described above, and starved overnight in EBM plus 1% FBS, before incubation with VEGF-A, VEGF-C and VEGF-C156S. The ratio of viable to dead cells was assessed 72 h later by manual counting in a Neubauer counting chamber, using the Trypan Blue exclusion test.
For haptotatic transwell migration assays, LECs were starved overnight in EBM supplemented with 0.2% BSA, 2 mM L-glutamine and penicillin-streptomycin. 30,000–40,000 cells were seeded in starvation medium on transwells (8-µm pore size, Costar) coated on the bottom side with type I collagen (50 µg/ml) (Shin et al., 2006). After 4 h, non-migrated cells were removed with cotton swaps from the upper side of the transwell. Migrated cells were visualized with Hoechst 33342 (Life Technologies). Five pictures were taken from each membrane with an Axiovert 200M Zeiss microscope at 5× magnification and the cell numbers were determined with ImageJ (NIH). Each condition was analyzed in triplicates.
Tubular morphogenesis assays were performed essentially as described previously (Cueni and Detmar, 2009). In brief, LECs were seeded on collagen-coated 24-well plates (8×104/well) and grown to confluency. Cells were incubated for 8 h in EBM plus 5% FBS and subsequently overlaid with 500 µl of a collagen hydrogel (1 mg/ml). Endothelial-cord-like structures were imaged 16 h later using an inverted microscope (Zeiss) and analyzed using ImageJ.
In vitro permeability assays
For macromolecular permeability measurements, LECs were seeded on collagen-coated 0.4 µm transwell inserts and cultured in complete medium for 48 h to form monolayers. Cells were serum-starved overnight in EBM plus 1% FBS before addition of 70-kDa FITC–Dextran to the inner well (final concentration 2.5 mg/ml). Samples of the lower well were analyzed on a SpectraMax Reader (Molecular Devices) in triplicates at the indicated time points. To determine trans-endothelial resistance, 8×104 LECs were seeded on collagen-coated 8W1E gold electrode slides (ibidi) in triplicates. Resistance was measured using the ECIS system (Applied Biophysics) over a period of 48 h.
Statistical analyses
Experiments were performed at least three times, if not stated differently. Statistical analysis was performed with GraphPad Prism software. If not stated differently, Student's t-test as used to compare selected groups. In case of significantly different variances between groups, Welsh's correction was used. Differences with P<0.05 were considered statistically significant.
Supplementary Material
Acknowledgements
We would like to thank all members of the FANTOM5 consortium (http://fantom.gsc.riken.jp/home/people/) for contributing to generation of samples and analysis of the data set and thank GeNAS for data production. The authors would like to thank Kari Alitalo and Michael Jeltsch (Vihuri Research Center, Helsinki, Finland) for providing VEGF-C156S, Jeannette Scholl and Heidi Baumberger for technical assistance, and Martina Vranova and Cornelia Halin (ETH Zurich, Switzerland) for advice with electric resistance measurements. We thank Dora Pak for management support provided to A.M. and W.W.W. FANTOM5 was made possible by a research grant for RIKEN Omics Science Center from Ministry of Education, Culture, Sports, Science, and Technology (MEXT) to Y.H. and a grant of the Innovative Cell Biology by Innovative Technology (Cell Innovation Program) from the MEXT, Japan to Y.H. It was also supported by research grants for RIKEN Preventive Medicine and Diagnosis Innovation Program (RIKEN PMI) to Y.H. and RIKEN Centre for Life Science Technologies, Division of Genomic Technologies (RIKEN CLST (DGT)) from the MEXT, Japan.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
S.K. and L.C.D. designed and performed experiments, analyzed and interpreted the data, and wrote the manuscript. A.M. and W.W.W. performed in silico analyses and interpreted the data. C.C., A.S.-P., and Y.-K.H. performed experiments and analyzed data. J.W.S. designed experiments. T.L. was responsible for all data processing pipelines and developed the mapping algorithm. M.I. was responsible for CAGE data production. H.K. managed the data handling. M.L. carried out CAGE data analysis on the HOXD10 overexpression and knockdown samples. E.A. carried out quality control on the libraries. P.C., Y.H., C.O.D. and A.R.R.F., were responsible for the FANTOM5 phase2 management. M.D. designed experiments, interpreted the data and wrote the manuscript.
