

Abstract
The transcription factor ΔFosB accumulates in response to chronic insults such as drugs of abuse, L-3,4-dihydroxyphenylalanine (l-DOPA) or stress in specific regions of the brain, triggering long lasting neural and behavioral changes that underlie aspects of drug addiction, dyskinesia, and depression. Thus, small molecule chemical probes are urgently needed to investigate biological functions of ΔFosB. Herein we describe the identification of a novel phenanthridine analogue ZL0220 (27) as an active and promising ΔFosB chemical probe with micromolar inhibitory activities against ΔFosB homodimers and ΔFosB/JunD heterodimers.
Keywords: ΔFosB homodimer, ΔFosB/JunD heterodimer, Chemical probes, High-throughput screening, Fluorescence polarization assay
The ΔFosB protein forms a transcription factor activator protein 1 (AP-1) complex.1 The AP-1 family consists of homodimers and heterodimers of Jun, Fos, ATF and MAF protein families which have highly conserved dimeric basic leucine zipper (bZIP) DNA-binding domains.2,3 ΔFosB is comprised of a large N-terminal domain which likely recruits different proteins involved in transcriptional regulation, a bZIP domain that interacts with DNA and a short C-terminal domain, while it lacks the 101 amino acid C-terminal transactivation domain of FosB.4 ΔFosB dimerizes with other bZIP domain-containing proteins via the leucine zipper of its bZIP domain to form a coiled-coil such as ΔFosB/ΔFosB homodimers and ΔFosB/JunD heterodimers.5–7 The coiled-coil brings the basic regions from the two partners together so that they can interact with DNA like a pair of forceps at AP-1 consensus sites within the promoters of specific target genes (e.g., cyclin-dependent kinase 5 and AMPA glutamate receptor subunit 2) and regulate their expression.8–11 Chronic administration of stimuli (e.g., drugs of abuse or l-DOPA) increases ΔFosB protein levels in specific regions of the brain.12–14 Altered expression of key target genes through the binding of ΔFosB to relevant promoter regions is thought to mediate neural, and behavioral changes.15 ΔFosB represents a promising therapeutic target to treat a variety of central nervous system (CNS) disorders, including drug addiction, depression, dyskinesias, and Alzheimer’s disease.16–19 Nevertheless, its function is very complicated and remains unclear up to date. Depending on the length of time, induction level, different stimulus, and the experimental condition, it can work as both an activator and repressor even for the same target gene.20–22 Small molecule chemical probes are therefore urgently needed to facilitate delineating the biological functions of ΔFosB.
We conducted high throughput (HTS) screening using a fluorescence polarization (FP)-assay to detect the specific binding of ΔFosB to a TAMRA-labeled cdk5 oligonucleotide carrying the CDK5 AP-1 consensus site by taking advantage of a 54,498 compounds library at the center for Chemical Genomics, University of Michigan.23 Compound C1 (2, Fig. 1) and C2 (3, Fig. 1) disrupted the binding of ΔFosB to DNA in the low micromolar range and altered in vitro ΔFosB-mediated transcription. Recently, we have identified another HTS hit STK893803 (4, Fig. 1) with an IC50 value of 28.9 pM against ΔFosB homodimers and 13.1 pM against ΔFosB/JunD heterodimers, respectively. Herein, we report our medicinal chemistry effort based on these hits and the discovery of a novel phenanthridine analogue ZL0220 (27) as an active and promising ΔFosB chemical probe and a new lead for further optimization.
Fig. 1.

Chemical structures of several compounds known to target AP-1 transcription factors, including T5224 (c-Fos/c-Jun), C1 and C2 (ΔFosB homodimers and ΔFosB/JunD heterodimers), as well as a new lead STK89303 and its development into the chemical probe ZL0220 (27) as described in this work.