Funding
This work was supported by the Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Swiss National Science Foundation) [grant numbers 310030B_147087]; European Research Council [grant LYVICAM]; Oncosuisse; and a Lymph Vessels in Obesity and Cardiovascular Disease grant from the Krebsliga Schweiz and Fondation Leducq (Zurich and Leducq Foundation Transatlantic Network of Excellence) [grant number 11CVD03] (all to M.D.). A.M. and W.W.W. are supported by a large-scale applied research grant from Genome Canada [grant number 174CDE]. Funding has been provided to A.M. by the BC Children's Hospital Foundation; and the Child and Family Research Institute, Vancouver, Canada.
Data availability
This work is part of the FANTOM5 project (Arner et al., 2015; Forrest et al., 2014). Data downloads, genomic tools and co-published manuscripts are summarized here: http://fantom.gsc.riken.jp/5/. In particular, the timecourse CAGE RNA seq data (LEC stimulated with VEGF-C156S) is available at http://fantom.gsc.riken.jp/5/datafiles/phase2.0/extra, and CAGE RNA seq data of LEC overexpressing or depleted of HOXD10 at http://fantom.gsc.riken.jp/5/suppl/Klein_et_al_2015/
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.186767.supplemental
References
- Arner E., Daub C. O., Vitting-Seerup K., Andersson R., Lilje B., Drablos F., Lennartsson A., Ronnerblad M., Hrydziuszko O., Vitezic M. et al. (2015). Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010-1014. 10.1126/science.1259418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau N., Andrews C., Srebrow A., Ravanpay A. and Cheresh D. A. (1997). Induction of the angiogenic phenotype by Hox D3. J. Cell Biol. 139, 257-264. 10.1083/jcb.139.1.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin J. W., Yuan S. Y. and Wu M. H. (2007). VEGF-C alters barrier function of cultured lymphatic endothelial cells through a VEGFR-3-dependent mechanism. Lymphat. Res. Biol. 5, 105-114. 10.1089/lrb.2007.1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brint E., O'Callaghan G. and Houston A. (2013). Life in the Fas lane: differential outcomes of Fas signaling. Cell. Mol. Life Sci. 70, 4085-4099. 10.1007/s00018-013-1327-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton J. B., Priceman S. J., Sung J. L., Brakenhielm E., An D. S., Pytowski B., Alitalo K. and Wu L. (2008). Suppression of prostate cancer nodal and systemic metastasis by blockade of the lymphangiogenic axis. Cancer Res. 68, 7828-7837. 10.1158/0008-5472.CAN-08-1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho M. A., Yashar P., Kim S. K., Noh T., Gillam M. P., Lee E. J. and Jameson J. L. (2008). HoxD10 gene delivery using adenovirus/adeno-associate hybrid virus inhibits the proliferation and tumorigenicity of GH4 pituitary lactotrope tumor cells. Biochem. Biophys. Res. Commun. 371, 371-374. 10.1016/j.bbrc.2008.04.085 [DOI] [PubMed] [Google Scholar]
- Cromer W. E., Zawieja S. D., Tharakan B., Childs E. W., Newell M. K. and Zawieja D. C. (2014). The effects of inflammatory cytokines on lymphatic endothelial barrier function. Angiogenesis 17, 395-406. 10.1007/s10456-013-9393-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueni L. N. and Detmar M. (2009). Galectin-8 interacts with podoplanin and modulates lymphatic endothelial cell functions. Exp. Cell Res. 315, 1715-1723. 10.1016/j.yexcr.2009.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich L. C., Klein S., Mathelier A., Sliwa-Primorac A., Ma Q., Hong Y.-K., Shin J. W., Hamada M., Lizio M., Itoh M. et al. (2015). DeepCAGE transcriptomics reveal an important role of the transcription factor MAFB in the lymphatic endothelium. Cell Rep. 13, 1493-1504. 10.1016/j.celrep.2015.10.002 [DOI] [PubMed] [Google Scholar]
- Dumont D. J. (1998). Cardiovascular failure in mouse embryos deficient in VEGF Receptor-3. Science 282, 946-949. 10.1126/science.282.5390.946 [DOI] [PubMed] [Google Scholar]
- Forrest A. R. R., Kawaji H., Rehli M., Baillie J. K., de Hoon M. J. L., Haberle V., Lassmann T., Kulakovskiy I. V., Lizio M., Itoh M. et al. (2014). A promoter-level mammalian expression atlas. Nature 507, 462-470. 10.1038/nature13182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro I., Casaca A., Nunes A., Monteiro S., Novoa A., Ferreira R. B., Bom J. and Mallo M. (2012). Regulatory role for a conserved motif adjacent to the homeodomain of Hox10 proteins. Development 139, 2703-2710. 10.1242/dev.081448 [DOI] [PubMed] [Google Scholar]
- Hakami F., Darda L., Stafford P., Woll P., Lambert D. W. and Hunter K. D. (2014). The roles of HOXD10 in the development and progression of head and neck squamous cell carcinoma (HNSCC). Br. J. Cancer 111, 807-816. 10.1038/bjc.2014.372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamers A. A. J., Uleman S., van Tiel C. M., Kruijswijk D., van Stalborch A.-M., Huveneers S., de Vries C. J. M. and van ‘t Veer C. (2014). Limited role of nuclear receptor Nur77 in Escherichia coli-induced peritonitis. Infect. Immun. 82, 253-264. 10.1128/IAI.00721-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada K., Yamazaki T., Iwata C., Yoshimatsu Y., Sase H., Mishima K., Morishita Y., Hirashima M., Oike Y., Suda T. et al. (2009). Identification of targets of Prox1 during in vitro vascular differentiation from embryonic stem cells: functional roles of HoxD8 in lymphangiogenesis. J. Cell Sci. 122, 3923-3930. 10.1242/jcs.052324 [DOI] [PubMed] [Google Scholar]
- Hayashi M., Majumdar A., Li X., Adler J., Sun Z., Vertuani S., Hellberg C., Mellberg S., Koch S., Dimberg A. et al. (2013). VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nat. Commun. 4, 1672 10.1038/ncomms2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Kozaki K.-I., Karpanen T., Koshikawa K., Yla-Herttuala S., Takahashi T. and Alitalo K. (2002). Suppression of tumor lymphangiogenesis and lymph node metastasis by blocking vascular endothelial growth factor receptor 3 signaling. J. Natl. Cancer Inst. 94, 819-825. 10.1093/jnci/94.11.819 [DOI] [PubMed] [Google Scholar]
- Hirakawa S., Hong Y.-K., Harvey N., Schacht V., Matsuda K., Libermann T. and Detmar M. (2003). Identification of vascular lineage-specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am. J. Pathol. 162, 575-586. 10.1016/S0002-9440(10)63851-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa S., Kodama S., Kunstfeld R., Kajiya K., Brown L. F. and Detmar M. (2005). VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J. Exp. Med. 201, 1089-1099. 10.1084/jem.20041896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa S., Brown L. F., Kodama S., Paavonen K., Alitalo K. and Detmar M. (2007). VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood 109, 1010-1017. 10.1182/blood-2006-05-021758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa S., Detmar M. and Karaman S. (2014). Lymphatics in nanophysiology. Adv. Drug Deliv. Rev. 74, 12-18. 10.1016/j.addr.2014.01.011 [DOI] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T. and Lempicki R. A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1-13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T. and Lempicki R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44-57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Huggenberger R. and Detmar M. (2011). The cutaneous vascular system in chronic skin inflammation. J. Investig. Dermatol. Symp. Proc. 15, 24-32. 