To establish a meaningful structure-activity relationship (SAR) and obtain more potent ΔFosB modulators disrupting either ΔFosB homodimers or ΔFosB/JunD heterodimers, we explored the species and positions of substituents on the phenyl ring, optimized the secondary amine, and introduced diverse scaffolds to the acid based on HTS hit 4 (Series A, Fig. 2). Meanwhile, we noticed that at least three chiral centers existed in this molecule, which may lead to challenges for isomer separation and limit the usefulness as pharmacological probes. We thus aromatized the tetrahydroquinoline scaffold (Series B) and replaced the cyclopentene moiety with phenyl ring to generate Series C to eliminate the chiral centers.
Fig. 2.
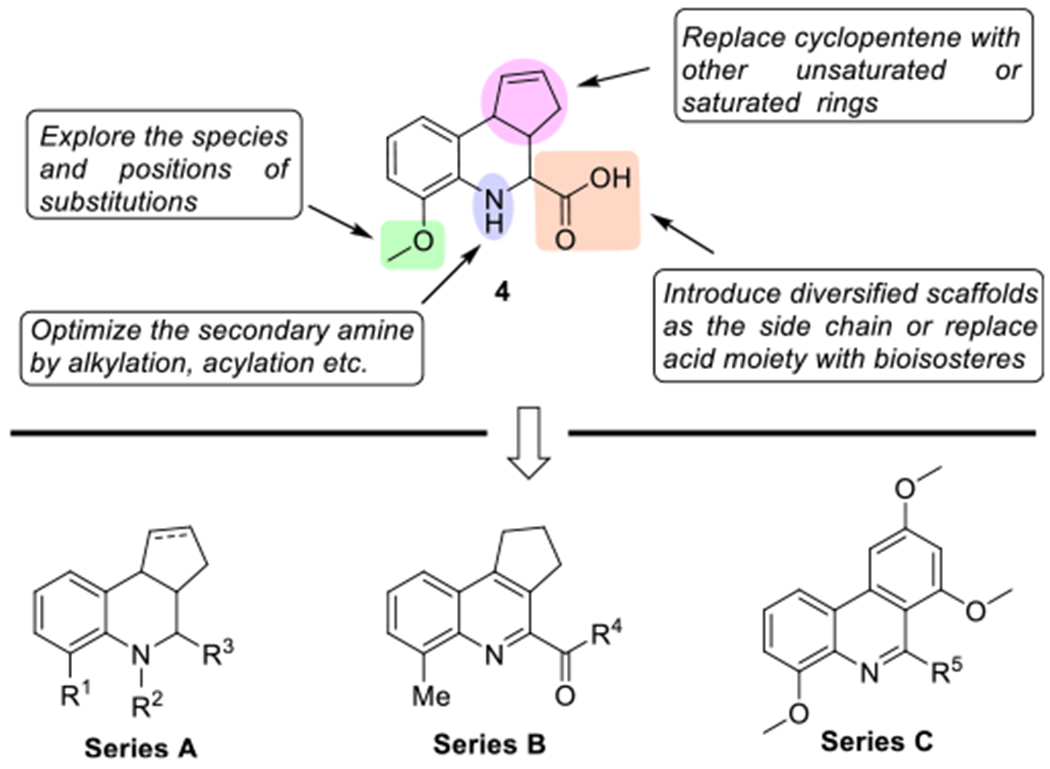
Optimization strategy on HTS hit 4 to generate three Series A–C of novel compounds as potential chemical probes.
As outlined in Scheme 1, the starting materials 2-methoxyaniline (5), ethyl 2-oxoacetate (6) and cyclopenta-1,3-diene (7) were commercially available. Direct synthesis of imine followed by a Diels-Alder reaction in one pot proceeded well in the presence of Cu(OTf)2 producing 8 in a yield of 59%. Hydrolysis of 8 by NaOH (aq.) led to 4, and coupling 4 with different amines in the presence of HBTU gave compounds 9 and 10 in yields of over 73%. Reduction of 8 with NaBH4 yielded compound 11. Acetylation of 8 following by hydrolysis generated compound 13 in a yield of 85% for two steps. Compound 14 was obtained via catalytic hydrogenation of 4 in the atmosphere of H2. Compounds 16 and 17 were prepared according to the similar procedure for synthesis of 4.
Scheme 1.