10.1038/jidsymp.2011.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggenberger R., Ullmann S., Proulx S. T., Pytowski B., Alitalo K. and Detmar M. (2010). Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J. Exp. Med. 207, 2255-2269. 10.1084/jem.20100559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggenberger R., Siddiqui S. S., Brander D., Ullmann S., Zimmermann K., Antsiferova M., Werner S., Alitalo K. and Detmar M. (2011). An important role of lymphatic vessel activation in limiting acute inflammation. Blood 117, 4667-4678. 10.1182/blood-2010-10-316356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultin S., Zheng Y., Mojallal M., Vertuani S., Gentili C., Balland M., Milloud R., Belting H.-G., Affolter M., Helker C. S. M. et al. (2014). AmotL2 links VE-cadherin to contractile actin fibres necessary for aortic lumen expansion. Nat. Commun. 5, 3743 10.1038/ncomms4743 [DOI] [PubMed] [Google Scholar]
- Iolyeva M., Aebischer D., Proulx S. T., Willrodt A.-H., Ecoiffier T., Haner S., Bouchaud G., Krieg C., Onder L., Ludewig B. et al. (2013). Interleukin-7 is produced by afferent lymphatic vessels and supports lymphatic drainage. Blood 122, 2271-2281. 10.1182/blood-2013-01-478073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov V., Sorsa T., Kumar V., Jeltsch M., Claesson-Welsh L., Cao Y., Saksela O., Kalkkinen N. and Alitalo K. (1997). Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J. 16, 3898-3911. 10.1093/emboj/16.13.3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov V., Kumar V., Sorsa T., Arighi E., Weich H., Saksela O. and Alitalo K. (1998). A recombinant mutant vascular endothelial growth factor-C that has lost vascular endothelial growth factor receptor-2 binding, activation, and vascular permeability activities. J. Biol. Chem. 273, 6599-6602. 10.1074/jbc.273.12.6599 [DOI] [PubMed] [Google Scholar]
- Kanamori-Katayama M., Itoh M., Kawaji H., Lassmann T., Katayama S., Kojima M., Bertin N., Kaiho A., Ninomiya N., Daub C. O. et al. (2011). Unamplified cap analysis of gene expression on a single-molecule sequencer. Genome Res. 21, 1150-1159. 10.1101/gr.115469.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman S. and Detmar M. (2014). Mechanisms of lymphatic metastasis. J. Clin. Invest. 124, 922-928. 10.1172/JCI71606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkkainen M. J., Haiko P., Sainio K., Partanen J., Taipale J., Petrova T. V., Jeltsch M., Jackson D. G., Talikka M., Rauvala H. et al. (2004). Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 5, 74-80. 10.1038/ni1013 [DOI] [PubMed] [Google Scholar]
- Koch S., Tugues S., Li X., Gualandi L. and Claesson-Welsh L. (2011). Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 437, 169-183. 10.1042/BJ20110301 [DOI] [PubMed] [Google Scholar]
- Kunstfeld R., Hirakawa S., Hong Y.-K., Schacht V., Lange-Asschenfeldt B., Velasco P., Lin C., Fiebiger E., Wei X., Wu Y. et al. (2004). Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood 104, 1048-1057. 10.1182/blood-2003-08-2964 [DOI] [PubMed] [Google Scholar]
- Kwon A. T., Arenillas D. J., Hunt R. W. and Wasserman W. W. (2012). oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 2, 987-1002. 10.1534/g3.112.003202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Kang J., Yoo J., Ganesan S. K., Cook S. C., Aguilar B., Ramu S., Lee J. and Hong Y.-K. (2009). Prox1 physically and functionally interacts with COUP-TFII to specify lymphatic endothelial cell fate. Blood 113, 1856-1859. 