Synthetic route of compounds 4, 8–14, 16 and 17. Reagents and conditions: a) Cu(OTf)2, CH3CN, rt, 6 h, 59% yield for 8, 40% yield for 16; b) NaOH, CH3OH/H2O, rt, 1 h, 83% yield for 4, 80% yield for 17; c) For 9, CH3NH2, HBTU, DIEA, DCM, rt, overnight, quant.; For 10, aniline, HBTU, DIEA, DCM, rt, overnight, 73%. d) NaBH4, LiCl, CH3OH, rt, 2 d, 24%; e) CH3COCl, Et3N, DCM, rt, 4 h, Quant.; f) LiOH, CH3OH/H2O, rt, overnight, 85%; (g) 5% Pd/C, H2, rt, overnight, 69%.
The synthesis of Series B compounds was outlined in Scheme 2. Intermediate 19 was produced following a procedure similar to that of compound 8 in the presence of BF3·Et2O. Compound 19 was subsequently oxidized by DDQ to give 20 in a yield of 74%, which was then hydrolyzed to produce compound 21 in a yield of 85%.
Scheme 2.

Synthetic procedure of compounds 20 and 21. Reagents and conditions: a) Na2SO4, BF3·Et2O, toluene, N2, −78 °C, 1 h, 12%; b) DDQ, ClCH2CH2Cl, rt, 2 min, 74%; c) NaOH, CH3OH/H2O, rt, 1 h, 85%.
As depicted in Scheme 3, Series C compounds were started from commercially available 2-bromo-6-methoxyaniline (22) and 3,5-dimethoxyphenyl boronic acid (23). Palladium-catalyzed Suzuki crosscoupling reaction proceeded smoothly by using Pd(PPh3)4 and K2CO3 in a yield of 90% leading to intermediate 24. Compounds 25, 26 and 29 were synthesized through a Pictet–Spengler reaction in the presence of Lewis acid propanephosphonic acid cyclic anhydride (T3P). Compound 26 was further deprotected by trifluoroacetic acid (TFA) to produce 27, which was acetylated to generate 28 in a yield of 65%. Structures and the purity of synthesized compounds were confirmed by 1H NMR, 13C NMR and HRMS as well as HPLC analysis.24 The structure of phenanthridine derivative 27 was also unambiguously determined by X-ray crystallographic analysis (CCDC number: 1960003) (Scheme 3).
Scheme 3.

Synthetic route of 25–29. Reagents and conditions: a) Pd(PPh3)4, K2CO3, dioxane, 100 °C, 3 h, 90%; b) For 25, i. ethyl 3-oxopropanoate, T3P, EtOAc, 65 °C, overnight, 38%; ii. LiOH, CH3OH/H2O, rt, 3 h, 56%; for 26, tert-butyl 4-formylpiperidine-l-carboxylate, T3P, EtOAc, 65 °C, overnight, 77%; for 29, propionaldehyde, T3P, EtOAc, 65 °C, overnight, 46%. c) TFA, DCM, rt, 85%; d) CH3COCl, K2CO3, CH3CN, rt, 65%.
The newly designed and synthesized small molecules were evaluated using the FP-assay25 (Tables 1 and 2). The FP-assay measured the extent to which a compound could disrupt the binding of ΔFosB/ΔFosB homodimers or ΔFosB/JunD heterodimers to a TAMRA-labeled cdk5 AP-1 19-mer oligonucleotide (probe). As the probe was liberated from the transcription factor upon increasing compound concentration, its tumbling increased in solution and its FP signal decreased concomitantly. For the first series of compounds (Series A), our initial optimization was focused on changing the acid moiety in STK893803 structure into different fragments, such as ethyl ester (8), N-methylformamide (9), N-phenylformamide (10) and CH2OH (11), while the commercially available STK893803 (4) was tested as a positive control (Table 1). Unfortunately, none of the newly synthesized compounds had significant activity (IC50 values of over 100 μM). Replacement of the hydrogen of compound 4 at the nitrogen with Ac or reduction of double bond led to compounds 13 and 14, which also lacked significant activity. Changing OMe of compound 4 into Me gave a compound (17) that interfered with the FP-assay, directly affecting the tumbling of the TAMRA-labeled oligonucleotide even in absence of protein.
Table 1.
In vitro activity of Series A compounds, disrupting the binding of ΔFosB homodimers and ΔFosB/JunD heterodimers to a TAMRA-labeled cdk5 AP-1 oligonucleotide.
 | |||||
|---|---|---|---|---|---|
| Cmpd ID | R1 | R2 | R3 | ΔFosB homodimer (IC50, μM)a | ΔFosB/JunD heterodimer (IC50, μM)a |
| 4 | OCH3 | H | CO2H | 28.9 ± 4.4 | 13.1 ± 4.5 |
| 8 | OCH3 | H | CO2Et | > 100 | > 100 |
| 9 | OCH3 | H | CONHCH3 | > 100 | > 100 |
| 10 | OCH3 | H | CONHPh | > 100 | > 100 |
| 11 | OCH3 | H | CH2OH | > 100 | > 100 |
| 13 | OCH3 | Ac | CO2h | > 100 | > 100 |
| 14 | OCH3 | H | CO2h | > 100 | > 100 |
| 17 | CH3 | H | CO2h | –b | –b |
IC50 values are reported as the mean ± SD derived from two FP-assays carried out on two independent days. For each FP-assay, an increasing concentration of compound (10 different concentrations, 0–200 μM) was tested in quadruplicate and the disruption of transcription factor binding to DNA quantitated.
Interferes with assay.
Table 2.
In vitro activities of compounds from Series B and Series C disrupting the binding of ΔFosB homodimers and ΔFosB/JunD heterodimers to a TAMRA-1abeled cdk5 AP-1 oligonucleotide.
 |
IC50 values are reported as the mean ± SD derived from two FP-assays carried out on two independent days. For each FP-assay, an increasing concentration of compound (10 different concentrations, 0–200 μM) was tested in quadruplicate and the disruption of transcription factor binding to DNA quantitated.
Interferes with assay.
Based on the results of Series A, we aromatized the tetrahydroquinoline scaffold (Series B) and replaced the cyclopentene moiety with a phenyl ring (Series C) to eliminate the chiral centers. Compounds 20–21 and 25–29 with new scaffolds were also pharmacologically evaluated. As presented in Table 2, compounds 20 and 21, containing a quinoline scaffold, showed little activity even at 100 μM or higher. Next, the five-membered ring was substituted with an electron-rich aromatic ring to yield the phenanthridine derivative 25, which also lacked activity. However, further replacement of the carboxyl group of compound 25 with piperidine (26–28) and ethyl (29) led to a novel phenanthridine analogue ZL0220 (27) as an active and promising ΔFosB chemical probe with moderate inhibitory activities against ΔFosB homodimers and ΔFosB/JunD heterodimers with IC50 values of 58.0 ± 0.5 μM and 55.7 ± 1.9 μM, respectively.
The dose response curves for ZL0220 (27) were shown in Fig. 3 and revealed a dose-dependent disruption of transcription factor binding to DNA, disrupting ΔFosB homodimers (Fig. 3A) and ΔFosB/JunD heterodimers (Fig. 3B) to a similar extent.
Fig. 3.

Activity of ZL0220 (27). A) representative dose–response curve with ΔFosB; B) representative dose–response curve with ΔFosB/JunD. Shown are protein + TAMRA-cdk5 AP-1 oligonucleotide (solid line); TAMRA-cdk5 AP-1 oligonucleotide alone (dotted line); and as controls in absence of any compound, TAMRA-labeled cdk5 AP-1 oligonucleotide (red circle, representing 100% inhibition) and protein + TAMRA-labeled cdk5 AP-1 oligonucleotide (green square, representing 0% inhibition). Ten concentrations were tested in the range 0–200 μM in quadruplicate, error bars depict the standard error of the mean (SEM).