10.1182/blood-2008-03-145789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I., Huber W. and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkinen T., Veikkola T., Mustjoki S., Karpanen T., Catimel B., Nice E. C., Wise L., Mercer A., Kowalski H., Kerjaschki D. et al. (2001). Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 20, 4762-4773. 10.1093/emboj/20.17.4762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandriota S. J., Jussila L., Jeltsch M., Compagni A., Baetens D., Prevo R., Banerji S., Huarte J., Montesano R., Jackson D. G. et al. (2001). Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J. 20, 672-682. 10.1093/emboj/20.4.672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A., Zhao X., Zhang A. W., Parcy F., Worsley-Hunt R., Arenillas D. J., Buchman S., Chen C.-Y., Chou A., Ienasescu H. et al. (2014). JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42, D142-D147. 10.1093/nar/gkt997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M., Nishiyori-Sueki H., Kojima-Ishiyama M., Carninci P., Hayashizaki Y. and Itoh M. (2014). Detecting expressed genes using CAGE. Methods Mol. Biol. 1164, 67-85. 10.1007/978-1-4939-0805-9_7 [DOI] [PubMed] [Google Scholar]
- Myers C., Charboneau A. and Boudreau N. (2000). Homeobox B3 promotes capillary morphogenesis and angiogenesis. J. Cell Biol. 148, 343-352. 10.1083/jcb.148.2.343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers C., Charboneau A., Cheung I., Hanks D. and Boudreau N. (2002). Sustained expression of homeobox D10 inhibits angiogenesis. Am. J. Pathol. 161, 2099-2109. 10.1016/S0002-9440(10)64488-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen K., Zhang G., Ridgway J. B., Chen H., Kolumam G., Siebel C. W. and Yan M. (2011). The Notch1-Dll4 signaling pathway regulates mouse postnatal lymphatic development. Blood 118, 1989-1997. 10.1182/blood-2010-11-319129 [DOI] [PubMed] [Google Scholar]
- Roberts N., Kloos B., Cassella M., Podgrabinska S., Persaud K., Wu Y., Pytowski B. and Skobe M. (2006). Inhibition of VEGFR-3 activation with the antagonistic antibody more potently suppresses lymph node and distant metastases than inactivation of VEGFR-2. Cancer Res. 66, 2650-2657. 10.1158/0008-5472.CAN-05-1843 [DOI] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J. and Smyth G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139-140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salameh A., Galvagni F., Bardelli M., Bussolino F. and Oliviero S. (2005). Direct recruitment of CRK and GRB2 to VEGFR-3 induces proliferation, migration, and survival of endothelial cells through the activation of ERK, AKT, and JNK pathways. Blood 106, 3423-3431. 10.1182/blood-2005-04-1388 [DOI] [PubMed] [Google Scholar]
- Sharpe D. J., Orr K. S., Moran M., White S. J., McQuaid S., Lappin T. R., Thompson A. and James J. A. (2014). POU2F1 activity regulates HOXD10 and HOXD11 promoting a proliferative and invasive phenotype in head and neck cancer. Oncotarget 5, 8803-8815. 10.18632/oncotarget.2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J. W., Min M., Larrieu-Lahargue F., Canron X., Kunstfeld R., Nguyen L., Henderson J. E., Bikfalvi A., Detmar M. and Hong Y.-K. (2006). Prox1 promotes lineage-specific expression of fibroblast growth factor (FGF) receptor-3 in lymphatic endothelium: a role for FGF signaling in lymphangiogenesis. Mol. Biol. Cell 17, 576-584. 10.1091/mbc.E05-04-0368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki T., Kondo S., Katayama S., Waki K., Kasukawa T., Kawaji H., Kodzius R., Watahiki A., Nakamura M., Arakawa T. et al. (2003). Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA 100, 15776-15781. 10.1073/pnas.2136655100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobe M., Hawighorst T., Jackson D. G., Prevo R., Janes L., Velasco P., Riccardi L., Alitalo K., Claffey K. and Detmar M. (2001). Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 7, 192-198. 10.1038/84643 [DOI] [PubMed] [Google Scholar]