Based on our recent crystal structure of the ΔFosB/JunD bZIP heterodimer bound to DNA,6 we performed a molecular docking study to predict the binding mode of ZL0220 (27) bound to ΔFosB/JunD in the DNA-bound form (with DNA removed).26 As illustrated in Fig. 4, ZL0220 was found to bind to ΔFosB/JunD at a site close to the DNA binding region (grey ribbon). The methoxy group at 4-position of phenanthridine ring formed an H-bond with C172 of ΔFosB, and the heterocyclic ring of ZL0220 interacted with R176 of ΔFosB through a itcation interaction. ZL0220 also interacted with JunD via hydrophobic interaction. The modeling results suggest that assembling a Michael acceptor at methoxy group position may provide an opportunity to form a covalent interaction with ΔFosB C172 over time improving its apparent binding affinity to the protein.
Fig. 4.

Predicted docked pose of ZL0220 (27) in the ΔFosB/JunD bZIP heterodimer (PDB ID: 5VPE; DNA removed). ΔFosB/JunD is depicted in ribbon representation and ZL0220 is displayed in CPK representation. Color scheme (FosB: red; JunD: blue; DNA: grey; DNA binding region motif: grey; ZL0220: magenta). Left panel is a zoomed view of the proposed binding site. ZL0220 and key interacting residues are shown in sticks. Hydrogen-bonds are depicted as purple dotted lines, and π-cation interactions as blue dotted line.
In summary, the novel phenanthridine analogue ZL0220 (27) has been identified as an active and promising ΔFosB chemical probe lead with micromolar inhibitory activities against ΔFosB homodimers and ΔFosB/JunD heterodimers. Docking studies using our crystal structure of ZL0220 in the crystal structure of the ΔFosB/JunD bZIP revealed several potential binding sites for their modes of action. Further pharmacological investigation of ZL0220 as a chemical probe, lead optimization that also includes a covalent bond formation strategy, and determination of the co-crystal structure of the transcription factor in complex with ZL0220 are now underway to determine more potent and selective chemical probes and understand their exact interactions with the target proteins.
Acknowledgements
This work was supported by the NIH/NIDA Grant R01 DA040621 (G.R. and J.Z.), and the John D. Stobo, M.D. Distinguished Chair Endowment Fund (J.Z.) at UTMB. We would like to thank Dr. Eric J. Nestler at Icahn School of Medicine at Mount Sinai and Dr. Alfred J. Robison at the Department of Physiology, Michigan State University for helpful discussions. We gratefully acknowledge Dr. Clifford Stephan and Ivy Nguyen at the Institute of Biosciences and Technology, Texas A&M University for the access of the HTS facility and experimental assistance in testing compounds.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Ye N, Ding Y, Wild C, Shen Q, Zhou J. J. Med Chem 2014;57(16):6930–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hai TW, Liu F, Allegretto EA, Karin M, Green MR. Genes Dev. 1988;2(10):1216–1226. [DOI] [PubMed] [Google Scholar]
- 3.Angel P, Karin M. Biochim. Biophys Acta 1991. ;1072(2–3): 129–157. [DOI] [PubMed] [Google Scholar]
- 4.Robison AJ, Nestler EJ. Nat Rev Neurosci 2011;12(ll):623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jorissen HJM, Ulery PG, Henry L, Gourneni S, Nestler EJ, Rudenko G. Biochemistry. 2007;46(28):8360–8372. [DOI] [PubMed] [Google Scholar]
- 6.Yin Z, Mac hi us M, Nestler EJ, Rudenko G. Nucleic Acids Res. 2017;45(19): 11425–11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin Z, Venkannagari H, Lynch H, et al. Curr. Opin. Struct Biol 2020;2(10):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nestler EJ. Philos Trans R Soc Lond B Biol Set 2008;363(1507):3245–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vialou V, Robison AJ, Laplant QC, Covington HE, et al. Nat Neurosci 2010;13(6):745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glover JN, Harrison SC. Nature. 1995;373(6511):257–261. [DOI] [PubMed] [Google Scholar]
- 11.Hess J, Angel P, Schorpp-Kistner M. J. Cell Sci 2004;117(Pt 25):5965–5973. [DOI] [PubMed] [Google Scholar]
- 12.Cao X, Yasuda T, Uthayathas S, et al. J. Neurosci 2010;30(21):7335–7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berton o, Guigoni C, Li Q, et al. Biol Psychiatry. 2009;66(6):554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cenci MA, Konradi C. Prog Brain Res. 2010;183:209–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ. Trends Neurosci 2010;33(6):267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruffle JK. Am J Drug Alcohol Abuse. 2014;40(6):428–437. [DOI] [PubMed] [Google Scholar]
- 17.Nestler EJ. Eur J Pharmacol 2015;753:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.You JC, Muralidharan K, Park JW, et al. Nat Med 2017;23(11): 1377–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corbett BF, You JC, Zhang X, et al. Cell Rep. 2017;20(2):344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersson M, Konradi C, Cenci MA. J. Neurosci 2001;21(24):9930–9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McClung CA, Nestler EJ. Nat Neurosci 2003;6(11):1208–1215. [DOI] [PubMed] [Google Scholar]
- 22.Zachariou V, Bolanos CA, Selley DE, et al. Nat Neurosci 2006;9(2): 205–211. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Cesena TI, Ohnishi Y, et al. ACS Chem. Neurosci 2012;3(7):546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Experimental procedures for the synthesis of 4,7,9-trimethoxy-6-(piperidin-4-yl) phenanthridine (27). To a solution of 2-bromo-6-methoxyaniline (750 mg, 3.7 mmol, 1.0 eq.) and (3,5-dimethoxyphenyl)boronie acid (1705 mg, 4.44 mmol, 1.2 eq.) in 20 mL dioxane, Pd(PPh3)4 (214 mg, 0.185 mmol, 0.05 eq.) and K2CO3 (1532 mg, 11.1 mmol, 3.0 eq.) in 4 mL H2O were added. After stirring for 3 h at 100 °C, the mixture was extracted with EtOAc, washed with saturated NaHCO3 and brine, dried over Na2SO4, filtered and concentrated to give a crude product. The residue was purified with silica gel column (Hexane/EtOAc = 10/1) to afford 24 (858 mg, 90% yield). 1H NMR (300 MHz, CDC13) δ 6.85-6.79 (m, 3H), 6.66 (d, J = 2.2 Hz, 2H), 6.49 (t, J = 2. 1 Hz, 1H), 4.04 (s, 2H), 3.93 (s, 3H), 3.85 (s, 6H). 13C NMR (75 MHz, CDC13) δ 161.05, 147.18, 141.45, 133.51, 127.38, 122.19, 117.48, 109.34, 106.97, 99.52, 55.69, 55.41. To a solution of 24 (194 mg, 0.75 mmol, 1.0 eq.) in 10 mL EtOAc, tert-butyl 4-formylpiperidine-l-carboxylate (240 mg, 1.125 mmol, 1.5 eq.) and T3P (360 mg, 1. 125 mmol, 1.5 eq.) were added. After stirring at 65 °C overnight, the mixture was extracted with EtOAc, washed with saturated NaHCO3 and brine, dried over Na2SO4, filtered and concentrated to give a crude product. The residue was purified with silica gel column (Hexane/EtOAc = 5/1) to afford 26 (261 mg, 77% yield). 