- Stadler R., Detmar M., Stephanek K., Bangemann C. and Orfanos C. E. (1989). A rapid fluorometric assay for the determination of keratinocyte proliferation in vitro. J. Invest. Dermatol. 93, 532-534. 10.1111/1523-1747.ep12284080 [DOI] [PubMed] [Google Scholar]
- Tacconi C., Correale C., Gandelli A., Spinelli A., Dejana E., D'Alessio S. and Danese S. (2015). Vascular endothelial growth factor C disrupts the endothelial lymphatic barrier to promote colorectal cancer invasion. Gastroenterology 148, 1438-1451.e8. 10.1053/j.gastro.2015.03.005 [DOI] [PubMed] [Google Scholar]
- Taniguchi Y. (2014). Hox transcription factors: modulators of cell-cell and cell-extracellular matrix adhesion. Biomed. Res. Int. 2014, 591374 10.1155/2014/591374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veikkola T., Jussila L., Makinen T., Karpanen T., Jeltsch M., Petrova T. V., Kubo H., Thurston G., McDonald D. M., Achen M. G. et al. (2001). Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. 20, 1223-1231. 10.1093/emboj/20.6.1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Kim C., Kim M.-J., Schwendener R. A., Alitalo K., Heston W., Kim I., Kim W.-J. and Koh G. Y. (2011). Soluble vascular endothelial growth factor receptor-3 suppresses lymphangiogenesis and lymphatic metastasis in bladder cancer. Mol. Cancer 10, 36 10.1186/1476-4598-10-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Zhou J., Mi J., Ma K., Fan Y., Ning J., Wang C., Wei X., Zhao H. and Li E. (2015). HOXD10 acts as a tumor-suppressive factor via inhibition of the RHOC/AKT/MAPK pathway in human cholangiocellular carcinoma. Oncol. Rep. 34, 1681-1691. 10.3892/or.2015.4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Zhang X., Kuzontkoski P. M., Jiang S., Zhu W., Li D. Y. and Groopman J. E. (2014). Slit2N and Robo4 regulate lymphangiogenesis through the VEGF-C/VEGFR-3 pathway. Cell Commun. Signal. 12, 25 10.1186/1478-811X-12-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha Y., Ding E., Yang L., Mao L., Wang X., McCarthy B. A., Huang S. and Ding H.-F. (2012). Functional dissection of HOXD cluster genes in regulation of neuroblastoma cell proliferation and differentiation. PLoS ONE 7, e40728 10.1371/journal.pone.0040728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D., Qin L., Bourbon P.-M., James L., Dvorak H. F. and Zeng H. (2011). Orphan nuclear transcription factor TR3/Nur77 regulates microvessel permeability by targeting endothelial nitric oxide synthase and destabilizing endothelial junctions. Proc. Natl. Acad. Sci. USA 108, 12066-12071. 10.1073/pnas.1018438108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W., Tammela T., Yamamoto M., Anisimov A., Holopainen T., Kaijalainen S., Karpanen T., Lehti K., Yla-Herttuala S. and Alitalo K. (2011). Notch restricts lymphatic vessel sprouting induced by vascular endothelial growth factor. Blood 118, 1154-1162. 10.1182/blood-2010-11-317800 [DOI] [PubMed] [Google Scholar]
- Zheng W., Aspelund A. and Alitalo K. (2014). Lymphangiogenic factors, mechanisms, and applications. J. Clin. Invest. 124, 878-887. 10.1172/JCI71603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q., Guo R., Wood R., Boyce B. F., Liang Q., Wang Y.-J., Schwarz E. M. and Xing L. (2011). Vascular endothelial growth factor C attenuates joint damage in chronic inflammatory arthritis by accelerating local lymphatic drainage in mice. Arthritis Rheum. 63, 2318-2328. 10.1002/art.30421 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.