1H NMR (300 MHz, CDCl3) δ 8.01 (d, J = 9.0 Hz, 1H), 7.58 (d, J = 2.3 Hz, 1H), 7.49 (t, J = 8.1 Hz, 1H), 7.12 (d, J = 9.0 Hz, 1H), 6.72 (d, J = 2.3 Hz, 1H), 4.42-4.26 (m, 2H), 4.22-4.13 (m, 1H), 4.11 (s, 3H), 4.04 (s, 3H), 4.03 (s, 3H), 2.97-2.94 (m, 2H), 2.15-1.95 (m, 4H), 1.51 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 161.4, 161.1, 159.5, 155.9, 154.9, 137.4, 135.2, 125.8, 124.0, 114.4, 112.5, 109.3, 99.4, 95.8, 79.1, 77.2, 56.8, 55.7, 55.5, 44.6, 31.9, 28.6. To a solution of 26 (120 mg, 0.265 mmol, 1.0 eq.) in 4 mL DCM, 1 mLTFA was added. After stirring for 2 h, the mixture was concentrated and purified by silica gel column (CH2Cl2/MeOH = 10/1) to afford 27 (79 mg, 85% yield) as a white solid. Mp 215-217 °C. 1H NMR (300 MHz, Methanol-d4) δ 8.14 (dd, J = 1.04,8.1 Hz, 1H), 7.69 (d, J = 2.1 Hz, 1H), 7.55 (t, J = 8.4 Hz, 1H), 7.24 (dd, J = 1.02, 7.91 Hz, 1H), 6.86 (d, J = 2.1 Hz, 1H), 4.40 (td, J = 4.5, 9.4 Hz, 1H), 4.07 (s, 3H), 4.06 (s, 3H), 4.05 (s, 3H), 3.65 (dt, J = 4.1,12.9 Hz, 2H), 3.25 (ddd, J = 3.9, 11.1, 12.5 Hz, 2H), 2.24-2.22 (m, 4H). 13C NMR (75 MHz, MeOD) δ 162.0, 159.4, 159.2, 155.4, 137.4, 134.4, 126.2, 123.9, 114.5, 111.7, 108.9, 99.4, 95.9, 55.4, 55.1, 54.8, 44.0, 40.8, 28.2. HR ESI-MS (M + H)+ m/z = 353.1855 (calcd for C21H25N2O3: 353.1865).
- 25.Recombinant (His)6-ΔFosB and (His)6-JunD proteins were produced in insect cells as described by Wang et al. 2012. The sense and antisense oligonucleotides of the 19-mer TMR-cdk5 (5′-CGTCGGTGACTCAAAACAC-3′) (HPLC purified, TAMRA labeled at 5′-end of both strands) were purchased from Sigma Aldrich. Oligos were annealed as described (see ref.5) and stored as 50 μM stocks in annealing buffer (10 mM Tris pH 8, 50 mM NaCl) at −20 °C. The fluorescence polarization (FP) assay was carried out based on ref.23 with slight modifications. Briefly, a serial dilution of compound (0-200 μM in quadruplicate) was loaded into black 384-well microtiter plates using an Acoustic Liquid Transfer Machine Echo 550 (Beckman) or manually.; Subsequently, TAMRA cdk5 AP-1 oligonucleotide (25 nM) in 20 mM HEPES pH 7.5, 50 mM NaCl (for ΔFosB) or 20 mM HEPES pH 7.5, 150 mM NaCl (for ΔFosB/JunD) was added to each well and incubated for 5 minutes. ΔFosB (320 nM with respect to monomers) or ΔFosB/JunD (280 nM with respect to monomers) was added to the relevant wells and the micro titer plates incubated for an additional 15 minutes. In parallel, as a control for compounds interfering with the assay by directly affecting the DNA, compound in presence of oligonucleotide but no protein, was taken along. The FP signal was measured on a BioTek, Synergy Neo2 plate reader (excitation 530 nm, emission 590 nm) or Pherastar plate reader, BMG LabTeeh (excitation 540 nm, emission 590 nm, 100 flashes per well, and the target set to 30 mP by adjusting the gain on a well with oligonucleotide in the absence of protein). The data was processed with the GraphPadPrism6 software using a dogfinhibition) vs response-variable slope (four parameters)’ algorithm model. For compounds with weak activity, data processing and calculation of IC50s were improved by constraining the top and bottom plateaus or the bottom plateau of the data.
- 26.The docking study was performed with Schrodinger Small-Molecule Drug Discovery Suite using the transcription factor FosB/JunD bZIP domain in complex with cognate DNA, type-I crystal structure (PDB ID: 5VPE). ZL0220 was prepared with LigPrep and the initial lowest energy conformation was calculated. DNA was removed and the ΔFosB/JunD heterodimer was prepared with Protein Prepared Wizard. We then carried a SiteMap calculation on the prepared protein. One druggable site (near the DNA binding region) was identified as the possible ligand binding site. Glide grid was generated on this predicted site with a box in size of 24 × 24 × 24 Å. Ligand docking was employed with Glide using SP precision. Docking poses were incorporated into Maestro for a visualization of ligand-receptor interactions. The DNA was added to the docked pose for an overlay analysis.